

Abstract
Amyotrophic Lateral Sclerosis (ALS) is characterized by the degeneration of upper and lower motor neurons. Clinical heterogeneity is a well‐recognized feature of the disease as age of onset, site of onset and the duration of the disease can vary greatly among patients. A number of genes have been identified and associated to familial and sporadic forms of ALS but the majority of cases remains still unexplained. Recent breakthrough discoveries have demonstrated that clinical manifestations associated with ALS‐related genes are not circumscribed to motor neurons involvement. In this view, ALS appears to be linked to different conditions over a continuum or spectrum in which overlapping phenotypes may be identified. In this review, we aim to examine the increasing number of spectra, including ALS/Frontotemporal Dementia and ALS/Myopathies spectra. Considering all these neurodegenerative disorders as different phenotypes of the same spectrum can help to identify common pathological pathways and consequently new therapeutic targets in these incurable diseases.
Keywords: amyotrophic lateral sclerosis, distal myopathy, fronto temporal dementia, inclusion body myopathy, mitochondrial diseases
Introduction
Amyotrophic Lateral Sclerosis (ALS) is a devastating neurodegenerative disease leading to paralysis and respiratory failure within 3–5 years after symptoms begin. The clinical picture of ALS is a stereotypical one, resulting from a combination of signs secondary to dysfunction of upper motor neurons (UMN) in the cerebral cortex and of lower motor neurons (LMN) located in the brainstem and the spinal cord. However, clinical heterogeneity is a well‐recognized feature of the disease as age of onset, site of onset and duration of the disease can vary greatly among patients 93, 103. The variable mix of UMN and LMN signs is an additional major contributor to clinical heterogeneity of ALS 39, 91, 92. Classic ALS, in which predominant LMN signs combine with slight to moderate pyramidal syndrome, is the most frequent form. Other less frequent phenotypes include Progressive Muscular Atrophy (PMA), with pure LMN involvement, and Primary Lateral Sclerosis (PLS), with isolated UMN involvement. PMA and PLS are the two extremities of the lower and upper motor neuron involvement spectrum, where intermediate phenotypes may be identified, including UMN‐Dominant ALS 91, 92, 103 (Figure 1).
Figure 1.
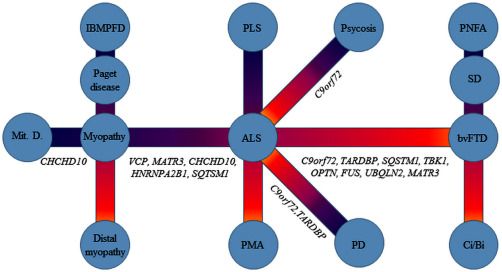
ALS is a part of several clinical spectra. Intermediate/overlapping phenotypes may occur in each spectrum. IBMPFD = inclusion‐body myopathy with Paget disease of the bone and fronto temporal dementia; PD = Parkinson Disease; PLS = primary lateral sclerosis; PMA = progressive muscle atrophy; PNFA = progressive nonfluent aphasia; SD = semantic dementia; Ci/Bi = mild cognitive (language, executive) or behavioral impairment); bvFTD = frontotemporal dementia behavioral variant; Mit.D.= mitochondrial diseases.
Since the discovery of SOD1 mutations in familial ALS cases (fALS) in 1993 89, genetic research has revealed that ALS is not a single entity but rather a syndrome in which a constellation of causative genes have been discovered and new ones are expected to be discovered in the next few years, most likely.
A limited number of genes, including C9orf72, SOD1, TARDBP, FUS and TBK1, are responsible for a significant proportion of familial and sporadic ALS cases. Conversely, several genes are increasingly being recognized, which are responsible for a small number of cases or even for isolated ALS families 93. The etiologic fragmentation of ALS, driven by the genetic discoveries, contrasts with an opposite view in which near all ALS subtypes appear to be unified by a single pathological signature, namely the presence of abnormal accumulation of the transactive response DNA binding protein (TDP‐43) in the cytoplasm of neuronal and glial cells 74.
Notably, in the light of the recent genetic discoveries some conceptual aspects regarding ALS are going to be revisited.
ALS: Part of Several Spectra Rather than a Single Entity
While ALS has long been considered a paradigm of pure motor neuron disorder, there is now evidence that the majority of ALS‐related genes are pleiotropic, as they give raise to disparate clinical manifestations. Importantly, clinical manifestations associated with single gene defects appear to vary over a continuum or spectrum with frequent overlapping phenotypes. The discovery of new ALS‐related genes is expanding the number of conditions linked to ALS, suggesting that the clinical syndrome called ALS should be reconsidered from the perspective of the “spectrum paradigm.”
ALS/ Frontotemporal Dementia Spectrum
The most common and well‐established spectrum is the ALS/Frontotemporal Dementia (FTD) spectrum, connecting ALS with FTD, the second cause of presenile dementia due to degeneration of the frontal and temporal lobes in the brain 60.
The link between these two conditions is supported by several clinical, pathological and genetic observations. From a clinical point of view, patients with ALS may exhibit cognitive abnormalities ranging from impaired frontal executive dysfunction to overt FTD. In a population‐based study, it was showed that co‐morbid dementia occurs in approximately 14% of patients with a new diagnosis of ALS 83. Cognitive impairment without overt dementia, mainly in the form of executive dysfunction, occurs in more than 40% of ALS patients. Conversely, 15% of FTD patients develop a motor neuron disease. The description of ALS patients with relatives affected by pure ALS or pure FTD or ALS/FTD strongly supports the concept that ALS and FTD are closely related conditions 60. On pathological ground, neuronal inclusions positive for TDP‐43, detected in ALS are also present in FTD patients 74. Finally, the decisive evidence was the identification of an etiologic link. Many genetic defects, including mutations in VCP, TARDBP, SQTSM1, FUS and UBQLN2 underlay this spectrum but the identification of C9orf72 gene was the biggest advance in this field. The large hexanucleotide (GGGGCC) repeat expansion in the first intron of C9orf72, located on chromosome 9p21, is the most common mutation detected so far in patients with familial ALS (50%–72% of fALS) as well as in patients with familial FTD (11.7%–17.8% of cases) 16, 26, 40, 68, 84, 86, 98, 99, 101. Interestingly, the frequency of cognitive impairment is 40%–50% in ALS patients carrying the C9orf72 expansion, compared with 8%–9% of ALS cases without C9orf72 mutation 26, 40, 98.
Newly discovered genes have been related to ALS/FTD spectrum. One of these is MATR3, encoding a nuclear matrix protein, identified by exome sequencing in 2014. A missense mutation in MATR3 was found in a family of European ancestry with several members affected by ALS and dementia or cognitive impairment 48. However, no cognitive impairment has been reported in the two other mutated MATR3 cases described, a Taiwanese patient with A72T mutation 65 and an Italian patient with C85S variant 76. MATR3 mutations appear to be rare in ALS patients as they were not found in ALS and ALS/FTD series from France 69 and Australia 35. Further cohorts need to be screened to assess the contribution of MATR3 in ALS/FTD.
