

Abstract
Seizures are a prominent clinical feature of encephalitis. Recent data suggest the adaptive as well as innate immune system to be involved directly in the pathomechanism of epileptogenesis. Cytotoxic T‐cells and antibody‐mediated complement activation are major components of the adaptive immune system, which can induce neurodegeneration, thereby probably contributing to epileptic encephalitis. The innate immune system operates via interleukin‐1 and toll‐like receptor‐associated mechanisms and was shown to play a direct role in epileptogenesis. Here, we review neuropathology hallmarks of various encephalitis conditions such as Rasmussen encephalitis (RE) but also introduce the more recently discovered antibody‐associated voltage‐gated potassium channel complex (VGKC), N‐methyl‐D‐aspartate receptor (NMDAR) or glutamic acid decarboxylase (GAD) 65 encephalitides. Neuropathological investigations are used to determine specific cellular components and molecular mechanisms used by the immune system to provoke neurodegeneration and to promote epileptogenesis. Based on recent findings, we propose concepts for the stratification of epileptic encephalitis. Knowledge of the role of the innate immunity has already translated into clinical treatment strategies and may help to discover novel drug targets for these epileptic disorders.
Keywords: antibodies, brain, cytotoxic T‐cells, encephalitis, interleukin‐1, microglial cells, neuropathology, seizures, toll‐like receptors
THE ROLE OF THE ADAPTIVE IMMUNE SYSTEM IN SEIZURE INDUCTION IN ENCEPHALITIS
Most pathomechanisms affecting the central nervous system (CNS) are capable to provoke seizures and to promote epileptogenesis (see reviews embedded into this mini‐symposium). Neurodegeneration, that is, Alzheimer's disease and other dementias 37, 70 as well as acute brain damage after stroke (68) is increasingly recognized as cause of epilepsy. Neurodegeneration, as a triggering event for seizures, can also be an immediate result of a direct and specific attack by the adaptive immune system during the clinical course of encephalitis (defined as brain inflammation caused by infiltration of blood‐derived cells). Many infectious (i.e. viral) and noninfectious encephalitides associate with seizures and this latter neuropathologic entity will be specifically addressed in this review as it includes clinically challenging variants such as Rasmussen encephalitis (RE), or recently described encephalitides associated with antibodies directed against neural antigens. These encephalitides occur either as paraneoplastic conditions in cancer patients but develop also in the absence of a tumor, most likely triggered by an aberrant autoimmune response. Examples of antibody‐mediated encephalitides comprise voltage‐gated potassium channel complex (VGKC), N‐methyl‐D‐aspartate receptor (NMDAR) or glutamic acid decarboxylase (GAD)65 encephalitides. RE typically occurs in children with seizure onset before the age of 10 in more than three quarters of affected individuals (7). Interestingly, antibody‐associated encephalitides are likely to occur more frequently in adults than in children or adolescents (Table 1). However, reliable epidemiological data is not yet available to verify this clinical notion and may differ between encephalitis types. NMDAR encephalitis for example was shown also in children and young adults with predominance in females (22).
Table 1.
Comparison of antibody‐associated encephalitides.
| Encephalitis | Demographical features | Affected CNS area | Clinical course | Neuronal loss | Possible cytotoxic mechanisms |
|---|---|---|---|---|---|
| Rasmussen encephalitis | Peak of onset 6 years, occasionally in about 10% adolescent or adult onset | One hemisphere, starts usually in peri‐insular area | Progression over approximately 1 year, then neurological stabilization with fluctuating seizure frequency | Yes | T‐cell cytotoxicity |
| Antibody‐associated encephalitis | |||||
| Intracellular antigens | |||||
| Anti‐Hu | Usually older adults, almost all have paraneoplastic disease | Mediotemporal (brain stem, cerebellum) | Either progression to death or chronic deterioration | Yes | T‐cell cytotoxicity |
| Anti‐Ma2 | Young men with testicular cancer or older people with other types of neoplasms. Almost all have paraneoplastic disease | Mediotemporal (brain stem) | Often better than in anti‐Hu | Yes | T‐cell cytotoxicity |
| Anti‐GAD65 | Mainly young female adults | Mediotemporal | Chronic, treatment resistant temporal lobe epilepsy | Yes | T‐cell cytotoxicity |
| Surface antigens | |||||
| Anti‐NMDAR | Mainly young females (peak early twenties). ¼ to ½ of the patients have ovarian teratomas. | Functionally: whole brain | Mostly complete or good remission after weeks to years, speeded up by tumor removal and immunotherapy | Little | Antibody‐mediated decrease of NR1 receptors |
| Anti‐VGKC complex | Usually older people, men two times more frequently than females | Mediotemporal | Complete or good remission within months to 2 years, speeded up by immunotherapy | Yes | Antibody‐mediated, complement activation, lysis |
Rasmussen encephalitis
In 1958, the Canadian neurosurgeon Theodore Rasmussen published a report on three pediatric patients with the title “Focal seizures due to chronic localized encephalitis”(78). Since then, this disorder is the “index condition” for the study of epilepsies caused by chronic inflammatory processes. Since the late 1980s, most researchers and clinicians have used the term Rasmussen encephalitis (RE) or Rasmussen syndrome for this condition 3, 75. RE is a pathophysiologically fascinating condition as both seizures and inflammation only are seen in one of two hemispheres. RE often starts with a “prodromal period” lasting for up to several years. This prodromal phase however is not obligatory. If it occurs, it is characterized by relatively minor symptoms such as mild hemiparesis or infrequent seizures. Thereafter (or as the initial disease manifestation), the patient enters the “acute stage”, which is characterized by more frequent intractable unilateral simple partial focal motor seizures, complex partial seizures or secondarily generalized seizures. During this disease course, inflammation seems to spread across the affected hemisphere, and other seizure semiologies, indicating newly recruited epileptogenic areas, can be observed. Epilepsia partialis continua (EPC), that is, unilateral myoclonic twitching of the distal extremities or the face for at least 1 h and with intervals of no more than 10 s (93), is observed in approximately half of the patients. Within a few months of the manifestation of epilepsy, progressive loss of neurological function associated with only one hemisphere starts, typically in the form of hemiparesis, hemianopia, cognitive deterioration and (if the dominant side is affected) aphasia. After some months and up to 1 year, the main decline is over, and the patient passes into the “residual stage” with a stable neurological deficit. Seizure frequency is still high and often variable but in general lower compared to the “acute stage”9, 31, 71.
