

Abstract
Causative treatment strategies for Parkinson's disease (PD) will have to address multiple underlying pathomechanisms to attenuate neurodegeneration. Additionally, the intrinsic regenerative capacity of the central nervous system is also an important factor contributing to restoration. Extracellular cues can limit sprouting and regrowth of adult neurons, but even aged neurons have a low intrinsic regeneration capacity. Whether this capacity has been lost or if growth inhibitory cues are increased during PD progression has not been resolved yet. In this study, we assessed the regenerative potential in the nigrostriatal system in post‐mortem brain sections of PD patients compared to age‐matched and young controls. Investigation of the expression pattern of the regeneration‐associated protein GAP‐43 suggested a lower regenerative capacity in nigral dopaminergic neurons of PD patients. Furthermore, the increase in protein expression of the growth‐inhibitory protein ROCK2 in astrocytes and a similar trend in microglia, suggests an important role for ROCK2 in glial PD pathology, which is initiated already in normal aging. Considering the role of astro‐ and microglia in PD pathogenesis as well as beneficial effects of ROCK inhibition on neuronal survival and regeneration in neurodegenerative disease models, our data strengthens the importance of the ROCK pathway as a therapeutic target in PD.
Keywords: astrogliosis, GAP‐43, microgliosis, Parkinson's disease, regeneration, rho kinase, ROCK, striatum, substantia nigra
Abbreviations
- 6‐OHDA
6‐hydroxydopamine
- amc
age‐matched control
- ANOVA
analysis of variance
- a.u.
arbitrary units
- BDNF
brain derived neurotrophic factor
- BSA
bovine serum albumin
- CI
intern capsule
- CNS
central nervous system
- DA
dopamine
- DAPI
4,6‐diamidino‐2‐phenylindole
- GAP −43
growth associated protein 43
- GDNF
glia cell derived neurotrophic factor
- IR
immunoreactivity
- ISH
in situ hybridization
- LBs
Lewy bodies
- MPTP
1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine
- NC
caudate nucleus
- ncl.
nucleus
- NR
red nucleus
- ON
over night
- PBS
phosphate‐buffered saline
- PC
cerebral peduncle
- PD
Parkinson's disease
- PET
Positron emission tomography
- PMI
post‐mortem interval
- PU
putamen
- ROCK
Rho Kinase
- RT
room temperature
- SNpc
substantia nigra pars compacta
- SPECT
single‐photon emission computed tomography
- TH
tyrosine hydroxylase
- VTA
ventral tegmental area
- yc
young control
Introduction
Parkinson's disease (PD) is a neurodegenerative disease characterized by neuronal cell death in the substantia nigra pars compacta (SNpc) and other parts of the brain. The SN normally provides the neuronal input to the neostriatum (constituted by the caudate nucleus and the putamen) via dopaminergic projections. Retrograde axonal degeneration from the striatum is suggested to be one of the initial events, which likely precedes neuronal cell death in the SN 10. SPECT imaging studies of the dopamine transporter (DAT) confirm that the amount of dopaminergic terminals is markedly reduced at time of symptom onset in PD patients compared to age‐matched controls 4, 18. However, evidence from post‐mortem studies in patients who have been treated with intraputaminal infusion of GDNF showed that even in highly dopamine‐depleted brains of patients with advanced PD a regeneration of dopaminergic nigrostriatal projections can be achieved and a reappearance of tyrosine hydroxylase (TH) can be detected 38. In mice, regeneration of the lesioned nigrostriatal system occurs even spontaneously, e.g., regrowth of TH‐immunoreactive fibers into the striatum can be observed several days after MPTP administration 43 and regenerative growth can be visualized by anterograde tracing after 6‐OHDA lesions 7, 19. Thus, the lesioned dopaminergic system generally has the ability to regenerate, but it is unclear whether compensatory regeneration takes place in PD brains or if this mechanism is impaired in pathology.
Regenerative sprouting and axon growth are regulated by the intrinsic growth potential of the neuron as well as by inhibitory environment, which is present in the myelinated adult central nervous system (CNS) 20. Neurons in a regenerative state show increased expression of the growth‐associated protein GAP‐43 (also known as neuromodulin), which therefore acts as an indicator for neuronal plasticity 3, 6, 32. Moreover, overexpression of GAP‐43 fosters regenerative growth of the nigrostriatal projections and is required for neuritic sprouting 1, 32. Conversely, the infiltration of glial cells into a lesioned area results in a glial scar, acting like a physiological barrier and hampering neuronal regrowth 20. Inhibitory components, such as Nogo, OMgp and MAG, which are expressed on adult oligodendrocytes, as well as ephrins and semaphorins, which are upregulated on the surface of astrocytes, can activate a growth inhibitory signaling pathway in neurons, which converges on rho kinase (ROCK) and finally results in regeneration failure 12, 35, 62, 68. In addition, microglial cells are important determinants for synaptic remodeling and neurodegeneration 15, 31. Activated by environmental stimuli, microglia promote an inflammatory state contributing to neuronal dysfunction and accelerating dopaminergic degeneration in PD 21, 40, 58.
ROCK regulates microglial motility, polarization and migration 61, 64 and may induce the formation of so‐called gliapses, which are neuron‐engulfing glia contacts that precede the phagocytic elimination of dopaminergic neurons 5. The impact of astrocytic malfunction on dopaminergic degeneration remains less clear and may also influence microglial responses 25, 40. ROCK has been shown to be widely distributed in the mammalian CNS and to increase in expression levels with age 27, 34, but its distribution in human brains has not been reported yet. Therefore, an increase in ROCK activity in PD might contribute to the progression of the pathophysiology, via regeneration block and deleterious function of micro‐ and astroglia. Previous studies from our group suggest that inhibition of ROCK improves regenerative axon growth and increases neuronal survival in models of optic nerve trauma and dopaminergic toxicity, e.g., MPTP‐ or 6‐OHDA lesions in the nigrostriatal system 33, 37, 52, 57, 60.
In this study, we evaluate the regenerative growth potential in patients with PD and control subjects. Because GAP‐43 and ROCK play reciprocal roles for regeneration and regenerative failure, alterations in their expression levels likely contribute to PD pathology. We investigated the expression of both proteins in different brain regions by immunohistochemical analysis and in situ hybridization on post‐mortem samples. Our data suggests that PD patients have an attenuated regenerative response and support the idea that ROCK may be a valuable therapeutic target in PD.
Material and Methods
Post‐mortem tissue
Brain tissue for immunohistochemistry was obtained from the Department of Neuropathology and Prion Research of the Ludwig‐Maximilians‐University in Munich, Germany. The diagnosis was based on the UK Brain Bank Criteria, substantiated by assessment of the Unified Parkinson's Disease Rating Scale and post‐mortem confirmed by ascertaining the formation of neurofibrillary tangles 2 and Lewy body (LB) staging 9, 41. Seventy‐one percent of the PD patients, whose brains were used for immunohistochemistry, had a disease duration of 9 or more years, while for the remaining cases disease duration was not announced.
Formalin‐fixed, paraffin–embedded brain sections (4 µm) were stored at room temperature (RT) until processed for immunohistochemistry. Sections from five young control subjects (yc) (five males, average age 55.4 ± 5.2 years), six age‐matched control subjects (amc) (4 males + 2 females, average age 78.3 ± 8.5 years) and from seven PD patients (PD) (4 males+ 3 females, average age 75.7 ± 4.2 years) were examined (see Supporting Information Table 1). From all cases we included unilateral sections from the region of the midbrain containing the SN and the anterior putaminal striatum (Figure 1A). From two subjects no putaminal sections were available. Because the putamen and the caudate nucleus form a functional entity and are similarly affected by atrophy and dopamine receptor decline during disease progression 48, 51, 56, we included sections of the head of the caudate nucleus in the analysis for these two patients. In the text of this manuscript the putamen and caudate are thus summarized as striatum.
Figure 1.