In 2014, the CHCHD10 gene, encoding a mitochondrial protein of the intermembrane space, was first associated to familial ALS/FTD 4. The S59L mutation was identified in a French family in which one member had isolated ALS and the remaining had FTD associated with ALS, ataxia or myopathy 4. Screening of CHCHD10 in French cohorts, including 26 familial FTD/ALS, 85 sporadic FTD/ALS and 9 familial FTD, resulted in the detection of the previously identified S59L mutation in 1/26 (3.8%) of familial ALS/FTD and of a novel P34S variant in 2/85 (2.3%) of sporadic ALS/FTD 14. Notably, patients with P34S presented with behavior disorders and later developed motor neuron disease. Among an Italian cohort of 224 SALS patients, of whom 31 also had FTD, the P34S variant was detected in two cases with isolated ALS 17. An additional Italian patient with isolated ALS and P34S variant has been described 88. The pathogenicity of P34S is controversial: the variant did not always segregate with the disease, it was found in healthy controls and some bioinformatics prediction programs suggested its nonpathogenicity 1, 30, 67, 113, 124. Other CHCHD10 variants have been identified later by whole exome sequencing (WES), consisting of R15L in 2/102 German cases, and of G66V in a Finnish patient out of 26 Nordic FALS subjects 71. All of them had isolated ALS without dementia. The R15L variant was also described in three additional families and in one additional sporadic patient with isolated ALS 47, 56, 125. In addition, the P80L and P12S variants were identified in patients with classic ALS, and the P23T, A35D and Q82X variants in patients with sporadic FTD 31, 125.
Thus, CHCHD10 mutations may be associated with different phenotypes and variable age of onset and disease duration. The higher frequency of CHCHD10 mutations in sporadic than in familial cases may be explained by incomplete penetrance.
Another new gene linking ALS to FTD is TUBA4A, encoding a component of microtubules, namely an α‐tubulin 100. By analyzing exomes of patients and controls, an excess of rare damaging variants in TUBA4A was found in fALS patients. Data were supported by functional studies, demonstrating that some of these variants disrupt microtubule dynamics and stability, at least in in vitro models 100. The screening was extended to a large cohort of sporadic cases with different geographic origin and to a smaller cohort of ALS patients with concomitant FTD. Further mutations, with predicted deleterious effects, were described, supporting the role of this gene in ALS and the hypothesis that defects in neuronal cytoskeleton architecture can lead to neurodegeneration 80. At least in three cases, TUBA4A mutations were associated to ALS/FTD: a patient with ALS/FTD carried the R215C (and his mother, not available for the test, had FTD as well), an ALS patient carried the T145P variant and reported a family history of FTD 100 and finally an ALS patient with D438N had also cognitive impairment 80. Further studies are needed to assess the impact of TUBA4A in these disorders, especially in ALS/FTD, to test the segregation of the mutations and to clarify the presence of variants in healthy controls. Conversely, all the mutations found only in patients affect a conserved region in exon 4, suggesting that they can cause the disruption of a crucial functional domain of the protein, as suggested by in vitro experiments 100.
The most recent ALS‐related gene is TANK‐binding kinase 1 (TBK1) 18, 36. Frequency of dominant TBK1 loss‐of‐function mutations was 4% in a cohort of 252 genetically unexplained fALS patients 18. Importantly, 50% of patients with TBK1 mutations showed cognitive impairment or overt FTD, a proportion that is similar to that observed in the affected carriers of C9orf72 expansion 18, 26, 40, 98. Eight different loss‐of‐function mutations in TBK1 were described in familial ALS/FTD patients of European origin 36. Two additional studies confirmed the role of TBK1 in ALS/FTD. Among 104 pathologically confirmed Frontotemporal Lobar Degeneration‐TDP patients, negative for C9orf72 and GRN mutations, three were found to carry a heterozygous missense mutation in TBK1 and one a loss‐of‐function mutation (R117*) in TBK1 associated with deletion of exons 13–15 of OPTN (G538Efs*27) 82. Of interest, an additional patient had a compound heterozygous mutations in OPTN 82. More recently, a frameshift mutation (L399fs), likely resulting in a truncated protein, has been found in a fALS patient of Chinese origin 121.
These findings make TBK1 a major gene causing ALS/FTD spectrum, add OPTN to the list of genes underlying this spectrum and stress the involvement of the autophagy pathway in the pathogenesis of these diseases.
ALS/ Myopathy Spectrum
Genetic findings along with clinical and pathological observations indicate that ALS may be linked to different forms of muscular disorders.
ALS/inclusion body myopathies spectrum
Inclusion Body Myopathies (IBMs) are a heterogeneous group of muscular disorders including two main categories: sporadic inclusion body myositis (sIBM) and hereditary inclusion body myopathy (hIBM). sIBM occurs sporadically in families while hIBM shows Mendelian inheritance with autosomal recessive or dominant transmission 29.
Muscle biopsy in both entities reveals distinctive abnormalities, including (i) myopathic changes such as variability of muscle fiber diameter and central nuclei, (ii) rimmed vacuoles, (iii) cytoplasmic inclusions. Endomysial inflammatory exudates surrounding and invading non‐necrotic muscle fibers are present in sIBM while are not a feature of hIBM.
However the distinction between the “inflammatory” and the “degenerative” components in IBMs is not well established. Clinically, sIBM is usually refractory to immunotherapies and shows a slowly progressive, degenerative‐like course leading to disability, though life expectancy is not affected. The degenerative pathophysiology of sIBM is further supported by the identification in muscle fibers of protein aggregates generally associated with other neurodegenerative diseases, including TDP‐43 and p62, amyloid‐β, hyperphosphorylated tau, ubiquitin, neurofilament heavy chain, presenilin and parkin 7, 29.
ALS/sIBM
sIBM is the most frequent acquired myopathy among the elderly. It is characterized by weakness and atrophy involving mainly the quadriceps and deep finger flexor muscles in the forearm, followed by involvement of more proximal muscle groups. ALS and sIBM may resemble each other in several aspects as to make differential diagnosis challenging. Clinically, some features of sIBM overlap to a large extent with those observed in ALS. In fact, in sIBM patients muscular involvement may be asymmetric, dysphagia occurs in 50%–70% of cases and the course of the disease is progressive. Importantly, EMG examination in sIBM demonstrates fibrillation potentials in the majority of cases, and motor unit action potentials show mixed myopathic and neuropathic changes, with the latter being likely due to reinnervation of denervated and split muscle fibers. In some cases, the neurogenic pattern may overshadow the myopathic changes leading to misdiagnosis of ALS. Muscle biopsy and imaging are useful tools to diagnosis 105. Though ALS and sIBM remain separate entities, TDP‐43 accumulation represents an important link between these conditions but its significance remains to be elucidated 7, 29. Interestingly, genetic components in sIBM are starting to be explored more systematically. Besides the known association with the major histocompatibility loci, rare variants in VCP and SQSTM1 genes as well as in genes involved in mitochondrial DNA maintenance have been reported in sIBM patients 37, 119.
ALS/hIBM
Three main clinical‐genetic conditions are included in the hIBM group: hereditary inclusion‐body myopathy with Paget's disease of the bone (PDB) and frontotemporal dementia (IBMPFD); GNE myopathy caused by mutations in the UDP‐Nacetylglucosamine 2‐epimerase/N‐acetylmannosamine kinase (GNE); hereditary inclusion‐body myopathy with congenital joint contractures and external ophthalmoplegia associated with mutations in Myosin Heavy Chain IIa gene (MyHC‐IIa).
Mutations in valosin‐containing protein (VCP) were identified in a proportion of IBMPFD 52, 115, 116, 118. The gene was identified using a candidate gene approach in 13 families linked to chromosome 9, where 82% of individuals had myopathy, 49% had PDB and 30% had early‐onset FTD 115. The majority of mutations were found within the N‐terminal domain of the protein, with a mutational hot‐spot at codon 155, where different amino acid changes were described (R155H, R155P, R155C) 115. The recurrent R155H missense variant was then identified in an Italian family where three carriers presented with progressive IBM and FTD and only one developed PDB. Muscle biopsy showed atrophic fibres, rimmed vacuoles and congophilic amyloid deposits, which were described also in a VCP mouse model 114. In a review of 122 VCP related IBMPFD cases, approximately 50% showed osteolytic bone lesions consistent with PDB and only one third developed FTD. Muscular weakness was present in about 90% of patients, with a mean onset of 45 years of age, and it was the presenting symptom in more than 50% of patients. Muscle involvement was an isolated symptom in 30% of patients 52.