The neuropathological hallmarks of RE are, besides the inflammation, neuronal loss, the presence of microglia activation, microglial nodules and astrogliosis (Figure 1). These characteristics are present in most cases and have been described in detail 28, 73, 83. In addition, we recently showed that, besides the astrogliosis, also areas exist in which astrocytes are degenerating and lost (5). Some reports mention the presence of a dual pathology, that is, encephalitis and concomitant vascular abnormality, tuberous sclerosis or focal cortical dysplasia 72, 90. The question arises whether the dual pathology of RE patients with focal cortical dysplasia (FCD) is really a dual pathology or whether these small group of patients in fact could be FCD type II patients, which recently also have been described to possess inflammatory infiltrates (45). The pathological involvement of the adaptive part of the immune system in the induction of seizures and the neuronal degeneration is still not completely clear. Reports in the 1990s focused on supposedly pathogenic antibodies against the glutamate receptor 3 (GluR3), which were found in serum of RE patients (84). These antibodies were suggested to kill neurons via an antibody‐ or complement‐mediated attack (109). Plasmapheresis in RE patients seemed to diminish progression, but recovery was limited to a short period of time (4). Moreover, anti‐GluR3 antibodies were absent in a number of patients (110) and reports also showed that some patients did not improve clinically after plasmapheresis (32). In our study from 2002 (8), we studied complement C9neo deposition (as a marker for a potential antibody‐dependent pathway, the classical complement cascade) in RE patients but did not find evidence for a complement‐mediated destruction of neurons. Instead, however, we could demonstrate the presence of cytotoxic T lymphocytes in close apposition to neurons as well as astrocytes. These cells were seen to be polarized with cytotoxic granules facing the neuronal or astrocytic membrane. This and reports showing that the CNS of RE patients contains restricted T‐cell populations that expanded from a few, to discrete antigenic epitopes responding, precursor T‐cells 53, 87 strongly suggest that cytotoxic T‐cells are involved in neurodegeneration and astrocyte loss in RE. In how far the inflammation is involved in epileptogenesis itself is not completely known. However, we learned some points from RE. First, it has been shown that inflammation can precede the occurrence of seizures rather than the opposite in which the seizures induce inflammation with influx of lymphocytic cell 11, 49. In addition, magnetic resonance imaging (MRI) studies indicate that inflammatory disease activity in RE may occur outside of the epileptic network, suggesting that the inflammation can be independent of epileptic activity (36). Further evidence arguing against a direct effect of infiltrating immune cells on seizure induction comes from therapeutic studies with inflammation suppressing tacrolimus. In RE patients, tacrolimus had a positive effect on conservation of motor and cognitive functions and on brain tissue, but had no effect on seizure frequency (10). In contrast to the inflammation, the loss of astrocytes in RE however may have an influence on seizure induction in RE. Astrocytes support neurons by many different functions, such as maintenance of potassium homeostasis and regulation of gamma‐aminobutyric acid (GABA) and glutamate, neurotransmitters critically involved in epileptic processes 26, 98. Therefore, astrocytes are indispensable for normal functioning of neurons. The presence of astrogliosis and loss of astrocytes in RE are interesting with respect to seizure induction as several reports showed that hypertrophic astrocytes can aggravate or induce seizures in epilepsy 47, 94. Both the presence of hypertrophic astrocytes as well as their loss therefore may have a serious impact on the seizure induction in RE.
Figure 1.
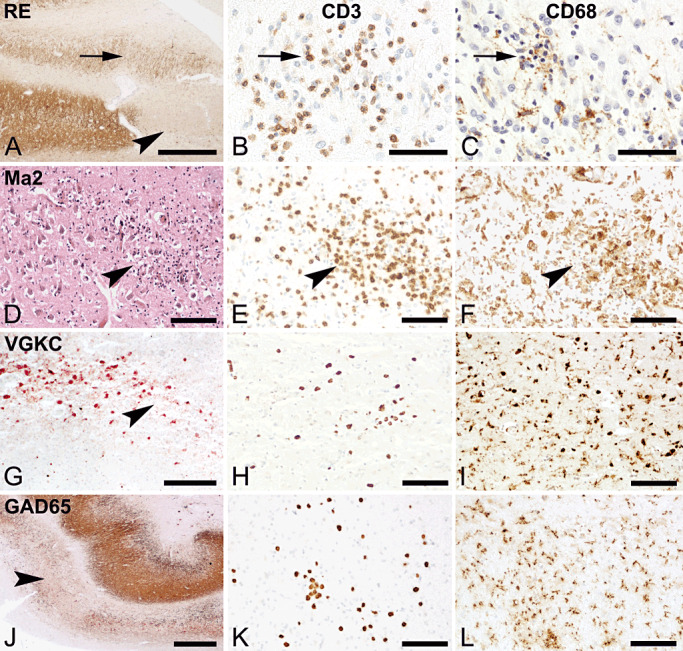
Pathology in the hippocampus of various forms of encephalitis. Left column shows neurodegeneration (HE or MAP‐2 staining), middle column shows T‐cell infiltration by staining for CD3. Right column shows microglia activation and/or macrophage infiltration by staining for CD68. (A, bar 500 µm) Hippocampus of RE patient shows complete neuronal loss in the CA3 region (arrowhead). The arrow shows at the area in the dentate gyrus, which contains inflammatory cells as shown in (B, bar: 50 µm) and activated microglial cells (C, bar: 50 µm). D, E and F. Hippocampus of a paraneoplastic MA2 case. (D, bar: 100 µm) HE staining shows loss of cells (arrowhead) and inflammatory cells. Staining for CD3 (E, bar:100 µm) and CD68 (F: bar:100 µm) show infiltration of T‐cells and activated phagocytic microglial cells. G, H, I. Hippocampus of a nonparaneoplastic anti‐VGKC complex case. (G, bar 200 µm) Loss of neurons in CA2 indicated by the arrowhead, staining for MAP‐2. The staining for CD3 shows low numbers of infiltrating T‐cells (H, bar:100 µm) while CD68 shows strongly activated microglial cells (I, bar:100 µm). J, K and L. Nonparaneoplastic anti‐GAD65 case. (J, bar:200 µm) Staining for MAP‐2 shows severe loss in the CA2 region. The area harbors moderate numbers of CD3+ T‐cells (K, bar:100 µm) and activated microglial cells (L, bar:100 µm).
Antibody‐associated encephalitis
A second group of encephalitides with prominent epileptic seizures are antibody‐associated encephalitides. These can be divided in the paraneoplastic (PE) and nonparaneoplastic limbic encephalitis (NPE). Paraneoplastic neurological disorders can affect different parts of the nervous system. Some affect only a single area (eg, limbic encephalitis or brainstem encephalitis) or a single cell type [for instance the Purkinje cells of the cerebellum in paraneoplastic cerebellar degeneration (PCD)]. In many cases however, multiple areas of the CNS are involved. Many patients with PE have antibodies in their serum that react with both the nervous system and the underlying primary cancer. In recent years, it has become clear that the neurological deficits are associated to the specificity of the autoimmune antibodies, rather than to the underlying tumor. This can also be seen by the increasing number of reports, which show encephalitic patients with comparable neurological deficits, including seizures, in which extensive diagnostic tests and follow‐up fail to reveal an underlying cancer. These cases, which can be grouped under the name of nonparaneoplastic encephalitis (NPE), have serum antibodies against a variety of neuronal antigens.
In recent years, another way of organizing these conditions with regard to the location of the antibody targets has acquired relevance. There are antibodies that are directed against intracellular antigens (such as GAD65 or amphiphysin) or even intranuclear antigens (such as Hu, Yo and Ma2), the latter being typical onconeural antigens. Secondly, antibodies can be directed to surface antigens such as the VGKC complex, various glutamate receptors (NMDA or α‐amino‐3‐hydroxy‐5‐methy‐4‐isoxazole propionate receptors or metabotropic glutamate receptor subunits) or GABA‐B receptor. Most of these antibody‐associated encephalitides can occur with or without an underlying neoplasm. The presence of these antibodies is diagnostically relevant. Whether these antibodies have a pathogenic role however is still a matter of ongoing investigations. Generally, it is assumed that antibodies against intracellular antigens themselves are not pathogenic (34) as it is hard to imagine how these antibodies, especially in normal brain, can reach the intracellular neural antigens. Exceptions may be antibodies to GAD65 (58) or to amphiphysin (29).
