

Abstract
Patients with mild cognitive impairment (MCI) or Alzheimer's disease (AD) might develop olfactory dysfunction that correlates with progression of disease. Alteration of olfactory neuroepithelium associated with MCI may be useful as predictor of cognitive decline. Biomarkers with higher sensitivity and specificity would allow to understand the biological progression of the pathology in association with the clinical course of the disease. In this study, magnetic resonance images, apolipoprotein E (ApoE) load, Olfactory Connecticut test and Montreal Cognitive Assessment (MoCA) indices were obtained from noncognitive impaired (NCI), MCI and AD patients. We established a culture of patient‐derived olfactory stromal cells from biopsies of olfactory mucosa (OM) to test whether biological properties of mesenchymal stromal cells (MSC) are concurrent with MCI and AD psychophysical pathology. We determined the expression of amyloid Aβ peptides in the neuroepithelium of tissue sections from MCI and AD, as well as in cultured cells of OM. Reduced migration and proliferation of stromal (CD90+) cells in MCI and AD with respect to NCI patients was determined. A higher proportion of anosmic MCI and AD cases were concurrent with the ApoE ε4 allele. In summary, dysmetabolism of amyloid was concurrent with migration and proliferation impairment of patient‐derived stem cells.
Keywords: amyloid Aβ, biomarkers, mesenchymal stromal cells, mild cognitive impairment, olfactory mucosa
Introduction
The majority of diagnostic tools for brain neurodegenerative disorders are based on expensive and time‐consuming methodologies. Early detection of proteins as amyloid‐Aβ or tau biomarkers of AD has become essential in order to evaluate whether preclinical diagnosed subjects progress to an established disease, or treatment with disease‐modifying therapeutic agents may affect progression of the pathology 13.
Considering behavioral and neuropathological complexities of AD, an appropriate approach for early diagnosis would incorporate risk factors and common co‐occurring markers associated to the pathology. Deficit in olfactory sensitivity, odor discrimination and identification of odorants appear to be the earliest detectable functional alterations in a high percentage of patients with AD and other neurodegenerative disorders 16, 17, 18, 20. The olfactory threshold is strongly related to sensory capability, whereas identification and discrimination of olfaction stimuli are closely associated with higher cognitive functions. Establishing olfactory sensitivity, identification and discrimination has resulted to be useful as predictors of mild cognitive impairment (MCI) to cognitive deficit in AD 1, 27, 28, 30, 34.
Early diagnosis for neurodegenerative diseases such as AD has been hindered by difficulties in access to the source of tissue to be sampled as the brain. Based on evidence that metabolism of amyloid precursor protein 36 and oxidative stress 5 in the olfactory epithelium from postmortem autopsied AD brain was different compared with control, we examined OM patient‐derived adult stromal cells from biopsies of NCI, MCI and AD subjects. The OM comprises the superficial epithelium and the underlying lamina propria separated by a basement membrane 3, 12, 26. Physiologically, the olfactory epithelium maintains resident multipotent adult stem cell known in rodents as horizontal and globose basal cells, which sustains a continuous renewal of the complete neuroepithelium throughout life 14, 15, 21, 22. The mesenchymal stromal population from the OM has been extensively characterized in rodents 14, 15 and humans 3, 4; it supports subpopulations of cells recognized by membrane common markers with bone marrow stromal cells such as CD29, CD73, CD90 and NESTIN 3, 4. In addition, it is one of the few places in the nervous system accessible under local anesthetic and noninvasive endoscopic surgical procedure 4, 12, 22. Here, we use immuno techniques to determine in biopsies of OM from NCI, MCI and AD patients the expression of amyloid‐Aβ as indicative of dysmetabolic amyloid precursor protein. In parallel, we also have established a culture system of explants to generate epithelial and mesenchymal stromal cells (MSCs), in order to ask whether anosmia, cognitive deficit and amyloid concomitantly converged and conditioned cell migration and proliferation of olfactory population of cells.
We showed that amyloid‐Aβ accumulates in cell compartments of the OM and MSCs. Our culture system is capable of demonstrating a reduced capability of migration and proliferation of stromal cells as well as a significant increment of astroglial subpopulation of cells in culture of OM cells from MCI and AD patients with respect to NCI subjects.
Materials and Methods
Patients who came for consultation for olfactory disability or cognitive deficit were examined independently by neurologist and otorhinolaryngologist; further evaluation of patients for the purpose of this study was performed as indicated by the flow chart (Figure 1). Patients with cerebral and nasosinusal tumors, rhinosinusitis, rhinitis, infection of the upper respiratory tract, schizophrenia, major depression or multifactorial dementia were excluded in the study. Magnetic resonance imaging (MRI) analysis FLAIR sequence was performed in Sigma 0.5 T (GE) with T1 and T2 axial and T1 sagittal protocol, with emphasis in nasal and paranasal structures, cerebral cortices and hippocampal formation. All patients had determination of ApoE load, olfactory and cognitive testing. Patients performed the Connecticut Olfactory Test 32 to establish the threshold and identification of a standardized battery of scents. Cognitive status was determined by MoCA test 23. All demographics, clinical information and behavioral index values obtained are summarized in Table 1. The ApoE load was determined briefly as follows: cubital blood samples were collected by phlebotomist into blood collection tubes. The DNA was isolated from leukocytes, and ApoE genotype was determined using polymerase chain reaction amplification and restriction enzyme digestion as described previously 6. All patients were diagnosed as follows: five NCI, three MCI and two clinically diagnosed as having AD. All subjects or their legal representatives gave their informed consent to participate in the study. The protocol was approved by the institutional review board at Instituto Venezolano de Investigaciones Cientificas (IVIC) at Caracas, Venezuela.
Figure 1.
