

Abstract
Alzheimer's disease is the most common form of progressive dementia, typified initially by short term memory deficits which develop into a dramatic global cognitive decline. The classical hall marks of Alzheimer's disease include the accumulation of amyloid oligomers and fibrils, and the intracellular formation of neurofibrillary tangles of hyperphosphorylated tau. It is now clear that inflammation also plays a central role in the pathogenesis of the disease through a number of neurotoxic mechanisms. Microglia are the key immune regulators of the CNS which detect amyloidopathy through cell surface and cytosolic pattern recognition receptors (PRRs) and respond by initiating inflammation through the secretion of cytokines such as interleukin‐1β (IL‐1β). Inflammasomes, which regulate IL‐1β release, are formed following activation of cytosolic PRRs, and using genetic and pharmacological approaches, NLRP3 and NLRP1 inflammasomes have been found to be integral in pathogenic neuroinflammation in animal models of Alzheimer's disease. Therefore, the inflammasomes are very promising novel pharmacological targets which merit further research in the continued endeavor for efficacious therapeutics for Alzheimer's disease.
Keywords: Alzheimer's disease, inflammation, inflammasomes
Introduction
Alzheimer's disease (AD) is the most common form of progressive dementia representing 60%–80% of dementia cases and affects 26 million people worldwide 7. It is characterized by memory loss and a gradual decline of cognitive function which leads to complete dependence on care, with death occurring an average of 5–7 years from diagnosis 166. Currently there are only symptom modifying interventions for AD which do not alter the progression of the disease 28. Therefore, new treatments are desperately needed 7.
Histological investigation provided the first insights into the underlying causes of AD. Over a century ago Alois Alzheimer described the pathological hall marks of the disease of large insoluble plaques and neurofibrillary tangles 6. The plaques are composed of aggregates of amyloid‐β (Aβ) peptide, while neurofibrillary tangles are caused by the accumulation of insoluble filaments of hyper‐phosphorylated tau. Subsequent histological studies have identified neuroinflammatory responses by astrocytes and microglia as another characteristic of AD. However, research is on‐going into whether these histological markers of the disease represent pathological drivers, unrelated by‐products or unsuccessful repair mechanisms.
Neuroimaging and biomarker studies have established that amyloid changes occur prior to tau pathology and this supports the most widely accepted description of the underlying pathology of AD which is the amyloid cascade hypothesis 119. This states that AD is caused by disruptions in amyloid processing and/or clearance leading to an accumulation of monomer amyloid peptides which oligomerize into soluble toxic oligomers and insoluble fibrils, the major constitute of plaques 54. This amyloid pathology then interacts with a number processes, including tau physiology and inflammation, to eventually cause neuronal death and cognitive decline 54.
Genetic evidence supports the amyloid hypothesis; mutations in amyloid precursor protein (APP) or amyloid processing enzymes are the only known causes of autosomal dominant inheritable familial AD 12. No mutations in tau have been found to cause AD. However, genome wide association studies have identified a number of other gene variants which confer an increased risk in the development of sporadic AD and these variants have been found to be involved in a variety of physiological processes including lipid transport and autophagy, such as APOE4 (apolipoprotein E4) and PICALM (phosphatidylinositol‐binding clathrin assembly protein), respectively reviewed by Tosto et al 160. Of interest to this review is that a number of variants of genes involved in regulating innate immune function confer a greater risk of developing AD 58, 128. Examples include loss/reduction of function mutations in the anti‐inflammatory/phagocytosis TREM‐2 (triggering receptor expressed on myeloid cells 2) gene 52; variants of promoter regions of inflammation modulating cytokines interleukin‐10 (IL‐10) and TNFα (tumor necrosis factor α) 129; loss/reduction of function of the anti‐inflammatory/phagocytosis receptor CD33 gene 20; and gene variants of the complement receptor 1 (CR1), which may be integral to the phagocytosis of opsonized amyloid oligomers 62. The number and range of risk genes that are related to immune function demonstrate the integral role inflammation may play in the pathogenesis of AD.
Neuroinflammation and AD
Inflammation is a beneficial immune‐vascular response to damage and infection which involves the activation and recruitment of immune cells. This response is regulated by cytokine signaling molecules, of which interleukin‐1β (IL‐1β) is considered a central member. However, chronic or excessive inflammation can exacerbate tissue damage and contribute to disease. Neuroinflammation is primarily regulated by microglia, the resident immune cells of the brain. These cells make up 10%–15% of the cells of the brain and in a resting state exist in a ramified morphology with long processes which are continually monitoring the extracellular environment for perturbations in homeostasis, tissue damage or infection 124. Upon sensing a change to the extracellular environment, microglia become activated and develop an amoeboid morphology. An activated microglia can act in an anti‐ or pro‐inflammatory manner depending on the stimuli. Anti‐inflammatory activated microglia clear debris through phagocytosis, and secrete anti‐inflammatory cytokines such as IL‐4 and resolution growth factors including brain derived neurotrophic factor (BDNF) 114. A pro‐inflammatory activated microglia will release neurotoxic reactive oxygen species (ROS) and inflammatory cytokines, initiating a potentially damaging immune‐vascular response 17, 124. Astrocytes are also heavily involved in immune regulation in the brain with continued research supporting growing overlap in astrocyte and microglia function including phagocytosis 68, antigen presentation 31, cytokine secretion 22, ROS production 143 and vascular modulation 23, 149. Histology and PET imaging studies demonstrate that inflammatory phenotypes of astrocytes and microglia are a pathological hallmark of AD 14, 65.
Using traditional histological methods, clusters of activated microglia and astrocytes have been shown to occur in in AD patients 14, 65. These clusters appear in close proximity with amyloid plaques and larger plaques correlate with a greater number of associated microglia, suggesting that amyloid fibrils, or the relatively high concentrations of amyloid oligomers found in the peri‐plaque region, are inflammatory 65, 78. Microglia activation can be investigated using PET imaging with radiopharmaceutical tags. Studies using the activated microglia tags [11C](R)‐PK11195 and [11C]DAA1106, which recognize the 18 kDa translocator protein (TSPO) present on activated microglia, found that AD patients have elevated levels of activated microglia, and the level of activation correlates with the severity of AD 25, 171, 174. Furthermore, the second generation TSPO ligand [11C]DAA1106 has been used to demonstrate that inflammation is present in people with mild cognitive impairment (MCI) who then go on to develop AD, suggesting that inflammation is chronic and ongoing prior to the onset of AD 171. The correlation between inflammation and AD severity and the presence of inflammation prior to AD onset suggests a causal relationship between inflammation and AD. This is further supported by epidemiological evidence that known risk factors for AD have an inflammatory component including stroke 162, head trauma 104, diabetes 110, mid‐life obesity 168, aging 79, 123 and infection 120 (Figure 1).
Figure 1.
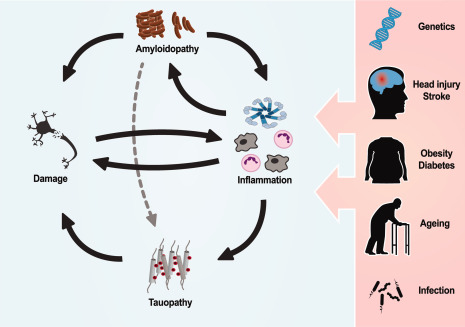
Inflammation has an integral role in the pathogenesis of AD and can be influenced through a number of genetic and environmental factors. Amyloidopathy has been demonstrated to induce neurotoxic inflammation which has been shown to cause and propagate tauopathy. Neuronal damage caused by these processes could result in further inflammation in an unresolved feedback pattern. Many risk factors for AD such as inflammatory gene variants, brain injury, midlife obesity, diabetes, ageing and infection all have an inflammatory component; this supports the critical role inflammation has in AD and highlights the therapeutic potential of targeting inflammation.
Mechanisms of inflammation induced neurodegeneration
Inflammation in the brain can cause neuronal dysfunction and death through a number of mechanisms. These can be grouped into the direct and indirect effects of inflammation on neurones. Direct effects are those in which immune cells engage in neurotoxic activities such as the production of digestive enzymes and ROS, and phagocytosis of healthy neurones. The indirect effects of neuroinflammation are caused by astrocytes and microglia not performing their role as homeostasis managing cells which results in neuronal death through perturbations in the intracellular and extracellular environments. Through these mechanisms it has been shown that neuroinflammation alone is enough to cause cognitive deficits and tauopathology; it is particularly interesting that brain regions most affected by AD, such as the hippocampus, are also the most vulnerable regions to neuroinflammation 61, 86.
