

Abstract
Pediatric cortical glioneuronal benign tumors mainly include gangliogliomas (GG) [differential diagnoses pilocytic astrocytomas (PA) and pleomorphic xanthoastrocytomas (PXA)] and dysembryoplastic neuroepithelial tumor (DNT). DNT include the specific form and the controversial non‐specific form that lack the specific glioneuronal element. Our aims were to search for BRAFV600E mutation and CD34 expression in DNT, PXA, GG and PA to correlate BRAFV600E mutation with BRAFV600E expression and to evaluate their diagnostic and prognostic values. Ninety‐six children were included. BRAFV600E mutation was studied by sequencing and immunohistochemistry; CD34 expression was analyzed by immunohistochemistry. BRAFV600E mutation was detected in PXA (60%), GG (38.7%), DNT (30%, including 3/11 specific and 3/9 non‐specific forms) and PA (12.5%). BRAFV600E expression was recorded in PXA (60%), GG (45.2%) and DNT (30%). CD34 expression was recorded in PXA (60%), GG (58.1%), DNT (25%) and PA (12.5%). Neither CD34 expression nor BRAFV600E status was predictive of prognosis, except for PA tumors where CD34 expression was associated with a shorter overall survival. In conclusion, DNT shared with PXA and GG, BRAFV600E mutation and/or CD34 expression, which represent molecular markers for these tumors, and we recommend searching for CD34 expression and BRAFV600E mutation in all DNT, especially the non‐specific forms.
Keywords: BRAFV600E expression, BRAFV600E mutation, CD34, dysembryoplastic neuroepithelial tumors (DNT), gangliogliomas (GG), pleomorphic xanthoastrocytomas (PXA)
Introduction
Cortical glioneuronal tumors of children and young adults include dysembryoplastic neuroepithelial tumors (DNT) and gangliogliomas (GG). DNT are benign lesions affecting young patients, clinically characterized by drug‐resistant partial seizures, normal neurologic examination and excellent prognosis 6. Neuroimaging typically shows a predominantly cortical and well‐demarcated non‐contrast‐enhancing lesion devoid of edema 11. Three histological forms have been described 16. The complex form is characterized by the association of specific glioneuronal elements (GNE) and glial nodules often associated with foci of cortical dysplasia, the simple form demonstrates only the GNE, and the non‐specific form does not show the GNE but displays the same clinical neuroimaging features as complex DNT. This peculiar subtype, although not recognized as a distinct DNT subtype, is discussed in the histopathology section in the World Health Organization (WHO) classification of tumors of the central nervous system (CNS) 7. Because non‐specific DNT mimics at histology any kind of glioma, this latter form is highly controversial. However, we have previously shown that specific form of DNT and intra‐cortical grade II gliomas share no isocitrate dehydrogenase (IDH) mutation in most cases, no p53 expression, normal comparative genomic hybridization analysis and same biological behavior 17, favoring the concept of non‐specific DNT. Interestingly, it is obvious that in contrast to DNT (specific and non‐specific), “true” diffuse grade II gliomas demonstrate a more infiltrative pattern and are usually not restricted to the cortex.
GGs share with DNT cortical location, epilepsy and good prognosis, but imaging features are different. They typically demonstrate a well‐demarcated cystic tumor with a contrast‐enhancing nodule. At pathological levels, diagnosis is easy if clusters of ganglionic cells are present, associated with a glial component often made of elongated, piloid cells, eosinophilic granular bodies and lymphocytic exudates. However, because ganglionic cells are focally distributed, they might be absent if surgical resection is not complete. In this case, diagnosis is challenging. Moreover, in some cases, bizarre, often multi‐nucleated cells might be encountered. Depending on the absence or presence of signs of anaplasia among the glial component, GG are grade I or III. A peculiar form, desmoplasic infantile ganglioglioma (grade I) is recorded in infants. The main differential diagnoses of GG are pilocytic astrocytomas (PA), when typical ganglionic cells are lacking, and pleomorphic xanthoastrocytomas (PXA). Interestingly, GG, PA and PXA share the same neuroradiological features. PXA is a rare tumor entity accounting for less than 1% of all astrocytic tumors. On pathological examination, it shows pleomorphic and lipidized astrocytic cells, eosinophilic granular bodies, a variably dense reticulin network, as well as neuronal marker expression, and inflammatory cells 16. PXA are classified as grade II. According to the WHO, lesions with significant mitotic activity (5 or more mitoses per 10 HPF) and/or with areas of necrosis are designed as “pleiomorphic xanthoastrocytoma with anaplastic features.”
Recently, BRAF alterations have been recorded in PA, PXA and GG. Although PA of the posterior fossa often demonstrated KIAA1549 : BRAF fusion 5, 12, extracerebellar PA, PXA and GG (whatever the grade) displayed BRAFV600E mutation in 33%, 65% and 18% of cases, respectively 23. BRAFV600E mutation was not recorded in the four DNT cases included in this series 23. Interestingly, an antibody directed against the specific BRAFV600E mutated antigen has recently been obtained with a very good correlation between immunohistochemistry and molecular biology in melanomas 4. In addition to BRAFV600E mutation, PXA, GG and DNT share the expression of CD34. This transmembrane protein expressed on the surface of hematopoietic progenitor cells and on vascular endothelium is also a stem cell marker transiently expressed during early neurulation but is not recorded in developing and adult brains 15, 25. CD34 is evenly known to be a common marker for glioneuronal lesions associated with chronic intractable epilepsy 2, 3, 8, 24.
Altogether, BRAFV600E recurrent mutation, CD34 expression and evidence for neuronal markers suggest that DNT, PXA, GG and some PA might belong to the same spectrum of tumors, although the diagnostic and prognostic values of these markers remain to be demonstrated. In contrast, these glial and glioneuronal tumors most often lack IDH mutation 17, 27.