The possible role of cytotoxic T lymphocytes is best documented in the “classical” paraneoplastic encephalitis cases with antibodies against oncogenes. In neuropathological studies of PE with anti‐Hu, anti‐Yo or anti‐Ma antibodies, a dominance of CD8 positive T‐cells in the infiltrates has been described 20, 30, 33, 46, 76, 99. Furthermore, a small number of studies have shown that these T‐cells possess cytotoxic granules and are in close apposition to neurons 33, 34, 35 suggesting that cytotoxic T lymphocytes play a role in neuronal cell death 6, 14, 91. Although these T‐cells are found in the neighborhood of neurons, no studies have actually shown to which antigens these cytotoxic T lymphocytes are directed. In addition, in pathological studies, evidence that these lymphocytes actually release cytotoxic substances and thereby are able to kill neurons is hard to show. Tanaka and colleagues however described that cytotoxic lymphocytes from a patient with anti‐Yo PE could lyse her own Yo protein‐expressing fibroblasts (92). Furthermore, comparable to those in RE studies, Voltz and colleagues investigated T‐cell responses and T‐cell receptor usage in PE with anti‐Hu antibodies and showed that an antigen‐driven oligoclonal cytotoxic T‐cell response might play a role in the pathogenesis of anti‐Hu‐associated PE (108). In patients suffering from paraneoplastic cerebellar degeneration, anti‐cdr2‐specific CD8 positive cytotoxic T‐cells are present in the cerebrospinal fluid. Treatment with tacrolimus decreases the number of these T‐cells in cerebrospinal fluid (CSF) but only has a limited effect on the clinical symptoms (1).
The pathology and the immune mechanisms responsible for the neurodegeneration and epileptic seizures in cases with antibodies to neuronal surface antigens are largely unknown. As the group of these antibody‐associated encephalitides is large and constantly new antibodies against known neural antigens are discovered, it is difficult to discuss all the individual antibody cases. Therefore, we selected a number of the more recently described encephalitides.
VGKC complex encephalitis
Antibodies against VGKC complex can be found both in paraneoplastic (16) but usually patients do have nonparaneoplastic (“idiopathic”) limbic encephalitis (77). Recently, it was shown that these antibodies are mostly not directed to the potassium channels themselves but to proteins complexed with them, such as contactin‐associated protein‐like 2 (Caspr2) (52) and leucine‐rich, glioma‐inactivated 1 (LGI1) 42, 50. Caspr2 is situated in the area of the juxtaparanodal loop of myelin‐surrounding axons. Antibodies against LGI1 are more often found than antibodies against Caspr2 (42). Clinically, these patients present with histories of memory loss, confusion, behavioral changes and seizures 19, 105. In addition, patients with LGI1 antibodies present with faciobrachial dystonic seizures preceding a limbic encephalitis (44). Pathologically, the anti‐VGKC complex cases reveal neuronal degeneration in the presence of infiltrating T‐cells and perivascular B‐cells 25, 48, 74, 105. Patients with antibodies specific for LGI1 or caspr2 show inflammation and severe degeneration in the hippocampus (13). Importantly, antibody‐lowering treatments like plasma exchange have been found to improve the clinical deficits in these patients, suggesting that the antibodies to the VGKC complex are responsible for the clinical signs 105, 111. In our recent study of four nonparaneoplastic anti‐VGKC complex cases, we found T‐cell infiltrates in hippocampus and cortex. In addition, we also found the presence of human IgG as well as deposition of C9neo, the end complex of complement activation, associated with dying neurons in the hippocampus, thus indicating that an antibody‐mediated complement activation is responsible for cell death in these patients. Such antibody and complement deposition was not observed in brain specimens from patients with onconeural antibodies, GAD antibodies or NMDA receptor antibodies and appears thus specific for this type of encephalitides (13).
NMDAR encephalitis
The clinical syndrome of NMDAR encephalitis was first reported in 2005 in patients with paraneoplastic encephalitis resulting from ovarian teratomas harboring antibodies to hippocampal neuropil (2). Soon after, it was discovered that this particular syndrome was associated with antibodies against the NR1 subunit of the NMDAR (22). Anti‐NMDAR encephalitis later also was found in the absence of a tumor in a large number of patients 22, 43. The syndrome is in many ways remarkable. It mainly occurs in young females with a peak of age at onset between 19 and 24 years (23). Clinically, a prodromal stage with symptoms such as fever, nausea, vomiting or diarrhea may be found in retrospect (40). If present, after about 2 weeks, patients develop seizures (partial) status epilepticus, short‐term memory loss and, in addition, psychiatric symptoms such as anxiety, insomnia, fear, mania and paranoia. This phase, again shortly after, is followed by the initiation of abnormal movements of limb and trunk and oro‐lingual‐facial dyskinesias, a sudden spontaneous fall in consciousness and autonomic manifestations such as tachy‐ or bradycardia, hyperventilation and central hypoventilation 21, 23. At this stage, patients need to be managed in intensive care units. Remarkably however is that about three quarters of the patients, despite these severe clinical signs, recover completely (23). The pathology of patients with antibodies against the NMDA receptor differs from other antibody‐associated encephalitides. First of all, although the disease is called an “encephalitis,” in the brain parenchyma of PE patients, only relatively few inflammatory cells are found 18, 96. T‐cells, B‐cells and plasma cells are, however, present in the perivascular space of blood vessels (61). Moreover, the neuronal loss in these patients is remarkably mild. Neuronal loss in the hippocampus was detected in only one of four brains studied by Dalmau et al (21). In some reports, this syndrome therefore is referred to as a “encephalopathy” rather than encephalitis, thereby underlining the functional as opposed to structural character of the damage the disease causes (43). Concordantly, follow‐up MRI studies in most patients show absence of brain atrophy or even reversible atrophy (41). An exception to this generally favorable outcome is shown in a case report recently published in Neurology (95). This male patient died after a short disease course and, different from the other NMDAR cases, exhibited severe neuronal loss. The reason for this is unknown, but it might be related to the presence of a second antibody directed against the glycine receptor in the serum of this patient which, in addition to the symptoms related to the anti‐NMDAR encephalitis, also showed the symptoms of progressive encephalomyelitis with rigidity and myoclonus (PERM). A question remains whether the anti‐NMDAR antibodies themselves really have a pathogenic capacity. In the available presentations of NMDAR encephalitis, immunoglobulin G binding to hippocampal neurons is seen in the absence of complement 61, 96, which suggests that an antibody‐mediated complement activation is not present in these brains. Interestingly, in the hippocampus, a decrease in NMDAR however is found. Additional experimental evidence suggests that the NMDAR antibodies therefore in fact may act by reducing the density of NMDAR clusters by cross‐linking and subsequent internalization of the receptors, leading to a state of reversible NMDAR hypofunction (39). Further studies with CSF from anti‐NMDAR antibody encephalitis patients suppressed induction of long‐term potentiation (LTP) in mouse hippocampal slices (113), suggesting that these antibodies can act as an NMDAR antagonist and thus may be involved in amnesia. In our study (13), we studied various immune mechanisms in anti‐NMDAR cases with neocortical biopsy samples only. Like in other studies, we did not observe clear cell loss, signs of acute cell damage or atrophy on brain MRI. Furthermore, we found few infiltrating T‐cells, their numbers being lower than in other antibody‐defined subgroups (anti‐VGKC complex and anti‐GAD65 cases see also Figure 1). Although a few cytotoxic T‐cells were present, we also could not detect any targeting of neurons. This again differs from the above mentioned case with NMDAR as well as GlyR antibodies where the authors described strong inflammation and targeting of neurons by cytotoxic T‐cells (95). Taken together, even though NMDAR antibodies appear to be involved in the clinical disease process, there is no evidence in favor of a complement‐mediated or a cytotoxic T‐cell‐mediated neuronal cell death in this disease. An exclusive effect of the antibodies in reducing NMDAR expression in the hippocampus, however, is difficult to reconcile with the complex progression of the disease, which begins with features that could stem from medial temporal lobe dysfunction but progresses to a much broader clinical phenotype including subcortical and even infratentorial brain dysfunction 22, 43.
