

Abstract
Cellular and humoral inflammations play important roles in ischemic brain injury. The effectiveness of immunomodulatory therapies may critically depend on the chosen experimental model. Our purpose was to compare the post‐ischemic neuroinflammation among murine permanent and transient middle cerebral artery occlusion (MCAO) models. Permanent MCAO was induced by transtemporal electrocoagulation and 30 minutes or 90 minutes transient MCAO was induced by intraluminal filament in C57BL/6 mice. Infiltration of leukocyte subpopulations was quantified by immunohistochemistry and fluorescence‐activated cell sorting. Cerebral cytokine and adhesion molecule expression was measured by real‐time polymerase chain reaction (RT‐PCR). Neutrophil infiltration was noted at 24 h after transient MCAO, but did not further increase until 5 days in the permanent MCAO model. Few T cells were observed in both MCAO models at 24 h, but permanent MCAO demonstrated much more infiltrating T cells at 5 days. Pronounced microglial activation was evident at 24 h and 5 days after permanent but not after transient MCAO. The number of invading NK cells and expression of MHCII on CD11b+ cells did not differ among the three groups. Five days after MCAO, the expression of IL‐1, TNF‐α and IFN‐γ and of the adhesion molecules ICAM‐1 and VCAM‐1 was significantly higher in the permanent than in the transient MCAO groups. Cellular and humoral inflammation differs substantially among commonly used MCAO models. Neuroinflammation is more pronounced after permanent electrocoagulatory MCAO compared with 30 minutes and 90 minutes filament‐MCAO.
Keywords: infiltration of leukocyte, permanent cerebral ischemia, transient cerebral ischemia
Introduction
Brain damage after ischemic stroke results from a variety of parallel and sequential processes, including excitotoxicity, calcium dysregulation, oxidative stress, inflammation and pro‐apoptotic stimuli 26. Inflammatory mechanisms and the interaction of activated immune cells with the ischemic brain tissue are currently under intensive investigation because they are believed to contribute substantially to secondary brain damage and potential repair mechanisms 12. Moreover, their delayed kinetics makes them amenable to therapeutic interventions. Key features of the neuroimmunological response to brain ischemia are early microglial activation 21 and subsequent recruitment of circulating leukocytes to the ischemic brain 12, 22. Indeed, several cells residing physiologically in the brain, including microglia, astrocytes, neurons and endothelial cells, are activated after ischemia and secrete a plethora of pro‐inflammatory mediators, including IL‐1β and TNF‐α 3, 30. These mediators facilitate adhesion by endothelial activation and migration of circulating leukocytes to the ischemic brain 37.
The failure of multiple therapeutic strategies for stroke patients despite very promising preclinical data has prompted an intensive search for underlying reasons 9. An important possible explanation is that many key targets, including inflammatory mediators, had a different and sometimes even opposing impact on outcome in different phases after ischemia 28, 32, 41. Moreover, considerable discrepancies become evident when different animal models of ischemia are investigated. Because pathophysiological processes differ depending on early reperfusion of the ischemic tissue, the STAIR committee recommends that a new therapy is tested both in permanent and transient animal models 1.
Although modulation of immune cells either by depletion 6, 18, 39, systemic arrest or attenuated cerebral immigration 19, 36, 40 is a promising target for brain protection in experimental studies, the basic immunological processes in different ischemia models require a better description and understanding. For example, the effectiveness of endogenous and therapeutic immunomodulation depended at least in part on the animal model under investigation in two of our recent extensive studies 17, 19. Interestingly, anti‐inflammatory strategies were more effective in permanent than after prolonged transient focal ischemia. This may contradict the concept that the inflammatory response to tissue damage increases in case of reperfusion and correlates with the extent of tissue damage 20, 38. Notably, the majority of the previous preclinical trials have tested immunomodulatory strategies only after transient MCAO.
Therefore, the purpose of the present study was to compare the infiltration patterns of different systemic immune cells in commonly used mouse models of focal cerebral ischemia.
Materials and Methods
The study was conducted following national and international guidelines for use of experimental animals. The protocols were approved by the local and governmental committees responsible for animal care and use (Regierungspraesidium Karlsruhe, Germany). One hundred twenty‐two male mice (C57BL/6, Charles River Laboratories, 10–12 weeks old, body weight 20–25 g) were examined in the present study.
Permanent middle cerebral artery occlusion (pMCAO) model
Briefly, as previously described by Tamura et al 35, mice were anesthetized with 1.0%–2.0% halothane in 30%O2/70% N2O. After a 1 cm long skin incision between the left eye and ear, the temporal muscle was removed and a burr hole was drilled through the temporal skull. After removal of the dura mater and placing a laser Doppler probe 2 mm left and 0.5 mm caudal to the bregma [ie, above the cortical area supplied by the middle cerebral artery (MCA)], the MCA was occluded permanently using a bipolar electrocoagulation forceps (Modell ICC 50, Erbe GmbH, Tübingen, Germany) under a stereo‐microscope (Hund, Wetzlar, Germany). During the operation, body temperature was kept at 37°C using a feedback‐controlled heating pad. After suturing the skin lesion, mice were placed in a cage under an infrared heating lamp until recovery from anesthesia. Sham control mice underwent the same surgical procedure except MCA occlusion.