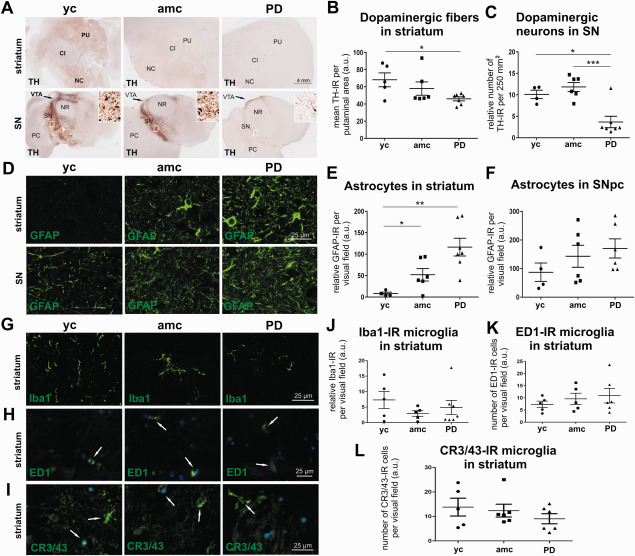
Immunohistochemical characterization of the investigated brain samples. Micrographs represent TH immunostaining and illustrate the analyzed areas of the striatum (A, upper panel) and the midbrain at the level of the substantia nigra (A, lower panel, additive window: detailed representation of SNpc) in young controls (yc), age‐matched controls (amc) and PD cases (PD). Abbreviations: CI = internal capsule, NC = caudate nucleus, NR = red nucleus, PC = cerebral peduncle, PU = putamen, SN = substantia nigra. Quantification of dopaminergic fiber density in striatum (B, n.s.; yc: n = 5; amc: n = 6; PD: n = 7) and TH‐IR cell number in SNpc (C, yc: n = 4; amc: n = 6; PD: n = 7) showed significantly reduced numbers in PD cases. Representative micrographs of GFAP‐IR cells in the striatum (D, upper panel) and SNpc (D, lower panel), quantified in E (striatum, yc: n = 4; amc: n = 6; PD: n = 7) and F (SN, n.s.; yc: n = 4; amc: n = 6; PD: n = 6). Iba1‐IR microglia immunohistochemistry in the striatum is represented for each group with micrographs in G, the quantification of Iba1‐IR cells is displayed in J (yc: n = 5; amc: n = 6; PD: n = 7). For the analysis of phagocytic active microglia exemplary images of ED1‐IR are shown in H and labeling of HLA‐DR with a CR3/43 antibody are illustrated in I, both denoted by white arrows. Quantitative evaluation showed no differences between the groups in both ED1‐IR (K, n.s.; yc: n = 5; amc: n = 5; PD: n = 6) and HLA‐DR expression (L, n.s; yc: n = 5; amc: n = 6; PD: n = 6). Data is given in averaged values from each case, bars represent means per group ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001 (One‐way ANOVA with Tukey Kramer test for independent comparison between groups for E, F, I and Kolmogorov–Smirnov test for B, C, J and L).
Brain tissue for in situ hybridization was provided by The Netherland Brain Bank, Harvard Brain Tissue Resource Center (Belmont, USA) and the Queen Square Brain Bank for Neurological Disorders, London. Cryosections from fresh frozen tissue were mounted on SuperFrost Plus microscope slides (Microm, Braunschweig, Germany) and stored at −20°C until processed for in situ hybridization. For the striatal analysis we included 10 age‐matched control cases (7 males + 3 females, average age = 78.5 ± 8.1 years) and 10 PD cases (seven males and three females, average age = 81.5 ± 6.8 years). To investigate the SN, midbrain sections from nine age‐matched controls (7 males + 2 females, average age = 74.9 ± 9.9 years) and seven PD cases (6 males + 1 females, average age = 75 ± 6.9 years) were included.
The characteristics of the investigated cases are summarized in Supporting Information Table 1. Detailed information on the disease duration and the neuropsychological function was only partially available and thus not included in the table.
Immunohistochemistry
Fluorescence immunostaining
Single or co‐fluorescent immunostainings were all performed according to the same protocol:
Paraffin‐embedded sections were deparaffinized with xylene (Sigma, Taufkirchen, Germany) and rehydrated in ethanol solutions of decreasing concentration: 100% ethanol for 5 minutes, followed by 99%, 96%, 90%, 70% and 50% ethanol, each incubated for 5 minutes and stored in distilled water until next steps. For antigen retrieval, slides were incubated in 50 mM glycine (Applichem, Darmstadt, Germany; pH 3.5) for 25 minutes at 90°C in water bath. After 1 h of cooling at RT, sections were treated with Sudan black B (Applichem; 0.3% in 70% ethanol) for 5 minutes at RT to eliminate auto‐fluorescence from lipids, followed by blocking and permeabilization with 5% normal donkey serum (NDS, Dianova, Hamburg, Germany), 1% free bovine serum albumin (BSA, immunoglobulin and protease free, Jackson Laboratory, Bar Harbor, Maine, USA) and 0.1% tritonX‐100 (Sigma) in PBS for 1 h at RT. Immediately after blocking, incubation with primary antibodies was performed for single and co‐stainings for 48 h at 4°C in 2.5% NDS in PBS. The following antibodies were used: anti‐tyrosine hydroxylase (anti‐TH rabbit, polyclonal, 1:1000, Zytomed, Berlin, Germany; anti‐TH mouse, monoclonal, 1:500, Sigma; anti‐TH goat, polyclonal, 1:500, Abcam, Cambridge, UK) anti‐ROCK2 (goat, polyclonal, 1:50, Santa Cruz, Heidelberg, Germany), anti‐GAP‐43 (rabbit, polyclonal, 1:400, Abcam), anti‐synaptophysin (rabbit, monoclonal, 1:100, Merck Millipore, Schwalbach, Germany), anti‐glial fibrillary acidic protein (anti‐GFAP rabbit, polyclonal, 1:300 DAKO, Hamburg, Germany), anti‐ionized calcium binding adapter molecule 1 (anti‐Iba1 rabbit, polyclonal, 1:300, Wako Pure Chemical Industries, Neuss, Germany), anti‐HLA‐DR (clone CR3/43, monoclonal, mouse, 1:50, Dako, Hamburg, Germany) and anti‐ED1 (CD68, monoclonal, mouse, 1:50, Serotec, Oxford, UK). Before incubation with the ED1 antibody, sections were pre‐treated with 0.05% trypsin in PBS for 20 minutes at 37°C to achieve optimal antigen retrieval.
All primary antibodies were detected by incubation with the appropriate species detecting secondary antibody for 1 h at RT. Fluorophore‐conjugated secondary antibodies used were DyLight 488, Alexa Fluor 488, Cy2, Cy3 (all 1:250, Dianova) or Cy5 (1: 250, Jackson Laboratories), respectively. For co‐staining the sections were incubated with the particular primary or secondary antibodies simultaneously. Nuclei were stained with 4,6‐diamidino‐2‐phenylindole (DAPI, 1 µg/mL, Sigma) and mounted with Mowiol® (Sigma). Negative control staining was performed without incubation in primary antibodies asserting the appropriate detection of the secondary antibodies. Because only insufficient information was available about the specificity in human tissue for the antibodies against ROCK2, GAP‐43 and synaptophysin, we re‐evaluated their specificity by protein blot using human, mouse and rat brain lysates (Supporting Information Figure S1).
Light stable immunostaining with 3,3′‐ diaminobenzidine (DAB)
For counting TH‐immunoreactive cells in the SNpc and to investigate the TH‐IR fiber density in the striatum, slides were deparaffinized as described above, endogenous peroxidases were blocked for 3 minutes with 3% hydrogen peroxide (H2O2, Applichem) and sections were incubated with anti‐TH antibody (rabbit, polyclonal, 1:1000, Zytomed) for 48 h at 4°C followed by treatment with a secondary biotinylated anti‐rabbit antibody (1:200, Dianova), at RT for 2 h. Incubation with VECTASTAIN ABC Peroxidase standard Kit (PK‐4000, Biozol, Eching, Germany) for 2 h at RT was performed to enhance detection signal. Finally, sections were stained with 3,3′‐diaminobenzidine (DAB peroxidase substrate Kit, SK‐4100, Vector Laboratories, Burlingame, USA) for 15 minutes and mounted with DPX (Sigma).
Stereological quantification of substantia nigra dopaminergic neurons and evaluation of the striatal dopaminergic fiber density
The number of TH‐immunoreactive (TH‐IR) neurons in the SN was assessed unilaterally using stereological methodology. One section through the SN per case was analyzed with Stereo Investigator software (Stereo Investigator 9.0, MicroBrightField Inc., Colchester, VT, USA, Zeiss microscope). The area of the SN was outlined manually using a 2.5× objective and a point grid was overlaid onto each section constituting areas of the counting frames (counting frames: 500 × 500 µm). Immunostained cells were counted by the optical fractionator method (10× objective) and the total number of TH‐IR cells in the unilateral SN was estimated per section by the software. Stereologically aided counting was performed by investigators blinded to the groups.
To analyze the TH‐IR fiber density, sections were acquired with a 40× objective using the Stereo Investigator software which assembled single acquired images to one picture via the Virtual Slice® module. TH‐IR was detected by measuring the mean grey value via ImageJ software 1.47v (N.I.H., Bethesda, USA).
Image acquisition and analysis of protein expression in the SNpc and striatum
Slides were imaged with an Axioplan microscope equipped with a 16‐bit grayscale CCD camera and AxioVision 4.6 software (Zeiss, Göttingen, Germany) using a 63× objective. Micrographs were taken at 15–20 randomly chosen visual fields per individual SNpc or striatum, respectively, containing TH‐IR cells or striatal neuropil. For a detailed analysis of the fluorescent signal of ROCK2, GAP‐43 or synaptophysin staining, an appropriate area of the TH‐IR soma and TH‐IR neurites, as well as of the adjacent neuropil was chosen to measure the mean grey value using the freehand selection from the ImageJ software.