The clinical phenotypes of VCP‐related myopathy are highly variable, including the limb girdle muscle phenotype, a scapulo‐humeral form, a distal myopathy and an axial form with head drop. The pattern of muscle involvement is frequently focal, while symmetric distribution is less common. The characteristic rimmed vacuoles are described in 39% of muscle biopsies 51, 52.
Patients carrying the same VCP mutation show different clinical phenotypes. For example, the missense variant R159H was reported in members of a Belgian family presenting with pure FTD, while patients from another unrelated Belgian family had FTD and PDB; no signs of IBM was observed in these families 111.
The identification of VCP mutations in a subset of sporadic and familial ALS cases 46 has established a consistent link between ALS and IBMPFD. Actually, in the original family with IBMPFD, in which a VCP mutation was later identified 52, 109, the disease was described as a “familial disorder of combined lower motor neuron degeneration and skeletal muscle disorganization,” suggesting coexistence of both a primarily neurogenic process and myopathy. The concept that ALS and IBMPFD are part of a spectrum of VCP proteinopathies is supported by the description of VCP mutated individuals from the same kindred presenting with different phenotypes, including ALS, IBM, FTD or myopathy 5, 38, 52, 55. Some specific VCP mutations have been reported to cause ALS, others only IBMPFD, while others have been described in association with both conditions 38. VCP mutations are rare in ALS patients, accounting for <1% of fALS and are more frequently associated with ALS/FTD than with a pure ALS phenotype 2, 44, 53, 59, 70, 107, 120, 126.
The presence of TDP‐43‐positive aggregates is a common phenomenon in ALS and myopathies associated with VCP mutation as well as in other forms of hereditary IBM 8, 24, 57, 75, 94, 117.
Importantly, ALS/IBMPFD spectrum is associated with other, though less frequent, genetic defects.
Mutations in HNRNPA2B1and HNRNPA1 have been identified in ALS and IBMPFD 50. In a family with IBMPFD, with no VCP mutations, WES allowed for detection of a variant in HNRNPA2B1 gene 50. Two affected members had myopathy and PDB, one had myopathy, PDB and cognitive impairment and two had motor neuron disease, associated with myopathy, PDB and cognitive impairment. Muscle biopsies from one patient with motor neuron dysfunction showed atrophic fibres, central nuclei and rimmed vacuoles with TDP‐43 accumulation. Conversely, a variant in HNRNPA1 was identified in a German family with PDB associated with a late‐onset autosomal dominant myopathy starting in the pelvic girdle at about age 40 years, with rapidly‐progressing course. Histology showed myopathic changes with rimmed vacuoles and inclusion bodies on muscle biopsy 50. Finally, a mutation in HNRNPA1 has been detected in cases with autosomal dominant ALS 50.
In another family with HNRNPA2B1 mutation, clinical manifestations were characterized by myopathy and PD in all four members, with associated neurogenic weakness in one individual and cognitive impairment in the remaining three 5.
The absence of known disease‐causing mutations in several sporadic and familial cases with the ALS/IBMPFD spectrum indicates that genetic heterogeneity of this condition is likely to expand over the next years 5, 63, 95.
Some authors proposed the term “Multisystem Proteinopathy (MSP)” to define the ALS/IBM spectrum 5, 106 where a combination of two or more signs among IBM, PDB and ALS/FTD was considered sufficient for the diagnosis of MSP.
ALS/Distal Myopathies
Distal myopathies are a group of muscle diseases characterized by predominant weakness in the feet and/or hands. More than 20 distinct subtypes have been identified, with both autosomal dominant and recessive inheritance and many of them yet lack genetic characterization 110. On muscle pathology, several forms of distal myopathies show the presence of vacuoles that are lined (rimmed) by red granules on modified Gomori trichrome staining called “rimmed vacuoles.” They represent accumulation of autophagic vacuoles, due to lysosomal dysfunction or to proteins accumulation 66.
At least three ALS‐related genes, including VCP, MATR3 and SQSTM1, have been also associated with distal myopathies. These genetic data, along with the observation of combined ALS/distal myopathy phenotypes in some individuals and the evidence of rimmed vacuoles with TDP‐43 inclusion on muscle biopsy, support the notion that ALS/distal myopathy with rimmed vacuoles may represent a subgroup of the ALS/myopathy spectrum.
Distal myopathy and VCP
As previously mentioned, almost 90% of reported patients with VCP mutations show proximal limb‐girdle myopathy. A new phenotype has been described in a Finnish family harboring the P137L mutation, where nine affected members had an autosomal dominant distal myopathy with onset after age 35 77. Three patients developed overt FTD several years after the onset of muscle weakness, but none of them had PDB. Muscle biopsy showed rimmed vacuoles with TDP‐43 and p62 positive inclusions. The same mutation has been detected in patients in whom weakness in hands or feet was the presenting symptom with later progression to proximal muscles 102.
Distal myopathy and MATR3
A mutation in MATR3 (S85C) was first identified as the cause of an autosomal dominant, distal, asymmetrical myopathy with vocal cord paralysis and pharyngeal weakness in a large North American multigenerational family 34 and in an unrelated Bulgarian family 96. Muscle biopsy disclosed a chronic noninflammatory myopathy with rimmed vacuoles, but features suggesting denervation were also detected, including presence of atrophic fibers. EMG examination detected myopathic changes in some members and neurogenic alteration in other relatives.
As mentioned before, in 2014 mutations in MATR3 were identified in ALS patients as well. The F115C variant in MATR3 was detected in a large ALS pedigree of European ancestry 48 and WES on additional 108 fALS cases revealed a T622A (exon11) mutation in one patient, corresponding to a cumulative frequency of MATR3 mutations in 2.75% of fALS. In addition, custom resequencing of genes linked to neurodegeneration in 96 British ALS cases identified a P154S missense variant in MATR3 in an individual diagnosed with sporadic disease 48. In the light of these findings, the American family with distal myopathy associated with S85C MATR3 mutation 34, 96 was re‐evaluated, leading to a reclassification of these cases as a form of slowly progressive ALS 48. The presence of the “split‐hand” sign, commonly observed in ALS patients 32 and of brisk reflexes in most cases was considered sufficient to diagnosis.
However, two recently published papers confirmed that the S85C MATR3 mutation is associated with a distal myopathy. In 16 German patients from 6 families, harboring the S85C MATR3 mutation, muscle biopsy showed only myopathic changes, including variation of fiber size, internal nuclei, minor fatty replacement of muscle fibers, rimmed vacuoles or end stage myopathic changes (eg, major fatty replacement of muscle tissue) 72. In an American family with the S85C MATR3 mutation no changes compatible with a neurogenic process were observed on both muscle pathology and electromyography 78. Muscle biopsy showed numerous rimmed‐vacuolated fibers with no neurogenic changes. Authors raised the possibility of minor upper motor neuron signs in four patients. Conversely, in a recently described Asian family with S85C MATR3 mutation a mixed myogenic and neurogenic pattern was detected on electrodiagnostic and muscle biopsy studies 122. Based on these results, there is evidence that the S85C MATR3 mutation is associated with both a pure distal myopathy phenotype and ALS/distal myopathy phenotype, in which combined myopathic and neurogenic features are present at clinical, EMG and muscle biopsy examination.
Mutations in MATR3 appear to be very rare in ALS, as only a few additional patients have been reported so far. One mutation (A72T) was found in one sporadic case from a cohort of 207 Taiwanese ALS patients, including 30 fALS and 177 sALS 65. The patient had a classic phenotype with bulbar onset and disease duration of 11 years. Exons 2, 11 and 12 of the MATR3 gene were studied in 200 Italian ALS patients, showing a R147W variant in one sporadic case with a slowly progressive disease with predominant lower motor neurons involvement 76. Three further variations have been identified in 3 sALS cases, 1 missense (V394M) and 2 spicing (c.48 + 1G>T, c.−339 + 2T>A), studying a total of 83 fALS and 164 SALS of French‐Canadian and European origins 64.
