

Abstract
The adenosine triphosphate‐binding cassette transport protein P‐glycoprotein (ABCB1) is involved in the export of beta‐amyloid from the brain into the blood, and there is evidence that age‐associated deficits in cerebral P‐glycoprotein content may be involved in Alzheimer's disease pathogenesis. P‐glycoprotein function and expression can be pharmacologically induced by a variety of compounds including extracts of Hypericum perforatum (St. John's Wort). To clarify the effect of St. John's Wort on the accumulation of beta‐amyloid and P‐glycoprotein expression in the brain, St. John's Wort extract (final hyperforin concentration 5%) was fed to 30‐day‐old male C57BL/6J‐APP/PS1 +/− mice over a period of 60 or 120 days, respectively. Age‐matched male C57BL/6J‐APP/PS1 +/− mice receiving a St. John's Wort‐free diet served as controls. Mice receiving St. John's Wort extract showed (i) significant reductions of parenchymal beta‐amyloid 1–40 and 1–42 accumulation; and (ii) moderate, but statistically significant increases in cerebrovascular P‐glycoprotein expression. Thus, the induction of cerebrovascular P‐glycoprotein may be a novel therapeutic strategy to protect the brain from beta‐amyloid accumulation, and thereby impede the progression of Alzheimer's disease.
Keywords: Alzheimer's disease, beta‐amyloid, blood–brain barrier, P‐glycoprotein, St. John's Wort
Introduction
Alzheimer's disease (AD) is a common, progressive neurodegenerative disorder that begins as mild short‐term memory disturbances and ends in total loss of cognition and executive functions. Histopathologically, AD is characterized by the so‐called “core” pathologies such as accumulation of amyloid‐β‐peptides (Aβ) and abnormal tau‐protein 9, 17. It has been proposed that the underlying cause of Aβ accrual in the sporadic, age‐related form of AD is a reduced clearance of Aβ from the brain via the blood–brain barrier (BBB) 4, 27, 30.
The adenosine triphosphate‐binding cassette (ABC) transporter P‐glycoprotein (ABCB1, P‐gp), which is encoded by the multidrug resistance‐1 (MDR1/ABCB1) gene, constitutes a critical component of the BBB 7, 19 and has been shown to mediate the transport of Aβ peptides 15, 16. In vivo and in vitro experiments as well as autopsy studies support the view that P‐gp is essential for the elimination of brain‐derived Aβ, thus playing a critical role in the pathogenesis of AD 3, 10, 28, 29.
To date, despite considerable scientific effort, there are no substantially effective treatments or preventives for AD. Given the fact that the activity of P‐gp can be modulated pharmacologically by a range of different compounds, including frequently used agents such as St. John's Wort, the antibiotic rifampin, or flavonoids 11, 20, 24, the upregulation of cerebral P‐gp could be a new therapeutic strategy for reducing the buildup of Aβ in the brain.
St. John's Wort (SJW; Hypericum perforatum), with its biologically active compound hyperforin, is a widely used herbal drug in patients with different types of depression 12. Recently, beneficial effects in animal models of AD have also been reported 2, 5. Because hyperforin has been shown to increase the expression of P‐gp 6, 8, 26, the aim of the present study was to investigate the effect of SJW treatment on cerebral Aβ deposition in a double transgenic AD mouse model. We hypothesized that, by enhancing the activity of P‐gp, SJW would promote the efflux of Aβ from the brain. Here we report that the administration of SJW significantly reduced soluble and aggregated Aβ levels in the brains of transgenic mice, indicating that upregulation of P‐gp may be a valuable approach to diminishing cerebral Aβ accumulation.
Materials and Methods
Chemicals
SJW extract containing 5% hyperforin was kindly donated by Dr. Wilmar Schwabe Pharmaceuticals (Karlsruhe, Germany). All other chemicals were purchased from commercial sources and were of analytical grade.