To allocate enhanced ROCK2 expression to specific cell types, 15–20 images (98.41 × 98.41 µm) from the individual SNpc or striatum, respectively, were acquired employing Leica Microsystems TCS SP5 confocal microscope (Mannheim, Germany) to correlate ROCK2‐IR to astrocytes or to Iba1‐IR microglia. Therefore co‐labeled GFAP‐ and ROCK2‐, or Iba1‐ and ROCK2‐IR cells were quantified using ImageJ cell counter plugin.
To detect ROCK2‐positive microglia, which display an activated phagocytic state, the number of CR3/43‐ and ED1 ‐/ROCK2 co‐labeled cells was quantified with ImageJ cell counter plugin on five randomly chosen micrographs (400 × 530 µm) per striatum taken with the AxioVision 4.6MosaiX module (Zeiss) with a 63× objective.
Oligonucleotide in situ hybridization and mRNA quantification
In situ hybridization was performed as described earlier 67 using radiolabeled oligonucleotide probes complementary to GAP‐43 and ROCK2. Briefly, 33P‐dATP labeled probes were hybridized overnight to cryosections, washed, dehydrated and exposed to autoradiographic films (Biomax, Kodak). A different set of sections was treated similar and dipped into photo‐emulsion, developed after three weeks and inspected under the microscope. The following oligonucleotides were used, targeting different exons of GAP‐43 (ref|NM 001130064.1| nt = 981 − 932; nt = 1027 − 978; nt = 1134 − 1085 and nt = 1496 − 1448) and ROCK2 (nt = 814 − 765; nt = 2190 − 2141; nt = 3055 − 3006 and nt = 3217 − 3168). All four nucleotides for GAP‐43 recognize both transcript variants.
To estimate the GAP‐43 mRNA expression in dopamine neurons of the substantia nigra, 20 neuromelanin‐containing neurons with a clearly visible cresyl‐violet‐stained nucleus were chosen close to the crus cerebri for each analyzed PD or control case. The amount of silver grains on top of each neuron was scored in comparison with the six examples depicted in Figure 3D and the distribution of signal scores as well as the sum of scores for each case are shown in Figure 3E,F, respectively (similar to data in Galter et al 23). Because no ROCK2 in situ signal was detectable in the SN, no quantification of ROCK2 mRNA was performed.
Figure 3.
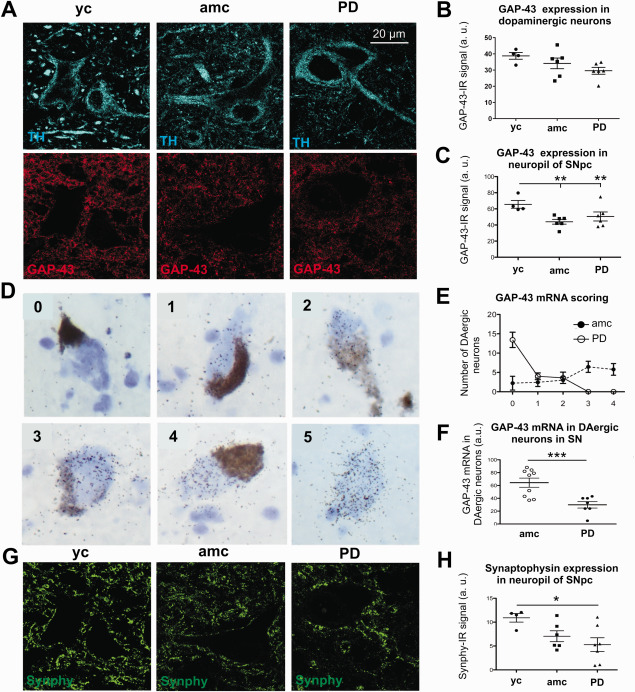
Gap‐43 expression in dopaminergic neurons and in the SN neuropil. Representative micrographs of the SNpc are illustrated in A (TH‐IR neurons, upper panel; GAP‐43‐IR, lower panel). Quantification of GAP‐43 protein expression in dopaminergic neuron somata (B) and the nigral neuropil (C) (yc: n = 4; amc: n = 6; PD: n = 6). Examples of cells showing increasing amounts (visual scores 0 – 4) of GAP‐43 mRNA (visual score 5 depicts a neuromelanin‐negative, non‐dopaminergic neuron) (D). The distribution of GAP‐43 mRNA scores for 20 neuromelanin‐containing cells per case is given in E and summary scores are shown in F (ctrl: n = 9; PD: n = 7). Synaptic density is demonstrated by representative micrographs of synaptophysin (synphy) protein expression in the SNpc (G). The quantification of synphy‐IR is visualized in H (yc: n = 4; amc: n = 6; PD: n = 7). Data is given in averaged values from each case, bars represent means per group ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001 (One‐way ANOVA with Tukey Kramer test for independent comparisons between groups for B, C, H and Student T test for F).
The striatal GAP‐43 and ROCK2 mRNA expression was analyzed on autoradiographic films by measuring the signal density with appropriate software (ImageJ, 1.47v). After background correction the data were statistically processed.
Statistical analysis
Statistical analyses of the protein expression were performed by testing data for normal distribution with the Shapiro–Wilk test and then by comparison of the groups (yc vs. amc, amc vs. PD or yc vs. PD) using one‐way ANOVA followed by the Tukey‐Kramer post‐hoc test (Kyplot software, KyensLab Incorporated, Tokyo, Japan). In case of not normally distributed values the data was tested for significance with the Kolmogorov–Smirnov test.
Levels of mRNA expression were compared between PD cases and controls with the Student T test (unpaired, two ways) using Prism (GraphPad Software Inc., La Jolla, CA, USA).
Data is presented as mean ± SEM. Differences were considered significant with *P < 0.05; **P < 0.01; ***P < 0.001.
Results
To answer the question how and to what extent intrinsic regenerative capacities in the nigrostriatal system may be altered in PD patients, we performed an immunohistochemical analysis of human brain tissue evaluating the expression levels of growth regulating proteins in PD patients and controls.
Dopaminergic and glial pathology in striatal and nigral regions of human PD brains
Midbrain and striatal brain sections of 5–7 cases from each group were first characterized by TH‐immunostaining (Figure 1A). There was a significant decrease of dopaminergic fibers by density measurement of TH‐immunoreactivity (TH‐IR) in PD cases (46.1 ± 2.3 a.u.) compared to the young controls (yc = 68.2 ± 8.2 a.u.), while some individuals from the age‐matched controls also show a trend to decreased TH‐IR fiber density, which was not significant in the mean (amc = 58.1 ± 7.8 a.u.) (Figure 1B). The relative number of dopaminergic neurons per individual was estimated by stereological counting of TH‐IR cells in one unilateral section through the SN. TH‐IR cell numbers confirmed the significant loss of ∼65% of dopaminergic neurons in PD patients to a mean of 3.7 ± 1.3 cells per counting frame (Figure 1C, declaring one outlier) compared to age‐matched and young controls (amc = 11.8 ± 1.1 and yc = 10.1 ± 1.0 cells per counting frame).
Additionally to the extent of dopaminergic degeneration we evaluated astrocytosis and microgliosis in the SNpc and striatum. Correlating to the dopaminergic degeneration in the nigrostriatal system, there is a strong increase of GFAP‐immunoreactivity (GFAP‐IR) in the striatum (Figure 1D, upper panel) in PD cases (PD = 116.4 ± 20.6 a.u.) in contrast to young controls (yc = 8.3 ± 3.1 a.u.) (Figure 1E). However, GFAP‐IR is also increased in the age‐matched control group, albeit less strongly (amc = 53.0 ± 14.4 a.u.). The analysis of GFAP immunoreactivity in the SNpc (Figure 1D, lower panel) showed a trend, but no significant change in PD patients (PD = 170.4 ± 33.6 a.u.) compared to controls (amc = 143.0 ± 38.4 a.u. and yc = 87.2 ± 32.4 a.u.) (Figure 1F). To assess the inflammatory response to dopaminergic degeneration in the nigrostriatal system in PD an immunohistochemical analysis of microglia infiltration was performed by quantification of Iba1 positive cells (Figure 1G,J). There was no significant difference in Iba1‐immunoreactivity (Iba1‐IR) in the striatum between the groups (Figure 1J), possibly caused by high inter‐individual variance in the young control group (yc = 7.3 ± 2.8 a.u. and amc = 2.9 ± 0.9 a.u.). Interestingly, except one case, all PD brains showed little Iba1‐IR (PD = 4.9 ± 2.3 a.u.). To quantify the number of microglia transformed into amoeboid shaped and phagocytic active microglia, we immunolabeled with ED1 (Figure 1H,K). The quantification of ED1‐IR cells revealed no significant differences in the amount of phagocytic active microglia neither in the SNpc (yc = 10.3 ± 0.7, amc = 16.6 ± 2.3 and PD = 14.4 ± 2.1 ED1‐IR cells per visual field) nor in the striatum of PD cases compared to controls (yc = 7.3 ± 0.6, amc = 9.6 ± 1.0 and PD = 10.9 ± 1.2 ED1‐IR cells per visual field) (Figure 1K). We also performed a staining for the major histocompatibility complex class II (MHC‐II) protein HLA‐DR with an antibody for CR3/43 to assess phagocytic activated microglia (Figure 1I). Similar to the results of the ED1 immunohistochemistry, the quantification of CR3/43‐immunoreactive (CR3/43‐IR) microglia revealed no significant differences in activated microglia numbers in the striatum of PD patients (PD = 9 ± 2 CR3/43‐IR cells per visual field) compared to age‐matched controls (amc = 12.4 ± 2.6 CR3/43‐IR cells per visual field) and young controls (yc = 13.8 ± 3.6 CR3/43 cells per visual field) (Figure 1L).