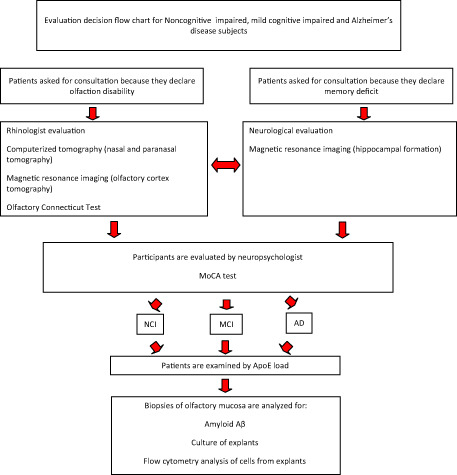
Evaluation decision flow chart for NCI, MCI and AD subjects. ApoE.
Table 1.
Demographic summary ApoE load, olfactory testing, cognitive evaluation, amyloid Aβ and MoCA index in NCI, MCI and AD patients
| Age | Gender | ApoE load | Olfactory Connecticut test * (threshold/identification/index score) | Amyloid Aβ expression | MoCA index | MRI | Cognitive evaluation |
|---|---|---|---|---|---|---|---|
| 67 | M | 3/3 | Bilateral anosmic (0/0/0) | − | 28 | Vasogenic leukoencephalopathy | NCI |
| 46 | M | 3/3 | Bilateral anosmic (0/0/0) | − | 27 | Demyelination in the frontal gyrus | NCI |
| 76 | M | 3/3 | Bilateral anosmic (0/0/0) | − | 26 | Vasogenic leukoencephalopathy | NCI |
| 56 | F | 3/4 | Hyperosmic normal (7/7/7)) | − | 28 | Lacunar infarcts | NCI |
| 62 | F | 3/4 | Bilateral anosmic (0/0/0) | − | 27 | Olfactory bulb | NCI |
| 57 | M | 3/3 | Slight hyposmic right (5.5/6/5.5) | + | 25 | Vasogenic leukoencephalopathy | MCI |
| 41 | M | 3/4 | Normosmic (7/7/7) | + | 25 | normal | MCI |
| 63 | M | 4/4 | Bilateral anosmic (0/0/0) | + | 24 | Vasogenic leukoencephalopathy | MCI |
| 77 | M | 3/4 | Bilateral anosmic (0/0/0) | +++ | 23 | Marked changes involutive hippocampal shrinkage | ADII |
| 74 | F | 4/4 | Bilateral anosmic (0/0/0) | ++ | 19 | Bilateral hippocampal atrophy | ADII |
AD = Alzheimer's disease; ApoE = apolipoprotein E; * = arbitrary intensity of fluorescence; MCI = mild cognitive impairment; MoCA = Montreal Cognitive Assessment; MRI = magnetic resonance imaging; NCI = noncognitive impaired
Human olfactory tissue
Isolation and culture of OM‐MSC
Briefly, biopsies from OM tissues were obtained from 10 subjects under nasal endoscopy 4. Samples once collected were immediately placed in Dulbecco's modified Eagle medium (DMEM‐F12 GIBCO, USA), with penicillin and streptomycin antibiotics added to the transport culture medium. Olfactory stromal cells started to migrate from the explant and became confluent 10–15 days in vitro. Then, cells were collected and reserved for further analysis. We determined a growing index for at least three explants per subject. The distance relative to the explant was measured with a calibrated scale in the ocular of the Axiovert D1 inverted microscope (Zeiss, Gottingen, Germany) every two other days at geographic coordinates. An arbitrary index of length was calculated by subtracting the values between days for each determination as a consequence of the cell migration per group of explants and patients as shown in Figure 6.
Figure 6.
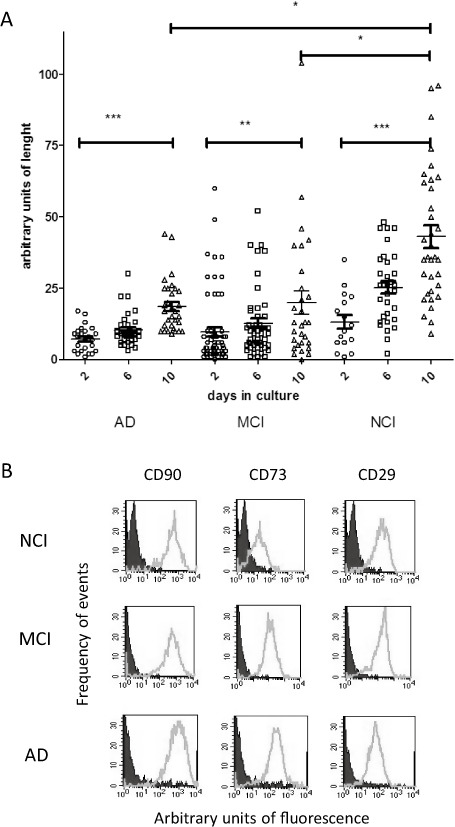
Migration of olfactory cells from tissue explants is reduced by A lzheimer's pathology. (A) Relative distance covered by cells from the explant was determined in at least three samples per patient. Migration was estimated from the net growth measured between days as indicated in the graph. Analysis of variance comparison was performed for each group of subjects [Alzheimer's disease (AD, two), mild cognitive impairment (MCI, three), noncognitive impaired (NCI, five)] and between groups from multiple measurements performed during days of observation as in the graph. *P < 0.05; **P < 0.01; ***P < 0.0001. (B) Flow cytometry analysis was performed for each group of NCI, MCI and AD subjects. The population of cells expressed mainly mesenchymal markers CD90, CD73 and CD29 as shown in (B).