Importantly, MATR3 encodes an RNA‐ and DNA‐binding protein that interacts with TDP‐43 48. TDP‐43 and other autophagic markers (p62 and SMI‐31) were components of the rimmed‐vacuolar pathology in S85C MATR3 myopathy 122.
These results indicate that the spectrum of diseases associated with MATR3 mutations includes ALS, distal myopathy with rimmed vacuoles, combined ALS/distal myopathy and possibly FTD.
Distal myopathy and SQSTM1
SQSTM1 mutations were already known to be associated with PDB 45, 61, accounting for 25%–50% of familial and 5%–10% of sporadic PDB patients 85. In the last few years SQSTM1 mutations have been found in patients with sALS and fALS 33, ALS/FTD, FTD/PD and pure FTD 9, 43, 54, 58, 62, 90, 97, 108, 112, 115. The latest discovery that SQSTM1 mutations can be associated with a distal myopathy with rimmed vacuoles indicates that SQSTM1 mutations contribute to the aetiology of ALS/IBMPFD spectrum 10. Interestingly, the variant associated to distal myopathy is a splice donor mutation (c.1165 + 1 G>A), resulting in the expression of two different isoforms of SQSTM1. This variant was found in one family and in one sporadic patient, all carriers having late‐onset distal lower extremity weakness with rimmed vacuoles with TDP‐43 and SQSTM1 inclusions on muscle biopsy 10.
ALS/Mitochondrial Disease Spectrum?
There are several works in literature supporting the role of mitochondrial dysfunction in the pathogenesis of ALS (for comprehensive reviews, see 6, 21, 22, 104). The evidence of swollen mitochondria in the early stages of disease in transgenic mice model carrying SOD1 mutations, indicates that mutant SOD1 toxicity may be mediated by damage to mitochondria in motor neurons 6, 25, 123.
However, pure motor neuron impairment is not included among clinical manifestations of Mitochondrial Diseases, although rare cases with motor neuron pathology have been described in patients with mutations in genes encoding for mitochondrial proteins 19, 27, 42, 87.
Mitochondrial diseases are phenotypically and genetically heterogeneous disorders caused by mutations in either the mitochondrial DNA (mtDNA) or the nuclear DNA (nDNA). nDNA encodes most of the respiratory‐chain proteins, which are needed for the proper assembly and function of respiratory chain complexes, for mtDNA maintenance and translation, phospholipid composition of the inner mitochondrial membrane or mitochondrial dynamics. Mutations in these factors affect mtDNA directly, either quantitatively (mtDNA‐depletion syndromes) or qualitatively (multiple deletion syndromes) 27, 28.
The recent identification of CHCHD10 mutations in a subset of ALS patients opened a new window on the relationship between ALS and Mitochondrial diseases 4. As previously mentioned, CHCHD10 encodes a coiled‐coil helix coiled‐coil helix protein, located in the intermembrane space of mitochondria, whose function is unknown. Patients in the pedigree reported by Bannwarth et al had reliable clinical, morphological and biochemical evidence of mitochondrial disease.
Clinically, affected individuals showed variable combinations of proximal myopathy, palpebral ptosis, ataxia, dementia and sensorineural deafness, all representing canonical signs of Mitochondrial Diseases. Muscle pathology showed typical mitochondrial alterations, consisting of cytochrome c oxidase (COX)‐negative fibres, ultrastructural mitochondrial abnormalities, impaired respiratory chain activity, and multiple deletions consistent with altered mtDNA maintenance.
The discovery of CHCHD10 mutations in a proportion of familial and sporadic cases of ALS/FTD‐ALS reinforces the hypothesis of a mitochondrial dysfunction in ALS and suggests the possibility of an ALS/mitochondrial diseases spectrum.
However, clinical signs reminiscent of Mitochondrial Diseases were never reported in all CHCHD10 mutated ALS cases reported so far. Furthermore, only few cases with CHCHD10 mutations underwent muscle pathology investigation. Severe histochemical COX deficiency with no evidence of mtDNA deletion or depletion was detected on muscle biopsy in a classic ALS patient harboring the P80L CHCHD10 mutation 23, 87. The same authors detected COX deficiency in additional 6 patients from a group of 50 sALS in which muscle biopsy was performed. Besides CHCHD10 mutation, genetic analysis showed SOD1 mutation in one patient and TARDBP mutation in another case 23. The association of CHCHD10 with mitochondrial disease was reinforced by the detection of a double missense mutation in CHCHD10 in cis (R15S and G58R) in a large Puerto Rican kindred with an autosomal dominant mitochondrial myopathy without sign of motor neuron involvement. Affected members had severe exercise intolerance, progressive proximal weakness and lactic academia presenting in the first decade of life 3. Muscle histochemistry showed ragged‐red fibers and electron microscopy showed globular mitochondrial inclusions 41.
Of note, CHCHD10 is involved in another group of motor neuron diseases. The missense G66V mutation was reported in 55 patients from 17 unrelated Finnish families with late onset spinal motor neuronopathy (LOSMoN/SMAJ) 81. On muscle pathology, chronic myopathic changes were found in SMAJ, with no mitochondrial abnormalities 49. Conversely, no mutations were detected in a cohort of 72 French Spinal Muscular Atrophy patients 73. Furthermore, the same G66V variant was observed to cause variable phenotypes within a Finnish family, ranging from axonal Charcot Marie Tooth neuropathy to spinal muscular atrophy with clinical signs leading to an initial diagnosis of ALS 79.
ALS/Other Neurological Disorders
Extrapyramidal, cerebellar, sensory or autonomic systems dysfunction may be sometimes identified in ALS patients [for review see 103]. Though these findings confirm the view that ALS may be part of a multisystem neurological disease, definite clinical, genetic and pathological data linking ALS with these conditions are still to be addressed.
Conclusions
ALS is a heterogeneous disorder. Genetic and pathological studies in familial forms of the disease are revealing that ALS is mechanistically linked with an increasing number of disorders. The spectrum paradigm represents a conceptual shift of our thinking about ALS, indicating that ALS should be considered as one of the different manifestations associated with defects in several pleiotropic genes.
The relevance of these considerations for the most common sporadic ALS remains to be elucidated. However, it should be considered that many clinical and genetic observations support the view that also fALS and sALS are linked over a spectrum. Family histories indicate that heritability of ALS varies over a continuum ranging from fALS with Mendelian segregation to sALS. In fact, different categories of fALS have been delineated, including definite, probable A, probable B and possible fALS 11, 12, 13, 20. Genetic information strongly supports this view as the frequency of mutations in currently known ALS genes decreases over a continuum from 60%–80% in definite fALS to 11%–28% in sALS (Figure 2) 13, 20. In this spectrum the same genetic variants may contribute to Mendelian forms and to apparently sporadic forms.
Figure 2.

The sporadic/familial spectrum. Definite fALS: patients with at least two first‐ or second‐degree relatives with ALS; Probable A fALS: patients with one affected direct‐line first‐degree‐relative (father/mother–child); Probable B fALS, patients with one affected sibling or second‐degree relative; Possible fALS: ALS patient with a distant relative with ALS (more distant than first‐ or second‐degree) or sporadic ALS with a relative affected by one of the other disorders of ALS spectra; sALS: sporadic ALS. Frequencies of mutation in major ALS genes in each category are described (*See Ref. 11; **See Ref. 20).