Transient middle cerebral artery occlusion (tMCAO) model
The surgical procedure was modified from the original description by Belayev 4. Mice were anesthetized using the same protocol as in the pMCAO model. A laser Doppler probe was placed 2 mm left and 0.5 mm caudal to the bregma over the cortical area supplied by the MCA. After neck dissection, the common carotid artery was incised between two ligations and a silicon coated 8‐0 monofilament was advanced through the internal carotid artery (ICA) until a resistance was felt. Successful occlusion of the origin of the MCA was assumed when a ≥70% reduction in relative perfusion values compared with baseline was documented by LDF. After MCAO (30 or 90 minutes), animals were anesthetized again and the filament was removed, allowing restoration of blood flow. During the operation, body temperature was kept at 37°C using a feedback‐controlled heating pad. After MCAO and reperfusion, mice were placed in a cage under an infrared heating lamp until recovery from anesthesia. Sham control mice were operated following the same protocol of 90 minutes MCAO without obstructing the MCA.
Immunohistology
Twenty‐four hours or 5 days after MCAO or the respective sham operation, mice were anesthetized with ketamine (100 mg/kg)/xylazine (10 mg/kg) i.p. and intracardially perfused with 20 mL saline. After decapitation, brain samples were collected and frozen in isopentane at −20°C. Cryosections (10 μm thick) were cut at the level of the anterior commissure. To identify infiltrating leukocyte subpopulations immunohistologically, we used myeloperoxidase for neutrophils, CD3 for T cells and Iba1 for activated microglia 13. After fixation with 4% PFA for 60 minutes, endogenous peroxidase was blocked by peroxidase blocking solution (Dako, Glostrup, Denmark) for 30 minutes. After being washed in phosphate‐buffered saline (PBS) containing 0.1% Tween‐20, the sections were incubated with primary antibody against CD3 (clone 3H698, Zytomed, Berlin, Germany), or myeloperoxidase (MPO, clone RP/053, Zytomed) or Iba1 (Wako, Richmond, VA, USA) for 60 minutes at 21°C. Then, sections were incubated with the corresponding secondary antibody for another 60 minutes and the immunoreactivity was visualized by an immunoenzyme polymer (Nichirei Biosciences, Tokyo, Japan) for CD3 and MPO according to the manufacturer's instruction and developed in diaminobenzidine (DAB). For Iba1, the immunoreactivity was visualized by avidin‐biotin complex and sections were developed in DAB.
For quantitative analysis of cell number in the peri‐infarct area and the corresponding area on the contralateral hemisphere, virtual slides were captured automatically with a 40× magnification (0.23 μm/pixel) on a Hamamatsu NanoZommer 2.0‐HT scansystem (Hamamatsu Photonics, Hamamatsu, Japan) and analyzed semi‐automatically with Matlab (R2011a, The Mathworks, Natick, MA, USA) using the “Image processing toolbox”. CD3 and MPO‐positive cells were determined separately in the total ischemic hemisphere of one section. Iba1‐positive cells were determined in six fields of interest (each 0.16 mm2) adjacent to the borderline of the infarct and in the corresponding areas of the non‐ischemic hemisphere.
RNA isolation and real‐time polymerase chain reaction (RT‐PCR)
Separated hemispheres were harvested from untreated naïve mice and 5 days after MCAO or sham operation, respectively. Samples were snap‐frozen in isopentane at 20°C. RNA was isolated from separated hemispheres using RNeasy kits (Qiagen, Hilden, Germany). Reverse transcription was performed with the High Capacity Complementary DNA Archive Kit (Applied Biosystems, Foster City, CA, USA) and RT‐PCR with SYBR‐Green assays (Applied Biosystems) on an ABI7500 RT‐PCR System (Applied Biosystems). Primers were purchased as ready‐to‐use primer sets for TNF‐α, INF‐γ, IL‐1, ICAM‐1 and VCAM‐1 (Super Array, Hilden, Germany). All assays were run in duplicate and the results for each individual gene were normalized to the level of the housekeeping gene encoding for peptidylprolyl isomerase A (cyclophilin).
Flow cytometric analysis
Five days after MCAO, additional mice were anesthetized again and perfused transcardially with 20 mL saline. Flow cytometric analysis of brain infiltrating cells was performed as previously reported 19. Briefly, both hemispheres were separately collected and mechanically homogenized in dissociation buffer. The cell suspension was overlaid on discontinous Percoll gradients of 1.03 and 1.088 g/mL density; leukocytes and microglia were collected from the interphase. Single cell suspensions from brain homogenates were gated for CD45 (Clone 30‐F11) expression and differentiated into the indicated subpopulation using different cell markers. CD3+ (clone 17A2) and B220+ (clone RA3‐6B2) cells were considered as T‐ and B‐lymphocytes, respectively. Neutrophils were identified by Gr‐1 (Ly‐6G, clone RB6‐8C5) staining, and natural killer (NK) cells were defined using NK1.1 monoclonal antibodies (clone PK136). Additionally, the expression of the Major Histocompatibility Complex II (MHCII) on antigen‐presenting CD11b+ cells as a marker of neuroinflammatory activation was determined using CD11b (Clone M1/70), MHC II (Clone M5/114.15.2) and the appropriate isotype control following the manufacturer's protocols (eBioscience, Frankfurt am Main, Germany). Flow cytometry was performed on a Becton Dickinson FACSCalibur (Heidelberg, Germany) and the results were analyzed by CellQuest Pro software (BD Biosciences, Heidelberg, Germany).
Statistical analysis
All values are presented as mean ± standard deviation. We analyzed infiltrating neutrophils, T cells and activated microglia by Student's t‐test between pMCAO and sham‐operated controls and one‐way analysis of variance (ANOVA) with post hoc Tukey's test for multiple comparisons among 30‐minute tMCAO, 90‐minute tMCAO and sham‐operated controls and among the three MCAO groups, respectively. All the analyses were performed using SPSS 13.0 (SPSS Inc., Chicago, IL, USA). A P‐value <0.05 was considered as statistically significant.