GAD65 encephalitis
GAD antibodies are associated with a broad spectrum of diseases. In low‐positive titers, they are found in patients with diabetes mellitus type 1 88, 89. Neurological diseases are associated with very high GAD antibody concentrations being two to three log ranks higher than in the diabetic population (65). The spectrum of neurological conditions associated with GAD antibodies ranges is broad (86). It ranges from stiff‐man syndrome (64) over cerebellar ataxia 38, 85 to limbic encephalitis (56) and pharmacoresistant temporal lobe epilepsy (54), which is probably the chronic form of GAD antibody‐associated limbic encephalitis (106). Some (not epilepsy related) experimental conditions have provided evidence that these antibodies might contribute to a loss of GABAergic inhibition (GAD being the rate limiting enzyme in the biosynthesis of GABA) 57, 58. We studied the brains of nonparaneoplastic GAD encephalitis patients by histopathology. In the hippocampi of these patients, we found clear neuronal loss and axonal dystrophy. Ig and complement deposition however was completely absent from these brains. As the antigen is intracellular, a T‐cell‐mediated pathology would be a likely mechanism (35). Therefore, we looked for signs of T‐cell cytotoxicity. In comparison with paraneoplastic anti‐Hu and anti‐Ma2 cases and cases with anti‐surface antibodies (anti‐NMDAR and anti‐VGKC complex antibodies) GAD antibody‐positive cases showed an “intermediate” ratio of CD8+/CD3+ T‐cells in between those of the onconeural cases and those with antibodies to surface antigens. Of note, the density of parenchymal T‐cells in the GAD antibody‐positive patients was lower than in the onconeural and surface antigen groups. On the other hand, however, we found clear appositions of multiple cytotoxic granzyme‐B+ T‐cells to hippocampal neurons (13). The reason for the low density of T‐cells is not clear. The absence of an underlying malignancy as an immunological stimulus may be a reason, something which also has been noted in nonparaneoplastic VGKC complex cases (48), but also the long disease duration in some of these patients may have an influence on this. To make things even more complicated, there have been observations of patients with GAD antibodies in combination with other antibodies—for example, to the GABA‐B‐receptor 15, 34, 51. Similarly, in sera from stiff person patients with GAD antibodies, additional cell surface antibodies have been observed (Chang, Vincent, in preparation). Such additional (pathogenic) antibodies to surface antigens might be an explanation for treatment responses to apheresis techniques 62, 63.
THE ROLE OF THE INNATE IMMUNE SYSTEM IN SEIZURE INDUCTION IN ENCEPHALITIS
The above discussed T‐cell cytotoxicity, antibody and complement‐mediated cytotoxicity in encephalitis are part of the adaptive immune system. These mechanisms play a role in degeneration of neural tissue, which may generate seizures secondarily. It is however the innate immune system, which may contribute directly to the induction of seizures. In the CNS, microglial and astroglial cells are the main cell types activated in the innate immune response. Upon activation, these cells can produce a large range of soluble inflammatory mediators, including cytokines such as interleukins (IL), chemokines, prostaglandins and complement factors (104). Additionally, glial cells, as well as neurons, can overexpress receptors for inflammatory molecules, including receptors for proinflammatory cytokines [eg, IL‐1β, IL‐6 and tumor necrosis factor (TNF)‐α], as well as toll‐like receptors (TLR) 82, 104. The involvement of cytokines in neuronal network excitability was initially suggested by the evidence that various convulsant drugs including kainic acid, bicuculline or electrical stimulation of seizure‐prone brain areas, increase mRNA and the related protein levels of various inflammatory and anti‐inflammatory molecules. The mechanisms by which innate immune proinflammatory pathways contribute to seizures have been studied in experimental models. Rapid effects of cytokines or prostaglandins on neuronal excitability have been reported to consist in posttranslational changes in receptor‐coupled or voltage‐dependent ion channels leading to increased glutamatergic neurotransmission, or reduced GABA‐mediated effects (107). Proinflammatory cytokines also can decrease glutamate reuptake by astrocytes, and can increase the release of excitatory gliotransmitters by activated glial cells, possibly also contributing to neuronal network hyperexcitability. Long‐term effects of inflammatory mediators involve gene transcription of proinflammatory genes, which may perpetuate inflammation in brain tissue, and play a role in alterations in blood–brain barrier (BBB) permeability properties. A compromised BBB in turn contributes to decrease seizure threshold by inducing ionic imbalance in the extracellular milieu, as well as astrocytes and microglia dysfunctions. The persistence of transcript and protein upregulation in brain is at least in part determined by the severity and frequency of seizure activity, as well as by the underlying neuropathology 24, 27, 66, 67, 69, 104. One of the predominant players investigated presently is IL‐1β. In the normal brain, this cytokine can be upregulated in glial cells by central or peripheral administration of bacterial lipopolysaccharides mimicking infectious processes, and plays an important role as mediator of endotoxin‐induced responses such as fever, sleep and anorexia 17, 97. In the absence of infection, this cytokine can also be induced in the brain by various injuries with proconvulsant or epileptogenic properties, such as trauma, stroke or seizure activity (104). In human brain with temporal lobe epilepsy (TLE) or malformations of cortical development (MCD), IL‐1β and its functional receptor type 1 (IL‐1R1) are upregulated, suggesting activation of the IL‐1 β signaling in these forms of epilepsy 79, 81. A neuropathological evaluation by Iyer et al shows that IL‐1β is upregulated in balloon cells, dysmorphic neurons as well as in activated microglia and astrocytes, in FCD type II but only to a minor extent in glia in FCD type I (45). Interestingly, this report also shows that FCD type II patients also have infiltration of cytotoxic T‐cells suggesting that the immune mechanisms between FCD type I and type II are largely different.
As part of the innate system, besides IL‐1β, also endogenous ligands for the TLRs and receptors for advanced glycation end products (RAGE), such as high‐mobility group box 1 (HMGB1), have been studied in epilepsy. HMGB1 is a chromatin‐bound factor that in physiological conditions enhances transcription of inflammatory genes in the nucleus of neurons, but upon cell damage or neuronal hyperexcitability this molecule translocates from the nucleus to the cytoplasm and can be released in the extracellular milieu to induce proinflammatory signals by stimulation of TLR4 or RAGE (55). In brain tissue from TLE or MCDs, HMGB1‐TLR4 signaling is upregulated as previously shown for IL‐1β‐IL‐1R1, and is also a hallmark of epileptic tissue in experimental models (59). In addition, TLR2 and RAGE upregulation have been found in developmental epileptogenic lesions (114).
Pharmacological studies of seizure susceptibility in experimental models have shown that the induction of proinflammatory innate immunity pathways plays a permissive role in seizure activity. Thus, suppression of acute and chronic seizures can be attained in mice or rats using specific inhibitors of IL‐1β biosynthesis, by IL‐1Ra or by blocking TLR4 60, 80, 102. In epilepsy models, injection of IL‐1β before induction of seizures enhances the recurrence of these seizures, whereas application of IL‐1Ra blocks this effect (100). Moreover, astrocytic overexpression of IL‐1Ra reduces mice susceptibility to bicuculline and kainic acid seizures (101). Accordingly, transgenic mice with impaired IL‐1β biosynthesis or lacking TLR4 function are intrinsically resistant to seizures (59). Inhibitors of the biologic actions of HMGB1, such as the peptide box A, also significantly reduce experimental seizures and delay their precipitation after a convulsant challenge (59). Furthermore, intracerebroventricular injection of recombinant HMGB1, by causing activation of TLR4 induces, induces significantly more injury after cerebral ischemia‐reperfusion than observed in TLR4 knockout mice. Interestingly, infiltrating macrophages were found to worsen brain injury in these ischemic brains (112). This is important information as it suggests that in those cases where macrophages from the periphery enter the brain (such as in viral encephalitides), in addition to the intrinsic microglial cells, can influence the course of epileptic seizures. An important aspect of these studies is the translational potential to the clinical application. The use of inhibitors of the IL‐1β signaling or TLRs inhibitors are already in clinical use for inflammatory disorders, and they might represent an alternative therapeutic strategy in those epileptic patients, which do not respond to the conventional anti‐epileptic drugs (103).