Animals
C57BL/6J‐APP/PS +/− mice 23, 25 were purchased from KOESLER (Rothenburg, Germany). Mice were housed in a 12‐h light/12‐h dark cycle at 23°C with free access to water.
Based on established protocols 18. mice were treated with SJW extract (1250 mg/kg body weight daily) containing 5% hyperforin. To avoid excessive stress on the animals because of the long feeding duration, SJW was administered with the food (SNIFF, Soest, Germany). Given a minimal food intake of 3 g/day per mouse, 4.166 mg SWJ were mixed with 1 g of food (Altromin, Lage, Germany). Mice were administered 3 g of the prepared food each day, and intake was monitored to ensure the required dosage.
Treatment started at the age of 30 days. Because, in this mouse model, sufficient plaques can be seen in the brain by the age of 90 days, the animals received SJW for either 60 or 120 days, respectively. C57BL/6J‐APP/PS1 +/− mice receiving food without added SJW served as controls.
Tissue preparation
At the age of 90 or 150 days, respectively, mice were sacrificed by cervical dislocation. The brains were removed and one hemisphere was snap‐frozen in liquid nitrogen and stored at −80°C for biochemical analysis. The other hemisphere was fixed in buffered 4% formalin and then paraffin‐embedded for immunohistochemistry.
Western blot analysis
For Western blot analysis, a crude membrane fraction was isolated by centrifugation of the brain homogenate at 100.000 × g at 4°C for 30 min. Thereafter, the pellet was resuspended in Tris/HCl buffer (5 mM, pH 7.4, supplemented with protease inhibitors) and protein concentration was determined by the bicinchoninic acid method. Next, 50 μg of membrane protein were loaded on a 7.5% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐Page) gel. Immunoblotting was performed using a tank blotting system (Bio‐Rad, Hercules, CA, USA). Primary antibodies were diluted in Tris‐buffered saline containing 0.05% Tween 20 to the following final concentrations: P‐gp (C219, Calbiochem, Darmstadt, Germany) 1:1000 and desmin (Sigma‐Aldrich, Munich, Germany) 1:500. The secondary horseradish peroxidase‐conjugated goat anti‐mouse antibody (Bio‐Rad) was used at a final concentration of 1:500. Signals were detected using an enhanced chemiluminescence detection system (ECL Plus Western Blotting Substrate, Pierce, Rockford, IL USA) and the ChemiDoc XRS system (Bio‐Rad) and then analysed by the QuantityOne 4.6.7 software using the mean intensity values of each band (Bio‐Rad). In the case of P‐gp detection the C219 antibody showed some cross reactivity, e.g. with a protein which migrates in the same position as myosin (about 200 kDa) resulting in additional unspecific bands. Here, only the P–gp‐specific signals (at about 180 kDa) were used for quantification.
Immunohistochemical analyses
For immunohistochemical quantification of Aβ accumulation and P‐gp expression 1‐μm thick sagittal sections were mounted onto slides and dried for 1 h at 58°C. Brain sections of all mice were processed simultaneously to obtain comparable staining intensity. Automated immunohistochemical staining was performed using the BOND‐MAX Autostainer (Leica Microsystems GmbH, Wetzlar, Germany).
For Aβ staining, serial brain sections were pretreated for 3 minutes with concentrated formic acid, and incubated with antibodies directed to Aβ 1–40 and Aβ 1–42, respectively (AB5074P, dilution 1:50, and AB5078P, dilution 1:50, both antibodies by Merck Millipore, Schwalbach, Germany). After antigen retrieval by incubating the slides for 30 minutes at 100°C, nonspecific binding sites were blocked with serum‐free Protein Block (DakoCytomation, Glostrup, Denmark). Cerebrovascular P‐gp was detected using the mouse monoclonal antibody C219 (dilution 1:25, pH 9.0, Enzo Life Sciences, Lörrach, Germany). Immunoreactivities were detected by appropriate secondary antibodies and the BOND‐MAX Bond Polymer Refine Detection Kit (Leica Microsystems GmbH).