Perhaps the best characterized mechanism of inflammation induced neurotoxicity is the production of ROS and reactive nitrogen species (RNS) 16, 42. ROS and RNS are highly reactive molecules which can cause auto‐catalytic oxidation of phospholipids resulting in the permeabilization of membranes, oxidation of proteins perturbing cellular function and DNA damage leading to disruption of protein production 16, 42. Ultimately, if ROS and RNS production overwhelms the antioxidant mechanisms of the cell, the build‐up of oxidative damage will lead to cell death. Amyloid has been shown to induce the production of ROS and RNS in microglia and astrocytes 2, 3, 66. Fibrillary Aβ induces the expression of the ROS producing enzymes nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and inducible nitric oxide synthase (iNOS). These enzymes produce the highly neurotoxic ROS species superoxide and nitric oxide, respectively 127, 161, 167, 169. Microglia cytokine secretion causes the recruitment of peripheral immune cells into the brain including neutrophils which produce the highly neurotoxic ROS hypochlorite 39, 172. In addition to ROS and RNS, neutrophils, microglia and astrocytes all secret neurotoxic proteases including neutrophil elastase, cathepsins and chymotrypsin‐like proteases 75, 76, 172. Independent from secreted neurotoxins, microglia induce neuronal death by direct phagocytosis of healthy neurones. Nanomolar concentrations of amyloid monomers, oligomers or fibrils induced microglial phagocytosis of healthy neurones through a membrane phosphatidylserine dependent mechanism 107, 170.
Microglia are essential to the functioning brain. Their role in synapse modulation, microenvironment maintenance and homeostasis is crucial to neuronal function. A seminal article by Parkhurst et al 114 demonstrated that depletion of microglia from the cortex of mice caused a significant impairment in learning and memory. Using selective deletion of BDNF from microglia, Parkhurst et al demonstrated that it is likely that BDNF production is one of the essential functions of microglia in a healthy brain 114. Microglia treated with amyloid have been shown to dramatically lower the production of BDNF while increasing the production of inflammatory cytokines 60. Additionally, chronically inflamed microglia fail to perform their role of protein uptake and degradation from the extracellular environment and this can lead to the build‐up of protein aggregates such as amyloid oligomers and fibrils. This is supported by research demonstrating that chronic inflammation induced by head trauma 26, 67, infection 44, obesity 77, 97 or bacterial toxins 121, 126 accelerates amyloid deposition and memory deficits 112 (Figure 1).
While it appears that amyloid pathology is a causal factor in neuroinflammation in AD, it remains unclear how amyloid is linked to the tau pathology and neurofibrillary tangles (Figure 1). There is growing evidence that neuroinflammation may be one of the critical linking factors (Figure 1). Overexpression of inflammatory cytokines has been shown to increase tau pathology 48. Furthermore, infection and bacterial toxins have been shown to exacerbate tau phosphorylation and aggregation in mouse models of AD and repeated mild head injury alone in wild‐type mice is enough to induce AD‐like tau pathology 83, 98, 156. There is also evidence that activated microglia cause tau pathology propagation through the secretion of phosphorylated tau in exosomes 8. Interestingly, tau pathology may be causal factor in neuroinflammation induced neurotoxicity with genetic deletion of tau providing protection from inflammatory stimuli in cultured neurones 94. Collectively, this evidence supports a model of AD where amyloid induces sustained inflammation which causes and propagates phosphorylated and aggregated tau species which contributes substantially to neuronal death in AD (Figure 1).
As the primary resident immune cell of the brain, microglia are equipped with a number of cell membrane and cytosolic pattern recognition receptors (PRRs) which initiate the inflammatory phenotype. The cell surface toll‐like receptor (TLR) family are a group of structurally similar PRRs expressed in adaptive and innate immune cells, as well as epithelial, endothelial and fibroblast cells. They are traditionally thought of as receptors which recognize pathogen associated molecular patterns (PAMPs) which upon activation initiate a range of responses including cytokine secretion, antigen presentation and proliferation; however it is now clear that several TLRs are integral to the neuroinflammatory response in AD. TLR2 can directly bind amyloid and initiate an inflammatory response through the transcription factor NFκB (nuclear factor kappa‐light‐chain‐enhancer of activated B cells) and JNKs (c‐Jun N‐terminal kinases) 32, 85, 89, 99 (Figure 2). Inhibition of TLR2 has been found to be therapeutic in mouse models of AD 99. TLR4 and its co‐receptor CD14, and scavenger receptor A and the Ca2+‐activated K+ channel (KCa3.1) have also been implicated in the detection of amyloid species in the extra‐cellular environment 92, 131 (Figure 2). The role of TLR4 signaling in AD pathology is supported by human genetic evidence. A rare variant in the TLR4 gene that causes a reduction in function has been found to dramatically decrease the risk of developing late onset AD 103. Amyloid is also phagocytosed by microglia through binding to the phagocytotic receptor complex that includes CD36, CD47, and α(6)β(1)‐integrin 11 (Figure 2). Inside the cell, amyloid may also affect cytosolic PRRs, such as NLRP3 (NLR family, pyrin domain containing 3), to activate inflammatory complexes called inflammasomes (Figure 2 , and see below). These have been found to be critical in AD associated inflammation through the release of the inflammatory cytokine IL‐1β 35, 56 (Figure 2).
Figure 2.
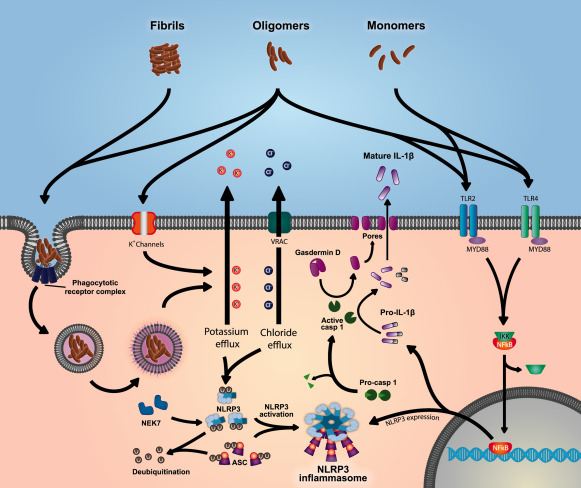
Amyloid oligomers and monomers cause the expression of NLRP3 and proIL‐1β through TLR mediated NFκB activation. The NLRP3 inflammasome is then activated by amyloid oligomers and fibrils through phagosomal disruption or cell surface K+ channels. Both pathways result in K+ efflux and cell swelling leading to Cl− efflux through VRAC. This, through an unknown mechanism, leads to deubiquitination of NLRP3 and ASC, and the binding of NEK7 to NLRP3 resulting in NLRP3 inflammasome activation. The NLRP3‐ASC speck then recruits and activates caspase‐1 which then cleaves gasdermin D and proIL‐1β into their active forms. The N‐terminus cleavage product of gasdermin D then forms pores in the cell membrane allowing the leaderless IL‐1β to leave the cell.
Interleukin‐1β
There is growing clinical and preclinical evidence that the inflammatory cytokine IL‐1β plays a central role in the induction of pathogenic neuroinflammation in AD 24, 148. The release of IL‐1β from immune cells facilitates the orchestration of an inflammatory response, mediating the increased expression of adhesion molecules, immune cell infiltration 165, and the production of further inflammatory cytokines 38. Because of the central role of IL‐1β in coordinating inflammatory responses it is regulated at multiple biological check points: expression, maturation, and secretion 142. IL‐1β is expressed as an inactive precursor, proIL‐1β, which is mediated through a NFκB‐dependent mechanism downstream of cell surface PRRs or IL‐1 receptor 1 (IL‐1R1) 157. For example, a well characterized method of inducing proIL‐1β expression is the activation of TLR4 by lipopolysaccharide (LPS) 27. ProIL‐1β is biologically inactive requiring proteolytic cleavage into its mature form which is mediated by caspase‐1, a pro‐inflammatory cysteine aspartate‐specific protease. Alongside IL‐1β cleavage caspase‐1 has additional essential roles, previously reviewed by Denes et al 36, of note: cleavage of proIL‐18 and initiating the inflammatory form of cell death, pyroptosis (Figure 2). During pyroptosis, gasdermin D is cleaved by caspase 1 and the N‐terminal fragment associates with the cell membrane facilitating membrane permeabilization, cell death and IL‐1β (and IL‐18) release 74, 145. Once cleaved, IL‐1β is secreted from cells through a non‐conventional pathway, bypassing the Golgi‐ER network, and has been demonstrated to be secreted by several mechanisms, including: the shedding of micro‐vesicles and cell membrane permeabilization. Secretion of IL‐1β from cells has not been fully elucidated, however it is largely accepted that the mode of secretion engaged by cells is a continuum dependent upon the strength stimulus, reviewed by Lopez‐Castejon and Brough 90. However, caspase‐1 is produced in cells as an inactive zymogen, procaspase‐1, and requires proximity‐induced self‐cleavage for activation. Homotypic interactions between death domains motifs between proteins facilitate the oligomerization of large multimeric protein structures which act as platforms to concentrate caspase‐1 and catalyze auto‐activation 81, 82, 132.