Results
Immunohistology
Recruitment of neutrophils
At 24 h after MCAO, both tMCAO groups showed significantly more invading neutrophils than the pMCAO model (30‐minute MCAO: 86 ± 48 cells/section; 90‐minute MCAO: 131 ± 69 cells/section, pMCAO 31 ± 18 cells/section, P < 0.05). In contrast, 5 days after MCAO, the number of infiltrating neutrophils increased significantly in the pMCAO model but declined markedly in the 30‐minute MCAO group (pMCAO: 171 ± 192 cells/section; 30‐minute MCAO 18 ± 23 cells/section, P < 0.05) and remained unaltered in the 90‐min MCAO group (87 ± 85 cells/section). Only very few MPO‐positive cells were observed in the sham controls of both stroke models (Figure 1A). MPO‐positive cells were detected predominantly in the peri‐ischemic area surrounding the infarct. The topographical distribution differed neither among the three groups, nor within the same model at different time points (Figure 1B).
Figure 1.
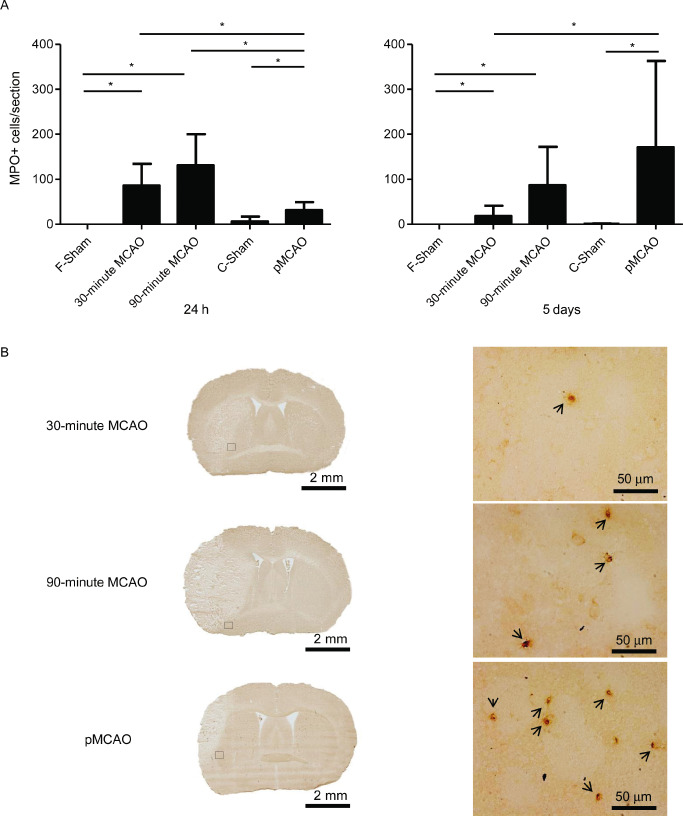
Comparison of neutrophil infiltration in the ischemic hemispheres among the permanent and transient MCAO (30 minutes and 90 minutes) models. (A) Analysis of the MPO‐positive cell counts per section at 24 h (left) and 5d (right) after MCAO (n = 10/group at each time point), *P < 0.05. (B) Representative immunohistochemically stained sections for neutrophils (MPO) shown as a histological overview (left) and at higher magnification (right) from the indicated region in the overview at 5 days after ischemia. MPO‐positive cells were stained by diaminobenzidine (brown). MPO‐positive cells are indicated by arrowheads. MCAO = middle cerebral artery occlusion; MPO = myeloperoxidase.
Influx of T lymphocytes
We observed only very few CD3‐positive T cells in the peri‐ischemic area in all three groups at 24 h after MCAO (30‐minute MCAO: 4 ± 4 cells/section; 90‐minute MCAO: 2 ± 3 cells/section and pMCAO: 4 ± 4 cells/section). In contrast, the number of infiltrating CD3‐positive cells increased substantially in the 30‐minute MCAO and the pMCAO groups while it remained very low in the 90‐minute MCAO group at 5 days (30‐minute MCAO: 15 ± 9 cells/section, P < 0.05; pMCAO:28 ± 23 cells/section, P < 0.05; 90‐minute MCAO: 3 ± 3 cells/section). Infiltration of T cells was negligible in both sham control groups (Figure 2A). Topographically, the infiltrating CD3‐positive cells accumulated, surrounding the infarct area in both models (Figure 2B).
Figure 2.
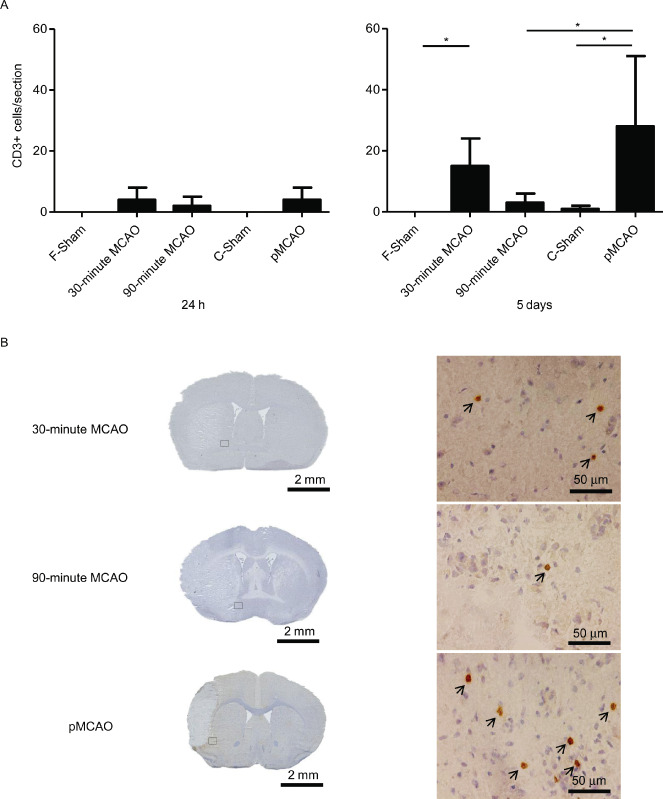
Comparison of infiltrating T cells in the ischemic hemispheres among the permanent and transient MCAO (30 minutes, 90 minutes) groups. (A) Analysis of the CD3‐positive cell counts per section at 24 h (left) and 5 days(right) after MCAO (n = 10/group at each time point), *P < 0.05. (B) Representative immunohistochemically stained sections for T cells (CD3) of the analyzed stroke models as histological overviews (left) and at higher magnification (right) at 5 days after ischemia. CD3‐positive cells were stained by diaminobenzidine (brown) and cell nuclei were counterstained by hematoxylin (blue). CD3‐positive cells are indicated by arrowheads. MCAO = middle cerebral artery occlusion; pMCAO = permanent middle cerebral artery occlusion.