The complexity of ALS is increasing but also our understanding of ALS pathogenesis is rapidly expanding. Genetic research is providing important insights into the cellular pathways and mechanisms underlying motor neuron degeneration. The identification of pathways involved in the disease and pathophysiological mechanisms will open new therapeutic perspectives for this spectrum of currently incurable disorders.
Acknowledgments
Work in our laboratory was supported by ICOMM‐onlus, AISLA (Associazione Italiana Sclerosi Laterale Amiotrofica) and FIGC (Federazione Italiana Gioco Calcio).
References
- 1. Abdelkarim S, Morgan S, Plagnol V, Lu CH, Adamson G, Howard R et al (2015) CHCHD10 Pro34Ser is not a highly penetrant pathogenic variant for amyotrophic lateral sclerosis and frontotemporal dementia. Brain 139:e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abramzon Y, Johnson JO, Scholz SW, Taylor JP, Brunetti M, Calvo A et al (2012) Valosin‐containing protein (VCP) mutations in sporadic amyotrophic lateral sclerosis. Neurobiol Aging 33:2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ajroud‐Driss S, Fecto F, Ajroud K, Lalani I, Calvo SE, Mootha VK et al (2015) Mutation in the novel nuclear‐encoded mitochondrial protein CHCHD10 in a family with autosomal dominant mitochondrial myopathy. Neurogenetics 16:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bannwarth S, Ait‐El‐Mkadem S, Chaussenot A, Genin EC, Lacas‐Gervais S, Fragaki K et al (2014) A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 137:2329–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Benatar M, Wuu J, Fernandez C, Weihl CC, Katzen H, Steele J et al (2013) Motor neuron involvement in multisystem proteinopathy: implications for ALS. Neurology 80:1874–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bendotti C, Calvaresi N, Chiveri L, Prelle A, Moggio M, Braga M et al (2001) Early vacuolization and mitochondrial damage in motor neurons of FALS mice are not associated with apoptosis or with changes in cytochrome oxidase histochemical reactivity. J Neurol Sci 191:25–33. [DOI] [PubMed] [Google Scholar]
- 7. Broccolini A, Mirabella M (2015) Hereditary inclusion‐body myopathies. Biochim Biophys Acta 1852:644–650. [DOI] [PubMed] [Google Scholar]
- 8. Broccolini A, Ricci E, Cassandrini D, Gliubizzi C, Bruno C, Tonoli E et al (2004) Novel GNE mutations in Italian families with autosomal recessive hereditary inclusion‐body myopathy. Hum Mutat 23:632. [DOI] [PubMed] [Google Scholar]
- 9. Boutoleau‐Bretonnière C, Camuzat A, Le Ber I, Bouya‐Ahmed K, Guerreiro R, Deruet AL et al (2015) A phenotype of atypical apraxia of speech in a family carrying SQSTM1 mutation. J Alzheimers Dis 43:625–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bucelli RC, Arhzaouy K, Pestronk A, Pittman SK, Rojas L, Sue CM et al (2015) SQSTM1 splice site mutation in distal myopathy with rimmed vacuoles. Neurology 85:665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Byrne S, Bede P, Elamin M, Kenna K, Lynch C, McLaughlin R, Hardiman O (2011) Proposed criteria for familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler 12:157–159. [DOI] [PubMed] [Google Scholar]
- 12. Byrne S, Walsh C, Lynch C, Bede P, Elamin M, Kenna K et al (2011) Rate of familial amyotrophic lateral sclerosis: a systematic review and meta‐analysis. J Neurol Neurosurg Psychiatry 82:623–627. [DOI] [PubMed] [Google Scholar]
- 13. Cady J, Allred P, Bali T, Pestronk A, Goate A, Miller TM et al (2015) Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann Neurol 77:100–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chaussenot A, Le Ber I, Ait‐El‐Mkadem S, Camuzat A, de Septenville A, Bannwarth S et al (2014) Screening of CHCHD10 in a French cohort confirms the involvement of this gene in frontotemporal dementia with amyotrophic lateral sclerosis patients. Neurobiol Aging 35:2884. [DOI] [PubMed] [Google Scholar]
- 15. Chen Y, Zheng ZZ, Chen X, Huang R, Yang Y, Yuan L et al (2014) SQSTM1 mutations in Han Chinese populations with sporadic amyotrophic lateral sclerosis. Neurobiol Aging 35:726. [DOI] [PubMed] [Google Scholar]
- 16. Chiò A, Borghero G, Restagno G, Mora G, Drepper C, Traynor BJ et al (2012) Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain 135:784–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chiò A, Mora G, Sabatelli M, Caponnetto C, Traynor BJ, Johnson JO et al (2015) CHCH10 mutations in an Italian cohort of familial and sporadic amyotrophic lateral sclerosis patients. Neurobiol Aging 36:1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS et al (2015) Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347:1436–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Comi GP, Bordoni A, Salani S, Franceschina L, Sciacco M, Prelle A et al (1998) Cytochrome c oxidase subunit I microdeletion in a patient with motor neuron disease. Ann Neurol 43:110–116. [DOI] [PubMed] [Google Scholar]
- 20. Conte A, Lattante S, Luigetti M, Del Grande A, Romano A, Marcaccio A et al (2012) Classification of familial amyotrophic lateral sclerosis by family history: effects on frequency of genes mutation. J Neurol Neurosurg Psychiatry 83:1201–1203. [DOI] [PubMed] [Google Scholar]
- 21. Cozzolino M, Carrì MT (2012) Mitochondrial dysfunction in ALS. Prog Neurobiol 97:54–66. [DOI] [PubMed] [Google Scholar]
- 22. Cozzolino M, Rossi S, Mirra A, Carrì MT (2015) Mitochondrial dynamism and the pathogenesis of Amyotrophic Lateral Sclerosis. Front Cell Neurosci 9:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Crugnola V, Lamperti C, Lucchini V, Ronchi D, Peverelli L, Prelle A et al (2010) Mitochondrial respiratory chain dysfunction in muscle from patients with amyotrophic lateral sclerosis. Arch Neurol 67:849–854. [DOI] [PubMed] [Google Scholar]
- 24. D'Agostino C, Nogalska A, Cacciottolo M, Engel WK, Askanas V (2011) Abnormalities of NBR1, a novel autophagy‐associated protein, in muscle fibers of sporadic inclusion‐body myositis. Acta Neuropathol 122:627–636. [DOI] [PubMed] [Google Scholar]
- 25. Dal Canto MC, Gurney ME (1994) Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am J Pathol 145:1271–1279. [PMC free article] [PubMed] [Google Scholar]
- 26. DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron 72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Di Mauro S, Schon EA (2003) Mitochondrial respiratory‐chain diseases. N Engl J Med 348:2656–2668. [DOI] [PubMed] [Google Scholar]
- 28. Di Mauro S, Schon EA, Carelli V, Hirano M (2013) The clinical maze of mitochondrial neurology. Nat Rev Neurol 9:429–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dimachkie MM, Barohn RJ (2014) Inclusion body myositis. Neurol Clin 32:629–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dobson‐Stone C, Shaw AD, Hallupp M, Bartley L, McCann H, Brooks WS et al (2015) Is CHCHD10 Pro34Ser pathogenic for frontotemporal dementia and amyotrophic lateral sclerosis? Brain 138:e385. [DOI] [PubMed] [Google Scholar]
- 31. Dols‐Icardo O, Nebot I, Gorostidi A, Ortega‐Cubero S, Hernández I, Rojas‐García R et al (2015) Analysis of the CHCHD10 gene in patients with frontotemporal dementia and amyotrophic lateral sclerosis from Spain. Brain 138:e400. [DOI] [PubMed] [Google Scholar]
