

Abstract
The “pediatric targeted therapy” (PTT) program aims to identify the presence and activity of druggable targets and evaluate the clinical benefit of a personalized treatment approach in relapsed or progressive tumors on an individual basis. 10 markers (HDAC2, HR23B, p‐AKT, p‐ERK, p‐S6, p‐EGFR, PDGFR‐alpha/beta, p53 and BRAFV600E) were analyzed by immunohistochemistry. Pediatric patients with tumors independent of the histological diagnosis, with relapse or progression after treatment according to standard protocols were included. N = 61/145 (42%) cases were eligible for analysis between 2009 and 2013, the most common entities being brain tumors. Immunohistochemical stainings were evaluated by the H‐Score (0–300). In 93% of the cases potentially actionable targets were identified. The expressed or activated pathways were histone deacetylase (HDACs; 83.0% of cases positive), EGFR (87.2%), PDGFR (75.9%), p53 (50.0%), MAPK/ERK (43.3%) and PI3K/mTOR (36.1%). Follow‐up revealed partial or full implementation of PTT results in treatment decision‐making in 41% of the cases. Prolonged disease stabilization responses in single cases were noticed, however, response rates did not differ from cases treated with other modalities. Further studies evaluating the feasibility and clinical benefit of personalized diagnostic approaches using paraffin material are warranted.
Keywords: brain tumors, pediatric oncology, personalized medicine, targeted therapy, relapsed childhood tumors, predictive markers
Introduction
The survival of pediatric tumor patients has improved significantly during the last decades, with 81% of the patients now surviving more than 15 years after diagnosis 11. However, certain tumor entities are still related to a very unfavorable prognosis and tend to progress or relapse despite multimodal treatment. The largest subgroup of solid childhood malignancies with adverse overall survival is the class of malignant brain tumors, which accounts for 27.9% of cancer‐related deaths in children 11. For example, in medulloblastoma and ependymoma, two of the most common malignant brain tumors in childhood, the survival rates differ strongly depending on molecular subgroup 13, 14, 26, and can be as low as 22% or 20%, respectively. Prognosis is even more unfavorable once relapse occurs and standard therapies are lacking. The current knowledge of mechanisms driving tumor development and progression opens up new venues for targeted therapies using novel targeted substances. Most tumor cells harbor more than one tumor‐propagating change within different cell signaling pathways 6. Moreover, progressive or relapsed tumors often display additional molecular changes mediating resistance to standard treatment regimens 3. With novel technologies such as next‐generation sequencing allowing for a more detailed analysis of every individual tumor, it is now becoming clear that every tumor is indeed unique and harbors an individual set of aberrations, as we know, for example, from medulloblastoma 20. The concept of personalized oncology postulates that the identification of molecular changes driving tumor growth in the individual patient allows the treating physician to select the most promising treatment for each cancer patient 15 and first steps have indeed been made in this respect in pediatric oncology 24, 27. Therapies targeting tumors in a specific manner have proven to be successful and warrant further clinical trials 2, 4, 19, 23. Studies investigating an individual targeted treatment approach found benefits both in terms of response as well as survival, most notably the program for personalized oncology at the MD Anderson cancer center, treating 1144 adult patients with a personalized and targeted approach 25.
This study aimed to identify molecular targets (such as HDAC2, p‐AKT, p‐ERK, p‐EGFR, PDGFR‐alpha and ‐beta, p53, BRAFV600E) or predictive markers (such as HR23B or p‐S6) in progressive or relapsed solid pediatric tumors on an individual basis, using immunohistochemistry (IHC) on formalin‐fixed paraffin‐embedded (FFPE) tissue. Using these markers we assessed the presence and/or activity of six actionable targets: HDAC, epidermal growth factor (EGF), platelet derived growth factor (PDGF), mitogen activated protein kinase (MAPK), p53 and PI3K/mTOR. A questionnaire‐based follow‐up was performed to evaluate the feasibility and clinical benefit of the information gained by the pediatric targeted therapy (PTT) analysis.
Materials and Methods
Patients and clinical samples
All patients suffering from a relapsed or progressive solid childhood tumor were eligible for enrollment in this study independent of histopathological diagnosis. Prior treatment according to at least one standard treatment protocol and the lack of a standard protocol for the treatment of relapse and progression were mandatory for inclusion. FFPE samples of the primary or the relapsed tumor were submitted by the treating physicians from pediatric oncology centers in Europe. Based on distinct marker expression, drug target identification was performed in an individualized manner for every patient. The results were reported back to the inquiring pediatric oncologists for clinical decision‐making (Supporting Information Figure 1). Approval to access clinical data was obtained by the Heidelberg University Ethics Committee. All patients/or legal representatives provided written informed consent before clinical data collection.
Study design
This study was designed as a retrospective clinical feasibility study. Thirty five pediatric oncology centers were included. The primary endpoint was the feasibility of IHC‐based detection of tumor pathway alterations in patients with relapsed or progressive solid childhood tumors. Secondary objectives included assessment of response to an experimental personalized treatment based on results of the PTT analysis and determination of the turnaround time from sample shipment to dispatch of the report to the inquiring center.