To quantitate cortical Aβ accumulation and P‐gp expression, hematoxylin/3,3′‐diaminobenzidine (DAB)‐stained slides were digitized using the Mirax Desk slide scanner (Carl Zeiss MicroImaging GmbH, Göttingen, Germany; Plan‐Apochromat 20×/0.8, pixel resolution: 0.36 μm) and saved as 24‐bit RGB‐tagged image format (tif) files. Image analysis was performed by Image J (version 1.43 q, Research Services Branch, National Institute of Mental Health/National Institutes of Health, Bethesda, MD, USA; http://rsb.info.nih.gov/ij) on high‐power image sections that were treated as ImageJ stacks. Hematoxylin/DAB color spaces were extracted by the color deconvolution plugin with predefined H DAB vectors. The individual background was subtracted with a rolling circle diameter of 64. To identify capillaries, the stack of DAB‐stained images was blurred via Gaussian Blur (sigma = 2). Aβ plaques and P–gp‐stained capillaries were identified as regions of interest (ROI) by thresholding the DAB channel. For each region of interest, DAB staining intensity and mean area were measured.
Quantification of soluble Aβ species
For quantification of soluble Aβ 1–40 and Aβ 1–42 peptides, human amyloid beta 1–40 or 1–42 brain enzyme‐linked immunosorbent assay (ELISA) kits (Merck Millipore) were used. Sample preparation was performed according to the manusfacturer's protocol. In brief, brain tissue was disrupted under frozen conditions using a ball mill. Tissue powder was scaled and 10× volume of lysis buffer was added. The resulting lysate was rotated for 2 h at 4°C and cleared by centrifugation at 13 000 rpm for 10 min at 4°C. Quantification of soluble Aβ 1–40 and Aβ 1–42 peptides by ELISA was done according to the manufacturer's instructions. The results were normalized to the amount of brain tissue used for the analysis.
Statistics
Mathematical calculations, statistical tests as well as data presentation were performed using GraphPad Prism 5 (GraphPad, La Jolla, CA, USA). For calculation of the ELISA data, standard curve data were fitted using a non‐linear regression model and Aβ concentrations within the samples were interpolated. When data are presented using box and whisker blots, the whiskers represent the 10–90 percentile. If not otherwise stated, the Mann–Whitney test was used for statistical analysis.
The experiments were approved by local authorities: Landesamt für Landwirtschaft, Lebensmittelsicherheit und Fischereiwesen (LALLF; LALLF M‐V/TSD/7221.3‐1.1‐008/08).
Results
Quantification of cerebral Aβ
The number of Aβ 1–40‐ and Aβ 1–42‐positive plaques was assessed using immunohistochemistry to demonstrate the amount of fibrillar and (in the case of diffuse plaques) protofibrillar Aβ. We found a significant reduction in plaque load after SJW administration for 60 or 120 days, respectively, for both Aβ 1–40 (P = 0.011 and P = 0.003) and Aβ 1–42 (P = 0.026 and P = 0.001) in comparison to controls (Figure 1). Furthermore, the size of Aβ 1–40 positive plaques was significantly smaller in SJW treated mice (P = 0.005 and P = 0.013). For Aβ 1–42 positive plaques there were no differences in plaque size after 60 days of SJW treatment but a highly significant decrease in plaque size after 120 days (P = 0.006) (Figure 2). Figure 3 shows representative examples of treated and control mice. In accordance with the characterization of the mouse model by Radde et al 23, no vascular Aβ accumulation was observed.
Figure 1.
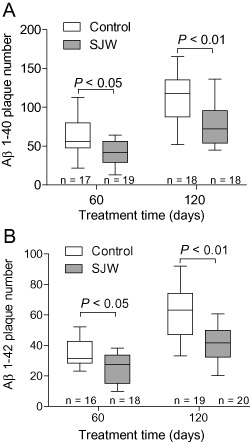
Aβ 1–40 (A) and Aβ 1–42 (B) Plaque number is significantly reduced after treatment with St. John's Wort (SJW).