- 32. Eisen A, Kuwabara S (2012) The split hand syndrome in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 83:399–403. [DOI] [PubMed] [Google Scholar]
- 33. Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W et al (2011) SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 68:1440–1446. [DOI] [PubMed] [Google Scholar]
- 34. Feit H, Silbergleit A, Schneider LB, Gutierrez JA, Fitoussi RP, Reyes C et al (1998) Vocal cord and pharyngeal weakness with autosomal dominant distal myopathy: clinical description and gene localization to 5q31. Am J Hum Genet 63:1732–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fifita JA, Williams KL, McCann EP, O'Brien A, Bauer DC, Nicholson GA, Blair IP (2015) Mutation analysis of MATR3 in Australian familial amyotrophic lateral sclerosis. Neurobiol Aging 36:1602. [DOI] [PubMed] [Google Scholar]
- 36. Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Müller K et al (2015) Haploinsufficiency of TBK1 causes familial ALS and fronto‐temporal dementia. Nat Neurosci 18:631–636. [DOI] [PubMed] [Google Scholar]
- 37. Gang Q, Bettencourt C, Houlden H, Hanna MG, Machado PM (2015) Genetic advances in sporadic inclusion body myositis. Curr Opin Rheumatol 27:586–594. [DOI] [PubMed] [Google Scholar]
- 38. González‐Pérez P, Cirulli ET, Drory VE, Dabby R, Nisipeanu P, Carasso RL et al (2012) Novel mutation in VCP gene causes atypical amyotrophic lateral sclerosis. Neurology 79:2201–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gordon PH, Cheng B, Katz IB, Mitsumoto H, Rowland LP (2009) Clinical features that distinguish PLS, upper motor neuron‐dominant ALS, and typical ALS. Neurology 72:1948–1952. [DOI] [PubMed] [Google Scholar]
- 40. Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G et al (2012) A C9orf72 promoter repeat expansion in a Flanders‐Belgian cohort with disorders of the frontotemporal lobar degeneration‐amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol 11:54–65. [DOI] [PubMed] [Google Scholar]
- 41. Heiman‐Patterson TD, Argov Z, Chavin JM, Kalman B, Alder H, Di Mauro S et al (1997) Biochemical and genetic studies in a family with mitochondrial myopathy. Muscle Nerve 20:1219–1224. [DOI] [PubMed] [Google Scholar]
- 42. Hirano M, Angelini C, Montagna P, Hays AP, Tanji K, Mitsumoto H et al (2008) Amyotrophic lateral sclerosis with ragged‐red fibers. Arch Neurol 65:403–406. [DOI] [PubMed] [Google Scholar]
- 43. Hirano M, Nakamura Y, Saigoh K, Sakamoto H, Ueno S, Isono C et al (2013) Mutations in the gene encoding p62 in Japanese patients with amyotrophic lateral sclerosis. Neurology 80:458–463. [DOI] [PubMed] [Google Scholar]
- 44. Hirano M, Nakamura Y, Saigoh K, Sakamoto H, Ueno S, Isono C et al (2015) VCP gene analyses in Japanese patients with sporadic amyotrophic lateral sclerosis identify a new mutation. Neurobiol Aging 36:1604. [DOI] [PubMed] [Google Scholar]
- 45. Hocking LJ, Lucas GJ, Daroszewska A, Mangion J, Olavesen M, Cundy T et al (2002) Domain‐specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget's disease. Hum Mol Genet 11:2735–2739. [DOI] [PubMed] [Google Scholar]
- 46. Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ et al (2010) Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68:857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Johnson JO, Glynn SM, Gibbs JR, Nalls MA, Sabatelli M, Restagno G et al (2014) Mutations in the CHCHD10 gene are a common cause of familial amyotrophic lateral sclerosis. Brain 137:e311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE et al (2014) Mutations in the matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci 17:664–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jokela M, Penttilä S, Huovinen S, Hackman P, Saukkonen AM, Toivanen J, Udd B (2011) Late‐onset lower motor neuronopathy: a new autosomal dominant disorder. Neurology 77:334–340. [DOI] [PubMed] [Google Scholar]
- 50. Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z et al (2013) Mutations in prion‐like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495:467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kimonis VE, Fulchiero E, Vesa J, Watts G (2008) VCP disease associated withmyopathy, Paget disease of bone and frontotemporal dementia: review of a unique disorder. Biochim Biophys Acta 1782:744–748. [DOI] [PubMed] [Google Scholar]
- 52. Kimonis VE, Mehta SG, Fulchiero EC, Thomasova D, Pasquali M, Boycott K et al (2008) Clinical studies in familial VCP myopathy associated with Paget disease of bone and frontotemporal dementia. Am J Med Genet A 146A:745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Koppers M, van Blitterswijk MM, Vlam L, Rowicka PA, van Vught PW, Groen EJ et al (2012) VCP mutations in familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging 33:837. [DOI] [PubMed] [Google Scholar]
- 54. Kovacs GG, van der Zee J, Hort J, Kristoferitsch W, Leitha T, Höftberger R et al (2016) Clinicopathological description of two cases with SQSTM1 gene mutation associated with frontotemporal dementia. Neuropathology 36:27–38. [DOI] [PubMed] [Google Scholar]
- 55. Kumar KR, Needham M, Mina K, Davis M, Brewer J, Staples C, et al (2010) Two Australian families with inclusion‐body myopathy, Paget's disease of bone and frontotemporal dementia: novel clinical and genetic findings. Neuromuscul Disord 20:330–334. [DOI] [PubMed] [Google Scholar]
- 56. Kurzwelly D, Krüger S, Biskup S, Heneka MT (2015) A distinct clinical phenotype in a German kindred with motor neuron disease carrying a CHCHD10 mutation. Brain 138:e376. [DOI] [PubMed] [Google Scholar]
- 57. Kusters B, van Hoeve BJ, Schelhaas HJ, Ter Laak H, van Engelen BG, Lammens M (2009) TDP‐43 accumulation is common in myopathies with rimmed vacuoles. Acta Neuropathol 117:209–211. [DOI] [PubMed] [Google Scholar]
- 58. Kwok CT, Morris A, de Belleroche JS (2014) Sequestosome‐1 (SQSTM1) sequence variants in ALS cases in the UK: prevalence and coexistence of SQSTM1 mutations in ALS kindred with PDB. Eur J Hum Genet 22:492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kwok CT, Wang HY, Morris AG, Smith B, Shaw C, de Belleroche J (2015) VCP mutations are not a major cause of familial amyotrophic lateral sclerosis in the UK. J Neurol Sci 349:209–213. [DOI] [PubMed] [Google Scholar]
- 60. Lattante S, Ciura S, Rouleau GA, Kabashi E (2015) Defining the genetic connection linking amyotrophic lateral sclerosis (ALS) with frontotemporal dementia (FTD). Trends Genet 31:263–273. [DOI] [PubMed] [Google Scholar]
- 61. Laurin N, Brown JP, Morissette J, Raymond V (2002) Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet 70:1582–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Le Ber I, Camuzat A, Guerreiro R, Bouya‐Ahmed K, Bras J, Nicolas G et al (2013) SQSTM1 mutations in French patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol 70:1403–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Le Ber I, Van Bortel I, Nicolas G, Bouya‐Ahmed K, Camuzat A, Wallon D et al (2014) hnRNPA2B1 and hnRNPA1 mutations are rare in patients with "multisystem proteinopathy" and frontotemporal lobar degeneration phenotypes. Neurobiol Aging 35:934. [DOI] [PubMed] [Google Scholar]