CONCLUSIONS
Antibody‐associated encephalitides are increasingly recognized in patients with early, as well as late, onset epilepsy. These epilepsies can be part of a paraneoplastic syndrome. Intriguingly however, such antibodies can be detected also in non‐tumor‐related encephalitides. The discovery of these new encephalitides variants explain a number of cases previously classified as epilepsy of unknown cause (12). The discovery of even more antibody‐associated encephalitis variants following improved testing of blood serum is to be expected. A better understanding of ictogenesis, as well as epileptogenesis in the encephalitic brain, remains challenging as it involves complex pathomechanisms and a variety of molecular signals. Recent data suggest seizure initiation by the innate immune system producing IL‐1β. At later stages, the adaptive immune system may indirectly enhance epileptogenesis by invasion of inflammatory cytotoxic T lymphocytes and antibody‐mediated complement activation, which destroys neurons and promotes neurodegeneration. In addition, aberrant cytokine production, failure to maintain potassium homeostasis and to buffer cytotoxic glutamate as pathogenic contribution of activated astrocytes, can also be considered to participate in the generation of seizures. This multifactorial induction of seizures and promotion of epileptogenesis makes it difficult to develop successful anti‐epileptogenic treatment strategies. The use of inhibitors of the IL‐1β signaling or TLRs inhibitors may, however, be a promising strategy. In addition, early reduction of inflammatory infiltrates, especially in those encephalitides with cytotoxic T lymphocytes, will be necessary to attenuate neurodegeneration and further induction of chronic seizures.
Christian G. Bien performs serum and CSF antibody testing. His employer, the Krankenhaus Mara gGmbH, charges sending institutions for this.
REFERENCES
- 1. Albert ML, Austin LM, Darnell RB (2000) Detection and treatment of activated T cells in the cerebrospinal fluid of patients with paraneoplastic cerebellar degeneration. Ann Neurol 47:9–17. [PubMed] [Google Scholar]
- 2. Ances BM, Vitaliani R, Taylor RA, Liebeskind DS, Voloschin A, Houghton DJ et al (2005) Treatment‐responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain 128:1764–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andermann F (1991) Chronic Encephalitis and Epilepsy. Rasmussen's Syndrome. Butterworth‐Heinemann: Boston. [Google Scholar]
- 4. Andrews PI, Dichter MA, Berkovic SF, Newton MR, McNamara JO (1996) Plasmapheresis in Rasmussen's encephalitis. Neurology 46:242–246. [DOI] [PubMed] [Google Scholar]
- 5. Bauer J, Elger CE, Hans VH, Schramm J, Urbach H, Lassmann H, Bien CG (2007) Astrocytes are a specific immunological target in Rasmussen's encephalitis. Ann Neurol 62:67–80. [DOI] [PubMed] [Google Scholar]
- 6. Bernal F, Graus F, Pifarre A, Saiz A, Benyahia B, Ribalta T (2002) Immunohistochemical analysis of anti‐Hu‐associated paraneoplastic encephalomyelitis. Acta Neuropathol (Berl) 103:509–515. [DOI] [PubMed] [Google Scholar]
- 7. Bien CG, Bauer J (2010) Rasmussen encephalitis. In: Inflammatory and Autoimmune Disorders of the Nervous System in Children. Dale RC, Vincent A (eds), pp. 207–226. Mac Keith Press: London. [Google Scholar]
- 8. Bien CJ, Bauer J, Deckwerth T, Wiendl H, Deckert M, Wiestler O et al (2002) Destruction of neurons by cytotoxic T cells: a new pathogenic mechanism in Rasmussen's encephalitis. Ann Neurol 51:311–318. [DOI] [PubMed] [Google Scholar]
- 9. Bien CG, Widman G, Urbach H, Sassen R, Kuczaty S, Wiestler OD et al (2002) The natural history of Rasmussen's encephalitis. Brain 125(Pt 8):1751–1759. [DOI] [PubMed] [Google Scholar]
- 10. Bien CG, Gleissner U, Sassen R, Widman G, Urbach H, Elger CE (2004) An open study of tacrolimus therapy in Rasmussen encephalitis. Neurology 62:2106–2109. [DOI] [PubMed] [Google Scholar]
- 11. Bien CG, Elger CE, Leitner Y, Gomori M, Ran B, Urbach H et al (2007) Slowly progressive hemiparesis in childhood as a consequence of Rasmussen encephalitis without or with delayed‐onset seizures. Eur J Neurol 14:387–390. [DOI] [PubMed] [Google Scholar]
- 12. Bien CG, Urbach H, Schramm J, Soeder BM, Becker AJ, Voltz R et al (2007) Limbic encephalitis as a precipitating event in adult‐onset temporal lobe epilepsy. Neurology 69:1236–1244. [DOI] [PubMed] [Google Scholar]
- 13. Bien CG, Vincent A, Barnett MH, Becker AJ, Blümcke M, Graus F et al (2012) Immunopathology of autoantibody‐associated encephalitides: clues for pathogenesis. Brain doi: 1093/brain/aws082. [DOI] [PubMed] [Google Scholar]
- 14. Blumenthal DT, Salzman KL, Digre KB, Jensen RL, Dunson WA, Dalmau J (2006) Early pathologic findings and long‐term improvement in anti‐Ma2‐associated encephalitis. Neurology 67:146–149. [DOI] [PubMed] [Google Scholar]
- 15. Boronat A, Sabater L, Saiz A, Dalmau J, Graus F (2011) GABA(B) receptor antibodies in limbic encephalitis and anti‐GAD‐associated neurologic disorders. Neurology 76:795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Buckley C, Oger J, Clover L, Tüzün E, Carpenter K, Jackson M, Vincent A (2001) Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol 50:73–78. [DOI] [PubMed] [Google Scholar]
- 17. Busbridge NJ, Dascombe MJ, Tilders FJ, van Oers JW, Linton EA, Rothwell NJ (1989) Central activation of thermogenesis and fever by interleukin‐1 beta and interleukin‐1 alpha involves different mechanisms. Biochem Biophys Res Commun 162:591–596. [DOI] [PubMed] [Google Scholar]
- 18. Camdessanche JP, Streichenberger N, Cavillon G, Rogemond V, Jousserand G, Honnorat J et al (2011) Brain immunohistopathological study in a patient with anti‐NMDAR encephalitis. Eur J Neurol 18:929–931. [DOI] [PubMed] [Google Scholar]
- 19. Chan D, Henley SM, Rossor MN, Warrington EK (2007) Extensive and temporally ungraded retrograde amnesia in encephalitis associated with antibodies to voltage‐gated potassium channels. Arch Neurol 64:404–410. [DOI] [PubMed] [Google Scholar]
- 20. Dalmau J, Gultekin SH, Voltz R, Hoard R, DesChamps T, Balmaceda C et al (1999) Ma1, a novel neuron‐ and testis‐specific protein, is recognized by the serum of patients with paraneoplastic neurological disorders. Brain 122(Pt 1):27–39. [DOI] [PubMed] [Google Scholar]