Analysis of growth‐associated and presynaptic proteins
To evaluate the regenerative potential of the nigrostriatal system we investigated the expression of GAP‐43 protein by immunofluorescence in the striatum. As illustrated in Figure 2A we did not find any significant changes in mean GAP‐43 protein expression in PD cases versus age‐matched or young controls (PD = 47.1 ± 3.0 a.u., amc = 26.3 ± 4.8 a.u. and yc = 27.1 ± 2.8 a.u.). This is in line with the quantification of the GAP‐43 mRNA‐signal revealed by in situ hybridization (Figure 2C,D; 1625 ± 195 a.u. vs. 1409 ± 198 a.u. in controls vs. PD cases, respectively). Furthermore, GAP‐43 mRNA expression was not correlated with PMI or age of the investigated samples (data not shown). In addition to GAP‐43 we studied the synaptic density in the striatum by immunostaining for the presynaptic vesicle protein synaptophysin (synphy) (Figure 2E). Similarly to the GAP‐43 analysis the quantification of the mean synphy‐immunoreactivity (synphy‐IR) signal in the striatum did not indicate any apparent differences in expression levels between the groups (yc = 10.9 ± 1.5 a.u., amc = 11.7 ± 1.0 a.u., PD = 8.5 ± 2.4 a.u., Figure 2F).
Figure 2.
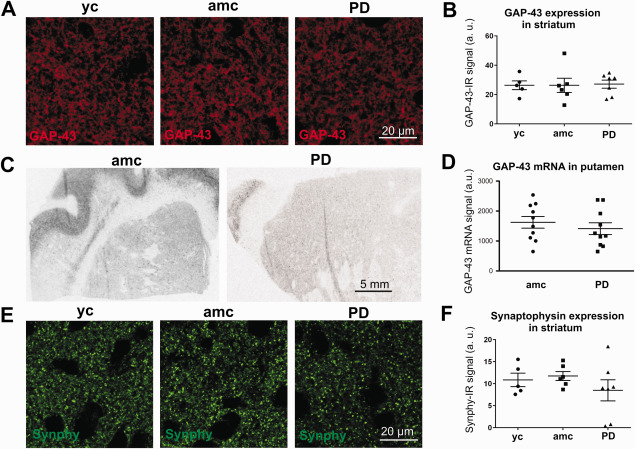
Analysis of striatal GAP‐43 expression and synaptic density. Representative micrographs of GAP‐43‐IR signal in the striatum of young controls (yc), age‐matched controls (amc) and PD cases (PD) (A). Evaluation of GAP‐43 protein expression in the striatum resulted in no differences between the groups (B, yc: n = 5; amc: n = 6; PD: n = 7; n.s.). C and D are depicting the striatal GAP‐43 mRNA signal (ctrl: n = 10 and PD: n = 10; n.s.). Acquisitions of striatal synaptophysin immunoreactivity (synphy–IR) (E), followed by statistical group comparison revealed no differences between the groups (F, yc: n = 5; amc: n = 6; PD: n = 7; n.s.). Data is given in averaged values from each case; bars represent means per group ± SEM (One‐way ANOVA for B, F and Students T test for D).
We then evaluated GAP‐43 expression in dopaminergic nigral neurons and the adjacent neuropil (Figure 3A). In dopaminergic neurons (Figure 3A, upper panel) we observed only minor GAP‐43‐IR in TH‐IR cells (Figure 3A, lower panel) with no significant differences between the groups. However, there was a distinct trend for less GAP‐43 expression in brains of PD cases compared to young controls (Figure 3B). In contrast to the dopaminergic cell somata of the SNpc, the adjacent neuropil showed strong GAP‐43 immunoreactivity (Figure 3A, lower panel) with significantly higher mean GAP‐43 expression in young controls (yc = 65.6 ± 4.8 a.u.), while in age‐matched controls and PD cases the GAP‐43 signal was less strong (amc = 44.0 ± 3.0 a.u. and PD = 50.6 ± 6.0 a.u., Figure 3C).
The GAP‐43 mRNA signal in the SNpc in situ was clearly detectable and analyzed in dopaminergic neurons, identified by their neuromelanin content (Figure 3D–F). To quantify the GAP‐43 mRNA amount in individual dopaminergic neurons, we adopted a visual rating scale based on the number of silver grains projecting on the cell body (very few (0) to very abundant (4)). The highest density of silver grains was detected only in non‐dopaminergic neurons in the red nucleus used as positive control (Figure 3D). Score analysis revealed that most neuromelanin‐positive cells from PD cases showed nearly no GAP‐43 mRNA signal (score 0), while there were virtually no dopaminergic cells with high amounts of GAP‐43 mRNA signal (score 4) (Figure 3E). On the contrary, the dopaminergic neurons from age‐matched controls featured scores from 0 to 4 and most of these cells showed high GAP‐43 mRNA signal compared to PD cases (PD = 29.9 ± 5.1 a.u. vs. amc = 64.3 ± 9.3 a.u., Figure 3F).
Similar to the finding of the decreased nigral GAP‐43 protein expression in the neuropil, the averaged synaptophysin immunoreactivity was also strongly reduced in the SNpc (Figure 3G) of PD cases (5.3 ± 1.5 a.u.), although inter‐individual synaptophysin expression was highly variable (Figure 3H). In age‐matched controls this decrease in presynaptic synphy‐IR was less pronounced (7.1 ± 1.1 a.u.), being only by trend different from synphy‐IR in young controls (10.9 ± 0.9 a.u.).
Expression of ROCK2 in the midbrain and striatum
As counterpart to the regeneration‐associated protein GAP‐43, we were additionally interested in the distribution of ROCK2 as intracellular signaling hub of growth‐inhibitory cues. One characteristic feature of active ROCK is to induce stress fiber formation and growth cone collapse, leading to inhibition of regenerative axon elongation. Immunohistochemical detection in the SNpc from PD cases and controls showed only a very weak expression of this protein. Interestingly, ROCK2 displayed a distinct punctate staining with a ubiquitous distribution, being less pronounced in TH‐IR somata as compared to the neuropil (Figure 4A). The evaluation of alterations in ROCK2 protein expression did not indicate any significant differences between the groups, neither in the dopaminergic somata nor in the neuropil of the SNpc (Figure 4B,C), although there were pronounced interindividual differences in protein expression in all groups. The ROCK2‐mRNA signal from in situ hybridization was of even lower intensity in dopaminergic neurons of the SNpc, which did not allow for a quantitative assessment (data not shown).
Figure 4.
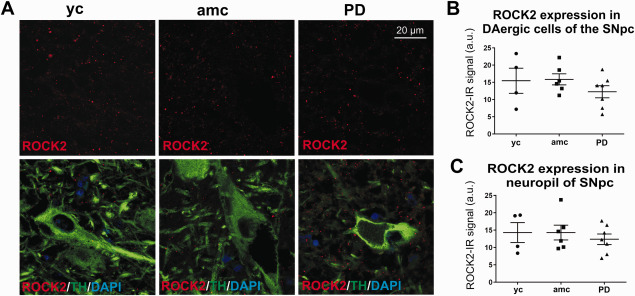
Analysis of ROCK2 expression in the SNpc of PD cases and controls. Representative micrographs of ROCK2‐IR (A, upper panel) and double immunoreactive ROCK2/TH‐co‐stainings (A, lower panel). ROCK2‐IR was evaluated in TH‐IR cells (B, yc: n = 4; amc: n = 6; PD: n = 7; n.s.) and in the adjacent neuropil (C, yc: n = 4; amc: n = 6; PD: n = 7; n.s.). Data is given in averaged values from each case; bars represent means per group ± SEM (One‐way‐ANOVA).
In the striatum, the ROCK2 protein was distributed in a similar pattern as in the SNpc. The overall ROCK2‐IR signal was low in all sections (Figure 5A). A quantification of the averaged ROCK2‐IR in all of the striatal structures by randomly chosen areas showed no clear expression changes between the groups (Figure 5B). The evaluation of ROCK2 mRNA signal in the striatum (Figure 5C) corresponded to the quantification of protein expression. Thus, no significant differences between the groups were observed (amc = 29.7 ± 5.3 a.u. vs. PD = 37.2 ± 6.4 a.u., Figure 5D). Furthermore, the ROCK2 mRNA signal was not correlated to the PMI or the age of investigated samples (data not shown).