In the present study, we have searched for BRAFV600E mutation and expression, CD34 and IDH1R132H expression in a series of 96 glial and glioneuronal pediatric tumors. Our aims were (i) to search for the incidence of BRAFV600E mutation in a series of pediatric epileptic cortical tumors including 20 DNT (11 specific and 9 non‐specific); (ii) to compare the incidence of BRAFV600E mutation in these tumors with other tumors known to express this mutation (PXA, GG); (iii) to correlate BRAFV600E mutation detected by DNA direct sequencing with BRAFV600E expression assessed by a specific monoclonal antibody; and (iv) to evaluate the diagnostic and prognostic values of these markers in this group of glial and glioneuronal tumors.
Materials and Methods
Patients and clinical data
Ninety‐six patients (age at diagnosis less than 20 years) with DNT (specific DNT, 11 patients; non‐specific DNT, 9 patients), PXA (5 patients), GG (31 cases), PA including classic pilocytic astrocytomas (34 cases) and PMA, (6 cases) were included in this retrospective study. In one case of complex DNT, cortical dysplasia was obvious, characterized by disorganized cortical lamination and numerous clusters of abnormal neurons, usually of small caliber and containing an abnormal amount of Nissl substance. Because neither eosinophilic granular bodies nor lymphocyte cuffing was recorded, it was difficult to assess whether this case might represent a composite GG and DNT as previously reported 19. All patients underwent surgery at our institution (Assistance Publique‐Hôpitaux de Marseille, La Timone Hospital, Marseille, France) between July 1986 and March 2011. For all patients, the following clinical data were collected: date of birth, age at diagnosis, gender, tumor location, extent of surgical resection [complete resection (CR); partial resection (PR)] and follow‐up: date of relapse, date of last medical examination and clinical status at last medical examination [free of disease (FOD); alive with stable disease (AWSD);, alive with progressive disease (AWPD); dead].
Pathology material
Tumor specimens were obtained according to a protocol approved by the local institutional review board, ethics committee and conducted according to the national regulations. Pathological material was available in all cases. Surgical specimens were fixed in formalin or Bouin's fixative (for the oldest ones, which included 5 PA) and embedded in paraffin. Some of them (17 cases) were also frozen in liquid nitrogen and stored in the AP‐HM tumor bank (authorization number 2008/70). Regarding PA, our series included 34 grade I (classic PA) and 6 grade II (PMA) selected because their KIAA1549 : BRAF fusion status was already determined by reverse‐transcription polymerase chain reaction (RT‐PCR) 5 and 31/40 (77.5%) demonstrated the fusion. For DNT, previous molecular data reported lack of p53 expression and lack of 1p19q co‐deletion in all cases, whereas 1/20 case (DNT15) demonstrated IDH1R132H expression and mutation 17.
Immunohistochemistry
BRAFV600E expression [VE1 clone kindly provided by D. Capper and A. Von Deimling 4], CD34 expression (QBEnd10 clone, Dako, Les Ulis, France) and IDH1R132H expression (H09 clone, Dianova, Hamburg, Germany) were analyzed in all cases. All analyses were performed on 5‐μm sections of formalin‐ (or Bouin‐) fixed paraffin embedded tissue by using an automated immunohistochemical procedure on Ventana Benchmark devices. The immunostaining was performed on the whole section for DNT and PXA, whereas we used tissue microarray (TMA) sections for PA and GG. In the second step, immunostaining was also performed on the whole section for cases showing discrepant results for BRAFV600E expression and mutation. BRAFV600E and IDH1R132H expression were recorded as positive or negative. Regarding CD34 expression, when present, it was classified as “solitary” (only focal immunostaining limited to few cells with intense ramification of processes), “bushy” (multifocal immunostaining) and “diffuse” (large areas of homogeneous immunostaining, where individual perikarya or cell processes could not be identified) according to previous reports 2, 8, 24. When present, peculiar attention was paid to the adjacent cortex to the tumor.
Genomic DNA extraction and BRAFV600 mutation analysis
Areas of viable and representative tumor following review of all blocks were marked by a pathologist (DFB). Then, tumor DNA was extracted from 4 × 5 μm thick sections of FFPE tissue after dewaxing (55 cases) or 50 mg of frozen tissue (17 cases). DNA were purified by automated extraction on Evo75® (Tecan, Lyon, France) according to the M&N protocol; NR (NucleoSpin® 96 Blood; NucleoSpin® 8 Viruses Binding stripes, Macherey‐Nagel, Hoerd, France). Elution was carried out in bovine serum albumin 500 μg/mL, and DNA samples were stored at −20°C until use. DNA were analyzed by a combination of polymerase chain reaction‐high resolution melting (PCR‐HRM) and direct sequencing, as previously described 21. Only the samples with no wild‐type HRM profiles were sequenced. Sample and mix (HRM Master Mix®, Roche Diagnostics, Meylan, France) distribution were automated and performed on 96 well plates (Evo75®, Tecan), and reactions were performed in duplicate. Mutated reference samples (cell lines), wild‐type DNA (placenta), dewaxing, extraction and PCR negative controls were included together with the samples to be analyzed. PCR was then carried out on LightCycler® 480 (Roche). The type of mutations defined as “not wild type” by PCR‐HRM was determined by sequencing Sanger (MixBigDye® 5X, Roche). The sequences were analyzed on 3500 or 3130 Dx Genetic Analyser® (Applied Biosystems, Saint Aubin, France).
Statistical analysis
This was performed using PASW Statistics version 17.02 (IBM SPSS Inc., Chicago, IL, USA) and MedCalc version 12 (MedCalc Software bvba, Ostend, Belgium). Continuous variables were expressed as median with interquartile range (IQR), and categorical variables were reported as count and percentages. Comparisons of mean values between two groups were performed using Student's t‐test or Mann–Whitney U‐test. Comparisons of percentages were performed using chi‐square test or Fisher's exact test, as appropriate. The overall survival (OS) and progression free survival (PFS) of patients were calculated. Survival was estimated using the Kaplan–Meier method and curves were compared using the log‐rank test. All the tests were two‐sided. The statistical significance was defined as P < 0.05.