Tumor pathways tested
The panel of pathways tested in this study was selected with the precondition that a pathway‐specific matching drug would be available as potential treatment option. Six marker, targets or tumor pathways were tested in this study for their presence and activity in the respective tumor specimen by IHC including HDAC, EGF, PDGF, MAPK, p53 and phosphatidylinositol‐3‐kinase/mammalian target of rapamycin (PI3K/mTOR) pathway. Specifically, for each pathway one to two associated markers were investigated: the activity of HDACs was tested by staining for the target HDAC2 and the predictive marker HR23B, which has been shown to govern the sensitivity of tumor cells to HDAC inhibitors via deregulation of proteasome activity 5 and is a predictive marker for HDACi sensitivity in cutaneous T‐cell lymphoma 12 and hepatocellular carcinoma 28. EGFR activation was investigated by determination of p‐EGFR. Expression of PDGFR‐alpha and ‐beta were used to determine PDGFR axis activation. p‐ERK and the BRAF V600E mutation were analyzed for MAPK/BRAF activation. p53 expression was determined to assess p53 activity and accumulation. The activity of PI3K/mTOR pathway was analyzed by IHC for p‐AKT and p‐S6 16.
Immunohistochemistry (IHC) and H‐Score
IHC was performed as reported previously 17 on 5‐μm thick FFPE sections. All slides submitted for analysis were specifically cut for the analysis. In case of external submission, slides were less than 2‐weeks‐old at time of staining, in case of internal submission (cases from the Neuropathology Department Heidelberg, Germany) slides were less than 1‐week‐old at time of staining. No significant regional heterogeneity was noted. Antibody staining was done using standard reagents provided by Ventana (Ventana, Tucson) and the OptiView DAB IHC Detection Kit (Ventana, Tucson). For antibodies see Supporting Information Table 1, for the BRAF V600E‐specific antibody see Capper et al 1. The following positive controls were used for the markers tested: HDAC2: breast carcinoma, HR23B: prostate, p‐AKT: glioblastoma, p‐ERK: pilocytic astrocytoma, p‐S6: normal prostate, p‐EGFR: breast adeno‐carcinoma, PDGFR‐alpha: glioblastoma, p53: glioblastoma, BRAFV600E: intracerebral melanoma metastasis. At least two experienced neuropathologists (A. Korshunov, D. Capper, F. Sahm, D. Reuss, C. Kölsche, A. von Deimling) evaluated the stained slides simultaneously on a multiheaded microscope, scoring was done in consensus of the two investigators. Especially for the phosphorylation specific antibodies, we repeatedly observed lower staining intensity in central tissue areas. In such cases, evaluation was performed in the stronger positive areas. The histological score (H‐Score) was used to quantify immunoreactivity 22. In brief, the score is obtained by the formula: H‐Score = 3 × percentage of strongly staining nuclei (%IHC3+ ×3) + 2 × percentage of moderately staining nuclei (%IHC2+ ×2) + percentage of weakly staining nuclei (%IHC1+ ×1), giving a range of 0–300. An H‐Score above 100 was considered positive for HDAC2 (nuclear), HR23B (nuclear or cytoplasmatic), p‐AKT (cytoplasmatic), p‐ERK (nuclear), p‐S6 (cytoplasmatic), p‐EGFR (cytoplasmatic or membrane), PDGFR‐alpha (cytoplasmatic or membrane) and PDGFR‐beta (cytoplasmatic or membrane). Any specific cytoplasmatic staining for BRAF V600E was scored as positive 1. The cutoff of the IHC for p53 was 10% stained nuclei, with any staining ≥10% considered positive. In a few cases, unspecific staining results were obtained and recorded accordingly.
Questionnaire‐based follow‐up and response assessment
Following approval by the University of Heidelberg ethics committee, a questionnaire‐based follow‐up was used to analyze how often and to which extent a targeted therapy was initiated by the inquiring centers, and if not for what reason a decision against targeted therapy was made. General clinical condition under applied treatment was recorded as well as individual response rates under personalized treatment. Response assessment by imaging was performed by the respective pediatric oncology centers using MRI, CT or sonography. The response rate was classified into progressive disease (PD), stable disease (SD), partial remission (PR) and complete remission (CR) by the local treating physician.
Results
Recruitment of patients
FFPE specimens of 145 patients with relapsed or progressive childhood tumors were submitted for analysis by 35 pediatric oncology centers between May 2009 and August 2013 (Figure 1). The material was obtained from standard of care biopsies, no biopsies were performed with the sole purpose of obtaining material for this study. n = 61 (42%) patients were eligible for enrollment in the study, while n = 84 (58%) patients could not participate in the study due to insufficient FFPE material, denial of consent or not returned questionnaires by the inquiring pediatric oncology center (Figure 1). Clinical and histopathological data of the n = 61 patients included in data analysis are summarized in Table 1. Two‐thirds of the study population were male (n = 42; 68.9%), and most patients were 4‐ to 18‐years‐old (n = 45; 73. 8%). The majority (n = 51; 82.0%) of enrolled patients suffered from progressive or relapsed tumors of the central nervous system, with anaplastic ependymoma (n = 15; 24.6%) and medulloblastoma (n = 11; 18.0%) being the most frequent diagnoses (Table 1). Most patients (n = 23; 37.7%) had previously been treated with a combination of chemotherapy, radiotherapy and surgery before inquiring for analysis, and a smaller group (n = 12; 19.7%) had already received second‐line or experimental treatment. The reason for request of the PTT analysis was either progression (n = 28; 45.8%) or relapse (n = 25; 41.0%) (no data: n = 8; 13.2%) (Table 1).
Figure 1.