Microglial activation
Twenty‐four hours after MCAO, significantly more Iba‐1‐positive cells were detected in the pMCAO model (pMCAO: 69 ± 13 cells/mm2; sham: 11 ± 13 cells/mm2, P < 0.05), and at 5 days after MCAO, their number increased considerably (pMCAO: 263 ± 88 cells/mm2, sham: 49 ± 39 cells/mm2, P < 0.05). In contrast, cell numbers of Iba‐1‐positive cells did not differ significantly between the tMCAO groups and the sham‐operated group and both analyzed time points after MCAO (24 h: 30‐minute MCAO: 50 ± 13 cells/mm2; 90‐minute MCAO: 35 ± 20 cells/mm2; sham: 40 ± 27 cells/mm2, 5 days: 30‐minute MCAO: 94 ± 57 cells/mm2, 90‐minute MCAO: 34 ± 20 cells/mm2, sham: 109 ± 42 cells/mm2, ANOVA P > 0.05) (Figure 3A). Topographically, in the pMCAO and the 90‐minute tMCAO groups, we detected marked expression of Iba‐1‐positive cells in the infarct border zone while no microglia was stained in the infarct core. In contrast, in the 30‐minute tMCAO model, Iba‐1‐positive cells were extensively distributed also in the lesion core area (Figure 3B).
Figure 3.
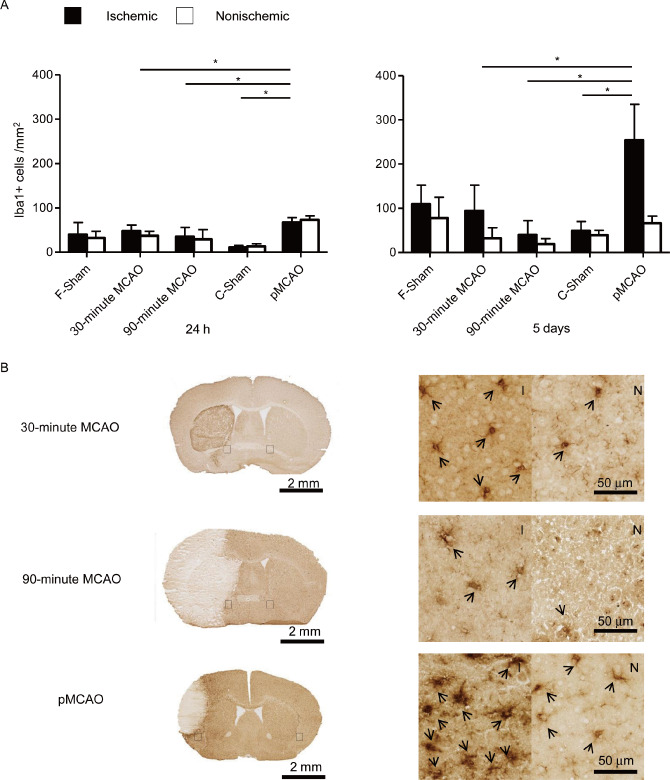
Comparison of microglial activation between the permanent and transient MCAO (30 minutes and 90 minutes) groups. (A). Analysis of the Iba‐1‐positive cell numbers in six fields of interest in the ischemic (black bars) and corresponding regions in the nonischemic (open bars) hemisphere per one section at 24 h (left) and 5 days (right) after MCAO (n = 10/group at each time point), *P < 0.05. (B). Representative immunohistochemically stained sections of the analyzed stroke models for activated microglia (Iba‐1) as a histological overview image (left) and at a higher magnification (right) at 5 days after ischemia. Iba‐1‐positive cells were stained by diaminobenzidine (brown) I: ischemic hemisphere and N: nonischemic hemisphere. Iba‐1‐positive cells are indicated by arrowheads. MCAO = middle cerebral artery occlusion; pMCAO = permanent middle cerebral artery occlusion.
Flow cytometric analysis of brain infiltrating immune cells
To validate the results obtained from the histological analysis shown above, to obtain quantitative results on cell number per total hemisphere instead of only per single section and to reflect a larger number of leukocyte subpopulations, we performed flow cytometric analysis of brain homogenates.
Five days after onset of ischemia, we measured significantly higher cell numbers of total lymphomononuclear cells (CD45+), T (CD3+) and B (B220+) cells in the pMCAO model than in both tMCAO groups. In contrast, no differences were observed for the numbers of invading NK cells and neutrophil granulocytes (Gr‐1+) among the three MCAO groups (Figure 4A,B). We additionally analyzed MHCII expression of CD11b+ monocytes to test for potential activation of cerebral antigen presenting cells.