Antibodies
Monoclonal anti‐amyloid Aβ (clone 6F3D; 1/400) and rabbit polyclonal anti‐glial fibrillary acidic protein (GFAP; 1/250) were from DAKO (Glopstrup, Denmark). Monoclonal anti‐β‐III tubulin (clone SDL.3D10; 1/250) from Sigma (St. Louis, MO, USA) and monoclonal anti‐cytokeratin 18 (CitK18; clone DA‐7; 1/100) from BioLegend (San Diego, CA, USA). Secondary antibodies Alexa fluor 488 (Invitrogen, Life Technologies, USA) or Dylight 594 (Vector Labs, USA) were used at 1:200 in phosphate‐buffered saline (PBS) solution. All other chemicals were of reagent grade and obtained commercially. Fluorescein isothiocyanate (FITC) or phycoerythrin (PE)‐conjugated monoclonal antibodies anti‐human: CD90, CD73, CD166, CD54, CD34 and CD45 were purchased from Becton Dickinson (San Diego, CA, USA); NESTIN, GFAP, DMEM‐F12 culture medium were from Gibco (NY, USA), and fetal bovine serum was from Sigma.
Immunohistochemistry of sections from OM
Paraffin embedded tissue sections (5 μm) were deparaffinated in xylene and alcohol series. Sections once free of paraffin were incubated in sodium phosphate buffer solution (0.1 M, pH 7.4) at RT for 10 minutes. Tissue slides were then treated with proteinase K (Invitrogen; NY, USA 20 μL/mL PBS solution for 15 minutes at RT) followed by PBS wash and subsequent incubation with formic acid 70% for 15 minutes at RT and rinsed with dH2O. Then, were permeabilized with PBS solution containing 0.1% Triton X‐100 at RT for 30 minutes. Nonspecific sites were blocked with 5% normal horse serum/PBS for 30 minutes. Slides were incubated with the primary antibody at 4°C overnight. Fluorescent secondary antibodies (Vector Labs, Burlingame, Cal, USA) at a dilution of 1:200 were applied for 2 h at RT. The immunoreactivity was detected under fluorescence using DAPI (Sigma, St Louis, Mo, USA) as counterstaining. Slides from NCI‐, MCI‐ and AD‐diagnosed cases were stained side by side in a single‐ or double‐label paradigm using the same batch of antibodies including slides lacking the primary antibody as negative control. We examined immunohistochemical labeling in OM immunoreactivity from one to three sections per brain sample. All histological and immunohistochemical images were acquired from a Zeiss Axiovert D1 microscope equipped with a Tucsen 5.0 ICE (Fujian, China) color video camera.
Flow cytometry analysis of OM cells
After two to five passages, cells were harvested and analyzed for the expression of mesenchymal stromal (CD90, CD73, CD29), olfactory precursor (horizontal basal marker) CD54 and neural progenitor cell (NESTIN, GFAP) markers. NESTIN and GFAP intracellular labeling was performed using cytofix/cytoperm Becton and Dickinson permeabilization kit protocol. Simultaneous negative control staining reactions were performed by incubating the cells with the FITC‐ and PE‐labeled immunoglobulin G isotype or secondary antibody when the primary was a nonconjugated antibody. Data collection and analysis of the fluorescent intensities were made using a FACSCalibur (Becton Dickinson, San Jose, CA, USA). Ten thousand events were acquired and analyzed using the Cell Quest software program.
Statistical analysis
All statistical calculations and/or graphic analyses were performed using GraphPad Prism version. Statistical analysis was performed using the Kruskal–Wallis nonparametric test to compare the equality of the medians and the nonparametric test for trend. Statistical analysis on EdU data was carried out using an independent two‐sample t‐test. All experiments were repeated at least three times and have documented reproducibility.
Results
MRI‐confirmed atrophy of hippocampal formation of anosmic AD subjects
Significant atrophy of the hippocampal formation was determined in the group of subjects with AD (Figure 2A), and hyperintensities were detected in the gray matter of parieto‐temporal cortices of the brain (not shown). No changes were detected in MRI neither in the hippocampi of MCI (Figure 2B) and NCI subjects (Figure 2C) nor in the olfactory cortex of MCI and AD with respect to NCI subjects.
Figure 2.
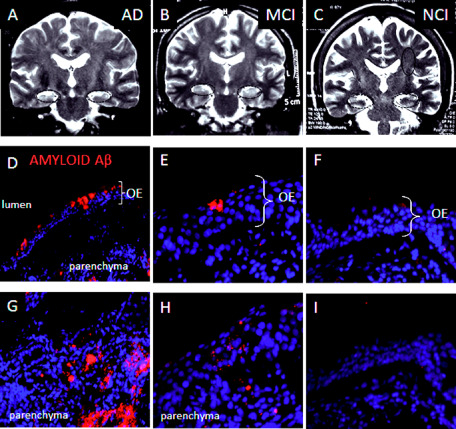
Magnetic resonance imaging ( MRI ) coronal sections and amyloid Aβ expression in olfactory mucosa from noncognitive impaired ( NCI ), mild cognitive impaired ( MCI ) and A lzheimer's disease ( AD ) patients. Bilateral atrophy of the hippocampi was detected in MRI coronal sections from AD (A, black dashed ellipses) but not in MCI (B) or NCI (C) subjects. Amyloid‐Aβ (40/42) expression immunolocalized with the 6F3D monoclonal antibody was detected in the olfactory epithelium. Accumulation of amyloid was observed in lumen‐associated cells in (D and E), and in the parenchyma (G and H) in tissue sections from AD (D, G) and in MCI (E, H), but not in NCI (F, I) subjects. Counterstaining with DAPI was performed to localize topographically amyloid peptide distribution. Immunohistochemistry is representative of two AD, three MCI and five NCI patients.