Figure 2.
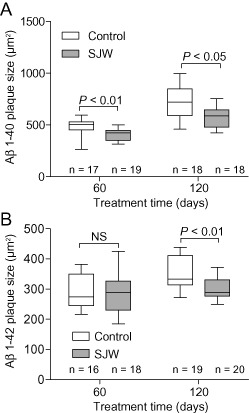
St. John's Wort (SJW) treatment leads to a marked decrease in the size of Aβ 1–40 immunopositive plaques (A). The size of Aβ 1–42 (B) positive plaques was unchanged after 60 days of SJW administration, but significantly reduced after a feeding duration of 120 days.
Figure 3.
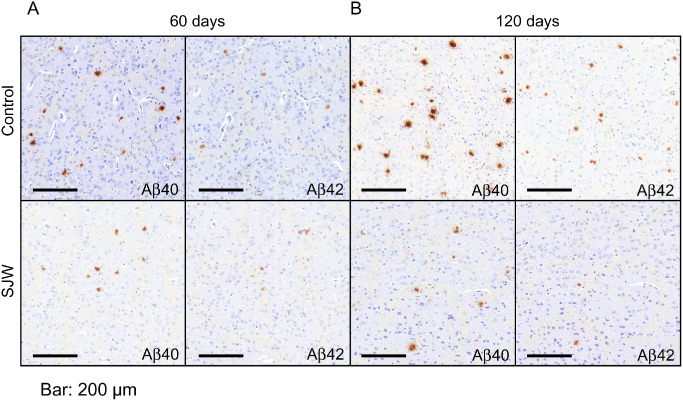
Representative images of brain sections immunostained for Aβ 1–40 and Aβ 1–42 in St. John's Wort (SJW)‐treated C57BL/6J‐APP/PS1+/− mice compared with controls (A: 60 days, B: 120 days). Original magnification: 100×, bar: 200 μm.
The observed reduction of the plaque load is in line with the finding of reduced levels of cerebral soluble Aβ in SJW‐treated animals measured by ELISA. Specifically, after 60 d of treatment no significant changes in Aβ 1–40 levels (10% reduction compared with control, P = 0.615) were detected, while Aβ 1–42 levels were significantly decreased (30% reduction, P = 0.020). After 120 days of SJW treatment, the effect was more pronounced for both peptides. For Aβ 1–40, we found a 47% reduction compared with controls; however, this was not statistically significant (P = 0.206). Soluble Aβ 1–42 showed a significant reduction of 53% (P = 0.006; Figure 4). There was no effect of treatment on food consumption and the mice did not show obvious differences in behavior during the treatment period.
Figure 4.
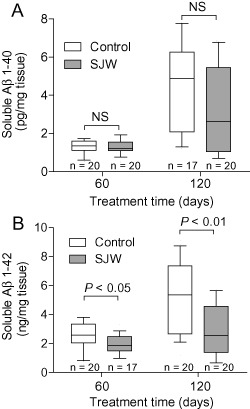
Quantification of soluble Aβ using enzyme‐linked immunosorbent assay. The levels of Aβ 1–40 did not differ significantly between the St. John's Wort (SJW)‐treated group and controls (A). Aβ 1–42 was significantly reduced in the SJW‐treated mice compared with controls (B).
Quantification of cerebrovascular P‐gp expression
According to our hypothesis, we also studied the inducing effect of SJW on P‐gp expression immunohistochemically. We found that cerebrovascular endothelial P‐gp expression was significantly augmented after 60 days (P = 0.0006) and 120 days (P = 0.0001), respectively, in SJW‐treated animals compared with controls (Figure 5A). In addition, when P‐gp expression was quantified by Western blot analysis after 120 days, P‐gp expression in the SJW‐treated animals was moderately, but significantly enhanced (P = 0.017; Figure 5B). Figure 6 shows representative examples of P‐gp immunostaining of treated and control mice.