Recruitment overview. FFPE samples were submitted and analyzed, and most of the samples (132/145 = 91%) were suitable for analysis. In case of successful IHC patients were contacted for their consent and included in the follow‐up. Number of cases indicated in brackets.
Table 1.
Overview of patients’ characteristics.
| Parameter | n (%) |
|---|---|
| Sex | |
| Male | 42 (68.9) |
| Female | 19 (31.1) |
| Age at enrollment (years) | |
| 0–1 | 4 (6.6) |
| 1–4 | 6 (9.8) |
| 4–18 | 45 (73.8) |
| >18 | 6 (9.8) |
| Diagnosis | |
| Brain tumors | 51 (82.0) |
| Anapl. Ependymoma WHO III | 15 (24.6) |
| Medulloblastoma | 11 (18.0) |
| Pilocytic Astrocytoma | 2 (3.3) |
| Ganglioglioma WHO I | 2 (3.3) |
| Astrocytoma WHO II | 3 (4.9) |
| Anaplastic Astrocytoma WHO III | 2 (3.3) |
| Glioblastoma WHO IV | 4 (6.6) |
| PNET | 2 (3.3) |
| AT/RT | 2 (3.3) |
| Ependymoblastoma | 1 (1.6) |
| Other brain tumor | 6 (9.8) |
| Non brain tumors | 10 (18.0) |
| Ewing sarcoma | 4 (6.6) |
| Ostesarcoma | 1 (1.6) |
| Neuroblastoma | 2 (3.3) |
| Desmoidtumor | 2 (3.3) |
| Nephroblastoma | 1 (1.6) |
| Other | 1 (1.6) |
| Previous treatment | |
| Chemotherapy only | 5 (8.2) |
| Surgery only | 2 (3.3) |
| Chemotherapy and radiation | 2 (3.3) |
| Chemotherapy and surgery | 4 (6.5) |
| Radiation and surgery | 3 (4.9) |
| Chemotherapy, radiation and surgery | 23 (37.7) |
| Second‐line experimental or relapse treatment | 12 (19.7) |
| No data | 10 (16.4) |
| Reason for request | |
| Relapse | 25 (41.0) |
| Progression | 28 (45.8) |
| No data | 8 (13.2) |
FFPE IHC uncovers actionable tumor pathway alterations in patient samples
Patients’ specimens were stained for the pathway markers described in Table 2, for IHC examples see Figure 2. Not every sample was investigated for the whole panel due to limited amount of material or material not suitable for phosphorylation specific antibody testing. Only two samples (3.3%) (Supporting Information Table 2) showed no aberration in any of the tested tumor pathways, while n = 52 (85.3%) of the tested samples displayed more than a single activated pathway, and the majority harbored equal to or more than three aberrations of different pathways (n = 43; 70.5%) (Supporting Information Table 2). The pathway tested positive with the highest frequency was EGF, with n = 41/47 (87.2%) tumors positive for p‐EGFR. HDAC was the second most commonly positive pathway with an overall of n = 49/59 (83.0%) of tested cases positive for HDAC2 and/or HR23B. The PDGF pathway tested positive in n = 41/54 (75.9%) of the cases, with PDGFR‐beta being positive in the majority of cases, as opposed to PDGF‐alpha. n = 26/60 (43.3%) of the samples tested positive for MAPK activation, as determined by positivity for p‐ERK and/or BRAF V600E. p53 was scored positive in n = 26/52 (50%) of the cases. PI3K/mTOR pathway activity was increased only in n = 21/61 (36.1%) of the FFPE samples as assessed by p‐AKT and p‐S6 IHC, thereby being the least frequent altered pathway of the panel.
Table 2.
Pathways and markers tested. Positivity of one pathway marker was sufficient to score for positivity of the respective pathway. H‐Score of 100 was used for positive interpretation of all markers except p53, where ≥10% positive cells were considered positive.
| Pathway | Patients tested | Patients not tested | Patients positive | Patients negative | Patients unspecific | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Marker | n | (% of all patients) | n | (% of all patients) | n | (% of patients tested) | n | (% of patients tested) | n | (% of patients tested) |
| HDAC | 59 | 96,7 | 2 | 3,3 | 49 | 83,1 | 9 | 15,3 | ||
| HDAC2 | 59 | 96,7 | 2 | 3,3 | 49 | 83,1 | 9 | 15,3 | 1 | 1,7 |
| HR23B | 50 | 82,0 | 11 | 18,0 | 44 | 88,0 | 2 | 4,0 | 4 | 8,0 |
| EGF | 47 | 77,0 | 14 | 23,0 | 41 | 87,2 | 5 | 10,6 | ||
| p‐EGFR | 47 | 77,0 | 14 | 23,0 | 41 | 87,2 | 5 | 10,6 | 1 | 2,1 |
| PDGF | 54 | 88,5 | 7 | 11,5 | 41 | 75,9 | 13 | 24,1 | ||
| PDGFR alpha | 54 | 88,5 | 7 | 11,5 | 5 | 9,3 | 49 | 90,7 | 0 | 0,0 |
| PDGFR beta | 54 | 88,5 | 7 | 11,5 | 39 | 72,2 | 14 | 25,9 | 1 | 1,9 |
| MAPK | 60 | 98,4 | 1 | 1,6 | 26 | 43,3 | 34 | 56,7 | ||
| p‐ERK | 60 | 98,4 | 1 | 1,6 | 24 | 40,0 | 36 | 60,0 | 0 | 0,0 |
| BRAF V600E | 13 | 21,3 | 48 | 78,7 | 2 | 15,4 | 11 | 84,6 | 0 | 0,0 |
| p53 | 52 | 85,2 | 9 | 14,8 | 26 | 50,0 | 26 | 50,0 | ||
| p53 | 52 | 85,2 | 9 | 14,8 | 26 | 50,0 | 26 | 50,0 | 0 | 0,0 |
| PI3K/m‐TOR | 61 | 100,0 | 0 | 0,0 | 21 | 34,4 | 39 | 63,9 | ||
| p‐AKT | 60 | 98,4 | 1 | 1,6 | 8 | 13,3 | 51 | 85,0 | 1 | 1,7 |
| p‐S6 | 19 | 31,1 | 42 | 68,9 | 19 | 100,0 | 0 | 0,0 | 0 | 0,0 |
Figure 2.