Inflammasomes—protein scaffolds for caspase‐1 activation
The large protein complexes which facilitate caspase‐1 activation are referred to as “inflammasomes” and are largely comprised of three core components: an inflammasome sensor molecule, an adaptor protein, apoptosis‐associated speck‐like protein containing a CARD (ASC), and caspase‐1 82, 142. Inflammasome sensing molecules are intracellular PRRs which sense inflammatory stimuli and oligomerise with ASC via pyrin (PYD) death domains. This initial ASC seeding triggers rapid recruitment of ASC dimers to form large protein specks 82. Subsequently, interactions between caspase activation and recruitment domains (CARD) present in ASC and procaspase‐1 recruit caspase‐1 to the inflammasome and initiate self‐cleavage 43, 125. Multiple sensor molecules have been identified which trigger inflammasome oligomerization, all maintaining a common basic organization but varying in formation, structure and activation.
The majority of inflammasome sensors that have been identified contain a NOD‐like receptor (NLR) domain, characterized by three distinct entities: a common NACHT domain; a leucine rich repeats (LRRs) domain and one or both death domains, PYD or CARD mediating ASC/caspase‐1 interaction 96. The first inflammasome to be identified was the NLRP1 (NOD‐, LRR‐ and pyrin domain‐containing 1) followed by the identification of additional NLR containing inflammasomes, including: NLRP3, NLRP6, NLRP7, NLRP12 and NLRC4 (NOD‐, LRR‐ and CARD‐containing 4) 82 which are activated in response to a broad range of molecular signals. For example, it has been demonstrated that murine NLRP1, NLRP7 and NLRC4 are activated in response to: anthrax toxin 19, bacterial LPS 74 and cytosolic flagellin 1, respectively. Another inflammasome, absent in melanoma 2 (AIM2), has been identified which contain a sensor molecule that contains a pyrin and HIN domain‐containing protein (PYHIN) domain 21. The HIN region has been shown to bind directly to cytosolic DNA to facilitate inflammasome formation and caspase‐1 activation. The identification and characterization of these inflammasome structures, their specific activators and independent mechanisms of activation demonstrates the immense complexity of the innate immune system and its ability to detect and respond to danger signals.
NLRP3 inflammasome
Canonical NLRP3 activation
The most extensively studied inflammasome is the NLRP3 inflammasome and has been strongly implicated in AD pathology 57. Despite being well studied the mechanisms underpinning NLRP3 activation have not been fully elucidated. Canonical NLRP3 activation, similar to IL‐1β maturation, requires two independent signals: (i) an initial NF‐κB activating signal to upregulate NLRP3 expression 13 and (ii) an additional activating signal which initiates a conformational change in NLRP3 and drives inflammasome assembly. Whilst a diverse range of molecules have been demonstrated to activate NLRP3, the molecular pathways which lead to its activation are incompletely understood. Various models of activation have been hypothesized, including: (i) formation of pores in the membrane and subsequent K+ efflux 45, 118; (ii) lysosomal rupture and release of cathepsins into the cytosol 64; (iii) mitochondrial dysfunction and the production of ROS 147 and (iv) post translational modifications, including deubiquitination 70, 91. In a landmark article, Muñoz‐Planillo et al 105 were able to demonstrate that the proposed hypotheses for NLRP3 activation converge on K+ efflux, leading to the acceptance of its pivotal role in triggering NLRP3 activation. Recent studies have also illustrated that volume regulated anion channels (VRAC) and subsequent Cl‐ efflux are also vital for inflammasome activation 35. Another landmark discovery in the field of NLRP3 activation is the identification of NEK7 (NIMA‐related kinase 7) as an essential upstream regulator of NLRP3 (Figure 2). Two groups independently demonstrated NEK7 directly interacting with NLRP3 and that this interaction is essential for ASC recruitment and inflammasome activation 141, 144. It is evident that the activation and regulation of NLRP3 is a rapidly expanding field and new discoveries are constantly being made identifying novel molecular pathways involved in its regulation (Figure 2).
A diverse range of activators have been identified including pathogenic, environmental and sterile molecules. Pathogenic activators which activate NLRP3, range across the microbial spectrum including: viruses, fungi and pore forming toxins produced from bacteria, including nigericin produced from Streptomyces hygroscopicus 117. Environmental pollutants, such as silica and asbestos 40, can also activate the inflammasome. Notably, NLRP3 inflammasome is activated to a diverse range of endogenous danger signals and consequently is implicated in the pathology of sterile inflammatory diseases. Sterile activators of NLRP3 can be largely grouped into two main categories: (i) molecules released from dying cells and (ii) extracellular particulates. An example of the former includes the release of ATP into the extracellular milieu from dying cells, which activates P2X7 ATP‐gated ion channels causing K+ efflux and NLRP3 activation 95. The latter encompasses large sterile particulate matter, including crystals of monosodium urate and calcium pyrophosphate dihydrate, central to gout and pseudogout pathology, respectively, and cholesterol crystals, involved in atherosclerosis 41. Moreover, Halle et al 53 identified fibrillary Aβ as an NLRP3 activator. Other sterile activators of the inflammasome that have been identified include elevated extracellular glucose 173, zinc deficiency 154 and changes in osmolality 30.
Non‐canonical NLRP3 activation
In addition to the canonical activation pathway, a non‐canonical pathway of activation has been identified. This pathway describes murine caspase‐11 or its human orthologues, caspase‐4 and caspase‐5 dependent NLRP3 activation, IL‐1β release and pyroptotic cell death in response to intra‐cellular Gram‐negative bacteria. The non‐canonical pathway was first described by Kayagaki et al 73, where they demonstrated NLRP3 activation by pathogen stimuli was caspase‐11 dependent yet caspase‐11 was not required for canonical NLRP3 inflammasome activation. It has since been discovered that intracellular LPS is the molecule which activates caspase‐11 through binding to caspase‐11 CARD domain and triggering oligomerization and activation 146. Further research elucidated K+ efflux as the trigger for NLRP3 activation in the non‐canonical pathway, identifying the point in which canonical and non‐canonical pathways converge 134. More recently, a novel pathway of activation has been described in human monocytes, the alternative pathway. Gaidt et al 46 discovered a novel pathway in human monocytes which leads to NLRP3 activation in response to LPS. Notably, activation via this pathway is independent of many of the hallmark features of canonical activation including K+ efflux and pyroptosis, and is mediated by a TLR‐4/caspase‐8 dependent pathway. Despite the identification and characterization of multiple inflammasomes adopting complex independent regulatory systems the end point remains unified: caspase‐1 activation and IL‐1β (and IL‐18) maturation.