Figure 5.
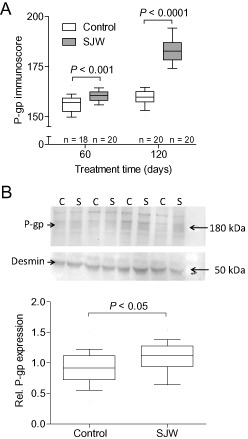
Immunohistochemical quantification of vascular P‐gp expression shows a significant induction of P‐gp in the St. John's Wort (SJW)‐treated group that was pronounced after administration of 120 days (A). Western blot analysis of P‐gp indicates a moderate, but significant upregulation of P‐gp in the brain of SJW‐treated (120 days) animals. The P‐gp‐specific band at about 180 kDa (marked with the arrow) was analyzed densitometrically and normalized to desmin (n = 20). An image of a representative Western blot of four controls (C) and four SJW (S)‐treated animals is shown in the insert. (B).
Figure 6.
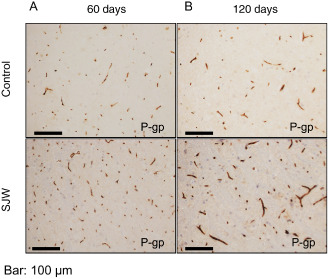
Representative images of brain sections immunostained for P‐gp in St. John's Wort (SJW)‐treated mice compared with controls (A: 60 days, B: 120 days). Original magnification: 200×, bar: 100 μm.
Discussion
Our results show that SJW treatment leads to a significant reduction of soluble Aβ 1–42 (representing mainly small oligomers and monomers) as well as Aβ 1–40 and Aβ 1–42 positive plaque number and size (representing mainly fibrillar and protofibrillar Aβ), while vascular P‐gp expression was increased in the brains of C57BL/6J‐APP/PS1 +/− mice. These findings support the view that the induction of P‐gp might augment the clearance of Aβ from the brain.
SJW, with its active component hyperforin, is a widely used herbal antidepressant that is thought to have potential beneficial effects in AD. Injection of Aβ fibrils together with hyperforin into the hippocampus of male Sprague‐Dawley rats resulted in a decrease of Aβ deposit formation, neuronal death and glial reaction as compared with controls 5. Cognitive testing of hyperforin‐injected animals revealed significant protection from the spatial memory loss induced by Aβ deposits. Furthermore, hyperforin protected against Aβ neurotoxicity in vitro using hippocampal neuronal cultures 5. Interestingly, incubation of amyloid fibrils with increasing concentrations of hyperforin led to almost complete disassembly of Aβ fibrils 5. Other in vitro experiments with a microglial cell line showed that pre‐treatment with SJW extract reduced the toxic influence of Aβ on microglial activation, proliferation and cell death in a dose‐dependent manner, suggesting that SJW might attenuate Aβ‐mediated toxicity in AD 14.
In addition to its several neurobiological effects (including neurotransmitter re‐uptake inhibition, the ability to increase intracellular sodium and calcium levels, canonical transient receptor potential 6 activation, or N‐methyl‐D‐aspartic acid receptor antagonism), SJW has antioxidant and anti‐inflammatory properties 1, 13 that might be beneficial in slowing the pathogenesis of AD. Furthermore, treatment of APP/PS1 double transgenic mice with the hyperforin derivate IDN5706 alleviated cognitive decline and affected the turnover of Aβ plaques 2.
In contrast to our results, Matheny et al did not find a significant induction of P‐gp in the brains of male FVB mice after SJW treatment for 5 days 18. Functionally, SJW and particularly hyperforin has been shown to be a potent inducer of P‐gp in a dose‐dependent manner 11, 22, 32. While some reports indicate that acute exposure to SJW results in a moderate inhibition of P‐gp function 21, 31, long‐term administration leads to a significant induction of P‐gp 31. SJW is known to induce P‐gp through pregnane X‐receptor (PXR) activation in several human tissues 6, 26. However, PXR exhibits considerable differences among mammalian species with respect to ligand binding leading to differing degrees of induction 33. In our mouse model, we found a moderate, but significant induction of cerebrovascular P‐gp at the protein level. This increase in protein might result from the long period of SJW administration in our experiments, during that P‐gp expression and/or function might be upregulated by indirect mechanisms rather than direct PXR interaction. Because SJW is a potent inducer of human PXR 33, one could speculate that the inductive effect of SJW on P‐gp would be stronger in humans than in mice.