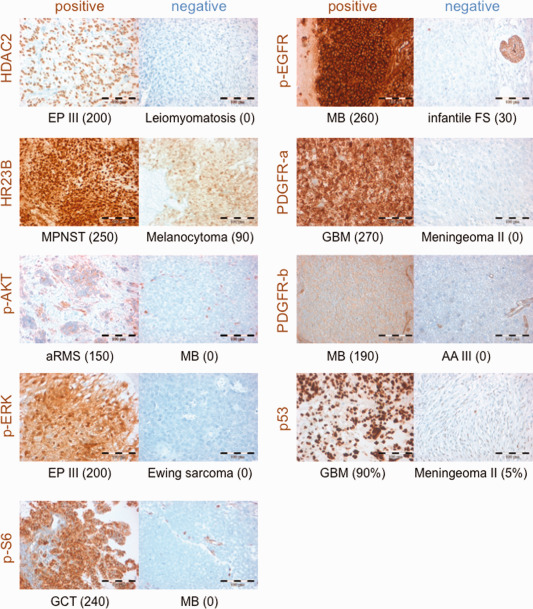
Examples of IHC staining for markers used in this study. Images depict samples with IHC stainings (brown color) scored positive (H‐Score ≥ 100) or negative (H‐Score < 100). The tumor histology is given below each image, in brackets the individual H‐Score is given, except for p53 where the percentage of positive nuclei is given. EP III: anaplastic ependymoma WHO III; MPNST: malignant peripheral nerve sheath tumor; aRMS: alveolar rhabdomyosarcoma; MB: medulloblastoma WHO IV; GCT: germ cell tumor of the pineal gland; infantile FS: infantile fibrosarcoma; GBM: glioblastoma WHO IV; AA III: anaplastic astrocytoma WHO III.
Altered tumor pathways as potential targets
Table 3 shows matching of tumor pathways with targeted drugs, which was the basis of the analysis report. According to the IHC results, most tumor specimens investigated in our series exhibited three or more actionable pathways. Consequently, the average number of matching drugs was n = 2.9. The results of IHC and drug mapping were included in the final PTT report. In n = 57/61 (93.0%) of the investigated cases a correlation between IHC findings and matching drugs was possible. No priority was given to any target‐drug combination, as the conclusion from the target analysis (ie, the choice of drug) remained the sole responsibility of the treating physician.
Table 3.
Overview of pathway‐drug matching.
| Patients positive | ||||
|---|---|---|---|---|
| Pathway | n | (% of patients tested) | Matching drug category | Matching drug example |
| HDAC | 49 | 83.1 | HDAC inhibitor | Valproic acid or vorinostat |
| EGF | 41 | 87.2 | EGFR inhibitor | Erlotinib or gefitinib |
| PDGF | 41 | 75.9 | PDGFR inhibitor | Imatinib or sunitinib |
| MAPK | 26 | 43.3 | BRAF inhibitor, MEK inhibitor | Sorafenib, trametinib |
| BRAF V600E | 2 | 15.4 | BRAF V600E inhibitor | Vemurafenib |
| p53 | 26 | 50.0 | p53 activiation | Low dose actinomycin D |
| PI3K/mTOR | 21 | 34.4 | PI3K/AKT/m‐TOR inhibitor | Everolimus or sirolimus |
Turnaround and follow‐up
The mean turnaround time between arrival of the tumor samples and final reporting of results was 5.7 weeks (arithmetic mean). It was left to the discretion of the local treating physician whether and how to use the report for his clinical decision‐making. A questionnaire‐based follow‐up was used to determine the proportion of treatments initiated by the results of the PTT analysis, as well as the general clinical condition and response of patients to personalized therapy according to the assessment of the local treating pediatric oncologist.
In n = 25 (41.0%) cases, a targeted treatment was initiated (Figure 3A). In n = 33 (54.0%) cases the results from the PTT report did not prompt the initiation of a targeted treatment, the major reasons being that a treatment other than based on the PTT results had already been initiated (SD under current treatment (n = 9; 27.5%), enrollment in another study (n = 9; 27.5%), cytotoxic chemotherapy (n = 2; 6%) and advanced disease at the time the PTT study results were available (n = 5; 15%) (Figure 3B).
Figure 3.