- 64. Leblond CS, Gan‐Or Z, Spiegelman D, Laurent SB, Szuto A, Hodgkinson A et al (2016) Replication study of MATR3 in familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging 37:e17–e21. [DOI] [PubMed] [Google Scholar]
- 65. Lin KP, Tsai PC, Liao YC, Chen WT, Tsai CP, Soong BW, Lee YC (2015) Mutational analysis of MATR3 in Taiwanese patients with amyotrophic lateral sclerosis. Neurobiol Aging 36:2005.e1–4. [DOI] [PubMed] [Google Scholar]
- 66. Malicdan MC, Noguchi S, Nishino I (2007) Autophagy in a mouse model of distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Autophagy 3:396–398. [DOI] [PubMed] [Google Scholar]
- 67. Marroquin N, Stranz S, Müller K, Wieland T, Ruf WP, Brockmann SJ et al (2015) Screening for CHCHD10 mutations in a large cohort of sporadic ALS patients: no evidence for pathogenicity of the p.P34S variant. Brain 139:e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Millecamps S, Boillée S, Le Ber I, Seilhean D, Teyssou E, Giraudeau M et al (2012) Phenotype difference between ALS patients with expanded repeats in C9ORF72 and patients with mutations in other ALS‐related genes. J Med Genet 49:258–263. [DOI] [PubMed] [Google Scholar]
- 69. Millecamps S, De Septenville A, Teyssou E, Daniau M, Camuzat A, Albert M et al (2014) Genetic analysis of matrin 3 gene in French amyotrophic lateral sclerosis patients and frontotemporal lobar degeneration with amyotrophic lateral sclerosis patients. Neurobiol Aging 35:2882. [DOI] [PubMed] [Google Scholar]
- 70. Miller JW, Smith BN, Topp SD, Al‐Chalabi A, Shaw CE, Vance C (2012) Mutation analysis of VCP in British familial and sporadic amyotrophic lateral sclerosis patients. Neurobiol Aging 33:2721. [DOI] [PubMed] [Google Scholar]
- 71. Müller K, Andersen PM, Hübers A, Marroquin N, Volk AE, Danzer KM et al (2014) Two novel mutations in conserved codons indicate that CHCHD10 is a gene associated with motor neuron disease. Brain 137:e309. [DOI] [PubMed] [Google Scholar]
- 72. Müller TJ, Kraya T, Stoltenburg‐Didinger G, Hanisch F, Kornhuber M, Stoevesandt D et al (2014) Phenotype of matrin‐3‐related distal myopathy in 16 German patients. Ann Neurol 76:669–680. [DOI] [PubMed] [Google Scholar]
- 73. Morel G, Rouzier C, Chaussenot A, Ait‐El‐Mkadem S, Bannwarth S, Genin EC et al (2015) CHCHD10 mutations are not a common cause of SMN1‐negative type III‐IV spinal motor atrophy. Ann Neurol 78:831. [DOI] [PubMed] [Google Scholar]
- 74. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT et al (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. [DOI] [PubMed] [Google Scholar]
- 75. Olive M, Janue A, Moreno D, Gamez J, Torrejon‐Escribano B, Ferrer I (2009) TAR DNA‐Binding protein 43 accumulation in protein aggregate myopathies. J Neuropathol Exp Neurol 68:262–273. [DOI] [PubMed] [Google Scholar]
- 76. Origone P, Verdiani S, Bandettini Di Poggio M, Zuccarino R, Vignolo M, Caponnetto C, Mandich P (2015) A novel Arg147Trp MATR3 missense mutation in a slowly progressive ALS Italian patient. Amyotroph Lateral Scler Frontotemporal Degener 16:530–531. [DOI] [PubMed] [Google Scholar]
- 77. Palmio J, Sandell S, Suominen T, Penttilä S, Raheem O, Hackman P et al (2011) Distinct distal myopathy phenotype caused by VCP gene mutation in a Finnish family. Neuromuscul Disord 21:551–555. [DOI] [PubMed] [Google Scholar]
- 78. Palmio J, Evilä A, Bashir A, Norwood F, Viitaniemi K, Vihola A et al (2015) Re‐evaluation of the phenotype caused by the common MATR3 p.Ser85Cys mutation in a new family. J Neurol Neurosurg Psychiatry. doi: 10.1136/jnnp-2014-309349. [DOI] [PubMed] [Google Scholar]
- 79. Pasanen P, Myllykangas L, Pöyhönen M, Kiuru‐Enari S, Tienari PJ, Laaksovirta H et al (2015) Intrafamilial clinical variability in individuals carrying the CHCHD10 mutation Gly66Val. Acta Neurol Scand. doi: 10.1111/ane.12470. [DOI] [PubMed] [Google Scholar]
- 80. Pensato V, Tiloca C, Corrado L, Bertolin C, Sardone V, Del Bo R et al (2015) TUBA4A gene analysis in sporadic amyotrophic lateral sclerosis: identification of novel mutations. J Neurol 262:1376–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Penttilä S, Jokela M, Bouquin H, Saukkonen AM, Toivanen J, Udd B (2015) Late onset spinal motor neuronopathy is caused by mutation in CHCHD10. Ann Neurol 77:163–172. [DOI] [PubMed] [Google Scholar]
- 82. Pottier C, Bieniek KF, Finch N, van de Vorst M, Baker M, Perkersen R et al (2015) Whole‐genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol 130:77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Phukan J, Elamin M, Bede P, Jordan N, Gallagher L, Byrne S et al (2012) The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population‐based study. J Neurol Neurosurg Psychiatry 83:102–108. [DOI] [PubMed] [Google Scholar]
- 84. Ratti A, Corrado L, Castellotti B, Del Bo R, Fogh I, Cereda C et al (2012) C9ORF72 repeat expansion in a large Italian ALS cohort: evidence of a founder effect. Neurobiol Aging 33:2528. [DOI] [PubMed] [Google Scholar]
- 85. Rea SL, Majcher V, Searle MS, Layfield R (2014) SQSTM1 mutations‐‐bridging Paget disease of bone and ALS/FTLD. Exp Cell Res 325:27–37. [DOI] [PubMed] [Google Scholar]
- 86. Renton AE, Majounie E, Waite A, Simón‐Sánchez J, Rollinson S, Gibbs JR et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron 72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ronchi D, Garone C, Bordoni A, Gutierrez Rios P, Calvo SE, Ripolone M et al (2012) Next‐generation sequencing reveals DGUOK mutations in adult patients with mitochondrial DNA multiple deletions. Brain 135:3404–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ronchi D, Riboldi G, Del Bo R, Ticozzi N, Scarlato M, Galimberti D et al (2015) CHCHD10 mutations in Italian patients with sporadic amyotrophic lateral sclerosis. Brain 138:e372. [DOI] [PubMed] [Google Scholar]
- 89. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62. [DOI] [PubMed] [Google Scholar]
- 90. Rubino E, Rainero I, Chiò A, Rogaeva E, Galimberti D, Fenoglio P et al (2012) SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology 79:1556–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sabatelli M, Madia F, Conte A, Luigetti M, Zollino M, Mancuso I et al (2008) Natural history of young‐adult amyotrophic lateral sclerosis. Neurology 71:876–881. [DOI] [PubMed] [Google Scholar]
- 92. Sabatelli M, Zollino M, Luigetti M, Grande AD, Lattante S, Marangi G et al (2011) Uncovering amyotrophic lateral sclerosis phenotypes: clinical features and long‐term follow‐up of upper motor neuron‐dominant ALS. Amyotroph Lateral Scler 12:278–282. [DOI] [PubMed] [Google Scholar]
- 93. Sabatelli M, Conte A, Zollino M (2013) Clinical and genetic heterogeneity of amyotrophic lateral sclerosis. Clin Genet 83:408–416. [DOI] [PubMed] [Google Scholar]