- 21. Dalmau J, Tüzün E, Wu HY, Masjuan J, Rossi JE, Voloschin A et al (2007) Paraneoplastic anti‐N‐methyl‐D‐aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 61:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M et al (2008) Anti‐NMDA‐receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol 7:1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dalmau J, Lancaster E, Martinez‐Hernandez E, Rosenfeld MR, Balice‐Gordon R (2011) Clinical experience and laboratory investigations in patients with anti‐NMDAR encephalitis. Lancet Neurol 10:63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. De Simoni MG, Perego C, Ravizza T, Moneta D, Conti M, Marchesi F et al (2000) Inflammatory cytokines and related genes are induced in the rat hippocampus by limbic status epilepticus. Eur J Neurosci 12:2623–2633. [DOI] [PubMed] [Google Scholar]
- 25. Dunstan EJ, Winer JB (2006) Autoimmune limbic encephalitis causing fits, rapidly progressive confusion and hyponatraemia. Age Ageing 35:536–537. [DOI] [PubMed] [Google Scholar]
- 26. Eid T, Williamson A, Lee TS, Petroff OA, de Lanerolle NC (2008) Glutamate and astrocytes—key players in human mesial temporal lobe epilepsy? Epilepsia 49(Suppl. 2):42–52. [DOI] [PubMed] [Google Scholar]
- 27. Eriksson C, Van Dam AM, Lucassen PJ, Bol JG, Winblad B, Schultzberg M (1999) Immunohistochemical localization of interleukin‐1beta, interleukin‐1 receptor antagonist and interleukin‐1beta converting enzyme/caspase‐1 in the rat brain after peripheral administration of kainic acid. Neuroscience 93:915–930. [DOI] [PubMed] [Google Scholar]
- 28. Farrell MA, Droogan O, Secor DL, Poukens V, Quinn B, Vinters HV (1995) Chronic encephalitis associated with epilepsy: immunohistochemical and ultrastructural studies. Acta Neuropathol (Berl) 89:313–321. [DOI] [PubMed] [Google Scholar]
- 29. Geis C, Weishaupt A, Hallermann S, Grunewald B, Wessig C, Wultsch T et al (2010) Stiff person syndrome‐associated autoantibodies to amphiphysin mediate reduced GABAergic inhibition. Brain 133:3166–3180. [DOI] [PubMed] [Google Scholar]
- 30. Giometto B, Marchiori GC, Nicolao P, Scaravilli T, Lion A, Bardin PG, Tavolato B (1997) Sub‐acute cerebellar degeneration with anti‐Yo autoantibodies: immunohistochemical analysis of the immune reaction in the central nervous system. Neuropathol Appl Neurobiol 23:468–474. [DOI] [PubMed] [Google Scholar]
- 31. Granata T (2003) Rasmussen's syndrome. Neurol Sci 24(Suppl. 4):S239–SS43. [DOI] [PubMed] [Google Scholar]
- 32. Granata T, Fusco L, Gobbi G, Freri E, Ragona F, Broggi G et al (2003) Experience with immunomodulatory treatments in Rasmussen's encephalitis. Neurology 61:1807–1810. [DOI] [PubMed] [Google Scholar]
- 33. Graus F, Ribalta T, Campo E, Monforte R, Urbano A, Rozman C (1990) Immunohistochemical analysis of the immune reaction in the nervous system in paraneoplastic encephalomyelitis. Neurology 40:219–222. [DOI] [PubMed] [Google Scholar]
- 34. Graus F, Saiz A, Lai M, Bruna J, Lopez F, Sabater L et al (2008) Neuronal surface antigen antibodies in limbic encephalitis: clinical‐immunologic associations. Neurology 71:930–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Graus F, Saiz A, Dalmau J (2010) Antibodies and neuronal autoimmune disorders of the CNS. J Neurol 257:509–517. [DOI] [PubMed] [Google Scholar]
- 36. Hauf M, Wiest R, Nirkko A, Strozzi S, Federspiel A (2009) Dissociation of epileptic and inflammatory activity in Rasmussen Encephalitis. Epilepsy Res 83:265–268. [DOI] [PubMed] [Google Scholar]
- 37. Hesdorffer DC, Hauser WA, Annegers JF, Kokmen E, Rocca WA (1996) Dementia and adult‐onset unprovoked seizures. Neurology 46:727–730. [DOI] [PubMed] [Google Scholar]
- 38. Honnorat J, Saiz A, Giometto B, Vincent A, Brieva L, de Andres C et al (2001) Cerebellar ataxia with anti‐glutamic acid decarboxylase antibodies: study of 14 patients. Arch Neurol 58:225–230. [DOI] [PubMed] [Google Scholar]
- 39. Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R et al (2010) Cellular and synaptic mechanisms of anti‐NMDA receptor encephalitis. J Neurosci 30:5866–5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Iizuka T, Sakai F, Ide T, Monzen T, Yoshii S, Iigaya M et al (2007) Anti‐NMDA receptor encephalitis in Japan. Long‐term outcome without tumor removal. Neurology 70:504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Iizuka T, Yoshii S, Kan S, Hamada J, Dalmau J, Sakai F, Mochizuki H (2010) Reversible brain atrophy in anti‐NMDA receptor encephalitis: a long‐term observational study. J Neurol 257:1686–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Irani SR, Alexander S, Waters P, Kleopa KA, Pettingill P, Zuliani L et al (2010) Antibodies to Kv1 potassium channel‐complex proteins leucine‐rich, glioma inactivated 1 protein and contactin‐associated protein‐2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia. Brain 133:2734–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Irani SR, Bera K, Waters P, Zuliani L, Maxwell S, Zandi MS et al (2010) N‐methyl‐D‐aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non‐paraneoplastic disorder of both sexes. Brain 133(Pt 6):1655–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Irani SR, Michell AW, Lang B, Pettingill P, Waters P, Johnson MR et al (2011) Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 69:892–900. [DOI] [PubMed] [Google Scholar]
- 45. Iyer A, Zurolo E, Spliet WG, van Rijen PC, Baayen JC, Gorter JA, Aronica E (2010) Evaluation of the innate and adaptive immunity in type I and type II focal cortical dysplasias. Epilepsia 51:1763–1773. [DOI] [PubMed] [Google Scholar]
- 46. Jean WC, Dalmau J, Ho A, Posner JB (1994) Analysis of the IgG subclass distribution and inflammatory infiltrates in patients with anti‐Hu‐associated paraneoplastic encephalomyelitis. Neurology 44:140–147. [DOI] [PubMed] [Google Scholar]
- 47. Kang TC, Kim DS, Kwak SE, Kim JE, Won MH, Kim DW et al (2006) Epileptogenic roles of astroglial death and regeneration in the dentate gyrus of experimental temporal lobe epilepsy. Glia 54:258–271. [DOI] [PubMed] [Google Scholar]
- 48. Khan NL, Jeffree MA, Good C, Macleod W, Al‐Sarraj S (2009) Histopathology of VGKC antibody‐associated limbic encephalitis. Neurology 72:1703–1705. [DOI] [PubMed] [Google Scholar]
- 49. Korn‐Lubetzki I, Bien CG, Bauer J, Gomori M, Wiendl H, Trajo L et al (2004) Rasmussen encephalitis with active inflammation and delayed seizures onset. Neurology 62:984–986. [DOI] [PubMed] [Google Scholar]
- 50. Lai M, Huijbers MG, Lancaster E, Graus F, Bataller L, Balice‐Gordon R et al (2010) Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 9:776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J et al (2010) Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 9:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lancaster E, Huijbers MG, Bar V, Boronat A, Wong A, Martinez‐Hernandez E et al (2011) Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol 69:303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li Y, Uccelli A, Laxer KD, Jeong MC, Vinters HV, Tourtellotte WW et al (1997) Local‐clonal expansion of infiltrating T lymphocytes in chronic encephalitis of Rasmussen. J Immunol 158:1428–1437. [PubMed] [Google Scholar]