Results
Patients, tumor characteristics and patient outcome
Our study included 56 boys and 40 girls. The median age at diagnosis for PA was 5.9 years [IQR (3.7 and 9.6)]. They were significantly younger (P = 0.001) than patients with PXA: 15.5 years (11 and 17.1), or patients with GG: 12.9 years (5.8 and 15.5) and patients with DNT: 11.6 years (9.3 and 14.8). There was no male or female predominance within each group. Two patients were diagnosed with neurofibromatosis type 1 (NF1): PA15 and GG14.
For the 40 PA and PMA, tumor location was the posterior fossa in 26 cases (65%), optic pathway in 8 cases (20%), supratentorial in 2 cases (5%) and spinal cord/brainstem in 5 cases (12.5%, 1/5 was also located in the optic pathway). All the PXAs were supratentorial tumors. For the 31 GG, 20 tumors were supratentorial (64.5%), 3 (9.6%) from the posterior fossa, 4 (12.9%) from the spinal cord/brainstem and 4 (12.9%) from the optic pathway. All DNTs were supratentorial (100%).
Follow‐up was available in 83/96 patients. Median follow‐up for the whole cohort was 60.7 months (IQR: 35.1–90.1). Median follow‐up for patients with PA was 50.3 months (IQR: 31.2–86.1), for patients with PXA, 64.1 month (IQR: 20.5–69.2), for patients with GG, 88.9 months (IQR: 50.3–105), and for patients with DNT, 60.9 months (IQR: 34.3–133.2).
Seventeen patients of 83 relapsed (20.5%); this included 8 PA (2 PMA), 1 PXA, 6 GG and 2 DNT. Among the patients alive at last follow‐up, clinical status was available in 57: 40 were free of disease (70.2%), 4 were alive with stable disease (7%) and 13 were alive with progressive disease (22.8%).
Five patients died: two with PXA (PXA1, PXA3) and three with optic pathway PA (PA4, PA22, PA40). All these data are summarized in Table 1.
Table 1.
Clinical and biological characteristics of the 96 pediatric tumors studied. Abbreviations: PA = pilocytic astrocytoma; PMA = pilomyxoid astrocytoma; PXA = pleomorphic xanthoastrocytoma; DNT = dysembryoplastic neuroepithelial tumor (S = specific form; NS = non‐specific form); M = male; F = female; H/C = hypothalamo‐chiasmatic; CR = complete resection; PR = partial resection; NA = not available; FOD = free of disease; AWSD = alive with stable disease; AWPD = alive with progressive disease; ND = not done
| Patient | Age at diagnosis (years) | Gender | Tumor location | Surgical resection | Clinical status at last medical examination | BRAFV600E mutation by IHC | BRAF mutation by direct sequencing | CD34 status by IHC |
|---|---|---|---|---|---|---|---|---|
| PA01 | 4 | M | Cerebellum | CR | AWSD | Negative | Negative | Negative |
| PA02 | 5 | F | Occipital | CR | FOD | Negative | Negative | Negative |
| PA03 | 4 | F | Optic nerve | CR | FOD | Negative | ND | Bushy |
| PA04 (PMA) | 1 | M | H/C region | PR | Dead | Negative | Negative | Bushy |
| PA05 | 3 | M | Cerebellum | CR | FOD | Negative | ND | Negative |
| PA06 | 5 | M | Temporal | CR | FOD | Negative | ND | Negative |
| PA07 | 3 | F | Cerebellum | CR | NA | Negative | ND | Negative |
| PA08 | 16 | M | Spinal cord | CR | FOD | Negative | Negative | Negative |
| PA09 | 4 | M | Cerebellum | CR | FOD | Negative | ND | Negative |
| PA10 | 8 | F | Cerebellum | PR | AWPD | Negative | ND | Negative |
| PA11 | 9 | M | Cerebellum | CR | FOD | ND | V600E | Negative |
| PA12 | 12 | M | Cerebellum | CR | FOD | Negative | Negative | Negative |
| PA13 | 8 | F | Optic nerve | PR | AWSD | Negative | ND | Negative |
| PA14 | 9 | M | Cerebellum | CR | FOD | Negative | Negative | Negative |
| PA15 | 12 | F | Spinal cord | PR | AWSD | Negative | ND | Negative |
| PA16 | 6 | M | H/C region | PR | AWSD | Negative | ND | Negative |
| PA17 | 13 | M | Cerebellum | PR | AWSD | Negative | ND | Negative |
| PA18 (PMA) | 9 | F | Cerebellum | CR | FOD | Negative | ND | Negative |
| PA19 | 3 | M | Cerebellum | NA | FOD | ND | V600E | Negative |
| PA20 | 14 | F | Brainstem | CR | FOD | Negative | ND | Negative |
| PA21 | 4 | M | Cerebellum | PR | AWPD | Negative | Negative | Negative |
| PA22 | 4 | M | H/C region | PR | dead | Negative | ND | Negative |
| PA23 | 7 | F | Cerebellum | CR | FOD | Negative | ND | Negative |
| PA24 | 7 | M | Cerebellum | CR | FOD | Negative | ND | Negative |
| PA25 | 2 | M | Brainstem | CR | FOD | Negative | ND | Negative |
| PA26 | 9 | F | Cerebellum | CR | FOD | Negative | ND | Negative |
| PA27 | 1 | F | Cerebellum | CR | FOD | Negative | ND | Negative |
| PA28 (PMA) | 14 | F | Cerebellum | PR | AWPD | Negative | ND | Negative |
| PA29 | 10 | M | Cerebellum | CR | FOD | Negative | Negative | Negative |
| PA30 | 9 | M | Cerebellum | CR | FOD | Negative | ND | Negative |
| PA31 (PMA) | 6 | M | Cerebellum | CR | FOD | Negative | Negative | Negative |
| PA32 | 5 | M | Cerebellum | PR | AWSD | Negative | ND | Negative |
| PA33 | 3 | M | Cerebellum | PR | AWSD | Negative | Negative | Negative |
| PA34 | 2 | F | Optic nerve | CR | FOD | Negative | ND | Solitary |
| PA35 | 4 | M | Cerebellum | CR | NA | Negative | Negative | Negative |
| PA36 | 12 | M | Cerebellum | CR | FOD | Negative | Negative | Negative |
| PA37 | 5 | F | Cerebellum | NA | FOD | Negative | Negative | Negative |