Expression of amyloid Aβ was immunolocalized in neuroepithelium and parenchyma of OM tissue sections
We examined whether amyloid Aβ expression occurred in biopsies of OM from the NCI, MCI and AD subjects. Amyloid Aβ peptide was expressed by lumen‐associated cells of AD (Figure 2D) and MCI (Figure 2E) patients. Furthermore, amyloid peptide was also found as fibrillar peptide and small deposits in the parenchyma of AD (Figure 2G) and MCI (Figure 2H) subjects, respectively. Accumulation of amyloid Aβ peptide was more frequent in the OM of AD‐diagnosed subjects compared with MCI patients (Table 1). The qualitative evaluation of amyloid Aβ deposits in biopsies from MCI and AD subjects was established from at least three independent immuno procedures staining side‐by‐side negative controls and NCI, MCI and AD sections from biopsy samples of OM. We found neither intracellular nor extracellular amyloid deposits in NCI patients (Figure 2F, I).
To examine the type of cells that accumulates the amyloid Aβ peptide in AD and MCI subjects, we performed double immunostaining in serial sections with either anti‐Aβ amyloid and CytK18 marker or Aβ and β‐III tubulin for sustentacular cells and olfactory sensory neurons, respectively, using mouse monoclonal antibodies. First, we established expression of CytK18 in the neuroepithelium and is strongly expressed by Bowman acini in the parenchyma (Figure 3). CytK18 was expressed by cells in the upper layer of cells of the olfactory epithelium (Figure 3A–C), also in basal cells of acini of the OM (Figure 3D–F). Otherwise, evidence of olfactory sensory neurons in biopsy samples was demonstrated using β‐III tubulin antibody; in the human OM, β‐III tubulin was located as cell component in deep layer of cells of the olfactory epithelium (Figure 3G–I), but not frequent in basal cells of acinus of Bowman (Figure 2J–L). When we test whether amyloid Aβ may be coexpressed by CytK18 olfactory cells, we observed amyloid Aβ‐immunopositive cells in Cytk18 cells from AD subjects ( Figure 4A–C).
Figure 3.
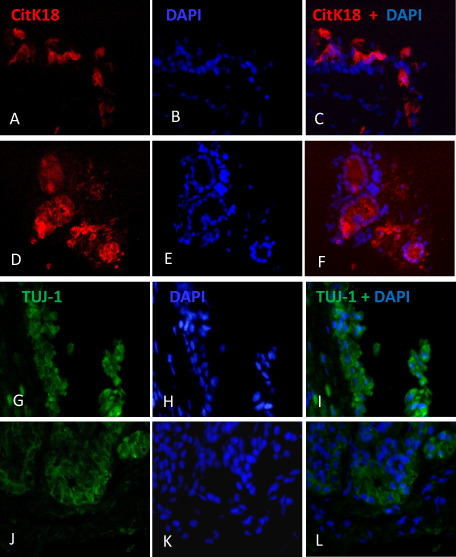
Evidence of C yt K 18 ( TROMA 1) and β III ‐tubulin ( TUJ ‐1) in human olfactory mucosa ( OM ) sections. Expression of CytK18 was found in the upper layer of cells of the olfactory epithelium (A–C) and in the basal cells of acini of the OM (D–F). Evidence of olfactory sensory neurons in biopsy samples was demonstrated using β‐III tubulin antibody; in the human OM, β‐III tubulin was located as cell component in deep layer of cells of the olfactory epithelium (G–I), but not frequent in basal cells of acinus of Bowman (J–L).
Figure 4.
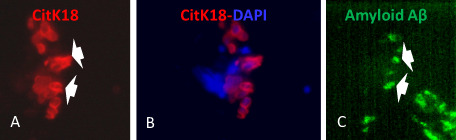
Evidence of amyloid A β in CitK 18 neuroepithelial cells of human olfactory mucosa ( OM ) A lzheimer's disease ( AD ) section biopsies. Serial sections 10 μm apart of OM biopsies from AD patients were immunolabeled with monoclonal anti‐amyloid Aβ and CytK18 antibodies as described in Methods section. CytK18 was expressed in cells from the upper layers of the OM (A). Amyloid Aβ was detected in CytK18 as shown in A and C (white arrows).
The frequency of the ApoE ε3 allele was significantly associated with NCI with respect to the ε4 in MCI and AD anosmic subjects
Taking into consideration a multiple methodology approach for detection of Alzheimer pathology, blood samples were also taken from all subjects in order to establish the ApoE load. A contingency analysis was performed for the distribution of frequencies of the ε3 and ε4 alleles among NCI, MCI and AD patients. Notably, a higher frequency of ε3 alleles, 8 over 10 alleles were found in the NCI group, whereas 3 over 6 alleles and 1 over 4 were determined for the MCI and AD group, respectively [chi‐square (3.98; 1) P < 0.0469; Figure 5 ]. Otherwise, the ApoE ε4 allele was determined in seven subjects having anosmia and 5 over 10 also showed cognitive deficit or presumptive diagnosis of AD.
Figure 5.
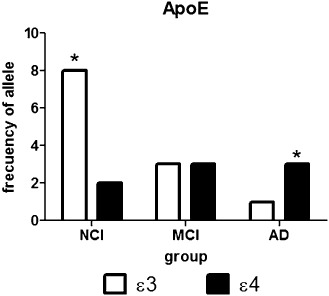
The frequency of the apolipoprotein E (ApoE) ε3 allele was significantly associated with noncognitive impaired (NCI) anosmic subjects. Blood samples were taken from patients under study, and ApoE genotyping was performed as described in Methods section. Contingency analysis was performed for the distribution of frequencies of the ε3 and ε4 alleles between NCI, mild cognitive impairment (MCI) and Alzheimer's disease (AD) patients. A higher frequency of ε3 alleles, 8 over 10 alleles were found in the NCI group, whereas 3 over 6 alleles and 1 over 4 were determined for the MCI and AD group, respectively [chi‐squared (3.98; 1) P < 0.0469]. Otherwise, the ApoE ε4 allele was determined in seven subjects having anosmia and 5 over 10 also showed cognitive deficit or presumptive diagnosis of AD.