- 54. Liimatainen S, Peltola M, Sabater L, Fallah M, Kharazmi E, Haapala AM et al (2010) Clinical significance of glutamic acid decarboxylase antibodies in patients with epilepsy. Epilepsia 51:760–767. [DOI] [PubMed] [Google Scholar]
- 55. Lotze MT, Tracey KJ (2005) High‐mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 5:331–342. [DOI] [PubMed] [Google Scholar]
- 56. Malter MP, Helmstaedter C, Urbach H, Vincent A, Bien CG (2010) Antibodies to glutamic acid decarboxylase define a form of limbic encephalitis. Ann Neurol 67:470–478. [DOI] [PubMed] [Google Scholar]
- 57. Manto M, Dalmau J, Didelot A, Rogemond V, Honnorat J (2010) Afferent facilitation of corticomotor responses is increased by IgGs of patients with NMDA‐receptor antibodies. J Neurol 258:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Manto MU, Laute MA, Aguera M, Rogemond V, Pandolfo M, Honnorat J (2007) Effects of anti‐glutamic acid decarboxylase antibodies associated with neurological diseases. Ann Neurol 61:544–551. [DOI] [PubMed] [Google Scholar]
- 59. Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM et al (2010) Toll‐like receptor 4 and high‐mobility group box‐1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med 16:413–419. [DOI] [PubMed] [Google Scholar]
- 60. Maroso M, Balosso S, Ravizza T, Iori V, Wright CI, French J, Vezzani A (2011) Interleukin‐1beta biosynthesis inhibition reduces acute seizures and drug resistant chronic epileptic activity in mice. Neurother 8:304–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Martinez‐Hernandez E, Horvath J, Shiloh‐Malawsky Y, Sangha N, Martinez‐Lage M, Dalmau J (2011) Analysis of complement and plasma cells in the brain of patients with anti‐NMDAR encephalitis. Neurology 77:589–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mata S, Muscas GC, Naldi I, Rosati E, Paladini S, Cruciatti B et al (2008) Non‐paraneoplastic limbic encephalitis associated with anti‐glutamic acid decarboxylase antibodies. J Neuroimmunol 199:155–159. [DOI] [PubMed] [Google Scholar]
- 63. Mazzi G, Roia DD, Cruciatti B, Mata S, Catapano R (2008) Plasma exchange for anti‐GAD associated non paraneoplastic limbic encephalitis. Transfus Apher Sci 39:229–233. [DOI] [PubMed] [Google Scholar]
- 64. Meinck HM, Thompson PD (2002) Stiff man syndrome and related conditions. Mov Disord 17:853–866. [DOI] [PubMed] [Google Scholar]
- 65. Meinck HM, Faber L, Morgenthaler N, Seissler J, Maile S, Butler M et al (2001) Antibodies against glutamic acid decarboxylase: prevalence in neurological diseases. J Neurol Neurosurg Psychiatry 71:100–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Minami M, Kuraishi Y, Yamaguchi T, Nakai S, Hirai Y, Satoh M (1990) Convulsants induce interleukin‐1 beta messenger RNA in rat brain. Biochem Biophys Res Commun 171:832–837. [DOI] [PubMed] [Google Scholar]
- 67. Minami M, Kuraishi Y, Satoh M (1991) Effects of kainic acid on messenger RNA levels of IL‐1 beta, IL‐6, TNF alpha and LIF in the rat brain. Biochem Biophys Res Commun 176:593–598. [DOI] [PubMed] [Google Scholar]
- 68. Nelson KB, Lynch JK (2004) Stroke in newborn infants. Lancet Neurol 3:150–158. [DOI] [PubMed] [Google Scholar]
- 69. Nishiyori A, Minami M, Takami S, Satoh M (1997) Type 2 interleukin‐1 receptor mRNA is induced by kainic acid in the rat brain. Brain Res Mol Brain Res 50:237–245. [DOI] [PubMed] [Google Scholar]
- 70. Noebels J (2011) A perfect storm: converging paths of epilepsy and Alzheimer's dementia intersect in the hippocampal formation. Epilepsia 52(Suppl. 1):39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Oguni H, Olivier A, Andermann F, Comair J (1991) Anterior callosotomy in the treatment of medically intractable epilepsies: a study of 43 patients with a mean follow‐up of 39 months. Ann Neurol 30:357–364. [DOI] [PubMed] [Google Scholar]
- 72. Palmer CA, Geyer JD, Keating JM, Gilliam F, Kuzniecky RI, Morawetz RB, Bebin EM (1999) Rasmussen's encephalitis with concomitant cortical dysplasia: the role of GluR3. Epilepsia 40:242–247. [DOI] [PubMed] [Google Scholar]
- 73. Pardo CA, Vining EP, Guo L, Skolasky RL, Carson BS, Freeman JM (2004) The pathology of Rasmussen syndrome: stages of cortical involvement and neuropathological studies in 45 hemispherectomies. Epilepsia 45:516–526. [DOI] [PubMed] [Google Scholar]
- 74. Park DC, Murman DL, Perry KD, Bruch LA (2007) An autopsy case of limbic encephalitis with voltage‐gated potassium channel antibodies. Eur J Neurol 14:e5–e6. [DOI] [PubMed] [Google Scholar]
- 75. Piatt JH, Jr , Hwang PA, Armstrong DC, Becker LE, Hoffman HJ (1988) Chronic focal encephalitis (Rasmussen syndrome): six cases. Epilepsia 29:268–279. [DOI] [PubMed] [Google Scholar]
- 76. Posner JB (1991) Paraneoplastic syndromes. Neurol Clin 9:919–936. [PubMed] [Google Scholar]
- 77. Pozo‐Rosich P, Clover L, Saiz A, Vincent A, Graus F (2003) Voltage‐gated potassium channel antibodies in limbic encephalitis. Ann Neurol 54:530–533. [DOI] [PubMed] [Google Scholar]
- 78. Rasmussen T, Olszewski J, Lloyd‐Smith D (1958) Focal seizures due to chronic localized encephalitis. Neurology 8:435–445. [DOI] [PubMed] [Google Scholar]
- 79. Ravizza T, Boer K, Redeker S, Spliet WG, van Rijen PC, Troost D et al (2006) The IL‐1beta system in epilepsy‐associated malformations of cortical development. Neurobiol Dis 24:128–143. [DOI] [PubMed] [Google Scholar]
- 80. Ravizza T, Lucas SM, Balosso S, Bernardino L, Ku G, Noe F et al (2006) Inactivation of caspase‐1 in rodent brain: a novel anticonvulsive strategy. Epilepsia 47:1160–1168. [DOI] [PubMed] [Google Scholar]
- 81. Ravizza T, Gagliardi B, Noe F, Boer K, Aronica E, Vezzani A (2008) Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis 29:142–160. [DOI] [PubMed] [Google Scholar]
- 82. Rivest S (2009) Regulation of innate immune responses in the brain. Nat Rev Immunol 9:429–439. [DOI] [PubMed] [Google Scholar]
- 83. Robitaille Y (1991) Neuropathologic aspects of chronic encephalitis. In: Chronic Encephalitis and Epilepsy Rasmussen's Syndrome. Andermann F (ed.), pp. 79–110. Butterworth‐Heinemann: Boston. [Google Scholar]
- 84. Rogers SW, Andrews PI, Gahring LC, Whisenand T, Cauley K, Crain B et al (1994) Autoantibodies to glutamate receptor GluR3 in Rasmussen's encephalitis. Science 265:648–651. [DOI] [PubMed] [Google Scholar]
- 85. Saiz A, Arpa J, Sagasta A, Casamitjana R, Zarranz JJ, Tolosa E, Graus F (1997) Autoantibodies to glutamic acid decarboxylase in three patients with cerebellar ataxia, late‐onset insulin‐dependent diabetes mellitus, and polyendocrine autoimmunity. Neurology 49:1026–1030. [DOI] [PubMed] [Google Scholar]