| PA38 | 19 | M | Cerebellum | NA | NA | Negative | ND | Negative |
| PA39 (PMA) | 9 | F | H/C region + brainstem + spinal cord | PR | AWSD | Negative | ND | Solitary |
| PA40 (PMA) | 5 | F | H/C region | PR | dead | Negative | Negative | Bushy |
| PXA01 | 12 | F | Temporal | CR | dead | 2+ | V600E | Negative |
| PXA02 | 16 | F | Frontal | PR | FOD | Negative | Negative | Negative |
| PXA03 | 18 | M | Temporal | CR | dead | 3+ | V600E | Diffuse |
| PXA04 | 8 | M | Temporal | PR | FOD | 3+ | V600E | Diffuse |
| PXA05 | 15 | F | Temporal | NA | FOD | Negative | Negative | Solitary |
| DNT01 (NS) | 11 | M | Temporal | CR | NA | Negative | Negative | Negative |
| DNT02 (NS) | 10 | M | Temporal | CR | FOD | Negative | Negative | Diffuse |
| DNT03 (S) | 15 | M | Frontal | CR | NA | Negative | Negative | Negative |
| DNT04 (S) | 13 | M | Temporal | CR | NA | Negative | Negative | Negative |
| DNT05 (NS) | 3 | F | Frontal | PR | NA | Negative | Negative | Negative |
| DNT06 (NS) | 10 | M | Temporal | CR | NA | Negative | Negative | Negative |
| DNT07 (S) | 9 | F | Temporal | CR | NA | Negative | Negative | Negative |
| DNT08 (NS) | 12 | F | Temporal | NA | FOD | 1+ | V600E | Bushy |
| DNT09 (NS) | 0 | M | Temporal | PR | FOD | 1+ | V600E | Negative |
| DNT10 (S) | 11 | M | Temporal | CR | NA | Negative | Negative | Negative |
| DNT11 (S) | 13 | M | Temporal | CR | NA | 2+ | V600E | Negative |
| DNT12 (S) | 12 | M | Temporal | CR | NA | 1+ | V600E | Bushy |
| DNT13 (NS) | 16 | M | Parietal | CR | FOD | Negative | Negative | Negative |
| DNT14 (S) | 10 | F | Occipital | CR | NA | Negative | Negative | Negative |
| DNT15 (NS) | 15 | M | Temporal | CR | FOD | Negative | Negative | Bushy |
| DNT16 (NS) | 17 | F | Temporal | PR | FOD | 2+ | V600E | Diffuse |
| DNT17 (S) | 5 | F | Temporal | CR | FOD | 2+ | V600E | Negative |
| DNT18 (S) | 7 | M | Temporal | NA | NA | Negative | Negative | Negative |
| DNT19 (S) | 15 | M | Frontal | CR | FOD | Negative | Negative | Negative |
| DNT20 (S) | 15 | F | Temporal | PR | NA | Negative | Negative | Negative |
| GG01 | 7 | M | Temporal | CR | NA | 1+ | V600E | Negative |
| GG02 | 13 | M | Frontal | CR | NA | 2+ | V600E | Negative |
| GG03 | 1 | M | Temporal | CR | NA | Negative | Negative | Bushy |
| GG04 | 11 | M | Temporal | CR | FOD | Negative | Negative | Diffuse |
| GG05 | 13 | F | Temporal | CR | NA | Negative | Negative | Solitary |
| GG06 | 6 | M | Spinal cord | CR | FOD | 2+ | V600E | Negative |
| GG07 | 1 | F | Brainstem | CR | NA | 1+ | Negative | Negative |
| GG08 | 18 | M | Temporal | NA | NA | 1+ | V600E | Solitary |
| GG09 | 1 | F | Brainstem | PR | NA | 2+ | Negative | Bushy |
| GG10 | 17 | M | Hemisphere | NA | NA | 2+ | V600E | Negative |
| GG11 | 14 | M | Frontal | CR | FOD | 2+ | V600E | Bushy |
| GG12 | 13 | M | Temporal | CR | FOD | Negative | Negative | Negative |
| GG13 | 16 | F | Temporal | CR | NA | 2+ | V600E | Bushy |
| GG14 | 12 | M | Temporal | PR | NA | Negative | Negative | Solitary |
| GG15 | 16 | F | H/C region | CR | NA | Negative | Negative | Solitary |
| GG16 | 12 | F | Frontal | CR | NA | Negative | Negative | Bushy |
| GG17 | 17 | F | Posterior fossa | PR | AWSD | 2+ | V600E | Negative |
| GG18 | 13 | F | Third ventricle | PR | AWSD | 2+ | V600E | Negative |
| GG19 | 6 | F | Posterior fossa | CR | NA | Negative | Negative | Bushy |
| GG20 | 17 | F | Parietal | NA | NA | Negative | Negative | Negative |
| GG21 | 12 | F | Parietal | CR | AWSD | Negative | Negative | Solitary |
| GG22 | 1 | M | Parietal + occipital | CR | NA | Negative | Negative | Negative |
| GG23 | 12 | M | Cerebellum + brainstem | PR | AWSD | Negative | Negative | Solitary |
| GG24 | 20 | M | Temporal | NA | NA | Negative | Negative | Bushy |
| GG25 | 5 | M | Temporal | NA | NA | Negative | Negative | Bushy |
| GG26 | 20 | M | H/C region | NA | NA | Negative | Negative | Bushy |
| GG27 | 1 | F | Temporal | CR | FOD | Negative | Negative | Negative |
| GG28 | 4 | M | Temporal | CR | NA | Negative | Negative | Negative |
| GG29 | 12 | F | H/C region | PR | AWPD | 2+ | V600E | Negative |
| GG30 | 14 | M | Brainstem + spinal cord | PR | AWSD | 1+ | V600E | Bushy |
| GG31 | 14 | F | Temporal | CR | FOD | 1+ | V600E | Diffuse |
Evidence for BRAFV600E expression and mutation in DNT, PXA, GG but not PA
BRAFV600E expression
The tumors that were positive for BRAFV600E expression were 6/20 cases of DNT (30%), including 3/11 cases of the specific form (Figure 1A,C) and 3/9 cases of the non‐specific form (Figure 2D,F); 3/5 cases of PXA (60%) (Figure 3D,F) and 14/31 cases of GG (45.2%) (Figure 3A,C). In the PA and PMA group, all cases tested (37/40) were negative. In the three remaining cases fixed in Bouin, immunostaining was unreliable (data not shown). In PXA, immunostaining was recorded in all tumor cells, although with variation in intensity from one cell to another. In GG, immunostaining was recorded in both atypical ganglion cells and glial cells, whereas in DNT, the immunostaining was intense and diffuse in the glial nodules and usually absent in floating neurons. Interestingly, in the case of complex DNT associated with cortical dysplasia, strong BRAF immunostaining was recorded in dysplastic neurons.