Figure 5.
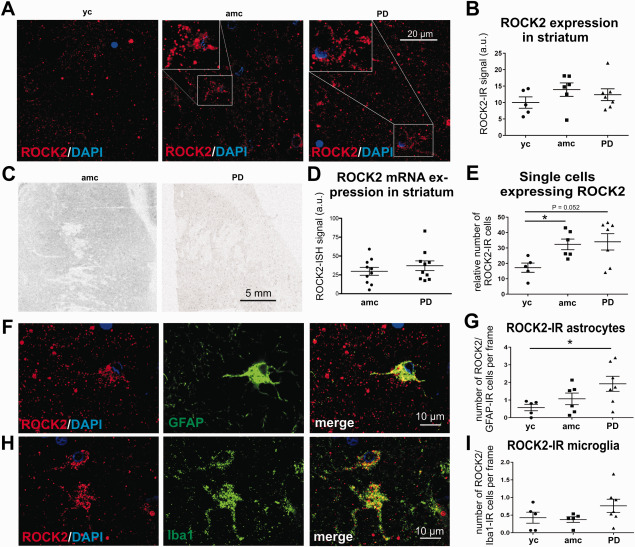
Quantification of ROCK2 expression in the striatum of PD cases and controls. Representative micrographs illustrate the ROCK2 protein expression in the striatum of young and age‐matched controls and PD cases (A). The evaluation of the general ROCK2 fluorescence signal resulted in no detectable differences between the groups (B, yc: n = 4; amc: n = 6; PD: n = 7; n.s.). In situ hybridization of striatal ROCK2 mRNA is depicted in C and quantification of ISH‐signal is shown in D (ctrl: n = 10 vs. PD: n = 10; n.s.). Details in A distinguish single cells with high ROCK protein‐IR, the numbers of which were significantly increased in age‐matched controls and strongly augmented in PD cases (E, yc: n = 5; amc: n = 6; PD: n = 7). Micrographs of double immunoreactivity for ROCK2 and GFAP are shown in F and for ROCK2 and Iba1 depicted in H. Quantification of these double‐IR cells resulted in significantly increased numbers of ROCK‐IR astroglia (G, yc: n = 5; amc: n = 6; PD: n = 7) and in a trend to increased ROCK2‐IR microglia (I, yc: n = 5; amc: n = 6; PD: n = 7) in PD cases. Data is given in averaged values from each case, bars represent means per group ± SEM. *P < 0.05 (One‐way ANOVA with Tukey Kramer test for independent comparison between groups for B, G, I and Student T test for D and Kolmogorov‐Smirnov test for E).
Interestingly, in the striata of PD cases and age‐matched controls, a number of single cells with an intensive ROCK2‐IR was observed (Figure 5A, detail), which we further quantified. This revealed a strong trend for an increase in ROCK2‐IR cells in PD cases as compared to young controls (34.0 ± 5.3 vs. 17.2 ± 3.0 ROCK‐IR cells/visual field in PD vs. yc, respectively, P = 0.052, Figure 5E). However, a significant increase to young controls was observed in age‐matched controls (32.3 ± 3.5 ROCK‐IR cells/visual field, Figure 5E).
Based on their morphology, ROCK2‐IR cells appeared potentially as glial cells, featuring multiple processes and smaller nuclei than neurons. With immunolabeling against GFAP and Iba1 in combination with ROCK2 (Figure 5F,H) the amount of ROCK2‐IR astrocytes and microglia was quantified. The analysis revealed that the number of ROCK2‐IR astrocytes was significantly higher in PD cases versus young controls (1.9 ± 0.4 vs. 0.6 ± 0.2 GFAP/ROCK2‐IR cells per visual field, respectively, Figure 5G). Age‐matched controls showed a tendency to increased numbers of ROCK2‐IR astrocytes (1.1 ± 0.3 GFAP/ROCK2‐IR cells per visual field, Figure 5G). There was only limited co‐labeling of Iba1 and ROCK2 in the different groups (Figure 5I). Interestingly, there were three cases with markedly higher numbers of ROCK‐IR microglia in the PD group, although the mean number of ROCK/Iba1‐IR cells was not significantly increased (PD = 0.8 ± 0.2; yc = 0.4 ± 0.2, and amc = 0.4 ± 0.1 ROCK2/Iba1‐IR cells per visual field) (Figure 5I). The combined immunolabeling of ROCK2 and the HLA‐DR protein CR3/43 resulted in only sporadic co‐localization, without differences of ROCK2 expression in microglia with activated morphology compared between the groups (yc = 3.6 ± 1.1, amc = 3.5 ± 0.6, PD = 3.7 ± 1.1 ROCK2/CR3/43‐IR cells per visual field).
Discussion
The dopaminergic system has intrinsic regenerative abilities, but it is not clear whether these abilities are impaired in PD and whether ROCK2 contributes to these effects. The latter would further advocate for an assessment of ROCK inhibition as a therapeutic approach in PD. We thus performed a comprehensive histological characterization of PD cases and controls to document the expression of the growth‐inhibitory ROCK2 protein and an indicator of regenerative growth, GAP‐43.
The immunohistochemical analysis of TH in the present brain sections confirmed a pronounced degeneration of dopaminergic neurons and their projecting axons to the striatum in the majority of the PD cases, representing a main pathoanatomical hallmark of PD. Although not significant in the mean, the TH signal in the striatum of several individuals from the age‐matched control group (non‐PD) also showed lower dopaminergic fiber density compared to young controls. This confirmed findings from other groups, who showed a linear decline in striatal dopaminergic projections during healthy ageing by studying the abundance of the DAT in the striatum by PET scan and SPECT imaging 63, 65. Conversely, our study did not show dopaminergic neuron loss in the SNpc in individuals of either the young or the age‐matched control group in contrast to PD cases corresponding to recent literature showing that nigral neuronal cell death is not a characteristic feature of healthy aging 44, 53, 55.
Another characteristic trait of neurodegeneration in the SNpc is the astrogliosis 39. Once CNS damage occurs, astrocytes are engaged to seclude the injury site from healthy tissue 54, which, however, constitutes simultaneously a major impediment to neuronal regeneration by building a physiological barrier 20, 68. Astrogliosis is characterized by an enlargement of the astrocyte cell body, a remodeling of processes at the level of asymmetric synapses and increased astrocyte numbers 13. This was similarly observed in this study, where the number of GFAP‐labeled astrocytes in the striatum was significantly increased in PD cases compared to young controls and less severely in age‐matched controls. Although we observed the same trend in the SNpc, statistical analysis failed to show intergroup differences, likely caused by the high interindividual spread resulting from the heterogeneous state of the disease at the point of death. Previous reports on astrogliosis in PD report conflicting data 16, 42, 59. As most analyses of PD tissue are based on autopsy material, the majority of the cases correspond to advanced stage of disease, where more pronounced changes occurring in the course of the disease may have already leveled off. Our analysis of young and age‐matched control groups illustrates that there is an age‐related astrocytic proliferation in the nigrostriatal system, which is reaching significant levels in the striatum and is further intensified in PD. These observations suggest that astrogliosis is a characteristic feature of PD contributing to the generation of a growth inhibitory environment.
Interestingly, the analysis of the microglial marker Iba1 showed no relevant differences in the number of microglia in the striatum of PD cases compared to controls. However, not only the amount of microglia is important, but even more their activation state 5. In the so called “resting state” microglia are engaged in surveillance of the adjacent environment, while in the “activated state” they follow noxious stimuli 15. PET studies revealed a correlation of dopaminergic cell loss and the increase of activated microglia in the basal ganglia of PD patients 47. Activated microglia, which can transform to phagocytic microglia may deliver trophic factors and remove cell debris in damaged tissue as a requirement for restoration 40. However, pro‐inflammatory cytokines decrease the beneficial phagocytic activity and commit microglia to an inflammatory response 26, 28. Furthermore, cytokines stimulate reactive astrocytes to an upregulation of inhibitory proteoglycans intensifying growth inhibition 20. By employing markers like ED1 and HLA‐DR we aimed to better characterize the activation state of the microglia, although the differentiation ability of both markers between M1 and M2 microglia is limited 66. Because the astroglial pathology was more pronounced in the striatum than in the SNpc and the marker ED1 for phagocytic active microglia did not show any alterations in the number of ED1‐IR cells, we performed an additional staining for CR3/43 in the striatum, which, however, also did not reveal any differences between the groups. As our analysis is based on post‐mortem tissue of patients with long disease duration, it is likely to reflect advanced pathology, which may not correspond to intermediate disease states, where transient changes of the highly variable microglia phenotypes and distribution as an response to acute neurodegeneration could be more obvious. Also, microglial alterations may show regional variability, which may have escaped our analysis of selected regions.