Figure 4.
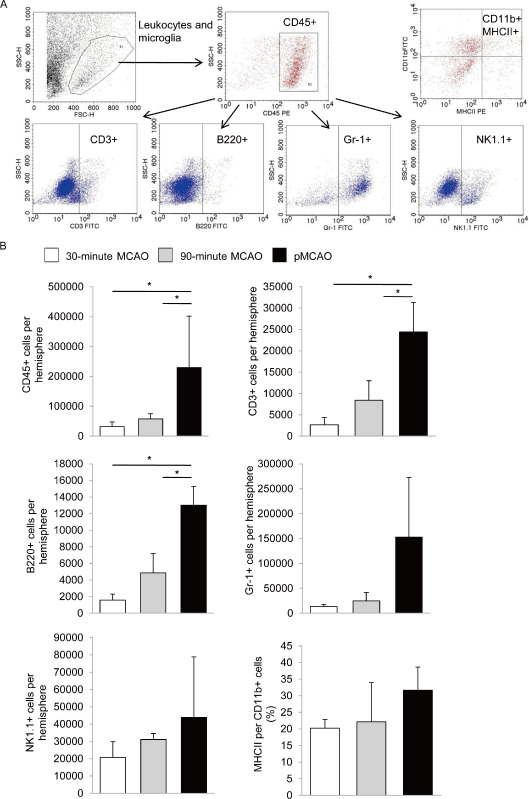
Flow cytometric analysis of cerebral leukocyte subpopulations 5 days after brain ischemia. (A) Representative gating strategy for absolute cell count analysis of leukocyte subpopulations in brain homogenates of the entire ischemic hemisphere. CD45+ leukocytes were gated for subpopulation analysis after using scattergram gates for viable leukocytes and quantification beads. (B) Flow cytometric analysis of absolute cell counts of granulocytes (Gr‐1+), B (B220+), NK (NK1.1+), total T cells (CD3+) and the ratio of MHC II presenting cells within CD11b+ cell population at 5 days after MCAO for the indicated MCAO groups (3 individual experiments per group, consisting of 2–3 pooled animals for each model), *P < 0.05. MCAO = middle cerebral artery occlusion; pMCAO = permanent middle cerebral artery occlusion.
Approximately 20%–30% of CD11b+ cells expressed MHCII without significant intergroup differences (Figure 4B).
Cerebral expression levels of pro‐inflammatory cytokines and adhesion molecules
Five days after MCAO, the expression of TNF‐α and IFN‐γ was significantly increased by 90‐minute tMCAO, and TNF‐α also by 30‐minute tMCAO, compared with the sham‐operated group. In contrast, IL‐1 expression was not altered by both transient occlusion models. Permanent MCAO induced a manifold higher increase of all three pro‐inflammatory cytokines. Expression of TNF‐α, INF‐γ and IL‐1 was significantly higher after pMCAO compared with its respective sham group and both tMCAO models (Figure 5A). These findings indicate a profound neuroinflammation correlating with the intergroup differences detected for cellular inflammation after MCAO.
Figure 5.
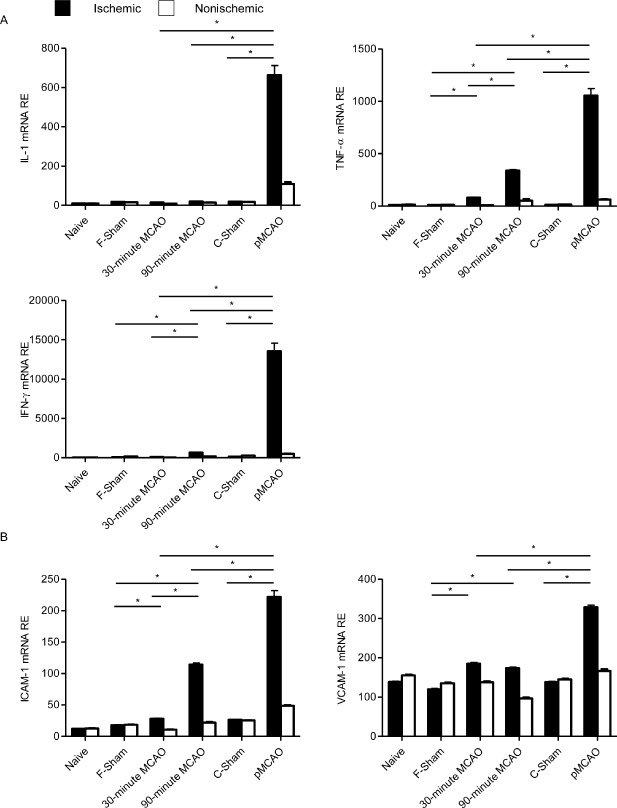
mRNA expression analyses of key proinflammatory cytokines and adhesion molecules. Relative expression (RE, mRNA expression values normalized to the housekeeping gene peptidylprolyl isomerase) of (A) IL‐1, TNF‐α, IFN‐γ and (B) ICAM‐1 and VCAM‐1 was assessed in ischemic (black bars) and nonischemic (open bars) hemispheres at 5 days after permanent or transient MCAO (30 minutes and 90 minutes) and of sham controls for both models (coagulation: C‐Sham; filament: F‐Sham) and naive control animals (n = 5/group), *P < 0.05. MCAO = middle cerebral artery occlusion; pMCAO = permanent middle cerebral artery occlusion.
We further analyzed the expression of the adhesion molecules ICAM‐1 and VCAM‐1 to investigate differences in adhesion molecule expression as a possible cause of differential cerebral leukocyte infiltration. Both 30‐minute and 90‐minute tMCAO induced a statistically significant but relatively minor increase of relative ICAM‐1 and VCAM‐1 mRNA expression. In contrast, permanent MCAO produced a significantly higher increase of both adhesion molecules compared with both tMCAO models and the sham‐operated control group (Figure 5B).