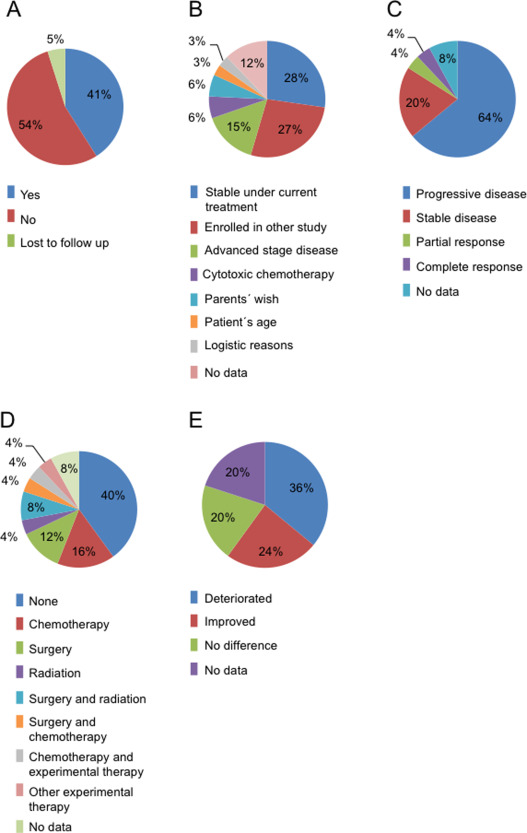
Results of personalized diagnostics can be used for treatment. A. In 41% of the cases PTT results were used by the treating physician for further therapeutic decision‐making (blue), in 54% the results were not used (red). B. Reasons for not using the PTT results for further therapy, as given by the treating physician. The most common reasons were a previous initiation of a treatment other than based on the PTT results (SD under current therapy, enrollment in a different study, classical therapy) or a disease that was advanced stage. C. Response of children treated at least partly based on the PTT results. 28% of the patients showed at least a SD (SD, PR and CR), while 64% of the patients did not respond to targeted therapy (PD). D. Additional treatment administered simultaneously with targeted therapy. While 40% of the patients received targeted therapy alone (blue), 52% of the patients were additionally treated with conventional therapy as indicated. E. Patient's subjective clinical condition under targeted therapy: 44% of the patients showed either no difference or improvement of their clinical condition, while 36% of the patients showed a deterioration of their clinical condition.
Table 4 summarizes all patients treated based on PTT results. Due to the limited sample size and pronounced heterogeneity of the cohort studied here in terms of both diagnoses and treatments, conclusions on therapy effects have to be drawn with extreme caution, if at all possible. Of the patients treated fully or partially based on the PTT results, n = 7 (28.0%) showed at least a SD (SD: n = 5, 20.0%; PR: n = 1, 4.0%; CR: n = 1, 4.0%), while n = 16 (64%) of the patients did not respond (PD). For n = 2 (8.0%) patients no data for response evaluation was available (Figure 3C). For example, PR under targeted therapy in addition to other modalities was seen in a patient with anaplastic ependymoma (PTT plus reresection) and CR in an AT/RT patient (PTT plus cytotoxic chemotherapy). In total n = 15 (60.0%) patients received additional conventional treatments including chemotherapy, radiation therapy, surgery or a combination thereof (Figure 3D). The response rates in patients treated with targeting agents did not differ from the response rate in the remaining patients: of n = 33 patients, who did not receive targeted therapy, n = 9 (27.2%) showed at least (SD: n = 6, 18.2%; PR: n = 0, 0.0%; CR: n = 3, 9.1%), while n = 13 (39.4%) showed PD.
Table 4.
Response under targeted therapy.
| Patient no. | Diagnosis | Reason for request | Pathways altered in patient | Targeted treatment | Other drugs administered | Duration of treatment (months) | Method of response assessment | Timepoint of response assessment (months) | Outcome |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Anaplastic ependymoma WHO III | Relapse | HDAC | Sorafenib | No | No data | MRI | 9 | PD |
| MAPK/ERK | |||||||||
| PIK3/mTOR | |||||||||
| 2 | Anaplastic ependymoma WHO III | Progression | MAPK/ERK | Sorafenib | Temozolomide | No data | MRI | 9 | PD |
| PIK3/mTOR | Sirolimus | ||||||||
| HDAC | Vorinostat | ||||||||
| 3 | Neuroblastoma | Relapse | HDAC | Vorinostat | No data | No data | MRI | 4 | SD |
| MAPK/ERK | |||||||||
| 4 | Desmoplastic small‐blue‐round‐cell tumor | Relapse | HDAC | Valproat | Topotecan | No data | CT | 11 | PD |
| p53 | Actinomycin D | ||||||||
| 5 | AT/RT | Progression | HDAC | Valproat | No | No data | – | 1 | PD |
| MAPK/ERK | |||||||||
| PDGF | |||||||||
| 6 | Anaplastic ependymoma WHO III | Progression | HDAC | Vorinostat | No data | No data | No data | No data | No data |
| MAPK/ERK | Sorafenib | ||||||||
| PIK3/mTOR | Sirolimus | ||||||||
| 7 | Ganglioglioma WHO I | Progression | HDAC | Vorinostat | No | 10 | MRI | 10 | SD |
| MAPK/ERK | Sorafenib | ||||||||
| PIK3/mTOR | Sirolimus | ||||||||
| p53 | Actinomycin D | ||||||||
| 8 | Anaplastic ependymoma WHO III | Relapse | MAPK/ERK | Sorafenib | No | 2 | MRI | 2 | PD |
| p53 | Actinomycin D | ||||||||
| EGF | gefitinib/erlotinib | ||||||||
| 9 | Ewing sarcoma | Relapse | HDAC | Desatinib | No | No data | MRI | No data | SD |
| p53 | |||||||||
| PDGF | |||||||||
| 10 | Diffuse astrocytoma WHO III | Gliomatosis cerebri | HDAC | Valproat | no | No data | MRI | No data | PD |
| p53 | |||||||||
| EGF | |||||||||