To determine the regenerative capacity of the nigrostriatal system, the expression level of the growth associated protein GAP‐43 was examined in the brains of PD cases, young and age‐matched controls. GAP‐43 is associated with neuronal development and is mainly present in neuronal growth cones as well as during neuronal sprouting, as it was observed during nigrostriatal regeneration after unilateral GDNF infusion in a PD patient with advanced disease 38. Overexpression of GAP‐43 resulted in axonal sprouting and terminal arborization in rodents suggesting that GAP‐43 is required for neuronal regrowth 1, 32. However, GAP‐43 levels decline precipitously by the completion of mature synaptic networks and during normal ageing 14, 22, 46.
Because the striatal TH‐labeling did not permit to identify single dopaminergic nerve fibers, but rather resulted in a diffuse tissue staining, we evaluated the total striatal GAP‐43 protein expression. Striatal protein levels did not show any differences between PD cases and controls, and this was also confirmed by mRNA in situ analysis. Dopaminergic degeneration thus seems to have no influence on GAP‐43 protein expression levels in the striatum of PD patients, indicating a failure of compensatory sprouting from adjacent neuronal tissue, eg, by glia‐mediated growth inhibition. Similar observations were described in the 6‐OHDA rat model, where striatal intoxication and dopaminergic degeneration were not followed by changes in the GAP‐43 protein expression 30. However, we cannot exclude that in the midstage of the disease GAP‐43 expression may have been altered, which leveled off at the analyzed advanced stages.
Striatal synaptophysin immunoreactivity (synphy‐IR), which was used as a marker for the presynaptic compartment, was not significantly altered in our PD cases, although there were high interindividual differences in this group. In two of the examined PD cases the drastic reduction of synphy‐IR could indicate a more severe degeneration of striatal terminals than in the other patients. The evaluation of GAP‐43 protein expression in the SN revealed less GAP‐43 in the adjacent neuropil of PD patients and age‐matched controls compared to young controls. This suggests an age‐related cessation of neuronal growth in neuronal terminals in ageing 11, 14. In PD patients, GAP‐43 expression in the nigral neuropil was similarly low, arguing for a lack of compensatory sprouting following degeneration of dopaminergic neurons in PD. Furthermore, we observed a significantly decreased synaptophysin expression in the SNpc of PD cases, reflecting nigral presynaptic pathology likely caused by the reduced number of dopaminergic nigral target neurons. Moreover, certain cell types appear more susceptible to environmental influences than others even during normal aging 11, 14, 44.
By analyzing the GAP‐43 protein expression in co‐labeled TH‐IR neurons, only a weak GAP‐43 immunofluorescent signal was observed, which was expected, because GAP‐43 is mainly expressed in the neuronal terminals. Nevertheless, the trend to decreased GAP‐43 in dopaminergic neurons of PD cases is in line with results from in situ hybridization that provided evidence for significantly decreased GAP‐43 mRNA expression levels in dopaminergic neurons of PD patients compared to age‐matched controls. In fact, most of the remaining dopaminergic neurons in our PD cases showed an absence of the GAP‐43 mRNA signal, while among the age‐matched controls most dopaminergic neurons showed moderate to high GAP‐43 mRNA signals. Supporting these findings, GAP‐43 mRNA levels in dopaminergic neurons were reported low after intrastriatal 6‐OHDA intoxication in rats, suggesting that the lack of GAP‐43 mRNA expression resulted from neuronal damage and restricted cellular functioning of the transcription machinery 30.
Taken together, our results suggest that PD patients fail to mount a compensatory regenerative answer in the adjacent neuropil in the striatum and the SN in response to dopaminergic neuron loss. Furthermore, the remaining dopaminergic neurons indeed showed reduced regenerative capacity reflected by reduced GAP‐43 mRNA expression. Furthermore, diminished synaptophysin expression in the nigral neuropil reflects an innervation deficit caused by neuronal degeneration in PD.
In this study, we visualized, for the first time to our knowledge, ROCK2 expression in the human nigrostriatal system. In both, the SN and the striatum, ROCK2‐IR was very weak and was mostly confined to intracellular puncta. Previous publications have documented the distribution of ROCK2 in punctate structures using the identical antibody as in our study, e.g., in hypoglossal motoneurons of rats and in primary rat retinal ganglion cells 24, 49. Due to the very low in situ hybridization signal, quantification was not possible in dopaminergic cells in the SNpc. Thus, at least at the time of death, neuronal ROCK2 mRNA and protein expression is very weak and not significantly affected by PD pathology. Our data thus does not support the hypothesis that regenerative failure in PD is accompanied by increased ROCK2 expression in dopaminergic neurons. In contrast to the weak overall ROCK2 expression, we found, however, single striatal cells, which showed a very pronounced ROCK2 expression. The number of these strongly ROCK2‐IR cells was significantly higher in age‐matched controls and a strong trend was also observed in PD cases. Interestingly, our analysis revealed that these cells corresponded mostly to GFAP‐IR astrocytes and Iba1‐IR microglia, contradicting the assumption that ROCK2 is predominantly expressed in neurons of the vertebrate brain 24. As ROCK2 is the predominantly expressed isoform in the brain, this study did not assess the expression of ROCK1 29. Because the age‐matched controls exhibited the same trend for increased ROCK2‐IR astrocytes, this phenomenon appears to be also age‐related, but intensified in PD. Increased ROCK2 expression in astrocytes and microglia thus correlates with the most important risk factor for PD—age—which argues for an involvement of increased ROCK expression in these cells in PD pathology.
Likewise, in an animal model of dopaminergic toxicity, the MPTP mouse model, ROCK activity and expression was increased and associated with an activation and morphological alteration of microglial cells 5, 8, 64. It has been reported that ROCK inhibition exerts beneficial effects on brain injuries by influencing glial responses 17, 36, 45, 69. Treatment with the ROCK inhibitor Fasudil reduced the number of deleterious intercellular contacts between microglia and dopaminergic neurons, so‐called gliapses, which act forming a microglial phagosome for damaged neurons. ROCK signaling affects microglial migration and shaping by regulating the organization of the actin cytoskeleton, thereby induce membrane ruffling and focal adhesion 50. Microglial migration and reshaping was blocked by ROCK inhibition and this improved the dopaminergic neuron survival in the MPTP mouse model of PD 5, 8, 64. Furthermore, recent investigations from our group revealed that Fasudil attenuated the release of inflammatory cytokines and chemokines from primary microglia on LPS stimulation 61, therefore converting primary microglia to a neuroprotective phenotype 70. In addition, we could show a significantly reduced astrogliosis in the anterior horn of SOD1‐G93A transgenic mice after oral treatment with Fasudil and this was accompanied by increased survival and improved behavior in this rodent ALS model 61. It was recently shown that Fasudil can regulate the astrocytic cytoskeleton and motility as well as induce transcriptomic changes, which thereby resulted in reduced astroglial scarring and increased support of neuronal function 36.
Upregulated ROCK expression therefore provides evidence for an exacerbated micro‐and astroglial activity in PD patients and represents a putative therapeutic target as has been shown in several models of neurodegeneration 52, 57, 60, 61.
In conclusion, the present data suggests a correlation between disease progression, the glial composition and regeneration failure mirroring advanced disease in a heterogeneous set of human samples. Progressive nigrostriatal dopaminergic degeneration is accompanied by an increased astrogliosis, while the microglial response may already be vanished at this time point. The inverse correlation in the expression pattern of the growth‐associated protein GAP‐43 and the growth‐inhibitory protein ROCK2 indicates a reduced regeneration potential, as well as the involvement of ROCK in glial PD pathophysiology. In this context, pharmacological manipulation of reactive micro‐and astrogliosis argues for ROCK2 as a target in future PD therapies.
Supporting information
Table S1. Summary of cases investigated either for immunohistochemistry or in situ hybridization. The mean ± SD is given below the group. Abbreviations: amc = age‐matched control, F = female, M = male, na = not announced, ncl. = nucleus, PD = Parkinson's disease, PMI = post mortem interval, SN = substantia nigra, yc = young control.
Figure S1. Western blots of ROCK2 (30 µg protein lysate /lane, 7% acrylamide gel), GAP‐43 (15 µg protein lysate /lane) and synaptophysin (5 µg protein lysate /lane, both 12% acrylamide gel) in human (H), mouse (M1, M2) and rat (R) brain with the calibrated molecular weight standard in the left lane (Bio‐Rad, Munich, Germany).
Acknowledgments
The authors thank Elisabeth Barski for expert technical assistance, the Max‐Planck‐Institute for Biophysical Chemistry in Göttingen for providing us the Leica TCS SP5 confocal microsystem and the Institute of Neuropathology of the University Medicine Göttingen for supplying the antibodies for ED1 and HLA‐DR (CR3‐43). Brain sections for immunohistochemistry were kindly provided by Prof. Hans Kretzschmar (†) from the Department of Neuropathology and Prion Research of the Ludwig‐Maximilians‐University in Munich (Germany). For in situ hybridization studies brain tissue was obtained from The Netherland Brain Bank (Amsterdam, NL), The Harvard Brain Tissue Resource Center (Belmont, USA) and the Queen Square Brain Bank for Neurological Disorders (London, UK). PL was supported by the Cluster of Excellence and DFG Research Center Nanoscale Microscopy and Molecular Physiology of the Brain, Göttingen, and the Else Kröner‐Fresenius Stiftung. DG was supported by the Swedish Brain Power, the Swedish Parkinson Foundation and the Swedish Research Council.