Figure 1.
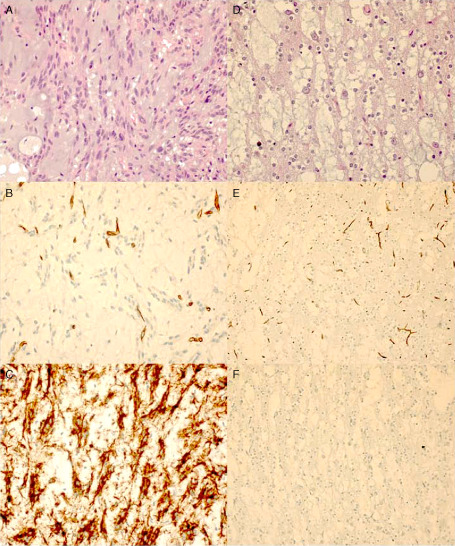
Photomicrographs showing the histopathological features of a specific form of a dysembryoplastic neuroepithelial tumor (DNT). A–C. Glial nodule of a complex DNT (DNT 11): A. hematoxylin‐eosin (HE) stain; B. negative CD34 immunostaining; and C. positive BRAFV600E immunostaining. D–F. Glioneuronal elements (DNT 3): D. HE stain; E. negative CD34 immunostaining; and F. negative BRAFV600E immunostaining. Original magnification: ×400 (A–D); ×200 (E, F).
Figure 2.
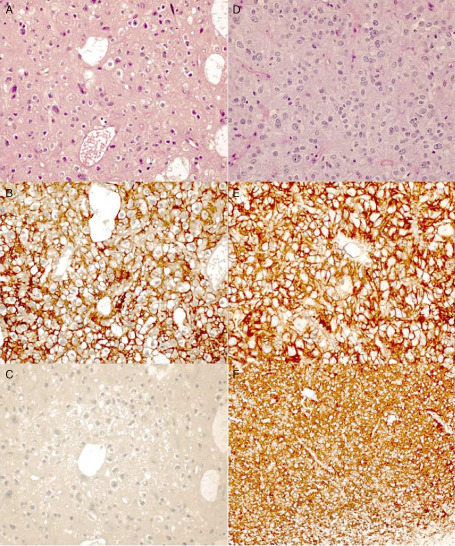
Photomicrographs showing the histopathological features of non‐specific forms of dysembryoplastic neuroepithelial tumor (DNT). A–C. Case DNT 2: A. hematoxylin‐eosin (HE) stain, oligodendroglioma‐like pattern; B. diffuse CD34 immunostaining; and C. negative BRAFV600E immunostaining. D–F. Case DNT 16: D. HE stain, oligodendroglioma‐like pattern; E. diffuse CD34 immunostaining; and F. positive BRAFV600E immunostaining. Original magnification: ×400 (A–E); ×200 (F).
Figure 3.
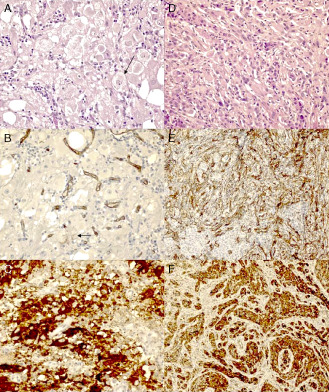
Photomicrographs showing the histopathological features of gangliogliomas (GGs) and pleomorphic xanthoastrocytomas (PXAs). A–C. GG 23: A. hematoxylin‐eosin (HE) stain (arrow: ganglionic cell); B. solitary CD34 immunostaining (arrow: positive cell); and C. positive BRAFV600E immunostaining. D–F. PXA 3. D. HE stain, pleomorphic cells of classic PXA; E. diffuse CD34 immunostaining; and F. positive BRAFV600E immunostaining. Original magnification: ×400 (A–C); ×200 (D–F).
BRAFV600 mutation
When BRAFV600 mutation was detected, it was always the V600E variant and it was recorded in 6/20 cases of DNT (30%), including 3/11 cases of the specific form and 3/9 cases of the non‐specific form; 3/5 cases of PXA (60%); 12/31 cases of GG (38.7%); and 2/16 cases of PA and PMA (12.5%), corresponding to two cases fixed in Bouin that were not tested in IHC. PA and PMA displayed significantly less BRAFV600E mutation than other tumors (P = 0.046).
Correlation between BRAFV600E expression and mutation
We recorded an excellent concordance between immunohistochemistry and DNA direct sequencing (κ = +0.93). At the end of the study, 2 out of the 69 cases tested with the two techniques were discordant, including 2 cases of GG with positive BRAFV600E expression and lack of mutation. It was worth noticing that we recorded first six discordant cases, including five cases positive by IHC on TMA and not mutated and one mutated case negative by IHC. In all these cases, immunostaining was repeated on the whole section and BRAFV600E mutation was also checked by direct sequencing. After control, we observed that the mutated case also showed BRAFV600E expression in a discrete part of the lesion, and among the five other cases, 3/5 were indeed negative, but two remained strongly positive. These two cases that were GG (fixed in formalin) represent unexplained discordance between the two techniques.