P‐gp is a major constituent of the BBB and is able to transport Aβ 1–40 as well as Aβ 1–42 actively 15, 16. Several lines of evidence suggest that P‐gp is involved in the clearance of Aβ from the brain, indicating that the age‐associated decline in P‐gp function could play a critical part in the pathogenesis of idiopathic AD 3, 10, 28, 29. The clearance of Aβ from the brain into the blood at the BBB comprises a two‐step mechanism involving the low‐densitiy lipoprotein receptor as a putative transcytosis mediator at the abluminal site of endothelial cells 27, and P‐gp and possibly other ABC transporters at the luminal side. However, the mechanism of transportation of Aβ from the brain is still not fully understood; thus, further studies are required to determine possible transporter/receptor interactions.
In conclusion, our study shows that long‐term administration of SJW results in a potent decrease of Aβ accumulation in the brains of double transgenic mice, and a moderate but significant upregulation of cerebrovascular P‐gp expression. Besides many neuroprotective effects of SJW, the induction of P‐gp by SJW thus might enhance Aβ clearance from the brain and thereby reduce the risk of developing AD. To test this hypothesis further studies are warranted to investigate the effects of long term SJW treatment on animal behaviour and memory. Our finding that SJW reduces Aβ load in a mouse model indicates that the induction of P‐gp is a promising therapeutic approach to the treatment and/or prevention of neurodegenerative diseases such as AD.
Author Contributions
AB and SV designed the study. AB, MG, GJ, AF, BS, ME, MK, MHG and SV performed the experiments and analyzed the data. All authors were involved in writing the paper and had final approval of the submitted and published versions.
Conflict of Interest
The study was financially supported by Dr. Wilmar Schwabe Pharmaceuticals (Karlsruhe, Germany).
Acknowledgments
We gratefully acknowledge helpful comments by Lary C. Walker (Emory University) and the excellent technical help of Cathrin Müller and Katrin Sokolowski, Department of Pathology, as well as of Tina Sonnenberger, Department of Pharmacology.
The study was supported by FP7‐REGPOT‐20081‐1 CSA Project ImpactG; Grant agreement no. 229750.
References
- 1. Cabrelle A, Dell'Aica I, Melchiori L, Carraro S, Brunetta E, Niero R et al (2008) Hyperforin down‐regulates effector function of activated T lymphocytes and shows efficacy against Th1‐triggered CNS inflammatory‐demyelinating disease. J Leukoc Biol 83:212–219. [DOI] [PubMed] [Google Scholar]
- 2. Cerpa W, Hancke JL, Morazzoni P, Bombardelli E, Riva A, Marin PP, Inestrosa NC (2010) The hyperforin derivative IDN5706 occludes spatial memory impairments and neuropathological changes in a double transgenic Alzheimer's mouse model. Curr Alzheimer Res 7:126–133. [DOI] [PubMed] [Google Scholar]
- 3. Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB et al (2005) P‐glycoprotein deficiency at the blood–brain barrier increases amyloid‐beta deposition in an Alzheimer disease mouse model. J Clin Invest 115:3285–3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Deane R, Bell RD, Sagare A, Zlokovic BV (2009) Clearance of amyloid‐beta peptide across the blood–brain barrier: implication for therapies in Alzheimer's disease. CNS Neurol Disord Drug Targets 8:16–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dinamarca MC, Cerpa W, Garrido J, Hancke JL, Inestrosa NC (2006) Hyperforin prevents beta‐amyloid neurotoxicity and spatial memory impairments by disaggregation of Alzheimer's amyloid‐beta‐deposits. Mol Psychiatry 11:1032–1048. [DOI] [PubMed] [Google Scholar]