References
- 1. Aigner L, Arber S, Kapfhammer JP, Laux T, Schneider C, Botteri F et al (1995) Overexpression of the neural growth‐associated protein gap‐43 induces nerve sprouting in the adult nervous system of transgenic mice. Cell 83:269–278. [DOI] [PubMed] [Google Scholar]
- 2. Alafuzoff I, Arzberger T, Al‐Sarraj S, Bodi I, Bogdanovic N, Braak H et al (2008) Staging of neurofibrillary pathology in alzheimer's disease: a study of the brainnet europe consortium. Brain Pathol 18:484–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Allegra Mascaro AL, Cesare P, Sacconi L, Grasselli G, Mandolesi G, Maco B et al (2013) In vivo single branch axotomy induces gap‐43 – dependent sprouting and synaptic remodeling in cerebellar cortex. PNAS 110:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ba F, Martin WRW (2015) Parkinsonism and related disorders dopamine transporter imaging as a diagnostic tool for parkinsonism and related disorders in clinical practice. Parkinsonism Relat Disord 21:87–94. [DOI] [PubMed] [Google Scholar]
- 5. Barcia C, Ros CM, Annese V, Carrillo‐de Sauvage MA, Ros‐Bernal F, Gómez A et al (2012) Rock/cdc42‐mediated microglial motility and gliapse formation lead to phagocytosis of degenerating dopaminergic neurons in vivo. Sci Rep 2:809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Benowitz LI, Routtenberg A (1997) Gap‐43: an intrinsic determinant of neuronal development and plasticity. Trends Neurosci 20:84–91. [DOI] [PubMed] [Google Scholar]
- 7. Blanchard V, Anglade P, Dziewczapolski G, Savasta M, Agid Y, Raisman‐vozari R (1996) Dopaminergic sprouting in the rat striatum after partial lesion of the substantia nigra. Brain Res 709:319–325. [DOI] [PubMed] [Google Scholar]
- 8. Borrajo A, Rodriguez‐Perez AI, Villar‐Cheda B, Guerra MJ, Labandeira‐Garcia JL (2014) Inhibition of the microglial response is essential for the neuroprotective effects of rho‐kinase inhibitors on mptp‐induced dopaminergic cell death. Neuropharmacology 85:1–8. [DOI] [PubMed] [Google Scholar]
- 9. Braak H, Tredici K Del, Rüb U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197–211. [DOI] [PubMed] [Google Scholar]
- 10. Burke RE, O'Malley K (2013) Axon degeneration in Parkinson's disease. Exp Neurol 246:72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burke SN, Barnes CA (2006) Neural plasticity in the ageing brain. Nat Rev Neurosci 7:30–40. [DOI] [PubMed] [Google Scholar]
- 12. Cafferty WBJ, Duffy P, Huebner E, Strittmatter SM (2010) Mag and omgp synergize with nogo‐a to restrict axonal growth and neurological recovery after spinal cord trauma. J Neurosci 30:6825–6837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Charron G, Doudnikoff E, Canron M‐H, Li Q, Véga C, Marais S et al (2014) Astrocytosis in parkinsonism: considering tripartite striatal synapses in physiopathology? Front Aging Neurosci 6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen YC, Chen QS, Lei JL, Wang SL (1998) Physical training modifies the age‐related decrease of gap‐43 and synaptophysin in the hippocampal formation in c57bl/6j mouse. Brain Res 806:238–245. [DOI] [PubMed] [Google Scholar]
- 15. Cherry JD, Olschowka JA, Banion MKO (2014) Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation 11:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Damier P, Hirsch EC, Zhang P, Agid Y (1993) Glutathione peroxidase, glial cells and Parkinson's disease. Neuroscience 52:1–6. [DOI] [PubMed] [Google Scholar]
- 17. Ding J, Li Q, Yu J, Wang X, Sun C, Lu CZ et al (2010) Molecular and cellular neuroscience fasudil, a rho kinase inhibitor, drives mobilization of adult neural stem cells after hypoxia/reoxygenation injury in mice. Mol Cell Neurosci 43:201–208. [DOI] [PubMed] [Google Scholar]
- 18. Fearnley JM, Lees AJ (1991) Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain 114:2283–2301. [DOI] [PubMed] [Google Scholar]
- 19. Finkelstein DI, Stanic D, Parish CL, Tomas D, Dickson K, Horne MK (2000) Axonal sprouting following lesions of the rat substantia nigra. Neuroscience 97:99–112. [DOI] [PubMed] [Google Scholar]
- 20. Fitch MT, Silver J (2008) CNS injury, glial scars, and inflammation: inhibitory extracellular matrices and regeneration failure. Exp Neurol 209:294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Frank‐Cannon TC, Alto LT, McAlpine FE, Tansey MG (2009) Does neuroinflammation fan the flame in neurodegenerative diseases? Mol Neurodegener 13:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Frey D, Laux T, Xu L, Schneider C, Caroni P (2000) Shared and unique roles of cap23 and gap43 in actin regulation, neurite outgrowth, and anatomical plasticity. J Cell Biol 149:1443–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. D Galter, S Buervenich, A Carmine, M Anvret, L Olson (2003) ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson's disease and in the ventral tegmental area in schizophrenia. Neurobiol Dis 14:637–647. [DOI] [PubMed] [Google Scholar]
- 24. Gonzalez‐Forero D, Montero F, Garcia‐Morales V, Dominguez G, Gomez‐Perez L, García‐Verdugo JM et al (2012) Endogenous rho‐kinase signaling maintains synaptic strength by stabilizing the size of the readily releasable pool of synaptic vesicles. J Neurosci 32:68–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Halliday GM, Stevens CH, Hons B (2011) Glia: initiators and progressors of pathology in Parkinson’ s disease. Mov Disord 26:6–17. [DOI] [PubMed] [Google Scholar]
- 26. Hanisch U‐K, Kettenmann H (2007) Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 10:1387–1394. [DOI] [PubMed] [Google Scholar]
- 27. Hashimoto R, Nakamura Y, Kosako H, Amano M, Kaibuchi K, Inagaki M, Takeda M (1999) Distribution of rho‐kinase in the bovine brain. Biochem Biophys Res Commun 263:575–579. [DOI] [PubMed] [Google Scholar]
- 28. Hunter RL, Dragicevic N, Seifert K, Choi DY, Liu M, Kim HC et al (2007) Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J Neurochem 100:1375–1386. [DOI] [PubMed] [Google Scholar]
- 29. Iizuka M, Kimura K, Wang S, Kato K, Amano M, Kaibuchi K, Mizoguchi A (2012) Distinct distribution and localization of rho‐kinase in mouse epithelial, muscle and neural tissues. Cell Struct Funct 37:155–175. [DOI] [PubMed] [Google Scholar]
- 30. Iwata S‐I, Nomoto M, Fukuda T (2001) Regulation of gap‐43 protein and mrna in nigrostriatal dopaminergic neurons after the partial destruction of dopaminergic terminals with intrastriatal 6‐hydroxydopamine. Synapse 39:16–22. [DOI] [PubMed] [Google Scholar]
- 31. Kettenmann H, Kirchhoff F, Verkhratsky A (2012) Perspective microglia: new roles for the synaptic stripper. Neuron 77:10–18. [DOI] [PubMed] [Google Scholar]
- 32. Klein RL, McNamara RK, King MA, Lenox RH, Muzyczka N, Meyer EM (1999) Generation of aberrant sprouting in the adult rat brain by gap‐43 somatic gene transfer. Brain Res 832:136–144. [DOI] [PubMed] [Google Scholar]
- 33. Koch JC, Tönges L, Barski E, Michel U, Bähr M, Lingor P (2014) Rock2 is a major regulator of axonal degeneration, neuronal death and axonal regeneration in the cns. Cell Death Dis 5:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Komagome R, Kimura K, Saito M (2000) Postnatal changes in rho and rho‐related proteins in the mouse brain. Jpn J Vet Res 47:127–133. [PubMed] [Google Scholar]
- 35. Kottis V, Thibault P, Mikol D, Xiao Z‐C, Zhang R, Dergham P, Braun PE (2002) Oligodendrocyte‐myelin glycoprotein (omgp) is an inhibitor of neurite outgrowth. J Neurochem 82:1566–1569. [DOI] [PubMed] [Google Scholar]
- 36. Lau CL, Perreau VM, Chen MJ, Cate HS, Merlo D, Cheung NS (2012) Transcriptomic profiling of astrocytes treated with the rho kinase inhibitor fasudil reveals cytoskeletal and pro‐survival responses. J Cell Physiol 227:1199–1211. [DOI] [PubMed] [Google Scholar]