CD34 expression in DNT, PXA, GG but not PA
CD34 expression
This was recorded in 5/20 DNT (25%). The staining was either bushy or diffuse. Among the positive cases we recorded, only 1/11 specific DNT but 4/9 non‐specific forms (Figure 2A,B,D,E). Interestingly, in non‐specific DNT, CD34 immunostaining was also recorded at the edges of the lesion in the surrounding cortex. It is worth noticing that the DNT15 case, mimicking at histology a grade II oligoastrocytoma and exhibiting IDH1R132H expression, also demonstrated a bushy pattern of CD34 expression. CD34 expression was recorded in 3/5 PXA (60%) (2 diffuse patterns, 1 solitary pattern) (Figure 3D,E) and in 18/31 GG (58.1%) (2 diffuse patterns, 10 bushy patterns and 6 solitary patterns) (Figure 3A,B). In contrast, CD34 expression was recorded in only 5/40 PA (12.5%) (3 bushy patterns, 2 solitary patterns) significantly less than other tumors (P < 0.001). It is worth noticing that all the CD34 expressing PA arose from the optic pathway. However, there remained three cases also located in the optic pathway that were negative for CD34 expression.
CD34 expression and/or BRAFV600E status (mutated if BRAFV600E mutation or BRAFV600E expression was recorded). In addition, 8/20 DNT cases (40%), 4/5 PXA (80%), 26/31 GG (83.8%) and 7/40 PA (17.5%) were BRAFV600E mutated and/or CD34 positive. The status BRAFV600E mutated and/or CD34 positive was less frequent in PA vs. other tumors (P = 0.001).
Prognostic factors
The univariate analysis was conducted in the four groups: DNT, PXA, GG and PA‐PMA. In the first step, statistical analysis was conducted in the whole cohort of patients because of the very low number of events recorded in some groups of patients. The following variables were searched for prognostic significance for OS and progression free survival PFS: histological subtype, tumor location, extent of surgical resection, age at surgical excision (<3 years then <10 years vs. others), BRAFV600E status, CD34 expression and/or BRAFV600E mutation/expression.
A shorter OS was significantly related to histological subtype (P < 0.001) (Figure 4A) and optic pathway location (P < 0.001) (Figure 4B). The other variables analyzed—age, extent of surgical resection, BRAFV600E status, CD34 expression and BRAFV600E mutation/expression and/or CD34 expression—were not related to OS.
Figure 4.
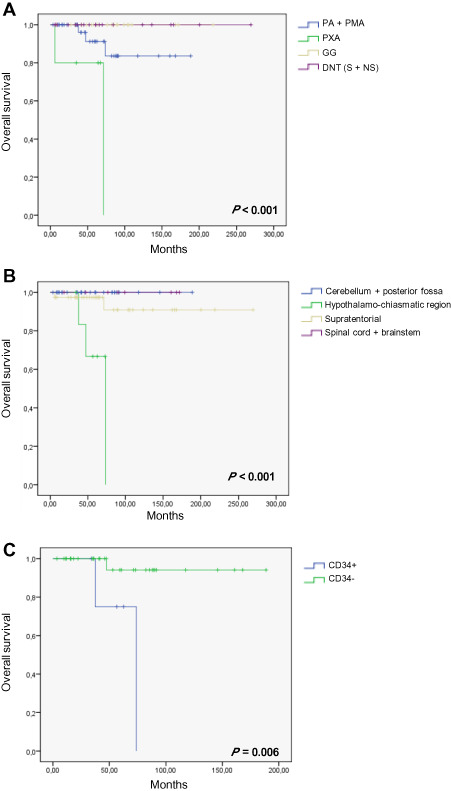
A. Overall survival (OS, in months) according to pathological variants: pilocytic astrocytomas (PAs) and pilomyxoid astrocytomas (PMAs), pleomorphic xanthoastrocytomas (PXAs), gangliogliomas (GGs) and dysembryoplastic neuroepithelial tumors (DNTs) (specific and non‐specific form: S + NS). B. OS according to tumor location whatever the pathological variant. C. OS according to CD34 expression in the PA‐PMA subgroup. Because no event was recorded in GG and DNT, the two curves are superimposed (A). Th same is recorded in (B) for cerebellum + posterior fossa and spinal cord + brainstem locations.
Regarding PFS, the extent of the surgical resection (CR vs. PR) was the only predictive factor (P = 0.02) (data not shown). The other variables analyzed—age, histological subtype, tumor location, BRAFV600E status, CD34 expression and BRAFV600E mutation/expression—were not related to PFS.
In the second step, we searched for prognostic significance of BRAFV600E status, CD34 expression and BRAFV600E mutation/expression and/or CD34 expression within each histological subgroup of patients, but this analysis was possible in the group of PA only because of the low number of PXA cases and the lack of event recorded in the group of DNT and GG. A shorter OS was related to PA with CD34 expression (P = 0.006) (Figure 4C).