- 6. Dürr D, Stieger B, Kullak‐Ublick GA, Rentsch KM, Steinert HC, Meier PJ, Fattinger K (2000) John's Wort induces intestinal P‐glycoprotein/MDR1 and intestinal and hepatic CYP3A4. Clin Pharmacol Ther 68:598–604. [DOI] [PubMed] [Google Scholar]
- 7. Fromm MF (2000) P‐glycoprotein: a defense mechanism limiting oral bioavailability and CNS accumulation of drugs. Int J Clin Pharmacol Ther 38:69–74. [DOI] [PubMed] [Google Scholar]
- 8. Gutmann H, Poller B, Büter KB, Pfrunder A, Schaffner W, Drewe J (2006) Hypericum perforatum: which constituents may induce intestinal MDR1 and CYP3A4 mRNA expression? Planta Med 72:685–690. [DOI] [PubMed] [Google Scholar]
- 9. Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353–356. [DOI] [PubMed] [Google Scholar]
- 10. Hartz AM, Miller DS, Bauer B (2010) Restoring blood–brain barrier P‐glycoprotein reduces brain amyloid‐beta in a mouse model of Alzheimer's disease. Mol Pharmacol 77:715–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Johne A, Brockmoeller J, Bauer S, Maurer A, Langheinrich M, Roots I (1999) Pharmacokinetic interaction of digoxin with an herbal extract from St John's wort (Hypericum perforatum). Clin Pharmacol Ther 66:338–345. [DOI] [PubMed] [Google Scholar]
- 12. Kasper S, Gastpar M, Müller WE, Volz HP, Dienel A, Kieser M, Möller HJ (2008) Efficacy of St. John's wort extract WS 5570 in acute treatment of mild depression: a reanalysis of data from controlled clinical trials. Eur Arch Psychiatry Clin Neurosci 258:59–63. [DOI] [PubMed] [Google Scholar]
- 13. Koeberle A, Rossi A, Bauer J, Dehm F, Verotta L, Northoff H et al (2011) Hyperforin, an anti‐inflammatory constituent from St. John's Wort, inhibits microsomal prostaglandin E(2) synthase‐1 and suppresses prostaglandin E(2) formation in vivo . Front Pharmacol 2:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kraus B, Wolff H, Heilmann J, Elstner EF (2007) Influence of Hypericum perforatum extract and its single compounds on amyloid‐beta mediated toxicity in microglial cells. Life Sci 81:884–894. [DOI] [PubMed] [Google Scholar]
- 15. Kuhnke D, Jedlitschky G, Grube M, Krohn M, Jucker M, Mosyagin I et al (2007) MDR1‐P‐Glycoprotein (ABCB1) Mediates Transport of Alzheimer's amyloid‐beta peptides–implications for the mechanisms of Abeta clearance at the blood–brain barrier. Brain Pathol 17:347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lam FC, Liu R, Lu P, Shapiro AB, Renoir JM, Sharom FJ, Reiner PB (2001) beta‐Amyloid efflux mediated by p‐glycoprotein. J Neurochem 76:1121–1128. [DOI] [PubMed] [Google Scholar]
- 17. Mandelkow EM, Mandelkow E (1998) Tau in Alzheimer's disease. Trends Cell Biol 8:425–427. [DOI] [PubMed] [Google Scholar]
- 18. Matheny CJ, Ali RY, Yang X, Pollack GM (2004) Effect of prototypical inducing agents on P‐glycoprotein and CYP3A expression in mouse tissues. Drug Metab Dispos 32:1008–1014. [PubMed] [Google Scholar]