Discussion
The major new findings of the present study are:
Twenty‐four hours after MCAO onset, both tMCAO groups showed more infiltration of neutrophil but less activation of resident microglia than the pMCAO model.
In contrast, 5 days after ischemia, infiltration of neutrophils and T cells as well as microglial activation were more pronounced after pMCAO than in the tMCAO groups.
The expression of key pro‐inflammatory cytokines and adhesion molecules was substantially more increased in the pMCAO model than in both tMCAO groups at 5 days after ischemia.
The present study compares cellular and humoral neuroinflammation in the most commonly used experimental stroke models. According to a recent review 27, 42% of all experimental stroke studies use the intraluminal filament MCAO and 25% use the coagulation model. In contrast to the filament‐induced transient MCAO model, the electrocoagulation model induces permanent MCAO and requires a craniotomy. Filament‐induced MCAO lasting longer than 45 minutes induces extensive infarction in mice with lesion volumes >50% of the ipsilateral hemisphere 10, 15, 24, 25 and results in high mortality. Permanent MCAO by electrocoagulation and 30‐min transient MCAO produce infarct lesions of approximately 15% of the hemisphere 35, have substantially less effects on physiological parameters (eg, temperature, loss of body weight) and cause only little depression of the systemic immune system compared with prolonged filament MCAO 16.
The kinetics of early cell infiltration found within the tMCAO and the pMCAO model in the present work is consistent with previous reports analyzing leukocyte brain infiltration in the respective stroke model 11, 17, 19, 31, 34. We focused on the 24 h and 5 days time points because the cell infiltration peaked between 3 and 7 days after ischemia onset in these studies. We observed that neutrophil granulocytes were the first leukocyte subpopulation to be recruited to the ischemic brain tissue. Extensive immigration of neutrophils occurred earlier after tMCAO than after pMCAO. Because infiltrating monocytes/macrophages may also contribute to immunohistological MPO staining, we additionally performed flow cytometrical analysis for the more specific neutrophil granulocyte marker Gr‐1 which confirmed our histological data.
We used Iba‐1 staining to detect activated microglia and infiltrating monocytes/macrophages. The number of Iba‐1‐positive cells increased considerably until 5 days after pMCAO. In contrast, there was no significant activation of microglia in both tMCAO groups compared with the sham‐operated control group in the peri‐infarcted region. Interestingly, we also detected pronounced Iba‐1 expression in the striatal ischemic lesion after short‐lasting (30 minutes) transient MCAO. This effect might be attributed to a more selective (neuron) apoptosis after short‐term ischemia associated with secondary glial activation within the ischemic lesions. This differential increase of Iba‐1‐positive cells between the used models was also reflected by the expression of IL‐1 and TNF‐α which are mainly expressed by microglia after ischemia 7, 17.
T cells have been shown to be the major leukocyte subpopulation mediating secondary inflammatory brain damage after experimental stroke 12. After MCAO, infiltrating T cells cannot only directly kill the damaged cells through release of perforin and various granzymes, but also secrete proinflammatory cytokines, such as IFN‐γ, which play key roles in the pathogenesis of cerebral ischemia 2. As three major cytokines in the postischemic inflammation, IL‐1, TNF‐α and IFN‐γ, induce the expression of chemokines and adhesion molecules, recruit immune cells into brain tissue, and activate the brain resident glial cells 14. At least in the early phase of ischemia, they promote neuronal death and exacerbate the brain damage related to cerebral ischemia 12. Consistent with our findings on cellular neuroinflammation, the expression of these cytokines was much stronger upregulated in the pMCAO than in both tMCAO groups at 5 days after ischemia. A limitation of our study is that we only examined cytokine expression at a single subacute time point.
Which mechanisms may underlie the differential infiltration of systemic immune cells in the different models? Technically, differences of surgical and anesthetic procedures may influence the inflammatory response in the brain 8. Yet the protocols were closely matched in the present study and controlled by respective sham‐operated groups. Alternatively, the lower leukocyte infiltration and cytokine expression after 90‐minute tMCAO may reflect the severely depressed state of the systemic immune system in this paradigm which includes a profound leukocytopenia 5, 23, 29. Moreover, this does not explain the differences in the cellular and humoral inflammation between 30‐minute tMCAO and pMCAO, because both models have comparable lesion volumes and similar effects on systemic immune cells 16. Instead, our findings suggest a more profound upregulation of factors in the brain in the pMCAO model that favor the immigration of systemic immune cells. Proinflammatory cytokines facilitate the infiltration of leukocytes into brain tissue by activating and inducing adhesion molecules on leukocytes and vascular endothelial cells. ICAM‐1 and VCAM‐1 are upregulated in endothelial cells following cerebral ischemia and sequentially involved in leukocyte diapedesis across the blood–brain barrier 33. The significantly higher levels of mRNA of both adhesion molecules were compatible with the more pronounced cellular inflammation in pMCAO at 5 days, while the less significant recruitment of leukocytes in both tMCAO groups may be attributed to the lower expression of ICAM‐1 and VCAM‐1.
In conclusion, the extent and kinetics of brain infiltration of leukocyte subpopulations differ substantially among commonly used murine experimental MCAO models in the first week after ischemia onset. Consideration of these differences in experimental studies on the one hand and a better description of immune cell infiltration after human stroke on the other hand are necessary for a valid translational evaluation of potential of immunomodulatory therapies for brain ischemia.
Funding Sources
This project was supported by the German Excellence program “FRONTIER” and Deutsche Forschungsgemeinschaft to R.V. (VE.196/3‐1).