Cells migrated differentially from explants of biopsies of OM from NCI, MCI and AD subjects
We assessed whether Alzheimer condition may impair migration of cells from tissue samples kept in vitro under culture conditions. Explants from NCI, MCI and AD subjects once seated in culture dishes started growing around day 7 after plating. Initially, predominantly bipolar cells migrated from the periphery of explants and generated a monolayer of cells that became 75%–90% confluent after 5–7 days. Cells were then collected and transferred to a new culture dish generating a stable population of cells for several passages. Multiple measurements of the surface area around explants occupied by cells at geographical coordinates were estimated as arbitrary units of length. Differences in amplitude were determined at several days of observation after plating. Over time, explants from NCI [P < 0.0001, Kruskal–Wallis test (3, 18.17)], MCI [P < 0.01, Kruskal–Wallis test (3, 4.53)] and AD subjects [P < 0.0001, Kruskal–Wallis test (3, 20.57)] grew significantly (Figure 6A). At day 10 of observation, the maximum amplitude occupied by cells that migrated from explants of NCI group was significantly higher [P < 0.0001, Kruskal–Wallis test (3, 16.48)] with respect to MCI and AD groups (P < 0.05). No differences were found between the MCI and AD group (P > 0.05).
MSCs were the predominant component determined by flow cytometry analysis from NCI, MCI and AD subjects
Once the adherent cells from the OM tissue explants were confluent, the phenotypic profile of the population was examined by extracellular or intracellular labeling by flow cytometry analysis. We identified mainly (95%–99%) MSC population recognized by the CD90, CD73 and CD29 antibodies (Figure 6B).
Based on the observation of adherent cells from NCI samples that migrated significantly faster with respect to MCI and AD tissue explants (Figure 6A), we asked whether AD condition may affect the proliferation capacity of mesenchymal, olfactory and neural progenitor cells. Cells collected from each subject and pathological condition were cultured for 24 h after plating and pulsed with the thymidine mimetic EdU for 24 h. Then, cells were collected and the EdU agent once incorporated to DNA was covalently linked to the fluorophore Alexa fluor 488. Subpopulation of cells that incorporated EdU was identified by flow cytometry analysis using multiple antibodies. Primarily, the viability of pulsed cells with the EdU label was not modified with respect to cells treated with vehicle (dimethyl sulfoxide) (results not shown). After EdU pulse, approximately 85%–90% of cells that incorporated EdU were successfully labeled.
First at all, co‐expression of EdU with CD90+, CD73+ and CD29+ phenotypic markers confirmed that MSCs are the main population that proliferated under our culture conditions in NCI, MCI and AD subjects. Second, co‐labeling of EdU proliferative marker with NESTIN was suggestive of a subpopulation of olfactory and neural precursor, which remained in the OM and became amplified under culture condition in MCI and AD subjects.
The percentage of EdU+, CD90+ cells indicative of the proliferative subpopulation of cells was different with respect to EdU−,CD90+ in the NCI group (P < 0.049) (Figure 7A). In contrast, although the percentage of EdU+, CD90+ was also elevated in the MCI and AD subjects, it was not different with respect to EdU−, CD90+ population of cells. These findings support the hypothesis that there is a reduced and probably defective proliferation capability of the MSC population in preclinical MCI and AD condition.
Figure 7.
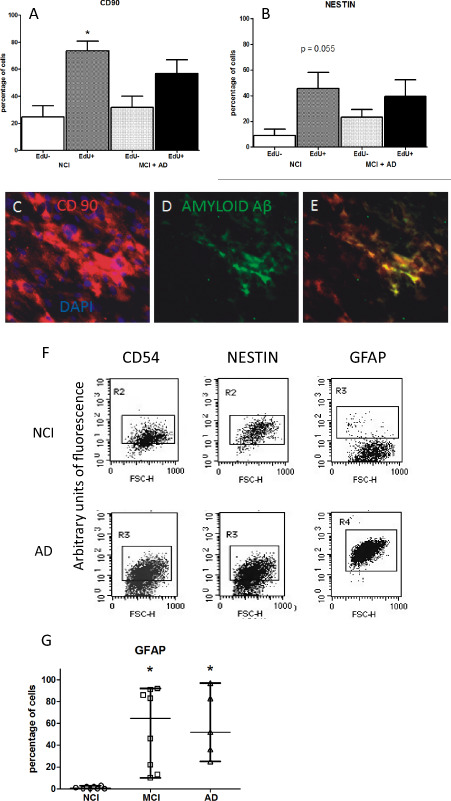
Amyloid A β expression may affect proliferation of EdU ‐labeled cells in A lzheimer's disease ( AD ) and is detected in mesenchymal stromal cells. Olfactory cells pulsed with EdU, a thymidine mimetic compound, evidenced that Alzheimer and mild cognitive impairment (MCI) condition precluded proliferation of CD90+ and NESTIN + mesenchymal stromal cells. The percentage of EdU +, CD90+ cells as indicative of proliferative subpopulation of cells was different with respect to EdU −,CD90+ in the noncognitive impaired (NCI) group (P < 0.049) (A). Although the percentage of EdU +, CD90+ was also elevated in the MCI and AD subjects, it was not different with respect to EdU −, CD90+ population of cells. The percentage of EdU −, NESTIN + population of cells in the MCI + AD group of subjects was not different with respect to EdU +, NESTIN + cells. A statistical value of P < 0.055 was determined when EdU +, NESTIN + with respect to EdU −, NESTIN + in the NCI group (B). No differences were found in the percentage of EdU −, NESTIN + population of cells between the NCI and MCI + AD group of subjects. Mesenchymal stromal cells (MSCs) were plated in coverslips by 7 days in vitro then fixed and examined by CD90 and amyloid Aβ immunostaining. A higher percentage of olfactory cells from AD subjects expressed CD90, a typical marker of MSCs (C). Amyloid Aβ peptide is immunolocalized in a subpopulation of CD90+ cells (D, E). Olfactory‐cultured cells from NCI, MCI and AD patients expressed markers of olfactory precursor (CD54) and neural progenitor cells [NESTIN and glial fibrillary acidic protein (GFAP)] (F). The median of GFAP olfactory stromal cells was different between AD, MCI and NCI groups of patients [Kruskall–Wallis test (P < 0.0008; 3, 14.35)] (G). The percentage of GFAP‐immunolabeled cells of AD and MCI was different with respect to NCI group (P < 0.05 Dunn's test). Cytograms are representative of multiple independent determinations.