Discussion
In this study which focused on glial and glioneuronal tumors in children, we have observed BRAFV600E mutation in 6/20 DNT (including 3/11 specific form and 3/9 non‐specific form). Moreover, in these cases, BRAFV600E mutation detected by DNA direct sequencing correlated exactly with the immunohistochemical detection of BRAFV600E expression. These results have diagnostic and histogenetic implications. They suggest that a search for BRAFV600E mutation should be performed in all children's cortical tumors with clinical and neuroradiological features of DNT, whatever the histopathological subtype, specific and non‐specific form especially if an oligodendroglial component is obvious. It is well recognized that in contrast to their adult counterparts, oligodendrogliomas in children usually do not demonstrate IDH mutation nor 1p19q co‐deletion 1, 17, 18, 20, suggesting that they are different in spite of common morphological features. Evidence for BRAFV600E mutation in some non‐specific histological forms of DNT, especially those mimicking at histology a grade II oligodendroglioma, is in keeping with that hypothesis. Although not all oligodendrogliomas in children are DNT, it is likely that some cortical tumors that mimic at histology grade II oligodendrogliomas share with DNT the same clinico‐radiological features, excellent prognosis and, for some of them, BRAFV600E mutation. Occurrence for BRAFV600E mutation in DNT extends the spectrum of CNS tumors expressing this marker, which also include GG, PXA and extracerebellar PA 10, 12, 23. BRAFV600E mutation was not recorded previously in DNT 10, 23, but the number of DNT included in each study did not exceed five cases. Interestingly, Schindler et al 23 also recorded BRAFV600E mutation in one grade II oligodendroglioma in a 39‐year‐old patient who suffered from a long history of temporal epilepsy and lacking IDH mutation or 1p19q co‐deletion. Previous studies have reported composite GG and DNT, suggesting a possible etiological relationship between these two entities 3, 19. Occurrence of BRAFV600E mutation in both tumor types offers a molecular basis for that hypothesis. In contrast to DNT, GG might demonstrate malignant tumoral transformation. However, we cannot determine from our study whether DNT cases harboring BRAFV600E mutation will demonstrate a tendency to malignant transformation, nor whether these cases represent under diagnosed GG, especially when partial resection was performed (as in cases 08 and 16 of the present study). Although our study was focused on children's glial and glioneuronal tumors, it has been previously recorded that BRAFV600E mutation was a common feature of PXA and GG, whatever the age of the patients 23, and could be used as a common genetic marker for these two entities. In our study, 3/5 PXA demonstrated BRAFV600E mutation, which was in keeping with previous studies that have analyzed BRAFV600E mutation in a high number of PXA, and recorded it in 65% of them 9, 10, 23. In our study, we observed BRAFV600E mutation/expression in 14/31 GG (45.2%), which is comparable to previous studies 10, 23, and in 2/40 PA‐PMA (5%) when we consider the sequencing and/or IHC technique, which is also comparable to the literature 14. Other teams have shown that up to 33% of diencephalic PA exhibited BRAFV600E mutation, and cerebellar and optic tract's PA rarely exhibited the mutation 23. Interestingly, two BRAFV600E mutated PA (PA11 and PA19) had concomitant KIAA1549 : BRAF fusion, as already described 13.
Except for two cases, concordance between BRAFV600E mutation and BRAFV600E expression was excellent with a κ coefficient of +0.93. For the cases that had benefited from the two techniques, discrepant results were recorded in 2/31 GG. We do not have clear explanation for these two discordant cases.
In addition to BRAFV600E mutation, we also searched for CD34 expression by immunohistochemistry in the same cohort of tumors. We recorded CD34 expression in 3/5 PXA (60%) and in 18/31 GG (58.1%). This was in accordance with previous results, which reported CD34 immunoreactivity in up to 74% of GG 2, 8 and to 84% of PXA 22. CD34 was also recorded in DNT that exhibited the typical GNE but the number of positive cases remained low (present study) 2, 8, 24. In contrast, CD34 expression was much higher in non‐specific and then in the so‐called diffused DNT, recorded in up to 100% of cases 3, 24. In accordance, we observed CD34 expression in 4/9 (44.4%) non‐specific DNT but in only 1/11 of specific DNT.
Taken together, CD34 expression and BRAFV600E expression are two markers that characterize glioneuronal tumors but they are not always associated. As an example, CD34 expression was recorded in one BRAFV600E mutated PXA and, conversely, was negative in one BRAFV600E mutated PXA. Interestingly, if we consider these two markers, 4/5 PXA, 26/31 GG, 8/20 DNT displayed CD34 expression or BRAFV600E mutation. In keeping with the previous report 2, PA rarely demonstrated CD34 expression. Interestingly, when CD34 expression was observed, it was always in PA arising from optic tract. Although we do not have a clear explanation for that, it further highlights that PA from the optic pathway have a distinctive genetic signature from cerebellar PA, as we have previously reported 26. In addition, because CD34 is a stem cell marker, it might also suggest that PA of the optic pathway are made by more immature cells in accordance with their putative origin from radial glial cells 26. The negative impact of CD34 expression on the prognosis is likely due to the optic pathway location of these tumors.
In conclusion, we have shown that some DNT exhibit BRAFV600E mutation/expression. BRAFV600E mutation/expression and/or CD34 expression are common features in PXA, GG and DNT. These tumors have to be distinguished from diffuse ordinary gliomas that usually demonstrate IDH mutations and from classic PA mainly arising from the cerebellum that demonstrate KIAA1549 : BRAF fusion. Although in our group of glial and glioneuronal tumors BRAFV600E mutation was devoid of prognostic value, BRAFV600E immunohistochemical detection is an easy diagnostic marker that can be used in routine practice and could potentially replace DNA sequencing.
Conflicts of Interest
The authors declare that they have no conflict of interest.
Acknowledgments
This work was supported by Institut National contre le Cancer (INCA) grants to DFB (Procan, SIRIC) and by the Groupement des Entreprises Françaises dans la Lutte contre le Cancer (GEFLUC). We thank the Association pour la Recherche sur les Tumeurs Cérébrales (ARTC‐Sud) and the Société Française de Lutte contre les Cancers et les Leucémies de l'Enfant et de l'Adolescent (SFCE) for their financial support. We are grateful to the patients and physicians for providing details on follow‐up. We thank Emeline Tabouret for her help in statistical analyses. Frozen samples were provided by the AP‐HM TumourBank (Authorization Number 2008‐70).