| 11 | Anaplastic ependymoma WHO III | Relapse | HDAC | Vorinostat | No data | 1 | MRI | 1 | PD |
| EGF | |||||||||
| 12 | Medulloblastoma | Progression | HDAC | Valproat | Etoposide | 5 | MRI | 5 | PD |
| p53 | Actinomycin D | Temozolomide | |||||||
| ERF | |||||||||
| 13 | AT/RT | Relapse | HDAC | Valproat | Trofosfamide/etoposide | No data | MRI | No data | CR |
| PIK3/mTOR | |||||||||
| p53 | |||||||||
| EGF | |||||||||
| 14 | PNET | Relapse | HDAC | Valproat | 6 | MRI | 6 | PD | |
| PDGF | |||||||||
| 15 | Ewing sarcoma | Relapse | HDAC | Vorinostat | No | No data | CT | 3 | SD |
| PDGF | |||||||||
| 16 | Endocrine pancreatic carcinoma | Progression | HDAC | Sunitinib | No | Sonography | 16 | PD | |
| EGF | |||||||||
| PDGF | |||||||||
| 17 | Medulloblastoma | Relapse | HDAC | Vorinostat | No data | 2 | MRI | No data | PD |
| EGF | |||||||||
| PDGF | |||||||||
| 18 | Anaplastic ependymoma WHO III | Progression | HDAC | Valproat | Etoposide | No data | MRI | 2.0 | PD |
| EGF | Erlotinib | ||||||||
| PDGF | Sunitinib | ||||||||
| 19 | Plexiform neurofibroma | No data | HDAC | VEGFi | No data | No data | MRI | 2.0 | SD |
| EGF | (Bevacizumab) | ||||||||
| PDGF | |||||||||
| 20 | Astrocytoma II | Malignisation | HDAC | Vorinostat | No | No data | MRI | No data | No data |
| (Astrocytoma III) | MAPK/ERK | ||||||||
| PIK3/mTOR | |||||||||
| EGF | |||||||||
| PDGF | |||||||||
| 21 | Medulloblastoma | Progression | HDAC | Imatinib | No data | No data | MRI | No data | PD |
| EGF | |||||||||
| PDGF | |||||||||
| 22 | Anaplastic ependymoma WHO III | Relapse | HDAC | Valproat | Etoposide | No data | MRI | No data | PR |
| PIK3/mTOR | Sirolimus | ||||||||
| EGF | Erlotinib | ||||||||
| PDGF | Sunitinib | ||||||||
| 23 | Medulloblastoma | Relapse | HDAC | Valproat | Other experimental | No data | MRI | No data | PD |
| PIK3/mTOR | Approach (MEMMAT‐protocol) | ||||||||
| EGF | |||||||||
| PDGF | |||||||||
| 24 | Medulloblastoma | Relapse | HDAC | Valproat | Sirolimus | No data | MRI | 3.0 | PD |
| EGF | |||||||||
| PDGF | |||||||||
| 25 | SHH‐medulloblastoma | Relapse | HDAC | LD225 | No | No data | MRI | No data | PD |
| MAPK/ERK | |||||||||
| PIK3/mTOR | |||||||||
| p53 | |||||||||
| EGF | |||||||||
| PDGF |
Under personalized treatment an improvement of general condition was seen in n = 6 (24.0%) patients, no difference in n = 5 (20.0%) and further deterioration under in n = 9 (36.0%) patients (Figure 3E). Again the results in patients not treated with matching drugs were similar: n = 5 (15.2%) patients showed improvement, n = 8 (24.2%) deterioration and n = 5 (15.2%) no difference of their general condition.
Discussion
We here report that identification of druggable molecular targets in samples of progressive or relapsed solid childhood tumors on an individual basis is feasible. The majority of cases (98.0%) of this multicenter cohort (n = 61) exhibited at least one actionable pathway. On the technical side, while the positive staining of at least one IHC marker in the vast majority of cases indicates sufficient general immunoreactivity of the samples, it cannot be entirely excluded that the two samples negative for all markers were impaired regarding immunoreactivity. As often most of the tissue used in this study was collected at the time of primary diagnosis, the samples investigated here may not display exactly the same biology as the progressing or relapsed tumors. Although certain molecular features such as, for example, medulloblastoma subgroup remain stable from first tumor to relapse 21, relapsed tumors may also acquire novel molecular alterations with relevance for optimal treatment regimes. Hill et al recently showed that combined MYC and p53 defects, which are not present at first diagnosis, can be detected in some relapsed medulloblastoma samples 7. In a case of progressing anaplastic ependymoma, we have previously shown that genetic alterations accumulate during cytotoxic treatment over time 18. According to Wolff et al 27, however, individual detection of altered tumor pathways in samples taken at any time in patient's history will likely display the tumor's biology more precisely than conventional histology. Nevertheless, it seems appropriate to preferably investigate the recurrent or progressive tumor instead of the primary tumor whenever possible.