- 94. Salajegheh M, Pinkus JL, Taylor JP, Amato AA, Nazareno R, Baloh RH, Greenberg SA (2009) Sarcoplasmic redistribution of nuclear TDP‐43 in inclusion body myositis. Muscle Nerve 40:19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Seelen M, Visser AE, Overste DJ, Kim HJ, Palud A, Wong TH et al (2014) No mutations in hnRNPA1 and hnRNPA2B1 in Dutch patients with amyotrophic lateral sclerosis, frontotemporal dementia, and inclusion body myopathy. Neurobiol Aging 35:1956. [DOI] [PubMed] [Google Scholar]
- 96. Senderek J, Garvey SM, Krieger M, Guergueltcheva V, Urtizberea A, Roos A et al (2009) Autosomal‐dominant distal myopathy associated with a recurrent missense mutation in the gene encoding the nuclear matrix protein, matrin 3. Am J Hum Genet 84:511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Shimizu H, Toyoshima Y, Shiga A, Yokoseki A, Arakawa K, Sekine Y et al (2013) Sporadic ALS with compound heterozygous mutations in the SQSTM1 gene. Acta Neuropathol 126:453–459. [DOI] [PubMed] [Google Scholar]
- 98. Simón‐Sánchez J, Dopper EG, Cohn‐Hokke PE, Hukema RK, Nicolaou N, Seelaar H et al (2012) The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain 135:723–735. [DOI] [PubMed] [Google Scholar]
- 99. Smith BN, Newhouse S, Shatunov A, Vance C, Topp S, Johnson L et al (2013) The C9ORF72 expansion mutation is a common cause of ALS+/‐FTD in Europe and has a single founder. Eur J Hum Genet 21:102–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Smith BN, Ticozzi N, Fallini C, Gkazi AS, Topp S, Kenna KP et al (2014) Exome‐wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 84:324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Stewart H, Rutherford NJ, Briemberg H, Krieger C, Cashman N, Fabros M et al (2012) Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol 123:409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Stojkovic T, Hammouda el H, Richard P, López de Munain A, Ruiz‐Martinez J, Camaño P et al (2009) Clinical outcome in 19 French and Spanish patients with valosin‐containing protein myopathy associated with Paget's disease of bone and frontotemporal dementia. Neuromuscul Disord 19:316–323. [DOI] [PubMed] [Google Scholar]
- 103. Swinnen B, Robberecht W (2014) The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 10:661–670. [DOI] [PubMed] [Google Scholar]
- 104. Tan W, Pasinelli P, Trotti D (2014) Role of mitochondria in mutant SOD1 linked amyotrophic lateral sclerosis. Biochim Biophys Acta 1842:1295–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Tasca G, Monforte M, De Fino C, Kley RA, Ricci E, Mirabella M (2015) Magnetic resonance imaging pattern recognition in sporadic inclusion‐body myositis. Muscle Nerve Mar 52:956–962. [DOI] [PubMed] [Google Scholar]
- 106. Taylor JP (2015) Multisystem proteinopathy: intersecting genetics in muscle, bone, and brain degeneration. Neurology 85:658–660. [DOI] [PubMed] [Google Scholar]
- 107. Tiloca C, Ratti A, Pensato V, Castucci A, Sorarù G, Del Bo R et al (2012) Mutational analysis of VCP gene in familial amyotrophic lateral sclerosis. Neurobiol Aging 33:630. [DOI] [PubMed] [Google Scholar]
- 108. Teyssou E, Takeda T, Lebon V, Boillée S, Doukouré B, Bataillon G et al (2013) Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol 125:511–522. [DOI] [PubMed] [Google Scholar]
- 109. Tucker WS Jr, Hubbard WH, Stryker TD, Morgan SW, Evans OB, Freemon FR, Theil GB (1982) A new familial disorder of combined lower motor neuron degeneration and skeletal disorganization. Trans Assoc Am Physicians 95:126–134. [PubMed] [Google Scholar]
- 110. Udd B (2012) Distal myopathies‐new genetic entities expand diagnostic challenge. Neuromuscul Disord 22:5–12. [DOI] [PubMed] [Google Scholar]
- 111. van der Zee J, Pirici D, Van Langenhove T, Engelborghs S, Vandenberghe R, Hoffmann M et al (2009) Clinical heterogeneity in 3 unrelated families linked to VCP p.Arg159His. Neurology 73:626–632. [DOI] [PubMed] [Google Scholar]
- 112. van der Zee J, Van Langenhove T, Kovacs GG, Dillen L, Deschamps W, Engelborghs S et al (2014) Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol 128:397–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. van Rheenen W, Diekstra FP, van den Berg LH, Veldink JH (2014) Are CHCHD10 mutations indeed associated with familial amyotrophic lateral sclerosis? Brain 137:e313. [DOI] [PubMed] [Google Scholar]
- 114. Viassolo V, Previtali SC, Schiatti E, Magnani G, Minetti C, Zara F et al (2008) Inclusion body myopathy, Paget's disease of the bone and frontotemporal dementia: recurrence of the VCP R155H mutation in an Italian family and implications for genetic counselling. Clin Genet 74:54–60. [DOI] [PubMed] [Google Scholar]
- 115. Watts GDJ, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D et al (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin‐containing protein. Nature Genet 36:377–381. [DOI] [PubMed] [Google Scholar]
- 116. Weihl CC, Dalal S, Pestronk A, Hanson PI (2006) Inclusion body myopathy‐associated mutations in p97/VCP impair endoplasmic reticulum‐associated degradation. Hum Mol Genet 15:189–199. [DOI] [PubMed] [Google Scholar]
- 117. Weihl CC, Temiz P, Miller SE, Watts G, Smith C, Forman M et al (2008) TDP‐43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosurg Psychiatry 79:1186–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Weihl CC, Pestronk A, Kimonis VE (2009) Valosin‐containing protein disease: inclusion body myopathy with Paget's disease of the bone and fronto‐temporal dementia. Neuromuscul Disord 19:308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Weihl CC, Baloh RH, Lee Y, Chou TF, Pittman SK, Lopate G et al (2015) Targeted sequencing and identification of genetic variants in sporadic inclusion body myositis. Neuromuscul Disord 25:289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Williams KL, Solski JA, Nicholson GA, Blair IP (2012) Mutation analysis of VCP in familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging 33:1488. [DOI] [PubMed] [Google Scholar]
- 121. Williams KL, McCann EP, Fifita JA, Zhang K, Duncan EL, Leo PJ et al (2015) Novel TBK1 truncating mutation in a familial amyotrophic lateral sclerosis patient of Chinese origin. Neurobiol Aging 36:3334. [DOI] [PubMed] [Google Scholar]
- 122. Yamashita S, Mori A, Nishida Y, Kurisaki R, Tawara N, Nishikami T et al (2015) Clinicopathological features of the first Asian family having vocal cord and pharyngeal weakness with distal myopathy due to a MATR3 mutation. Neuropathol Appl Neurobiol 41:391–398. [DOI] [PubMed] [Google Scholar]
- 123. Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA et al (1995) An adverse property of a familial ALS‐linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14:1105–1116. [DOI] [PubMed] [Google Scholar]
- 124. Wong CH, Topp S, Gkazi AS, Troakes C, Miller JW, de Majo M et al (2015) The CHCHD10 P34S variant is not associated with ALS in a UK cohort of familial and sporadic patients. Neurobiol Aging 36:2908. [DOI] [PubMed] [Google Scholar]
- 125. Zhang M, Xi Z, Zinman L, Bruni AC, Maletta RG, Curcio SA et al (2015) Mutation analysis of CHCHD10 in different neurodegenerative diseases. Brain 138:e380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Zou ZY, Liu MS, Li XG, Cui LY (2013) Screening of VCP mutations in Chinese amyotrophic lateral sclerosis patients. Neurobiol Aging 34:1519. [DOI] [PubMed] [Google Scholar]