Supporting information
Figure S1. Schematic overview of different MCAO models and resulting infarct demarcation on histological sections. (A) Left: in transient MCAO caused by the intraluminal filament, the origin of the MCAO is occluded. Right: transtemporal electrocoagulation of the MCAO is performed beyond the branching of the lenticulostriate arteries. Site of occlusion is marked in red. ACA = anterior cerebral artery; MCA = middle cerebral artery; PCA = posterior cerebral artery; BA = basilar artery; ICA = internal carotid artery. (B) Resulting infarcts depend on the site of occlusion and the duration of ischemia. Representative cresyl‐violett stained cryosection of the analyzed stroke models, infarct areas are encircled. Comparison of infarct volume (C) and mortality (D) at 24 h and 5 days among the MCAO models.
Figure S2. Schematic overview of the fields of interest (FOIs) for comparison of activated microglia; 6x FOIs (each 0.16 mm2) surrounding the infarct area in the ischemic hemisphere and corresponding areas in the nonischemic hemisphere were defined on the immunohistochemistry sections for Iba1 of the analyzed stroke models to evaluate the activated microglia.
References
- 1. Fisher M (1999) Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke 30:2752–2758. [DOI] [PubMed] [Google Scholar]
- 2. Arumugam TV, Granger DN, Mattson MP (2005) Stroke and T‐cells. Neuromolecular Med 7:229–242. [DOI] [PubMed] [Google Scholar]
- 3. Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN et al (1997) Tumor necrosis factor‐alpha: a mediator of focal ischemic brain injury. Stroke 28:1233–1244. [DOI] [PubMed] [Google Scholar]
- 4. Belayev L, Alonso OF, Busto R, Zhao W, Ginsberg MD, Hsu CY (1996) Middle cerebral artery occlusion in the rat by intraluminal suture: neurological and pathological evaluation of an improved model. Stroke 27:1616–1623. [DOI] [PubMed] [Google Scholar]
- 5. Chamorro Á, Urra X, Planas AM (2007) Infection after acute ischemic stroke. Stroke 38:1097–1103. [DOI] [PubMed] [Google Scholar]
- 6. Czech B, Pfeilschifter W, Mazaheri‐Omrani N, Strobel MA, Kahles T, Neumann‐Haefelin T et al (2009) The immunomodulatory sphingosine 1‐phosphate analog FTY720 reduces lesion size and improves neurological outcome in a mouse model of cerebral ischemia. Biochem Biophys Res Commun 389:251–256. [DOI] [PubMed] [Google Scholar]
- 7. Davies CA, Loddick SA, Toulmond S, Stroemer RP, Hunt J, Rothwell NJ (1999) The progression and topographic distribution of interleukin‐1[beta] expression after permanent middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab 19:87–98. [DOI] [PubMed] [Google Scholar]
- 8. Denes A, McColl BW, Leow‐Dyke SF, Chapman KZ, Humphreys NE, Grencis RK et al (2011) Experimental stroke‐induced changes in the bone marrow reveal complex regulation of leukocyte responses. J Cereb Blood Flow Metab 31:1036–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dirnagl U, Macleod MR (2009) Stroke research at a road block: the streets from adversity should be paved with meta‐analysis and good laboratory practice. Br J Pharmacol 157:1154–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Garcia JH, Liu K‐F, Ho K‐L (1995) Neuronal necrosis after middle cerebral artery occlusion in wistar rats progresses at different time intervals in the caudoputamen and the cortex. Stroke 26:636–643. [DOI] [PubMed] [Google Scholar]
- 11. Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe C‐U, Siler DA et al (2009) Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 40:1849–1857. [DOI] [PubMed] [Google Scholar]
- 12. Iadecola C, Anrather J (2011) The immunology of stroke: from mechanisms to translation. Nat Med 17:796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S (1998) Microglia‐specific localisation of a novel calcium binding protein, Iba1. Mol Brain Res 57:1–9. [DOI] [PubMed] [Google Scholar]
- 14. Konsman JP, Drukarch B, Van Dam AM (2007) (Peri)vascular production and action of pro‐inflammatory cytokines in brain pathology. Clin Sci (Lond) 112:1–25. [DOI] [PubMed] [Google Scholar]
- 15. Li Y, Chopp M, Jiang N, Yao F, Zaloga C (1995) Temporal profile of in situ DNA fragmentation after transient middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab 15:389–397. [DOI] [PubMed] [Google Scholar]
- 16. Liesz A, Hagmann S, Zschoche C, Adamek J, Zhou W, Sun L et al (2009) The spectrum of systemic immune alterations after murine focal ischemia. Stroke 40:2849–2858. [DOI] [PubMed] [Google Scholar]
- 17. Liesz A, Suri‐Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S et al (2009) Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med 15:192–199. [DOI] [PubMed] [Google Scholar]
- 18. Liesz A, Sun L, Zhou W, Schwarting S, Mracsko E, Zorn M et al (2011) FTY720 reduces post‐ischemic brain lymphocyte influx but does not improve outcome in permanent murine cerebral ischemia. PLoS ONE 6:e21312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liesz A, Zhou W, Mracsko E, Karcher S, Bauer H, Schwarting S et al (2011) Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain 134:704–720. [DOI] [PubMed] [Google Scholar]
- 20. Lo EH (2008) Experimental models, neurovascular mechanisms and translational issues in stroke research. Br J Pharmacol 153(S1):S396–S405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mabuchi T, Kitagawa K, Ohtsuki T, Kuwabara K, Yagita Y, Yanagihara T et al (2000) Contribution of microglia/macrophages to expansion of infarction and response of oligodendrocytes after focal cerebral ischemia in rats editorial comment. Stroke 31:1735–1743. [DOI] [PubMed] [Google Scholar]