- 19. Miller DS (2010) Regulation of P‐glycoprotein and other ABC drug transporters at the blood–brain barrier. Trends Pharmacol Sci 31:246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Molnár J, Engi H, Hohmann J, Molnár P, Deli J, Wesolowska O et al (2010) Reversal of multidrug resitance by natural substances from plants. Curr Top Med Chem 10:1757–1768. [DOI] [PubMed] [Google Scholar]
- 21. Ott M, Huls M, Cornelius MG, Fricker G (2010) St. John's Wort constituents modulate P‐glycoprotein transport activity at the blood–brain barrier. Pharm Res 27:811–822. [DOI] [PubMed] [Google Scholar]
- 22. Perloff MD, Störmer E, von Moltke LL, Greenblatt DJ (2003) Rapid assessment of P‐glycoprotein inhibition and induction in vitro . Pharm Res 20:1177–1183. [DOI] [PubMed] [Google Scholar]
- 23. Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L et al (2006) Abeta42‐driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 7:940–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rautio J, Humphreys JE, Webster LO, Balakrishnan A, Keogh JP, Kunta JR et al (2006) In vitro p‐glycoprotein inhibition assays for assessment of clinical drug interaction potential of new drug candidates: a recommendation for probe substrates. Drug Metab Dispos 34:786–792. [DOI] [PubMed] [Google Scholar]
- 25. Rupp NJ, Wegenast‐Braun BM, Radde R, Calhoun ME, Jucker M (2011) Early onset amyloid lesions lead to severe neuritic abnormalities and local, but not global neuron loss in APPPS1 transgenic mice. Neurobiol Aging 32:2324.e1–2324.e6. [DOI] [PubMed] [Google Scholar]
- 26. Schwarz UI, Hanso H, Oertel R, Miehlke S, Kuhlisch E, Glaeser H et al (2007) Induction of intestinal P‐glycoprotein by St John's wort reduces the oral bioavailability of talinolol. Clin Pharmacol Ther 81:669–678. [DOI] [PubMed] [Google Scholar]
- 27. Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B et al (2000) Clearance of Alzheimer's amyloid‐β 1‐40 peptide from brain by LDL receptor‐related protein‐1 at the blood–brain barrier. J Clin Invest 106:1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vogelgesang S, Cascorbi I, Schroeder E, Pahnke J, Kroemer HK, Siegmund W et al (2002) Deposition of Alzheimer's beta‐amyloid is inversely correlated with P‐glycoprotein expression in the brains of elderly non‐demented humans. Pharmacogenetics 12:535–541. [DOI] [PubMed] [Google Scholar]
- 29. Vogelgesang S, Warzok RW, Cascorbi I, Kunert‐Keil C, Schroeder E, Kroemer HK et al (2004) The role of P‐glycoprotein in cerebral amyloid angiopathy; implications for the early pathogenesis of Alzheimer's disease. Curr Alzheimer Res 1:121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vogelgesang S, Jedlitschky G, Brenn A, Walker LC (2011) The role of the ATP‐binding cassette transporter P‐glycoprotein in the transport of β‐amyloid across the blood–brain barrier. Curr Pharm Des 17:2778–2786. [DOI] [PubMed] [Google Scholar]
- 31. Wang Z, Hamman MA, Huang SM, Lesko LJ, Hall SD (2002) Effect of St John's wort on the pharmacokinetics of fexofenadine. Clin Pharmacol Ther 71:414–420. [DOI] [PubMed] [Google Scholar]
- 32. Xie R, Tan LH, Polasek EC, Hong C, Teillol‐Foo M, Gordi T et al (2005) CYP3A and P‐glycoprotein activity induction with St. John's Wort in healthy volunteers from 6 ethnic populations. J Clin Pharmacol 45:352–356. [DOI] [PubMed] [Google Scholar]
- 33. Zhou C, Verma S, Blumberg B (2009) The steroid and xenobiotic receptor (SXR), beyond xenobiotic metabolism. Nucl Recept Signal 7:e001. doi: 10.1621/nrs.07001. [DOI] [PMC free article] [PubMed] [Google Scholar]