The role of IL‐1β and IL‐18 in AD
Elevated IL‐1β levels in AD brains has been reported as early as 1989, and subsequent research has established a distinct role for IL‐1β in AD pathology 51. There is increased IL‐1β expression in microglia which cluster around amyloid plaques in the APPSwe/PS1deltaE9 (APP/PS1) mouse model of AD 148, and mice lacking IL‐1 receptor antagonist, an endogenous IL‐1 receptor 1 blocker, have increased microglial activation and neuronal damage after intracerebroventricular Aβ injection 34. Evidence also suggests that IL‐1β can directly affect both the amyloidgenesis and tauopathy that is central to AD pathogenesis. It has been shown that IL‐1β can upregulate APP and Aβ production in astrocytes 15 and can induce tau phosphorylation via the MAPK‐p38 pathway to form neurofibrillary tangles 50. Alongside IL‐1β, IL‐18 has been implicated in AD pathology; brains have increased mRNA and protein levels of IL‐18 that co‐localize with peri‐plaque neurones, astrocytes and microglia in human AD tissue 108. Preclinical studies have also demonstrated a link between IL‐18 and amyloidopathy and tauopathy. IL‐18 has been shown to upregulate components of the γ‐secretase complex accelerating Aβ production 155, and to elevate proteins associated with the hyperphosphorylation of tau, such as, glycogen synthase kinase 3β and cyclin dependent kinase‐5, in SH‐SY5Y neuroblastoma cells 109. Additionally, genetic analysis has identified polymorphisms in the IL‐18 promoter region to be associated with an increased risk in developing sporadic late onset AD in specific populations 18. Combined this research has shown that IL‐1β and IL‐18 have a pivotal role in AD and has thus provoked further research focusing on the molecular entities upstream of IL‐1β and IL‐18, investigating how inflammasome dysregulation may contribute to AD.
Inflammasome activation in AD
Following the discovery that fibrillar Aβ can activate NLRP3 53, further research has identified that all amyloid species, monomers, oligomers and fibrils, have effects on NLRP3 expression and activation (Figure 2). A seminal article published by Heneka et al 56 directly implicated NLRP3 activation in AD pathology. Heneka showed that APP/PS1/NLRP3−/− and APP/PS1/caspase‐1−/− mice have reduced neuroinflammation, decreased amyloid burden and notably were protected from AD associated memory deficits. Interestingly, the reduced amyloid burden was found not to be because of a decrease in APP processing but rather an increase in phagocytic activity from microglia. This suggests that activated NLRP3 contributes to AD pathogenesis two‐fold: generating toxic IL‐1β and propagating neuroinflammation, whilst impeding Aβ clearance resulting in plaque build‐up 49. Furthermore, research which crossed ASC−/− mice with the APPSwe,Flor,Lon, PSEN1, M146L, L286V (5xFAD) mouse model of AD found that 5xFAD/ASC+/− mice had reduced amyloid burden, increased astrocytic phagocytic activity and reduced memory deficits compared with the 5xFAD controls 33. The role of NLRP3 in AD has been further acknowledged in clinical studies, alongside a further inflammasome, NLRP1. Seresella et al 139 investigated gene expression and inflammasome activation in monocytes from patients diagnosed with severe AD, mild AD and MCI. NLRP3 and NLRP1 inflammasome components were upregulated compared with age matched healthy controls and there was an augmented response to LPS and Aβ stimulation. An additional mechanism in which NLRP3 can contribute to AD pathogenesis is in response to dying neurones releasing ATP. The release of ATP can activate P2X7 receptors on microglia to activate NLRP3 and consequently exacerbate inflammation and damage 59. Furthermore, there is evidence of P2X7 receptor upregulation in both preclinical and clinical AD research 100.
Unlike NLRP3 which is highly expressed in microglia, NLRP1 is mainly expressed in neurones 80 and its proposed role in AD pathogenesis is largely associated with neuronal death and axonal degeneration, although its exact role is not clearly defined. Tan et al 159 found that NLRP1 levels are upregulated in APP/PS1 mice and went on to show in vitro that silencing of NLRP1 reduced Aβ‐induced pyroptotic cell death. They also showed that silencing NLRP1 and caspase‐1 in APP/PS1 mice reduced cell death in the cortex and hippocampus, and improved spatial learning and memory in these animals. Therefore, proposing a role of NLRP1 in AD pathology via neuronal pyroptotic cell death, synaptic loss and subsequent cognitive decline. However, Kaushal et al 72 identified a novel NLRP1/caspase‐1/caspase‐6 pathway, demonstrating that activation of NLRP1 mediates caspase‐1 activation which: (i) cleaves IL‐1β, (ii) activates caspase‐6 and subsequent caspase‐6 associated axonal degeneration and, (iii) increases the ratio of Aβ42 to total Aβ proteins. Despite proposing different hypotheses both groups have identified an important role for NLRP1 activation in neurones and axonal degeneration in AD, further highlighting an area of interest to elucidate its exact role. It is important to note there are fundamental differences in NLRP1 between mice and humans. Rodents express three paralogous NLRP1 genes where as humans only express one, and there are structural differences in the death domains 163. Furthermore, a genetic association between NLRP1 and AD has been proposed because of the identification of four non‐synonymous polymorphisms in the NLRP1 gene which confer an increased risk for the development of AD 122.
A role for the NLRC4 inflammasome in AD pathology has been identified in response to the fatty acid palmitate in astrocytes. Lui et al 87 demonstrates that NLRC4 is activated and IL‐1β is secreted in palmitate treated primary astrocyte cultures, and furthermore NLRC4 and ASC are upregulated in AD brains. Lui et al 87 also showed that conditioned media from palmitate treated astrocytes increases the expression of BACE‐1 and production of Aβ42 in neurones. This is of significant interest because fatty acid metabolism has been identified as a risk factor for AD development 113 and there is a higher fatty acid content in AD brain compared with healthy controls 135.
Targeting The Inflammasome for AD
The processes involved in IL‐1β secretion and signaling can be pharmacologically targeted at a number of locations in the pathway [reviewed by Baldwin et al 10]. Recently, our group were the first to successfully pharmacologically target the NLRP3 inflammasome in animal models of AD 35. We screened NSAIDs for activity on NLRP3 activation in vitro and found that the fenamate subclass selectively inhibited NLRP3 inflammasome formation. The target was established to be the inhibition of the membrane ion channel VRAC. Treatment with the fenamate mefenamic acid was then found to abate memory deficits seen in a rat amyloid oligomer injection model and APPSwe, PS1M146V and tauP301L (3xTgAD) mouse model of AD 35. Previous research has shown that mefenamic acid can reduce amyloid toxicity in neuronal cultures and abate memory deficits in rats infused with amyloid monomers 69. Furthermore, the fenamate tolfenamic acid, which is structurally very similar to mefenamic acid, has been found to be therapeutic in the APPSwe R1.40 mouse model of AD, lowering plaque burden, tau pathology and cognitive deficits 4, 152, 153. It was proposed that tolfenamic acid was therapeutic through the inhibition of the gene regulator specificity protein 1 (SP1). However, similar therapeutic effects in similar animal models of AD were seen solely from the genetic deletion of the NLRP3 inflammasome and the inflammasome adapter molecule ASC, suggesting that inhibitory activity of fenamates on NLRP3 activation could exclusively explain their efficacy 33, 53, 56. Collectively, this evidence demonstrates that through NLRP3 inhibition and other potential mechanisms, fenamates have been found to be therapeutic in four animal models of AD and are therefore a promising potential therapeutic in AD.
Pharmacologically inhibiting cell surface receptors which induce IL‐1β expression may prove difficult in AD because of the diversity of the receptors involved. TLR2, TLR4, CD36 and IL‐1R1, have all been implicated in AD associated neuroinflammation. Therefore, a polypharmacy approach would be required, increasing the potential of off‐target effects, as discussed below. Downstream of these receptors is the intracellular adaptor molecule MyD88, which is essential for TLR2, TLR4 and IL‐1R signaling and may therefore be a promising target in AD. Genetic deletion of MyD88 has been found to reduce plaque load and abate neuroinflammation in the APP/PS1 mouse model of AD 84. However, MyD88 remains a controversial target for AD with further studies showing that MyD88−/+ mice having accelerated AD pathology and memory deficits in the APP/PS1 mouse model 102. This may be because of the MyD88 receptor family being integral to the beneficial phagocytotic response by microglia 47, 102, 133. The MyD88 receptor family induce transcriptional changes through transcription regulator of NFκB, however, targeting NFκB in AD is unlikely to be successful because of the broad range of processes and genes that NFκB regulates 111. For example NFκB expression and activation is upregulated during synaptic activity and this has been shown to be essential for long‐term potentiation (LTP), an essential process in learning and memory 5, 101. An additional problem for targeting TLRs, MyD88 and NFκB in AD is that these proteins are essential for host response to infection and therefore the chronic inhibition needed to treat AD may render the patient susceptible to infection 88, 130, 137, 158. Conversely, the NLRP3 inflammasome is primarily activated by sterile stimuli. Furthermore, the minimal effect of genetic deletion of NRLP3 on infection has led to the proposal that inflammasomes are largely redundant in vertebrate adapted pathogens 93. This suggests that chronic inhibition of inflammasomes, particularly the NLRP3 inflammasome, would not greatly affect the susceptibility of patients to infection, making inflammasomes an excellent target for AD.