- 37. Lingor P, Tönges L, Pieper N, Bermel C, Barski E, Planchamp V, Bähr M (2008) Rock inhibition and cntf interact on intrinsic signalling pathways and differentially regulate survival and regeneration in retinal ganglion cells. Brain 131:250–263. [DOI] [PubMed] [Google Scholar]
- 38. Love S, Plaha P, Patel NK, Hotton GR, Brooks DJ, Gill SS (2005) Glial cell line – derived neurotrophic factor induces neuronal sprouting in human brain. Nat Med 11:703–704. [DOI] [PubMed] [Google Scholar]
- 39. Maragakis NJ, Rothstein JD (2006) Mechanisms of disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol 2:679–689. [DOI] [PubMed] [Google Scholar]
- 40. McGeer PL, McGeer EG (2008) Glial reactions in Parkinson's disease. Mov Disord 23:474–483. [DOI] [PubMed] [Google Scholar]
- 41. McKeith IG, Dickson DW, Lowe J, Emre M, Brien JTO, Feldman H et al (2005) Diagnosis and management of dementia with lewy bodies third report of the dlb consortium. Neurology 65:1–10. [DOI] [PubMed] [Google Scholar]
- 42. Mirza B, Hadberg H, Thomsen P, Moos T (2000) The absence of reactive astrocytosis is indicative of a unique inflammatory process in Parkinson's disease. Neuroscience 95:425–432. [DOI] [PubMed] [Google Scholar]
- 43. Mitsumoto Y, Watanabe A, Mori A, Koga N (1998) Spontaneous regeneration of nigrostriatal dopaminergic neurons in mptp‐treated c57bl/6 mice. Biochem Biophys Res Commun 248:660–663. [DOI] [PubMed] [Google Scholar]
- 44. Morrison JH, Hof PR (1997) Life and death of neurons in the aging brain. Science 278:412–419. [DOI] [PubMed] [Google Scholar]
- 45. Mueller BK, Mack H, Teusch N (2005) Rho kinase, a promising drug target for neurological disorders. Nat Rev Drug Discov 4:387–398. [DOI] [PubMed] [Google Scholar]
- 46. Neve RL, Finch EA, Birdt ED, Benowitz LI (1988) Growth‐associated protein gap‐43 is expressed selectively in associative regions of the adult human brain. Proc Natl Acad Sci U S A 85:3638–3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T, Torizuka T (2005) Microglial activation and dopamine terminal loss in early Parkinson's disease. Ann Neurol 57:168–175. [DOI] [PubMed] [Google Scholar]
- 48. Pitcher TL, Melzer TR, Macaskill MR, Graham CF, Livingston L, Keenan RJ et al (2012) Reduced striatal volumes in Parkinson's disease: a magnetic resonance imaging study. Alzheimers Res Ther 1:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Planchamp V, Bermel C, Tönges L, Ostendorf T, Kügler S, Reed JC et al (2008) Bag1 promotes axonal outgrowth and regeneration in vivo via raf‐1 and reduction of rock activity. Brain 131:2606–2619. [DOI] [PubMed] [Google Scholar]
- 50. Riento K, Ridley AJ (2003) Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol 4:446–456. [DOI] [PubMed] [Google Scholar]
- 51. Rodriguez‐Oroz MC, Jahanshahi M, Krack P, Litvan I, Macias R, Bezard E, Obeso JA (2009) Initial clinical manifestations of Parkinson’ s disease: features and pathophysiological mechanisms. Lancet Neurol 8:1128–1139. [DOI] [PubMed] [Google Scholar]
- 52. Saal K‐A, Koch JC, Tatenhorst L, Szegő EM, Ribas VT, Michel U et al (2015) AAV.shRNA‐mediated downregulation of rock2 attenuates degeneration of dopaminergic neurons in toxin‐induced models of Parkinson's disease in vitro and in vivo. Neurobiol Dis 73:150–162. [DOI] [PubMed] [Google Scholar]
- 53. Seibyl JP, Marek JKL, Quinlan D, Sheff PK, Zoghbi SS, Zea‐Ponce Y et al (1995) Decreased single‐photon emission computed tomographc [123i]ß‐cit striatal uptake correlates with symptom severity in Parkinson's disease. Ann Neurol 38:589–598. [DOI] [PubMed] [Google Scholar]
- 54. Silver J, Miller JH (2004) Regeneration beyond the glial scar. Nat Rev Neurosci 5:146–156. [DOI] [PubMed] [Google Scholar]
- 55. Stark A, Pakkenberg B (2004) Histological changes of the dopaminergic nigrostriatal system in aging. Cell Tissue Res 318:81–92. [DOI] [PubMed] [Google Scholar]
- 56. Sterling NW, Du G, Lewis MM, Dimaio C, Kong L, Eslinger PJ et al (2013) Striatal shape in Parkinson's disease. Neurobiol Aging 34:2510–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tatenhorst L, Tönges L, Saal K‐A, Koch JC, Szegő EM, Bähr M, Lingor P (2014) Rho kinase inhibition by fasudil in the striatal 6‐hydroxydopamine lesion mouse model of Parkinson disease. J Neuropathol Exp Neurol 73:770–779. [DOI] [PubMed] [Google Scholar]
- 58. Teismann P, Schulz JB (2004) Cellular pathology of Parkinson's disease: astrocytes, microglia and inflammation. Cell Tissue Res 318:149–161. [DOI] [PubMed] [Google Scholar]
- 59. Tong J, Ang L‐C, Williams B, Furukawa Y, Fitzmaurice P, Guttman M et al (2015) Low levels of astroglial markers in Parkinson's disease: relationship to α‐synuclein accumulation. Neurobiol Dis 82:243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tönges L, Frank T, Tatenhorst L, Saal K‐A, Koch JC, Szego ÉM et al (2012) Inhibition of rho kinase enhances survival of dopaminergic neurons and attenuates axonal loss in a mouse model of Parkinson's disease. Brain 135:3355–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tönges L, Günther R, Suhr M, Jansen J, Balck A, Saal KA et al (2014) Rho kinase inhibition modulates microglia activation and improves survival in a model of amyotrophic lateral sclerosis. Glia 62:217–232. [DOI] [PubMed] [Google Scholar]
- 62. Tönges L, Koch J‐C, Bähr M, Lingor P (2011) Rocking regeneration: rho kinase inhibition as molecular target for neurorestoration. Front Mol Neurosci 4:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Van Dyck CH, Malison RT, Laruelle M, Zoghbi SS, Baldwin RM, Innis RB (2002) Age related decline in dopamine transporters. analysis of striatal subregions, nonlinear effects, and hemispheric asymmetries. Am J Geriatr Psychiatry 10:36–43. [PubMed] [Google Scholar]
- 64. Villar‐Cheda B, Dominguez‐Meijide A, Joglar B, Rodriguez‐Perez AI, Guerra MJ, Labandeira‐Garcia JL (2012) Involvement of microglial rhoa/rho‐kinase pathway activation in the dopaminergic neuron death. Role of angiotensin via angiotensin type 1 receptors. Neurobiol Dis 47:268–279. [DOI] [PubMed] [Google Scholar]
- 65. Volkow ND, Ding Y, Fowler JS, Wang G, Logan J, Gatley SJ et al (1996) Dopamine transporters decrease with age. J Nucl Med 37:554–559. [PubMed] [Google Scholar]
- 66. Walker DG, Lue L (2015) Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res Ther 7:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Westerlund M, Belin AC, Olson L, Galter D (2008) Expression of multi‐drug resistance 1 mRNA in human and rodent tissues: reduced levels in Parkinson patients. Cell Tissue Res 334:179–185. [DOI] [PubMed] [Google Scholar]
- 68. Yiu G, He Z (2009) Glial inhibition of cns axon regeneration. Nat Rev Neurosci 7:617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yu J‐Z, Ding J, Ma C‐G, Sun C‐H, Sun Y‐F, Lu CZ, Xiao BG (2010) Therapeutic potential of experimental autoimmune encephalomyelitis by fasudil, a rho kinase inhibitor. J Neurosci Res 88:1664–1672. [DOI] [PubMed] [Google Scholar]
- 70. Zhang H, Li Y, Yu J‐Z, Guo M, Meng J, Liu C et al (2013) Rho kinase inhibitor fasudil regulates microglia polarization and function. Neuroimmunomodulation 20:313–322. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of cases investigated either for immunohistochemistry or in situ hybridization. The mean ± SD is given below the group. Abbreviations: amc = age‐matched control, F = female, M = male, na = not announced, ncl. = nucleus, PD = Parkinson's disease, PMI = post mortem interval, SN = substantia nigra, yc = young control.
Figure S1. Western blots of ROCK2 (30 µg protein lysate /lane, 7% acrylamide gel), GAP‐43 (15 µg protein lysate /lane) and synaptophysin (5 µg protein lysate /lane, both 12% acrylamide gel) in human (H), mouse (M1, M2) and rat (R) brain with the calibrated molecular weight standard in the left lane (Bio‐Rad, Munich, Germany).