Of note, we found more than one pathway alteration per tumor in n = 52 (85.3%) patients. This finding stands in contrast to results of the personalized medicine phase I clinical trials program of the MD Anderson cancer center initiative where Tsimberidou et al found no aberration in 59.8% and only one aberration in 33.1% of all investigated adult cancer patients 25. Other than completely different tumor entities in both studies, two reasons might be responsible for the difference in these findings. First, we found two pathways positive in most of the cases, EGF and HDAC2 with 87.2% and 83.0%, respectively. This could indicate that these markers are weakly or noninformative as used under these distinct conditions. Most likely, the arbitrary H‐Score cutoff of 100 used in our study is not optimal for all pathways studied. H‐score cutoffs, therefore, have to be re‐evaluated and determined separately for every marker. Second, Tsimberidou et al screened for a panel of distinct mutations in oncogenes and losses of tumor suppressors, while we screened for expression or activation of tumor pathways. Activation of ERK, for example, can depend on many causes in the upstream MAPK‐signaling pathway and, therefore, this marker reflects a whole set of different activating events. Pilocytic astrocytoma, for example, is known to be most often dependent on KIAA1549:BRAF‐fusions 9 but can also be caused by BRAF‐V600E mutations, FGFR1 mutations, KRAS mutations or NF1 mutations 8. The downstream target p‐ERK would be activated in any of these cases. Targeted screening for single mutations in this context may underestimate druggable pathway alterations, but on the other hand might also reveal more precise information of distinct targets (eg, type of mutation). In a study by Jones et al, it was found that sequencing for targetable mutations indeed seems to be not only a clinically feasible approach, but also discovers actionable targets in the majority of cases 10. The study by Jones et al furthermore highlighted the importance of sequencing both tumor and normal DNA, as illustrated by detection of germline mutations in 3% of the cases, conversely, and a high rate of false positive alterations identified if only tumor DNA was sequenced 10.
Our observation that most tumors show activation of several druggable signaling pathways implies the need for a multitargeting approach, while at the same time indicating the need for an exact characterization of pathway and marker alterations in the individual tumor. In future personalized medicine programs next‐generation diagnostics such as whole genome sequencing and methylation arrays will be implemented as well and will give more precise information about every tumor's alterations, hopefully resulting in a better informed selection of targeted drugs.
Only 42% of the patients were eligible for our study, the vast majority of the noneligible patients were excluded because of missing consent, either because it was denied or because of no reply at all (see Figure 1). The retrospective nature of this study can explain the large number of missing consents. This highlights the need for future prospective studies, which will need to obtain consent upfront, as is usually mandatory. Only a small fraction (n = 13/145 = 8.97%) of patients was excluded because of insufficient tissue for IHC. It is possible that a small number of patients will be excluded because of insufficient material in future studies, however, it is likely that the number of these cases will be smaller in prospective designs.
Our follow‐up revealed that treatment suggestions based on our study's results were translated into clinical decision‐making in 41% of the cases, demonstrating feasibility. 24.0% of patients treated with targeted agents showed at least a SD. Due to the retrospective and descriptive nature of this study, this has to be interpreted with caution however. Advanced stage disease was the third most frequent reason why personalized therapy was not administered. With particular emphasis on this finding and taking into account that the mean turnaround time from sample submission to target reporting was 5.7 weeks in this study, a shortened turnaround time could increase the percentage of cases where targeted therapy is applied in future trials.
In conclusion, we show that personalized diagnostics in pediatric oncology is clinically feasible, yields actionable results in the majority of cases, and leads to informed clinical decision‐making. Most patients display more than one signaling pathway aberration, suggesting the need of multitargeted approaches in interventional trials in relapsed pediatric solid tumors. However, improvements are needed for future personalized therapy programs to convey measurable benefits for the patients: (i) for accuracy of target detection and molecular subgrouping molecular diagnostics such as targeted or whole genome sequencing (including both tumor and normal DNA), and whole genome methylation analysis should be added to the diagnostic repertoire; (ii) the analysis and reporting process has to be accelerated significantly to reach a reasonably short turnaround time from sample submission to report, which is crucial for fast clinical implementation of molecular results; and finally (iii) thorough clinical follow‐up of targeted treatment based on individualized analysis is needed to evaluate the clinical benefit of these novel approaches for the patients.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher≈s web‐site:
Table S1. Antibodies used in this study.
Table S2. Number of pathway aberrations per patient.
Figure S1.
Acknowledgments
This work was supported by the German Childhood Cancer Foundation (Deutsche Kinderkrebsstiftung) and the Parents Initiative DLFH‐Pfalz e.V. JE was supported by Talents in Medicine program of University Hospital Heidelberg and by The German National Academic Foundation (Studienstiftung des deutschen Volkes), IO was supported by the H.W.&J. Hector Stiftung, Weinheim (#M71) and the DFG (OE 542/2‐1).
Conflict of interest
OW receives clinical trial support from MSD/Merck and Novartis, research grant funding from Bayer Healthcare and served as a scientific advisory board member for Astra Zeneca, Novartis, MSD/Merck. Under a licensing agreement between Ventana Medical Systems, Inc., Tucson, Arizona and the German Cancer Research Center, Dr. Capper and Dr. von Deimling are entitled to a share of royalties received by the German Cancer Research Center on the sales of VE1 antibody. The terms of this arrangement are being managed by the German Cancer Research Center in accordance with its conflict of interest policies.