We also analyzed the percentage of EdU+, NESTIN+ population of cells in the NCI and MCI + AD groups of subjects. A marginal statistical value of P < 0.055 (Figure 7B) was determined when the percentage of EdU+, NESTIN+ cells was compared with the percentage of EdU−, NESTIN+ in the NCI group. Otherwise, a P > 0.05 was calculated in the percentage of EdU−, NESTIN+ cells with respect to EdU+, NESTIN+ in the MCI + AD group. As a consequence, it seems that slight differences in proliferation capability of neural progenitor cells as suggested by NESTIN+‐immunopositive cells were present in NCI with respect to MCI + AD patients.
We asked whether differences observed in migration as we noticed in olfactory stromal cells from MCI and AD subjects could be associated to dysmetabolic amyloid Aβ expression. Then, we examined if amyloid Aβ peptide can be detected in adherent cells of cultured OM. We found that the amyloid Aβ was colocalized with CD90+ cells from AD subjects (Figure 7C–D). We did not detect the amyloid‐Aβ peptide in CD90+‐cultured cells from MCI or NCI subjects.
Under our culture conditions, MSCs also expressed phenotypic markers of horizontal basal cells, an olfactory precursor recognized by CD54 immunolabeling. The percentage of CD54+ cells was highly variable in the sampled population. In addition, findings of NESTIN+‐ and GFAP+‐immunolabeled cells that were suggestive of MSCs also coexpressed neural progenitor phenotypic markers in the OM population (Figure 4F). The percentage of CD90+, CD29+, CD73+, CD54+ and NESTIN+ was not different in NCI with respect to MCI or AD subjects (results not shown).
Finally, we observed zero to low percentage of the GFAP‐immunolabeled cells in NCI subjects. Interestingly, an elevated percentage of GFAP+ was significantly found in MCI (P < 0.05) and AD subjects (P < 0.05) with respect to NCI subjects [Kruskal–Wallis test (P < 0.0004; 3;15.43)] (Figure 7G).
Discussion
Here we have studied biopsies of OM from anosmic and NCI, MCI and AD subjects in an in vitro experimental protocol. This approach has allowed us to dissect and generate either histological sections to be examined for histopathological landmarks of AD or explants for the purpose of establishing stable cell cultures of MSCs.
To our knowledge, no previous studies have established expression of amyloid Aβ peptides in the neuroepithelium and parenchyma of biopsies of OM from MCI or AD subjects. Findings of amyloid Aβ expression in cells of the olfactory epithelium and also in the parenchyma of the OM of MCI and AD subjects were suggestive that the anosmia and cognitive deficit may be functionally associated with the dysmetabolism of amyloid precursor protein. These results support this proposal and strengthen the fact that OM is a source of potential biomarkers for early AD neuropathology.
It is probable that anosmia and preclinical AD or slight cognitive deficit may be associated with amyloid dysmetabolism and occur in parallel with amyloid deposition in limbic structures of the brain. Recent evidence have found that subjects with reduced olfactory sensitivity associated to MCI may have demonstrated AD pathology in the brain 33. Accumulation of amyloid Aβ in subpopulations of cells of the olfactory neuroepithelium and in the parenchyma, as we found, supports the proposal that Alzheimer pathology may occur in the olfactory system as an early event.
Lack of evidence of dystrophic neurites and abnormal tau protein in biopsies of OM from clinically mild‐to‐moderate AD patients as previously reported suggested that cytoskeletal changes and tau pathology in the olfactory epithelium may occur in late stages of the disease 7. Furthermore, progressive accumulation of tau pathology as AD disease progress in contrast with an exacerbated amyloid Aβ expression independent of duration or onset disease was suggestive that amyloid dysmetabolism and fibrillogenesis are independent events 8.
Loss of the olfactory function in multiple pathologies emphasizes the relevance of common circuits in the process of this sensorial information. Deficit in olfactory sensitivity, odor discrimination and identification of odorants appear to be the earliest detectable functional alterations in a high percentage of patients with AD and other neurodegenerative disorders 19, 21.
We hypothesized that anosmia and preclinical cognitive deficit may be associated with dysregulated metabolism of amyloid Aβ peptide. Accumulation of amyloid‐Aβ may impair turnover of olfactory precursor cells and neurogenesis in the olfactory niche and as a consequence would reduce their regenerative potential to replenish the olfactory neuroepithelium. We established anosmia or hiposmia in more than 90% of subjects after application of the Connecticut Olfactory Test. The olfactory evaluation was followed by MRI studies that detected atrophy of the hippocampus in advanced diagnosed AD but not in MCI subjects. Lack of anatomical changes in the hippocampus of MCI, in contrast with accumulation of amyloid in OM, supported the hypothesis that the dysmetabolism of amyloid in OM may precede impairment of the hippocampal circuitry; this finding is coherent with the hypothetical model of dynamic biomarkers where amyloid Aβ precedes large volumetric changes of the brain and when they occur a transition stage from the MCI stage progress to dementia 31.