- 22. Macrez R, Ali C, Toutirais O, Le Mauff B, Defer G, Dirnagl U, Vivien D (2011) Stroke and the immune system: from pathophysiology to new therapeutic strategies. The Lancet Neurology 10:471–480. [DOI] [PubMed] [Google Scholar]
- 23. Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U (2005) Central nervous system injury‐induced immune deficiency syndrome. Nat Rev Neurosci 6:775–786. [DOI] [PubMed] [Google Scholar]
- 24. Memezawa H, Minamisawa H, Smith ML, Siesjö BK (1992) Ischemic penumbra in a model of reversible middle cerebral artery occlusion in the rat. Exp Brain Res 89:67–78. [DOI] [PubMed] [Google Scholar]
- 25. Memezawa H, Smith M, Siesjo B (1992) Penumbral tissues salvaged by reperfusion following middle cerebral artery occlusion in rats. Stroke 23:552–559. [DOI] [PubMed] [Google Scholar]
- 26. Moskowitz MA, Lo EH, Iadecola C (2010) The science of stroke: mechanisms in search of treatments. Neuron 67:181–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. O'Collins VE, Macleod MR, Donnan GA, van der Horky LL, Worp BH, Howells DW (2006) 1,026 experimental treatments in acute stroke. Ann Neurol 59:467–477. [DOI] [PubMed] [Google Scholar]
- 28. Page‐McCaw A, Ewald AJ, Werb Z (2007) Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol 8:221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Prass K, Meisel C, Höflich C, Braun J, Halle E, Wolf T et al (2003) Stroke‐induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1–like immunostimulation. J Exp Med 198:725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rothwell N, Allan S, Toulmond S (1997) The role of interleukin 1 in acute neurodegeneration and stroke: pathophysiological and therapeutic implications. J Clin Invest 100:2648–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I et al (2009) Pivotal role of cerebral interleukin‐17‐producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med 15:946–950. [DOI] [PubMed] [Google Scholar]
- 32. Shohami E, Ginis I, Hallenbeck JM (1999) Dual role of tumor necrosis factor alpha in brain injury. Cytokine Growth Factor Rev 10:119–130. [DOI] [PubMed] [Google Scholar]
- 33. Steiner O, Coisne C, Cecchelli R, Boscacci R, Deutsch U, Engelhardt B, Lyck R (2010) Differential roles for endothelial ICAM‐1, ICAM‐2, and VCAM‐1 in shear‐resistant T cell arrest, polarization, and directed crawling on blood–brain barrier endothelium. J Immunol 185:4846–4855. [DOI] [PubMed] [Google Scholar]
- 34. Stevens SL, Bao J, Hollis J, Lessov NS, Clark WM, Stenzel‐Poore MP (2002) The use of flow cytometry to evaluate temporal changes in inflammatory cells following focal cerebral ischemia in mice. Brain Res 932:110–119. [DOI] [PubMed] [Google Scholar]
- 35. Tamura A, Graham DI, McCulloch J, Teasdale GM (1981) Focal cerebral ischaemia in the rat: 1. Description of technique and early neuropathological consequences following middle cerebral artery occlusion. J Cereb Blood Flow Metab 1:53–60. [DOI] [PubMed] [Google Scholar]
- 36. Vemuganti R, Dempsey RJ, Bowen KK (2004) Inhibition of Intercellular adhesion molecule‐1 protein expression by antisense oligonucleotides is neuroprotective after transient middle cerebral artery occlusion in rat. Stroke 35:179–184. [DOI] [PubMed] [Google Scholar]
- 37. Wang Q, Tang XN, Yenari MA (2007) The inflammatory response in stroke. J Neuroimmunol 184:53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Woodruff T, Thundyil J, Tang S‐C, Sobey C, Taylor S, Arumugam T (2011) Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol Neurodegener 6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yilmaz G, Arumugam TV, Stokes KY, Granger DN (2006) Role of T Lymphocytes and interferon‐γ in ischemic stroke. Circulation 113:2105–2112. [DOI] [PubMed] [Google Scholar]
- 40. Zhang RL, Chopp M, Li Y, Zaloga C, Jiang N, Jones ML et al (1994) Anti‐ICAM‐1 antibody reduces ischemic cell damage after transient middle cerebral artery occlusion in the rat. Neurology 44:1747–1751. [DOI] [PubMed] [Google Scholar]
- 41. Zhao B‐Q, Tejima E, Lo EH (2007) Neurovascular proteases in brain injury, hemorrhage and remodeling after stroke. Stroke 38:748–752. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Schematic overview of different MCAO models and resulting infarct demarcation on histological sections. (A) Left: in transient MCAO caused by the intraluminal filament, the origin of the MCAO is occluded. Right: transtemporal electrocoagulation of the MCAO is performed beyond the branching of the lenticulostriate arteries. Site of occlusion is marked in red. ACA = anterior cerebral artery; MCA = middle cerebral artery; PCA = posterior cerebral artery; BA = basilar artery; ICA = internal carotid artery. (B) Resulting infarcts depend on the site of occlusion and the duration of ischemia. Representative cresyl‐violett stained cryosection of the analyzed stroke models, infarct areas are encircled. Comparison of infarct volume (C) and mortality (D) at 24 h and 5 days among the MCAO models.
Figure S2. Schematic overview of the fields of interest (FOIs) for comparison of activated microglia; 6x FOIs (each 0.16 mm2) surrounding the infarct area in the ischemic hemisphere and corresponding areas in the nonischemic hemisphere were defined on the immunohistochemistry sections for Iba1 of the analyzed stroke models to evaluate the activated microglia.