References
- 1. Capper D, Preusser M, Habel A, Sahm F, Ackermann U, Schindler G, et al (2011) Assessment of BRAF V600E mutation status by immunohistochemistry with a mutation‐specific monoclonal antibody. Acta Neuropathol 122:11–19. [DOI] [PubMed] [Google Scholar]
- 2. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364:2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al (2012) Clonal evolution in relapsed acute myeloid leukaemia revealed by whole‐genome sequencing. Nature 481:506–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, et al (2007) Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T‐cell lymphoma (CTCL). Blood 109:31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fotheringham S, Epping MT, Stimson L, Khan O, Wood V, Pezzella F, et al (2009) Genome‐wide loss‐of‐function screen reveals an important role for the proteasome in HDAC inhibitor‐induced apoptosis. Cancer Cell 15:57–66. [DOI] [PubMed] [Google Scholar]
- 6. Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674. [DOI] [PubMed] [Google Scholar]
- 7. Hill RM, Kuijper S, Lindsey JC, Petrie K, Schwalbe EC, Barker K, et al (2014) Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jones DT, Gronych J, Lichter P, Witt O, Pfister SM (2012) MAPK pathway activation in pilocytic astrocytoma. Cell Mol Life Sci 69:1799–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, Collins VP (2008) Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68:8673–8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jones S, Anagnostou V, Lytle K, Parpart‐Li S, Nesselbush M, Riley DR, et al (2015) Personalized genomic analyses for cancer mutation discovery and interpretation. Sci Transl Med 7:283ra53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kaatsch P, Spix C (2014) German Childhood Cancer Registry ‐ Report 2013/14 (1980‐2013). Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI), University Medical Center of the Johannes Gutenberg University Mainz.
- 12. Khan O, Fotheringham S, Wood V, Stimson L, Zhang C, Pezzella F, et al (2010) HR23B is a biomarker for tumor sensitivity to HDAC inhibitor‐based therapy. Proc Natl Acad Sci USA 107:6532–6537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kool M, Korshunov A, Remke M, Jones DT, Schlanstein M, Northcott PA, et al (2012) Molecular subgroups of medulloblastoma: an international meta‐analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, group 3, and group 4 medulloblastomas. Acta Neuropathol 123:473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Korshunov A, Witt H, Hielscher T, Benner A, Remke M, Ryzhova M, et al (2010) Molecular staging of intracranial ependymoma in children and adults. J Clin Oncol 28:3182–3190. [DOI] [PubMed] [Google Scholar]
- 15. La Thangue NB, Kerr DJ (2011) Predictive biomarkers: a paradigm shift towards personalized cancer medicine. Nat Rev Clin Oncol 8:587–596. [DOI] [PubMed] [Google Scholar]
- 16. Magnuson B, Ekim B, Fingar DC (2012) Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J 441:1–21. [DOI] [PubMed] [Google Scholar]
- 17. Milde T, Hielscher T, Witt H, Kool M, Mack SC, Deubzer HE, et al (2012) Nestin expression identifies ependymoma patients with poor outcome. Brain Pathol 22:848–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Milde T, Kleber S, Korshunov A, Witt H, Hielscher T, Koch P, et al (2011) A novel human high‐risk ependymoma stem cell model reveals the differentiation‐inducing potential of the histone deacetylase inhibitor Vorinostat. Acta Neuropathol 122:637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mosse YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, et al (2013) Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large‐cell lymphoma: a children's oncology group phase 1 consortium study. Lancet Oncol 14:472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Northcott PA, Jones DT, Kool M, Robinson GW, Gilbertson RJ, Cho YJ, et al (2012) Medulloblastomics: the end of the beginning. Nat Rev Cancer 12:818–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ramaswamy V, Remke M, Bouffet E, Faria CC, Perreault S, Cho YJ, et al (2013) Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol 14:1200–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rizzardi AE, Johnson AT, Vogel RI, Pambuccian SE, Henriksen J, Skubitz AP, et al (2012) Quantitative comparison of immunohistochemical staining measured by digital image analysis versus pathologist visual scoring. Diagn Pathol 7:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shaw AT, Yeap BY, Solomon BJ, Riely GJ, Gainor J, Engelman JA, et al (2011) Effect of crizotinib on overall survival in patients with advanced non‐small‐cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol 12:1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shukla N, Schiffman J, Reed D, Davis IJ, Womer RB, Lessnick SL, et al (2013) Biomarkers in Ewing sarcoma: the promise and challenge of personalized medicine. a report from the children's oncology group. Front Oncol 3:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tsimberidou AM, Iskander NG, Hong DS, Wheler JJ, Falchook GS, Fu S, et al (2012) Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clin Cancer Res 18:6373–6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Witt H, Mack SC, Ryzhova M, Bender S, Sill M, Isserlin R, et al (2011) Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell 20:143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wolff JE, Brown RE, Buryanek J, Pfister S, Vats TS, Rytting ME (2012) Preliminary experience with personalized and targeted therapy for pediatric brain tumors. Pediatr Blood Cancer 59:27–33. [DOI] [PubMed] [Google Scholar]
- 28. Yeo W, Chung HC, Chan SL, Wang LZ, Lim R, Picus J, et al (2012) Epigenetic therapy using belinostat for patients with unresectable hepatocellular carcinoma: a multicenter phase I/II study with biomarker and pharmacokinetic analysis of tumors from patients in the Mayo Phase II Consortium and the Cancer Therapeutics Research Group. J Clin Oncol 30:3361–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher≈s web‐site:
Table S1. Antibodies used in this study.
Table S2. Number of pathway aberrations per patient.
Figure S1.