References
- 1. Balss J, Meyer J, Mueller W, Korshunov A, von Hartmann C, Deimling A (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 116:597–602. [DOI] [PubMed] [Google Scholar]
- 2. Blumcke I, Giencke K, Wardelmann E, Beyenburg S, Kral T, Sarioglu N et al (1999) The CD34 epitope is expressed in neoplastic and malformative lesions associated with chronic, focal epilepsies. Acta Neuropathol 97:481–490. [DOI] [PubMed] [Google Scholar]
- 3. Bodi I, Selway R, Bannister P, Doey L, Mullatti N, Elwes R, Honavar M (2012) Diffuse form of dysembryoplastic neuroepithelial tumour: the histological and immunohistochemical features of a distinct entity showing transition to dysembryoplastic neuroepithelial tumour and ganglioglioma. Neuropathol Appl Neurobiol 38:411–425. [DOI] [PubMed] [Google Scholar]
- 4. Capper D, Preusser M, Habel A, Sahm F, Ackermann U, Schindler G et al (2011) Assessment of BRAF V600E mutation status by immunohistochemistry with a mutation‐specific monoclonal antibody. Acta Neuropathol 122:11–19. [DOI] [PubMed] [Google Scholar]
- 5. Colin C, Padovani L, Chappé C, Mercurio S, Scavarda D, Loundou A et al (2012) Outcome analysis of childhood pilocytic astrocytomas: a retrospective study of 148 cases at a single institution. Neuropathol Appl Neurobiol. doi: 10.1111/nan.12013. [DOI] [PubMed] [Google Scholar]
- 6. Daumas‐Duport C, Scheithauer BW, Chodkiewicz JP, Laws ER Jr, Vedrenne C (1988) Dysembryoplastic neuroepithelial tumor: a surgically curable tumor of young patients with intractable partial seizures. Report of thirty‐nine cases. Neurosurgery 23:545–556. [DOI] [PubMed] [Google Scholar]
- 7. Daumas‐Duport C, Hawkins C, Shankar S (2007) Dysembryoplastic neuroepithelial tumor. In: WHO Classification of Tumors of the Central Nervous System, 4th edn. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds), pp. 99–102. IARC: Lyon. [Google Scholar]
- 8. Deb P, Sharma MC, Tripathi M, Sarat Chandra P, Gupta A, Sarkar C (2006) Expression of CD34 as a novel marker for glioneuronal lesions associated with chronic intractable epilepsy. Neuropathol Appl Neurobiol 32:461–468. [DOI] [PubMed] [Google Scholar]
- 9. Dias‐Santagata D, Lam Q, Vernovsky K, Vena N, Lennerz JK, Borger DR et al (2011) BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. PLoS One 6:e17948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dougherty MJ, Santi M, Brose MS, Ma C, Resnick AC, Sievert AJ et al (2010) Activating mutations in BRAF characterize a spectrum of pediatric low‐grade gliomas. Neuro Oncol 12:621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fernandez C, Girard N, Paz Paredes A, Bouvier‐Labit C, Lena G, Figarella‐Branger D (2003) The usefulness of MR imaging in the diagnosis of dysembryoplastic neuroepithelial tumor in children: a study of 14 cases. AJNR Am J Neuroradiol 24:829–834. [PMC free article] [PubMed] [Google Scholar]
- 12. Forshew T, Tatevossian RG, Lawson AR, Ma J, Neale G, Ogunkolade BW et al (2009) Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol 218:172–181. [DOI] [PubMed] [Google Scholar]
- 13. Hawkins C, Walker E, Mohamed N, Zhang C, Jacob K, Shirinian M et al (2011) BRAF‐KIAA1549 fusion predicts better clinical outcome in pediatric low‐grade astrocytoma. Clin Cancer Res 17:4790–4798. [DOI] [PubMed] [Google Scholar]
- 14. Jones DT, Kocialkowski S, Liu L, Pearson DM, Ichimura K, Collins VP (2009) Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene 28:2119–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin A, Rodriguez FJ, Karajannis MA, Williams SC, Legault G, Zagzag D et al (2012) BRAF alterations in primary glial and glioneuronal neoplasms of the central nervous system with identification of 2 novel KIAA1549:BRAF fusion variants. J Neuropathol Exp Neurol 71:66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A et al (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Padovani L, Colin C, Fernandez C, Maues de Paula A, Mercurio S, Scavarda D et al (2012) Search for distinctive markers in DNT and cortical grade II glioma in children: same clinicopathological and molecular entities? Curr Top Med Chem 12:1683–1692. [DOI] [PubMed] [Google Scholar]
- 18. Paugh BS, Qu C, Jones C, Liu Z, Adamowicz‐Brice M, Zhang J et al (2010) Integrated molecular genetic profiling of pediatric high‐grade gliomas reveals key differences with the adult disease. J Clin Oncol 28:3061–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Prayson RA (1999) Composite ganglioglioma and dysembryoplastic neuroepithelial tumor. Arch Pathol Lab Med 123:247–250. [DOI] [PubMed] [Google Scholar]
- 20. Raghavan R, Balani J, Perry A, Margraf L, Vono MB, Cai DX et al (2003) Pediatric oligodendrogliomas: a study of molecular alterations on 1p and 19q using fluorescence in situ hybridization. J Neuropathol Exp Neurol 62:530–537. [DOI] [PubMed] [Google Scholar]
- 21. Reed GH, Kent JO, Wittwer CT (2007) High‐resolution DNA melting analysis for simple and efficient molecular diagnostics. Pharmacogenomics 8:597–608. [DOI] [PubMed] [Google Scholar]
- 22. Reifenberger G, Kaulich K, Wiestler OD, Blumcke I (2003) Expression of the CD34 antigen in pleomorphic xanthoastrocytomas. Acta Neuropathol 105:358–364. [DOI] [PubMed] [Google Scholar]
- 23. Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold‐Mende C et al (2011) Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra‐cerebellar pilocytic astrocytoma. Acta Neuropathol 121:397–405. [DOI] [PubMed] [Google Scholar]
- 24. Sung CO, Suh YL, Hong SC (2011) CD34 and microtubule‐associated protein 2 expression in dysembryoplastic neuroepithelial tumours with an emphasis on dual expression in non‐specific types. Histopathology 59:308–317. [DOI] [PubMed] [Google Scholar]
- 25. Sutherland DR, Stewart AK, Keating A (1993) CD34 antigen: molecular features and potential clinical applications. Stem Cells 11(Suppl. 3):50–57. [DOI] [PubMed] [Google Scholar]
- 26. Tchoghandjian A, Fernandez C, Colin C, El Ayachi I, Voutsinos‐Porche B, Fina F et al (2009) Pilocytic astrocytoma of the optic pathway: a tumour deriving from radial glia cells with a specific gene signature. Brain 132(Pt 6):1523–1535. [DOI] [PubMed] [Google Scholar]
- 27. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]