Taking into consideration a multiple methodology approach for detection of Alzheimer pathology, we established in the sampled population the ApoE load. Notably, in 6 over 10 subjects studied at least one ApoE ε4 allele was determined. As a consequence, anosmic MCI or AD subjects are concurrent with a higher frequency of ApoE ε4 allele. In contrast, unexplained anosmia associated with NCI occurred in larger significant proportion of subjects with the ApoE ε3 allele. We report here in agreement with previous studies that individuals at risk for AD also show dysfunctional olfaction that may be associated with pre‐symptomatic AD 11, 19, 25, 35.
This is also the first study that reports biological properties of olfactory MSCs from NCI, MCI and AD subjects. The intrinsic capacity of generate cells by explants of OM was reduced in AD and MCI with regard to NCI subjects. We developed an enriched culture of CD90+, CD73+, CD29+ (mesenchymal stromal), CD54+ (horizontal olfactory precursor), NESTIN+ and GFAP+ (neural progenitor) cells from explants of OM. The percentage of cells estimated by flow cytometry analysis suggested a representative phenotypic profile of mesenchymal stromal cells that co‐expressed olfactory precursor and neural progenitor phenotypic markers.
We detected several populations of OM under EdU in an active, semi‐active or quiescent state of proliferation identified by typical phenotypic markers. MSCs identified by co‐expression of CD90 and EdU were highly active in NCI and AD subjects, whereas NESTIN+ appears to be semi‐active and CD54+ and GFAP+ (not shown) labeled cells were in a quiescent state. However, it seems that under AD condition NESTIN+, CD54+ and GFAP+ changed their proliferative state. A reduced proliferation capacity of MSCs in AD as indicated by EdU+ CD90+ co‐expression may be explained by intracellular accumulation of amyloid Aβ in cultured cells as we reported in this study. As a consequence, we have established that biopsies of OM are a reliable reporter to be examined by histological methodology and primary cultures in order to determine surrogate markers of Alzheimer pathology such as amyloid Aβ. In addition, exploration of the biology of olfactory mesenchymal cells in the OM may lead to a standardized procedure to analyze samples from early AD.
Finding of a significant increase of immunolabeled GFAP cells in MCI and AD but not in cells from NCI groups that was suggestive of Alzheimer pathology may predispose to the OM to generate a specific subset of stromal cells. Modulation of GFAP expression during senescence and pathology is supported by the role of astrocytes as a prominent feature of AD associated with neuropathological landmarks 9, 25. For instance, histological observations have showed both reactive astrocytes and activated microglia surrounding dense core amyloid plaques 2, 8. A better understanding of how astroglial become reactive during the disease course and how they relate to the classic AD pathological landmarks is crucial to establishing the role of glial cells in AD pathophysiological features. Interestingly, the meaning of appearance of glial cells in a culture system of OM maybe associated with recent development of new positron emission tomographic radiotracers for activated glial cells as diagnostic and progressive biomarkers of disease 24, 29.
Increase of GFAP stromal cells from MCI and AD patients as we found in culture may be associated to an adaptive response of the olfactory niche to AD pathology. Multiple isoforms of GFAP in the brain are associated to development, senescence and pathology, respectively, which suggest a complex role of this kind of intermediate filament protein in cell function 10, 33. Findings of stromal cells labeled by GFAP and NESTIN, a recognized stem cell marker, are compelling evidence for an adaptive response sustained by neurogenic astrocytes in the olfactory niche.
References
- 1. Arnold SE, Lee EB, Moberg PJ, Stutzbach L, Kazi H, Han LY et al (2010) Olfactory epithelium amyloid‐beta and paired helical filament‐tau pathology in Alzheimer disease. Ann Neurol 67:462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ayala Grosso C, Crocker SF, Leslie R, Robertson GS (2004) Inmunolocalización del Receptor de Proliferación de Peroxisomas Activados subtipo‐γ (PPAR‐γ) en la corteza cerebral de pacientes con diagnóstico de la enfermedad de Alzheimer (AD). Rev Fac Farm 70:4–12. [Google Scholar]
- 3. Delorme B, Nivet E, Gaillard J, Häupl T, Ringe J, Devèze A et al (2010) The human nose harbors a niche of olfactory ectomesenchymal stem cells displaying neurogenic and osteogenic properties. Stem Cells Dev 19:853–866. [DOI] [PubMed] [Google Scholar]
- 4. Féron F, Perry C, McGrath JJ, Mackay‐Sim A (1998) New techniques for biopsy and culture of human olfactory epithelial neurons. Arch Otolaryngol Head Neck Surg 124:861–866. [DOI] [PubMed] [Google Scholar]
- 5. Ghanbari HA, Ghanbari K, Harris PL, Jones PK, Kubat Z, Castellani RJ et al (2004) Oxidative damage in cultured human olfactory neurons from Alzheimer's disease patients. Aging Cell 3:41–44. [DOI] [PubMed] [Google Scholar]
- 6. Hixson JE, Vernier DT (1990) Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res 31:545–548. [PubMed] [Google Scholar]
- 7. Hock C, Golombowski S, Müller‐Spahn F, Peschel O, Riederer A, Probst A et al (1998) Histological markers in nasal mucosa of patients with Alzheimer's disease. Eur Neurol 40:31–36. [DOI] [PubMed] [Google Scholar]
- 8. Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley‐Whyte ET, Frosch MP et al (2004) Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 62:925–931. [DOI] [PubMed] [Google Scholar]