There are multiple cell pathways that act as the secondary stimulus in inflammasome activation and these may provide an attractive target for pharmacological intervention in AD. The P2X7 receptor is activated by extracellular ATP which is released upon cell death and leads to NLRP3 inflammasome stimulation via K+ efflux. Evidence is building that amyloid mediated NLRP3 inflammasome activation is dependent on the P2X7 receptor 115, 138. This is supported by research which demonstrated that pharmacological intervention with P2X7 antagonists were found to be therapeutic in a rat amyloid injection model 138. Yet again there is an issue with off target effects because of the P2X7 receptor having a range of functions on a range of cell types including neurones, astrocytes and oligodendrocytes. However, evidence is building that activation of the P2X7 receptor is pathologically elevated in AD in multiple cell types which leads to amyloidogenic APP processing. This suggests that P2X7 inhibition remains an attractive target in AD with multiple therapeutic mechanisms 37, 150.
Phagosomal stress causes the release of cathepsin B into the cytosol where it activates the NLRP3 inflammasome. Amyloid fibrils have been shown to induce phagosomal stress causing NLRP3 activation through a cathepsin B dependent mechanism. There is evidence that cathepsin B's role in NLRP3 activation involves both the prototypical NLRP3 activation stimulus of K+ efflux 53, 105 as well as the cathepsin B dependent degradation of NLRP10 which acts as an inhibitor of NLRP3 activation 106. Targeting cathepsin B has been successful in animal models of AD with research demonstrating that administration of the cathepsin B inhibitor CA074Me is therapeutic in the APPLon mouse model of AD 63. However, there is evidence that cathepsins play an important role in amyloid degradation 164, therefore further research is required to evaluate the potential of cathepsins as a putative therapeutic target in AD.
Downstream from inflammasome activation there are a number of potential therapeutic targets including caspase‐1 activation and signaling at the IL‐1R1 receptor. Neither, caspase‐1 or IL‐1R1 have been pharmacologically targeted in animal models of AD. However, genetic deletion of caspase‐1 has been shown to increase amyloid phagocytosis in isolated microglia and reduce neuroinflammation following striatal amyloid injections in mice 53, 56. Therefore, there is some evidence that this approach is worth pursuing. Conversely, evidence for IL‐1R1 antagonists as therapeutic in AD does not appear promising. IL‐1R1 KO mice have cognitive deficits, suggesting that chronic inhibition of IL‐1R1 may have detrimental effects in AD 9. Possible causes for the cognitive effects of IL‐1R1 inhibition include: (i) the need for low levels of IL‐1 signaling to promote phagocytosis of extracellular debris 9, (ii) the critical role of neuronal IL‐1R1 signaling in LTP induction 136 and (iii) the role of IL‐1 signaling in synapse formation through IL1RAPL1 (interleukin‐1‐receptor accessory protein like 1) mediated JNK activation pathway 116. Targeting IL‐1R1 also has the additional drawback of having no effect on caspase‐1 dependent pyroptosis. Therefore, there will continue to be microglial death, resulting in the release of damaging cell contents, and fewer microglia to perform important functions independent of inflammation. Because of these limitations, inhibition of IL‐1R1 is not the preferred therapeutic strategy of inflammasome dependent AD pathology.
Targeting the molecular and physiological processing directly involved in inflammasome formation is the optimal approach for limiting the negative effects of IL‐1β signaling in AD. Inflammasome specific approaches would have limited side‐effects and would not greatly impact the patients’ resistance to disease. However, there are currently no drugs which have been conclusively shown to directly inhibit inflammasome formation. Several approaches could be taken in drug design including: (i) inhibiting NLRP3‐NEK7 binding 55, possibly by targeting NEK7 phosphorylation 144; (ii) targeting NLRP3 ubiquitination status by augmenting specific ubiquitin ligases activity or blocking deubiquitinases 70, 91, although this approach may have potential off target effects because of the many roles of the ubiquitin system; (iii) inhibiting the phosphorylation of the NLRP3 protein 151; (iv) or targeting the PRR‐ASC, ASC‐ASC or ASC‐caspase interaction sites directly 140. Currently, there are existing inflammasome inhibiting drugs available where the mechanism of action has not fully been elucidated and may involve targeting the processes mentioned above or an unknown regulatory system of inflammasome formation. The drugs include: (i) 3,4‐methylenedioxy‐β‐nitrostyrene (MNS) which has been shown to alter cysteines on the NLRP3 protein itself and this may alter NLRP3‐NEK7, NLRP3‐NLRP3 or NLRP3‐ASC associations; (ii) MCC950 (CP‐456773) is also a potent inhibitor of NLRP3 activation whose mechanism of action has been shown to be down stream of potassium efflux but does not alter NLRP3‐ASC or ASC‐ASC binding, possibly implicating NLRP3‐NEK7, NLRP3‐NLRP3, ubiquitination or phosphorylation as potential mechanisms of action 29. There are continuing efforts to develop novel inflammasome inhibitors using screening and structure based molecular modeling techniques to target inflammasome formation and these will provide a diverse set of tools to further investigate the role of inflammasomes in a range of diseases including AD.
Conclusion
It is now clear that inflammation plays a fundamental role in the pathophysiology of AD. Neuroinflammation in AD is mediated through a number of PRRs including cell surface receptors such as TLR2 and TLR4, as well as cytosolic receptors, of which the NLRP3 inflammasome has been found to be central. Consequently, inflammasomes are an attractive therapeutic target for AD and have multiple points in the activation pathway which can be inhibited. Because of non‐specific effects and complicated interactions with AD pathology targets upstream of inflammasome formation, such as TLR4 and cathepsin B, may not be preferable as a chronic pharmacological intervention strategy required for AD. Similarly, targeting IL‐1R1 may have negative effects of cognition and AD progression because of the essential role of basal IL‐1 signaling in brain parenchyma maintenance. However, the NLRP3 inflammasome is an attractive pharmacological target as inhibition would specifically abate pathological inflammation without altering basal microglia function or leaving the patient overly susceptible to infection. No drugs have currently been established to directly bind and inhibit the NLRP3 inflammasome, however, the essential processes for NLRP3 activation of VRAC activation has been targeted using currently indicated fenamate NSAIDs and these were found to be therapeutic in four separate animal models of AD 35, 68, 152. However, fenamate NSAIDs are also COX inhibitors and potentially have other effects on APP expression and cleavage 71. The challenge for the field now is to develop non‐toxic and specific inflammasome inhibitors to fully elucidate the therapeutic potential of targeting this pathway in AD.