- 86. Saiz A, Blanco Y, Sabater L, Gonzalez F, Bataller L, Casamitjana R et al (2008) Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain 131:2553–2563. [DOI] [PubMed] [Google Scholar]
- 87. Schwab N, Bien CG, Waschbisch A, Becker A, Vince GH, Dornmair K, Wiendl H (2009) CD8+ T cell clones dominate brain infiltrates in Rasmussen encephalitis and persist in the periphery. Brain 132:1236–1246. [DOI] [PubMed] [Google Scholar]
- 88. Solimena M, Folli F, Denis Donini S, Comi GC, Pozza G, De Camilli P, Vicari AM (1988) Autoantibodies to glutamic acid decarboxylase in a patient with stiff‐man syndrome, epilepsy, and type I diabetes mellitus. N Engl J Med 318:1012–1020. [DOI] [PubMed] [Google Scholar]
- 89. Striano P, Perruolo G, Errichiello L, Formisano P, Beguinot F, Zara F, Striano S (2006) Glutamic acid decarboxylase antibodies in idiopathic generalized epilepsy and type 1 diabetes. Ann Neurol 63:127–128. [DOI] [PubMed] [Google Scholar]
- 90. Takei H, Wilfong A, Malphrus A, Yoshor D, Hunter JV, Armstrong DL, Bhattacharjee MB (2010) Dual pathology in Rasmussen's encephalitis: a study of seven cases and review of the literature. Neuropathology 30:381–391. [DOI] [PubMed] [Google Scholar]
- 91. Tanaka K, Tanaka M, Inuzuka T, Nakano R, Tsuji S (1999) Cytotoxic T lymphocyte‐mediated cell death in paraneoplastic sensory neuronopathy with anti‐Hu antibody. J Neurol Sci 163:159–162. [DOI] [PubMed] [Google Scholar]
- 92. Tanaka M, Tanaka K, Shinozawa K, Idezuka J, Tsuji S (1998) Cytotoxic T cells react with recombinant Yo protein from a patient with paraneoplastic cerebellar degeneration and anti‐Yo antibody. J Neurol Sci 161:88–90. [DOI] [PubMed] [Google Scholar]
- 93. Thomas JE, Reagan TJ, Klass DW (1977) Epilepsia partialis continua. A review of 32 cases. Arch Neurol 34:266–275. [DOI] [PubMed] [Google Scholar]
- 94. Tian GF, Azmi H, Takano T, Xu Q, Peng W, Lin J et al (2005) An astrocytic basis of epilepsy. Nat Med 11:973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Turner MR, Irani SR, Leite IM, Nithi K, Vincent A, Ansorge O (2011) Glycine & NMDA receptor antibodies in progressive encephalomyelitis with rigidity & myoclonus. Neurology 77:414–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Tüzün E, Zhou L, Baehring JM, Bannykh S, Rosenfeld MR, Dalmau J (2009) Evidence for antibody‐mediated pathogenesis in anti‐NMDAR encephalitis associated with ovarian teratoma. Acta Neuropathol (Berl) 118:737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Van Dam AM, Bauer J, Tilders FJ, Berkenbosch F (1995) Endotoxin‐induced appearance of immunoreactive interleukin‐1 beta in ramified microglia in rat brain: a light and electron microscopic study. Neuroscience 65:815–826. [DOI] [PubMed] [Google Scholar]
- 98. Vernadakis A (1996) Glia‐neuron intercommunications and synaptic plasticity. Prog Neurobiol 49:185–214. [DOI] [PubMed] [Google Scholar]
- 99. Verschuuren J, Chuang L, Rosenblum MK, Lieberman F, Pryor A, Posner JB, Dalmau J (1996) Inflammatory infiltrates and complete absence of Purkinje cells in anti‐Yo‐associated paraneoplastic cerebellar degeneration. Acta Neuropathol (Berl) 91:519–525. [DOI] [PubMed] [Google Scholar]
- 100. Vezzani A, Conti M, De Luigi A, Ravizza T, Moneta D, Marchesi F, De Simoni MG (1999) Interleukin‐1beta immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: functional evidence for enhancement of electrographic seizures. J Neurosci 19:5054–5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Vezzani A, Moneta D, Conti M, Richichi C, Ravizza T, De Luigi A et al (2000) Powerful anticonvulsant action of IL‐1 receptor antagonist on intracerebral injection and astrocytic overexpression in mice. Proc Natl Acad Sci U S A 97:11534–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Vezzani A, Moneta D, Richichi C, Aliprandi M, Burrows SJ, Ravizza T et al (2002) Functional role of inflammatory cytokines and antiinflammatory molecules in seizures and epileptogenesis. Epilepsia 43(Suppl. 5):30–35. [DOI] [PubMed] [Google Scholar]
- 103. Vezzani A, Balosso S, Maroso M, Zardoni D, Noe F, Ravizza T (2010) ICE/caspase 1 inhibitors and IL‐1beta receptor antagonists as potential therapeutics in epilepsy. Curr Opin Investig Drugs 11:43–50. [PubMed] [Google Scholar]
- 104. Vezzani A, French J, Bartfai T, Baram TZ (2011) The role of inflammation in epilepsy. Nat Rev Neurol 7:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Vincent A, Buckley C, Schott JM, Baker I, Dewar BK, Detert N et al (2004) Potassium channel antibody‐associated encephalopathy: a potentially immunotherapy‐responsive form of limbic encephalitis. Brain 127(Pt 3):701–712. [DOI] [PubMed] [Google Scholar]
- 106. Vincent A, Bien CG, Irani SR, Waters P (2011) Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol 10:759–772. [DOI] [PubMed] [Google Scholar]
- 107. Viviani B, Gardoni F, Marinovich M (2007) Cytokines and neuronal ion channels in health and disease. Int Rev Neurobiol 82:247–263. [DOI] [PubMed] [Google Scholar]
- 108. Voltz R, Dalmau J, Posner JB, Rosenfeld MR (1998) T‐cell receptor analysis in anti‐Hu associated paraneoplastic encephalomyelitis. Neurology 51:1146–1150. [DOI] [PubMed] [Google Scholar]
- 109. Whitney KD, Andrews JM, McNamara JO (1999) Immunoglobulin G and complement immunoreactivity in the cerebral cortex of patients with Rasmussen's encephalitis. Neurology 53:699–708. [DOI] [PubMed] [Google Scholar]
- 110. Wiendl H, Bien CG, Bernasconi P, Fleckenstein B, Elger CE, Dichgans J et al (2001) GluR3 antibodies: prevalence in focal epilepsy but no specificity for Rasmussen's encephalitis. Neurology 57:1511–1514. [DOI] [PubMed] [Google Scholar]
- 111. Wong SH, Saunders MD, Larner AJ, Das K, Hart IK (2010) An effective immunotherapy regimen for VGKC antibody‐positive limbic encephalitis. J Neurol Neurosurg Psychiatry (in press). [DOI] [PubMed] [Google Scholar]
- 112. Yang QW, Lu FL, Zhou Y, Wang L, Zhong Q, Lin S et al (2011) HMBG1 mediates ischemia‐reperfusion injury by TRIF‐adaptor independent Toll‐like receptor 4 signaling. J Cereb Blood Flow Metab 31:593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Zhang Q, Tanaka K, Sun P, Nakata M, Yamamoto R, Sakimura K et al (2012) Suppression of synaptic plasticity by cerebrospinal fluid from anti‐NMDA receptor encephalitis patients. Neurobiol Dis 45:610–615. [DOI] [PubMed] [Google Scholar]
- 114. Zurolo E, Iyer A, Maroso M, Carbonell C, Anink JJ, Ravizza T et al (2011) Activation of Toll‐like receptor, RAGE and HMGB1 signalling in malformations of cortical development. Brain 134(Pt 4):1015–1032. [DOI] [PubMed] [Google Scholar]