- 9. Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D (1989) Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol 24:173–182. [DOI] [PubMed] [Google Scholar]
- 10. Kamphuis W, Mamber C, Moeton M, Kooijman L, Sluijs JA, Jansen AH et al (2012) GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS ONE 7:e42823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kowalewski J, Murphy C (2012) Olfactory ERPs in an odor/visual congruency task differentiate ApoE ε4 carriers from non‐carriers. Brain Res 1442:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leung CT, Coulombe PA, Reed RR (2007) Contribution of olfactory neural stem cells to tissue maintenance and regeneration. Nat Neurosci 10:720–726. [DOI] [PubMed] [Google Scholar]
- 13. Mackay‐Sim A (2013) Patient‐derived stem cells: pathways to drug discovery for brain diseases. Front Cell Neurosci 7:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mackay‐Sim A, Kittel P (1991) Cell dynamics in the adult mouse olfactory epithelium: a quantitative autoradiographic study. J Neurosci 11:979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mackay‐Sim A, Kittel PW (1991) On the life span of olfactory receptor neurons. Eur J Neurosci 3:209–215. [DOI] [PubMed] [Google Scholar]
- 16. Mesholam RI, Moberg PJ, Mahr RN, Doty RL (1998) Olfaction in neurodegenerative disease: a meta‐analysis of olfactory functioning in Alzheimer's and Parkinson's diseases. Arch Neurol 55:84–90. [DOI] [PubMed] [Google Scholar]
- 17. Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE (1997) Metabolic reduction in the posterior cingulate cortex in very early Alzheimer's disease. Ann Neurol 42:85–94. [DOI] [PubMed] [Google Scholar]
- 18. Morgan CD, Murphy C (2012) Individuals at risk for Alzheimer's disease show differential patterns of ERP brain activation during odor identification. Behav Brain Funct 8:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Morgan CD, Nordin S, Murphy C (1995) Odor identification as an early marker for Alzheimer's disease: impact of lexical functioning and detection sensitivity. J Clin Exp Neuropsychol 17:793–803. [DOI] [PubMed] [Google Scholar]
- 20. Murphy C (1999) Loss of olfactory function in dementing disease. Physiol Behav 66:177–182. [DOI] [PubMed] [Google Scholar]
- 21. Murrell W, Bushell GR, Livesey J, McGrath J, MacDonald KP, Bates PR et al (1996) Neurogenesis in adult human. Neuroreport 7:1189–1194. [DOI] [PubMed] [Google Scholar]
- 22. Murrell W, Féron F, Wetzig A, Cameron N, Splatt K, Bellette B et al (2005) Multipotent stem cells from adult olfactory mucosa. Dev Dyn 233:496–515. [DOI] [PubMed] [Google Scholar]
- 23. Nasreddine ZS, Phillips NA, Bédirian V, Charbonneau S, Whitehead V, Collin I et al (2005) The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 53:695–699. [DOI] [PubMed] [Google Scholar]
- 24. Okello A, Edison P, Archer HA, Turkheimer FE, Kennedy J, Bullock R et al (2009) Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology 72:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Olofsson JK, Rönnlund M, Nordin S, Nyberg L, Nilsson LG, Larsson M (2009) Odor identification deficit as a predictor of five‐year global cognitive change: interactive effects with age and ApoE‐epsilon4. Behav Genet 39:496–503. [DOI] [PubMed] [Google Scholar]
- 26. Packard A, Schnittke N, Romano RA, Sinha S, Schwob JE (2011) DeltaNp63 regulates stem cell dynamics in the mammalian olfactory epithelium. J Neurosci 31:8748–8759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reyes PF, Deems DA, Suarez MG (1993) Olfactory‐related changes in Alzheimer's disease: a quantitative neuropathologic study. Brain Res Bull 32:1–5. [DOI] [PubMed] [Google Scholar]
- 28. Reyes PF, Golden GT, Fagel PL, Fariello RG, Katz L, Carner E (1987) The prepiriform cortex in dementia of the Alzheimer type. Arch Neurol 44:644–645. [DOI] [PubMed] [Google Scholar]
- 29. Serrano‐Pozo A, Mielke ML, Gómez‐Isla T, Betensky RA, Growdon JH, Frosch MP, Hyman BT (2011) Reactive glia not only associates with plaques but also parallels tangles in Alzheimer's disease. Am J Pathol 179:1373–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sohrabi HR, Bates KA, Weinborn MG, Johnston AN, Bahramian A, Taddei K et al (2008) Olfactory discrimination predicts cognitive decline among community‐dwelling older adults. Transl Psychiatry 2:e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM et al (2011) Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 7:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Toledano A, Ruiz C, Navas C, Herráiz C, González E, Rodríguez G, Galindo AN (2009) Development of a short olfactory test based on the Connecticut Test (CCCRC). Rhinology 47:465–469. [DOI] [PubMed] [Google Scholar]
- 33. Van den Berge SA, Middeldorp J, Zhang CE, Curtis MA, Leonard BW, Mastroeni D et al (2010) Longterm quiescent cells in the aged human subventricular neurogenic system specifically express GFAP‐delta. Aging Cell 9:313–326. [DOI] [PubMed] [Google Scholar]
- 34. Wilson RS, Arnold SE, Schneider JA, Tang Y, Bennett DA (2007) The relationship between cerebral Alzheimer's disease pathology and odour identification in old age. J Neurol Neurosurg Psychiatry 78:30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wilson RS, Arnold SE, Schneider JA, Boyle PA, Buchman AS, Bennett DA (2009) Olfactory impairment in presymptomatic Alzheimer's disease. Ann N Y Acad Sci 1170:730–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wolozin B, Lesch P, Lebovics R, Sunderland T (1993) A.E. Bennett Research Award 1993. Olfactory neuroblasts from Alzheimer donors: studies on APP processing and cell regulation. Biol Psychiatry 34:824–838. [DOI] [PubMed] [Google Scholar]