References
- 1. Abdelaziz DH, Amr K, Amer AO (2010) Nlrc4/Ipaf/CLAN/CARD12: more than a flagellin sensor. Int J Biochem Cell Biol 42:789–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abramov AY, Canevari L, Duchen MR (2004) Beta‐amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci 24:565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Abramov AY, Jacobson J, Wientjes F, Hothersall J, Canevari L, Duchen MR (2005) Expression and modulation of an NADPH oxidase in mammalian astrocytes. J Neurosci 25:9176–9184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Adwan L, Subaiea GM, Zawia NH (2014) Tolfenamic acid downregulates BACE1 and protects against lead‐induced upregulation of Alzheimer's disease related biomarkers. Neuropharmacology 79:596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Albensi BC, Mattson MP (2000) Evidence for the involvement of TNF and NF‐kappaB in hippocampal synaptic plasticity. Synapse 35:151–159. [DOI] [PubMed] [Google Scholar]
- 6. Alzheimer A (1991) Über eigenartige Krankheitsfälle des späteren Alters: (On certain peculiar diseases of old age). Hist Psychiatry 2:74–101. [DOI] [PubMed] [Google Scholar]
- 7. Alzheimer's Association (2015) 2015 Alzheimer's disease facts and figures. Alzheimers Dement 11:332–384. [DOI] [PubMed] [Google Scholar]
- 8. Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T et al (2015) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18:1584–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Avital A, Goshen I, Kamsler A, Segal M, Iverfeldt K, Richter‐Levin G et al (2003) Impaired interleukin‐1 signaling is associated with deficits in hippocampal memory processes and neural plasticity. Hippocampus 13:826–834. [DOI] [PubMed] [Google Scholar]
- 10. Baldwin AG, Brough D, Freeman S (2016) Inhibiting the inflammasome: a chemical perspective. J Med Chem 59:1691–1710. [DOI] [PubMed] [Google Scholar]
- 11. Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE (2003) A cell surface receptor complex for fibrillar beta‐amyloid mediates microglial activation. J Neurosci 23:2665–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bateman RJ, Aisen PS, De Strooper B, Fox NC, Lemere CA, Ringman JM et al (2011) Autosomal‐dominant Alzheimer's disease: a review and proposal for the prevention of Alzheimer's disease. Alzheimers Res Ther 3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D et al (2009) Cutting edge: NF‐kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183:787–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Beach TG, Walker R, McGeer EG (1989) Patterns of gliosis in Alzheimer's disease and aging cerebrum. Glia 2:420–436. [DOI] [PubMed] [Google Scholar]
- 15. Blasko I, Veerhuis R, Stampfer‐Kountchev M, Saurwein‐Teissl M, Eikelenboom P, Grubeck‐Loebenstein B (2000) Costimulatory effects of interferon‐gamma and interleukin‐1beta or tumor necrosis factor alpha on the synthesis of Abeta1‐40 and Abeta1‐42 by human astrocytes. Neurobiol Dis 7(6 Pt B):682–689. [DOI] [PubMed] [Google Scholar]
- 16. Block ML, Zecca L, Hong JS (2007) Microglia‐mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8:57–69. [DOI] [PubMed] [Google Scholar]
- 17. Boche D, Perry VH, Nicoll JAR (2013) Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol 39:3–18. [DOI] [PubMed] [Google Scholar]
- 18. Bossù P, Ciaramella A, Moro ML, Bellincampi L, Bernardini S, Federici G et al (2007) Interleukin 18 gene polymorphisms predict risk and outcome of Alzheimer's disease. J Neurol Neurosurg Psychiatry 78:807–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boyden ED, Dietrich WF (2006) Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet 38:240–244. [DOI] [PubMed] [Google Scholar]
- 20. Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A et al (2013) CD33 Alzheimer's disease locus: altered monocyte function and amyloid biology. Nat Neurosci 16:848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H et al (2009) An orthogonal proteomic‐genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol 10:266–272. [DOI] [PubMed] [Google Scholar]
- 22. Burkert K, Moodley K, Angel CE, Brooks A, Graham ES (2012) Detailed analysis of inflammatory and neuromodulatory cytokine secretion from human NT2 astrocytes using multiplex bead array. Neurochem Int 60:573–580. [DOI] [PubMed] [Google Scholar]
- 23. Cabezas R, Avila M, Gonzalez J, El‐Bachá RS, Báez E, García‐Segura LM et al (2014) Astrocytic modulation of blood brain barrier: perspectives on Parkinson's disease. Front Cell Neurosci 8:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cacabelos R, Alvarez XA, Fernandez‐Novoa L, Franco A, Mangues R, Pellicer A et al (1994) Brain interleukin‐1 beta in Alzheimer's disease and vascular dementia. Methods Find Exp Clin Pharmacol 16:141–151. [PubMed] [Google Scholar]
- 25. Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE et al (2001) In‐vivo measurement of activated microglia in dementia. Lancet 358:461–467. [DOI] [PubMed] [Google Scholar]
- 26. Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH (2004) Long‐term accumulation of amyloid‐beta, beta‐secretase, presenilin‐1, and caspase‐3 in damaged axons following brain trauma. Am J Pathol 165:357–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F (1999) Toll‐like receptor‐4 mediates lipopolysaccharide‐induced signal transduction. J Biol Chem 274:10689–10692. [DOI] [PubMed] [Google Scholar]
- 28. Citron M (2010) Alzheimer's disease: strategies for disease modification. Nat Rev Drug Discov 9:387–398. [DOI] [PubMed] [Google Scholar]
- 29. Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz‐Planillo R, Inserra MC et al (2015) A small‐molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Compan V, Baroja‐Mazo A, López‐Castejón G, Gomez AI, Martínez CM, Angosto D et al (2012) Cell volume regulation modulates nlrp3 inflammasome activation. Immunity 37:487–500. [DOI] [PubMed] [Google Scholar]
- 31. Constantinescu CS, Tani M, Ransohoff RM, Wysocka M, Hilliard B, Fujioka T et al (2005) Astrocytes as antigen‐presenting cells: expression of IL‐12/IL‐23. J Neurochem 95:331–340. [DOI] [PubMed] [Google Scholar]
- 32. Costello DA, Carney DG, Lynch MA (2015) alpha‐TLR2 antibody attenuates the Abeta‐mediated inflammatory response in microglia through enhanced expression of SIGIRR. Brain Behav Immun 46:70–79. [DOI] [PubMed] [Google Scholar]
- 33. Couturier J, Stancu I‐C, Schakman O, Pierrot N, Huaux F, Kienlen‐Campard P et al (2016) Activation of phagocytic activity in astrocytes by reduced expression of the inflammasome component ASC and its implication in a mouse model of Alzheimer disease. J Neuroinflamm 13:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Craft JM, Watterson DM, Hirsch E, Van Eldik LJ (2005) Interleukin 1 receptor antagonist knockout mice show enhanced microglial activation and neuronal damage induced by intracerebroventricular infusion of human beta‐amyloid. J Neuroinflamm 2:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Daniels MJD, Rivers‐Auty J, Schilling T, Spencer NG, Watremez W, Fasolino V et al (2016) Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer's disease in rodent models. Nat Commun 7:12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Denes A, Lopez‐Castejon G, Brough D (2012) Caspase‐1: is IL‐1 just the tip of the ICEberg?. Cell Death Dis 3:e338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Diaz‐Hernandez JI, Gomez‐Villafuertes R, Leon‐Otegui M, Hontecillas‐Prieto L, Del Puerto A, Trejo JL et al (2012) In vivo P2X7 inhibition reduces amyloid plaques in Alzheimer's disease through GSK3beta and secretases. Neurobiol Aging 33:1816–1828. [DOI] [PubMed] [Google Scholar]
- 38. Dinarello CA (2009) Immunological and inflammatory functions of the interleukin‐1 family. Annu Rev Immunol 27:519–550. [DOI] [PubMed] [Google Scholar]
- 39. Dinkel K, Dhabhar FS, Sapolsky RM (2004) Neurotoxic effects of polymorphonuclear granulocytes on hippocampal primary cultures. Proc Natl Acad Sci U S A 101:331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J (2008) Innate immune activation through nalp3 inflammasome sensing of asbestos and silica. Science 320:674–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG et al (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464:1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Emerit J, Edeas A, Bricaire F (2004) Neurodegenerative diseases and oxidative stress. Biomed Pharmacother 58:39–46. [DOI] [PubMed] [Google Scholar]
- 43. Fernandes‐Alnemri T, Wu J, Yu J‐W, Datta P, Miller B, Jankowski W et al (2007) The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase‐1 activation. Cell Death Differ 14:1590–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Finch CE, Morgan TE (2007) Systemic inflammation, infection, ApoE alleles, and Alzheimer disease: a position paper. Curr Alzheimer Res 4:185–189. [DOI] [PubMed] [Google Scholar]
- 45. Franchi L, Muñoz‐Planillo R, Núñez G (2012) Sensing and reacting to microbes through the inflammasomes. Nat Immunol 13:325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid‐Burgk JL, Rapino F et al (2016) Human monocytes engage an alternative inflammasome pathway. Immunity 44:833–846. [DOI] [PubMed] [Google Scholar]
- 47. Gambuzza ME, Sofo V, Salmeri FM, Soraci L, Marino S, Bramanti P (2014) Toll‐like receptors in Alzheimer's disease: a therapeutic perspective. CNS Neurol Disord Drug Targets 13:1542–1558. [DOI] [PubMed] [Google Scholar]
- 48. Ghosh S, Wu MD, Shaftel SS, Kyrkanides S, LaFerla FM, Olschowka JA et al (2013) Sustained interleukin‐1beta overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer's mouse model. J Neurosci 33:5053–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gold M, El Khoury J (2015) β‐amyloid, microglia, and the inflammasome in Alzheimer's disease. Semin Immunopathol 37:607–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Griffin WST, Liu L, Li Y, Mrak RE, Barger SW (2006) Interleukin‐1 mediates Alzheimer and Lewy body pathologies. J Neuroinflammation 3:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ et al (1989) Brain interleukin 1 and S‐100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci 86:7611–7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E et al (2013) TREM2 variants in Alzheimer's disease. N Engl J Med 368:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T et al (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid‐beta. Nat Immunol 9:857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hardy J, Selkoe DJ (2002) The amyloid hypothesis of alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353–356. [DOI] [PubMed] [Google Scholar]
- 55. He Y, Zeng MY, Yang D, Motro B, Núñez G (2016) NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530:354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira‐Saecker A et al (2013) NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 493:674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Heneka MT, Kummer MP, Latz E (2014) Innate immune activation in neurodegenerative disease. Nat Rev Immunol 14:463. [DOI] [PubMed] [Google Scholar]
- 58. Heppner FL, Ransohoff RM, Becher B (2015) Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 16:358–372. [DOI] [PubMed] [Google Scholar]
- 59. Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang L, Means TK et al (2013) The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16:1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hjorth E, Frenkel D, Weiner H, Schultzberg M (2010) Effects of immunomodulatory substances on phagocytosis of aβ(1–42) by human microglia. Int J Alzheimers Dis 2010:798424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Holmin S, Mathiesen T (2000) Intracerebral administration of interleukin‐1beta and induction of inflammation, apoptosis, and vasogenic edema. J Neurosurg 92:108–120. [DOI] [PubMed] [Google Scholar]
- 62. Hong S, Beja‐Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S et al (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352:712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hook VY, Kindy M, Hook G (2008) Inhibitors of cathepsin B improve memory and reduce beta‐amyloid in transgenic Alzheimer disease mice expressing the wild‐type, but not the Swedish mutant, beta‐secretase site of the amyloid precursor protein. J Biol Chem 283:7745–7753. [DOI] [PubMed] [Google Scholar]
- 64. Hornung V, Latz E (2010) Critical functions of priming and lysosomal damage for NLRP3 activation. Eur J Immunol 40:620–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D (1989) Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol 24:173–182. [DOI] [PubMed] [Google Scholar]
- 66. Jantzen PT, Connor KE, DiCarlo G, Wenk GL, Wallace JL, Rojiani AM et al (2002) Microglial activation and β‐amyloid deposit reduction caused by a nitric oxide‐releasing nonsteroidal anti‐inflammatory drug in amyloid precursor protein plus presenilin‐1 transgenic mice. J Neurosci 22:2246–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Johnson VE, Stewart W, Smith DH (2010) Traumatic brain injury and amyloid‐β pathology: a link to Alzheimer's disease?. Nat Rev Neurosci 11:361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jones RS, Minogue AM, Connor TJ, Lynch MA (2013) Amyloid‐beta‐induced astrocytic phagocytosis is mediated by CD36, CD47 and RAGE. J Neuroimmune Pharmacol 8:301–311. [DOI] [PubMed] [Google Scholar]
- 69. Joo Y, Kim HS, Woo RS, Park CH, Shin KY, Lee JP et al (2006) Mefenamic acid shows neuroprotective effects and improves cognitive impairment in in vitro and in vivo Alzheimer's disease models. Mol Pharmacol 69:76–84. [DOI] [PubMed] [Google Scholar]
- 70. Juliana C, Fernandes‐Alnemri T, Kang S, Farias A, Qin F, Alnemri ES (2012) Non‐transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem 287:36617–36622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Katsouri L, Parr C, Bogdanovic N, Willem M, Sastre M (2011) PPARγ co‐activator‐1α (PGC‐1α) reduces amyloid‐β generation through a PPARγ‐dependent mechanism. J Alzheimers Dis 25:151–162. [DOI] [PubMed] [Google Scholar]
- 72. Kaushal V, Dye R, Pakavathkumar P, Foveau B, Flores J, Hyman B et al (2015) Neuronal NLRP1 inflammasome activation of Caspase‐1 coordinately regulates inflammatory interleukin‐1‐beta production and axonal degeneration‐associated Caspase‐6 activation. Cell Death Differ 22:1676–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J et al (2011) Non‐canonical inflammasome activation targets caspase‐11. Nature 479:117–121. [DOI] [PubMed] [Google Scholar]
- 74. Khare S, Dorfleutner A, Bryan NB, Yun C, Radian AD, de Almeida L et al (2012) An NLRP7‐containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity 36:464–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kim S, Ock J, Kim AK, Lee HW, Cho JY, Kim DR et al (2007) Neurotoxicity of microglial cathepsin D revealed by secretome analysis. J Neurochem 103:2640–2650. [DOI] [PubMed] [Google Scholar]
- 76. Klegeris A, McGeer PL (2005) Chymotrypsin‐like proteases contribute to human monocytic THP‐1 cell as well as human microglial neurotoxicity. Glia 51:56–64. [DOI] [PubMed] [Google Scholar]
- 77. Knight EM, Martins IVA, Gümüsgöz S, Allan SM, Lawrence CB (2014) High‐fat diet‐induced memory impairment in triple‐transgenic Alzheimer's disease (3xTgAD) mice is independent of changes in amyloid and tau pathology. Neurobiol Aging 35:1821–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Koffie RM, Meyer‐Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia‐Alloza M et al (2009) Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A 106:4012–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Krabbe KS, Pedersen M, Bruunsgaard H (2004) Inflammatory mediators in the elderly. Exp Gerontol 39:687–699. [DOI] [PubMed] [Google Scholar]
- 80. Kummer JA, Broekhuizen R, Everett H, Agostini L, Kuijk L, Martinon F et al (2007) Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site‐specific role in the inflammatory response. J Histochem Cytochem 55:443–452. [DOI] [PubMed] [Google Scholar]
- 81. Lamkanfi M, Dixit VM (2014) Mechanisms and functions of inflammasomes. Cell 157:1013–1022. [DOI] [PubMed] [Google Scholar]
- 82. Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13:397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lee DC, Rizer J, Selenica ML, Reid P, Kraft C, Johnson A et al (2010) LPS‐ induced inflammation exacerbates phospho‐tau pathology in rTg4510 mice. J Neuroinflamm 7:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lim JE, Kou J, Song M, Pattanayak A, Jin J, Lalonde R et al (2011) MyD88 deficiency ameliorates beta‐amyloidosis in an animal model of Alzheimer's disease. Am J Pathol 179:1095–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lin W, Ding M, Xue J, Leng W (2013) The role of TLR2/JNK/NF‐kappaB pathway in amyloid beta peptide‐induced inflammatory response in mouse NG108‐15 neural cells. Int Immunopharmacol 17:880–884. [DOI] [PubMed] [Google Scholar]
- 86. Liraz‐Zaltsman S, Alexandrovich AG, Trembovler V, Fishbein I, Yaka R, Shohami E et al (2011) Regional sensitivity to neuroinflammation: in vivo and in vitro studies. Synapse 65:634–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Liu L, Chan C (2014) IPAF inflammasome is involved in interleukin‐1β production from astrocytes, induced by palmitate; implications for Alzheimer's Disease. Neurobiol Aging 35:309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Liu S, Kielian T (2011) MyD88 is pivotal for immune recognition of Citrobacter koseri and astrocyte activation during CNS infection. J Neuroinflamm 8:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B et al (2012) TLR2 is a primary receptor for Alzheimer's amyloid beta peptide to trigger neuroinflammatory activation. J Immunol 188:1098–1107. [DOI] [PubMed] [Google Scholar]
- 90. Lopez‐Castejon G, Brough D (2011) Understanding the mechanism of IL‐1β secretion. Cytokine Growth Factor Rev 22:189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lopez‐Castejon G, Luheshi NM, Compan V, High S, Whitehead RC, Flitsch S et al (2013) Deubiquitinases regulate the activity of caspase‐1 and interleukin‐1β secretion via assembly of the inflammasome. J Biol Chem 288:2721–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Maezawa I, Zimin PI, Wulff H, Jin LW (2011) Amyloid‐beta protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J Biol Chem 286:3693–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Maltez VI, Miao EA (2016) Reassessing the evolutionary importance of inflammasomes. J Immunol 196:956–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Maphis N, Xu G, Kokiko‐Cochran ON, Cardona AE, Ransohoff RM, Lamb BT et al (2015) Loss of tau rescues inflammation‐mediated neurodegeneration. Front Neurosci 9:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose‐Girma M et al (2006) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440:228–232. [DOI] [PubMed] [Google Scholar]
- 96. Martinon F, Mayor A, Tschopp J (2009) The inflammasomes: guardians of the body. Annu Rev Immunol 27:229–265. [DOI] [PubMed] [Google Scholar]
- 97. Martins IVA, Rivers‐Auty J, Allan SM, Lawrence CB (2017) Mitochondrial abnormalities and synaptic loss underlie memory deficits seen in mouse models of obesity and Alzheimer's disease. J Alzheimer's Dis 55:915–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. McAteer KM, Corrigan F, Thornton E, Turner RJ, Vink R (2016) Short and long term behavioral and pathological changes in a novel rodent model of repetitive mild traumatic brain injury. PLoS One 11:e0160220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. McDonald CL, Hennessy E, Rubio‐Araiz A, Keogh B, McCormack W, McGuirk P et al (2016) Inhibiting TLR2 activation attenuates amyloid accumulation and glial activation in a mouse model of Alzheimer's disease. Brain Behav Immun 58:191–200. [DOI] [PubMed] [Google Scholar]
- 100. McLarnon JG, Ryu JK, Walker DG, Choi HB (2006) Upregulated expression of purinergic P2X(7) receptor in Alzheimer disease and amyloid‐beta peptide‐treated microglia and in peptide‐injected rat hippocampus. J Neuropathol Exp Neurol 65:1090–1097. [DOI] [PubMed] [Google Scholar]
- 101. Meberg PJ, Kinney WR, Valcourt EG, Routtenberg A (1996) Gene expression of the transcription factor NF‐kappa B in hippocampus: regulation by synaptic activity. Brain Res Mol Brain Res 38:179–190. [DOI] [PubMed] [Google Scholar]
- 102. Michaud JP, Richard KL, Rivest S (2011) MyD88‐adaptor protein acts as a preventive mechanism for memory deficits in a mouse model of Alzheimer's disease. Mol Neurodegener 6:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Minoretti P, Gazzaruso C, Vito C, Di Emanuele E, Bianchi M, Coen E et al (2006) Effect of the functional toll‐like receptor 4 Asp299Gly polymorphism on susceptibility to late‐onset alzheimer's disease. Neurosci Lett 391:147–149. [DOI] [PubMed] [Google Scholar]
- 104. Mortimer JA, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A et al (1991) Head trauma as a risk factor for Alzheimer's disease: a collaborative re‐analysis of case‐control studies. EURODEM Risk Factors Research Group. Int J Epidemiol 20 Suppl 2:S28–S35. [DOI] [PubMed] [Google Scholar]
- 105. Muñoz‐Planillo R, Kuffa P, Martínez‐Colón G, Smith BL, Rajendiran TM, Núñez G (2013) K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38:1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Murphy N, Grehan B, Lynch MA (2014) Glial uptake of amyloid beta induces NLRP3 inflammasome formation via cathepsin‐dependent degradation of NLRP10. Neuromolecular Med 16:205–215. [DOI] [PubMed] [Google Scholar]
- 107. Neniskyte U, Neher JJ, Brown GC (2011) Neuronal death induced by nanomolar amyloid beta is mediated by primary phagocytosis of neurons by microglia. J Biol Chem 286:39904–39913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ojala J, Alafuzoff I, Herukka S‐K, van Groen T, Tanila H, Pirttilä T (2009) Expression of interleukin‐18 is increased in the brains of Alzheimer's disease patients. Neurobiol Aging 30:198–209. [DOI] [PubMed] [Google Scholar]
- 109. Ojala JO, Sutinen EM, Salminen A, Pirttilä T (2008) Interleukin‐18 increases expression of kinases involved in tau phosphorylation in SH‐SY5Y neuroblastoma cells. J Neuroimmunol 205:86–93. [DOI] [PubMed] [Google Scholar]
- 110. Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM (1999) Diabetes mellitus and the risk of dementia: the Rotterdam Study. Neurology 53:1937–1942. [DOI] [PubMed] [Google Scholar]
- 111. Pahl HL (1999) Activators and target genes of Rel/NF‐kappaB transcription factors. Oncogene 18:6853–6866. [DOI] [PubMed] [Google Scholar]
- 112. Pan X, Zhu Y, Lin N, Zhang J, Ye Q, Huang H et al (2011) Microglial phagocytosis induced by fibrillar β‐amyloid is attenuated by oligomeric β‐amyloid: implications for Alzheimer's disease. Mol Neurodegener 6:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Di Paolo G, Kim T‐W (2011) Linking lipids to Alzheimer's disease: cholesterol and beyond. Nat Rev Neurosci 12:284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, 3rd Lafaille JJ et al (2013) Microglia promote learning‐dependent synapse formation through brain‐derived neurotrophic factor. Cell 155:1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Parvathenani LK, Tertyshnikova S, Greco CR, Roberts SB, Robertson B, Posmantur R (2003) P2X7 mediates superoxide production in primary microglia and is up‐regulated in a transgenic mouse model of Alzheimer's disease. J Biol Chem 278:13309–13317. [DOI] [PubMed] [Google Scholar]
- 116. Pavlowsky A, Zanchi A, Pallotto M, Giustetto M, Chelly J, Sala C et al (2010) Neuronal JNK pathway activation by IL‐1 is mediated through IL1RAPL1, a protein required for development of cognitive functions. Commun Integr Biol 3:245–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Pelegrin P, Surprenant A (2007) Pannexin‐1 couples to maitotoxin‐ and nigericin‐induced interleukin‐1beta release through a dye uptake‐independent pathway. J Biol Chem 282:2386–2394. [DOI] [PubMed] [Google Scholar]
- 118. Perregaux D, Gabel CA (1994) Interleukin‐1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem 269:15195–15203. [PubMed] [Google Scholar]
- 119. Perrin RJ, Fagan AM, Holtzman DM (2009) Multi‐modal techniques for diagnosis and prognosis of Alzheimer's disease. Nature 461:916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Perry VH (2004) The influence of systemic inflammation on inflammation in the brain: implications for chronic neurodegenerative disease. Brain Behav Immun 18:407–413. [DOI] [PubMed] [Google Scholar]
- 121. Philippens IH, Ormel PR, Baarends G, Johansson M, Remarque EJ, Doverskog M (2016) Acceleration of amyloidosis by inflammation in the amyloid‐beta marmoset monkey model of alzheimer's disease. J Alzheimers Dis 55:101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Pontillo A, Catamo E, Arosio B, Mari D, Crovella S (2012) NALP1/NLRP1 genetic variants are associated with Alzheimer disease. Alzheimer Dis Assoc Disord 26:277–281. [DOI] [PubMed] [Google Scholar]
- 123. Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP (2013) The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement 9:63–75.e2. [DOI] [PubMed] [Google Scholar]
- 124. Prinz M, Priller J (2014) Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci 15:300–312. [DOI] [PubMed] [Google Scholar]
- 125. Proell M, Gerlic M, Mace PD, Reed JC, Riedl SJ (2013) The CARD plays a critical role in ASC foci formation and inflammasome signalling. Biochem J 449:613–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Qiao X, Cummins DJ, Paul SM (2001) Neuroinflammation‐induced acceleration of amyloid deposition in the APPV717F transgenic mouse. Eur J Neurosci 14:474–482. [DOI] [PubMed] [Google Scholar]
- 127. Qin B, Cartier L, Dubois‐Dauphin M, Li B, Serrander L, Krause KH (2006) A key role for the microglial NADPH oxidase in APP‐dependent killing of neurons. Neurobiol Aging 27:1577–1587. [DOI] [PubMed] [Google Scholar]
- 128. Ramanan VK, Saykin AJ (2013) Pathways to neurodegeneration: mechanistic insights from GWAS in Alzheimer's disease, Parkinson's disease, and related disorders. Am J Neurodegener Dis 2:145–175. [PMC free article] [PubMed] [Google Scholar]
- 129. Ramos EM, Lin M‐T, Larson EB, Maezawa I, Tseng L‐H, Edwards KL et al (2006) Tumor necrosis factor alpha and interleukin 10 promoter region polymorphisms and risk of late‐onset alzheimer disease. Arch Neurol 63:1165–1169. [DOI] [PubMed] [Google Scholar]
- 130. Raval FM, Mishra R, Garcea RL, Welsh RM, Szomolanyi‐Tsuda E (2013) Long‐lasting T cell‐independent IgG responses require MyD88‐mediated pathways and are maintained by high levels of virus persistence. MBio 4:3–e00812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Reed‐Geaghan EG, Savage JC, Hise AG, Landreth GE (2009) CD14 and toll‐like receptors 2 and 4 are required for fibrillar A{beta}‐stimulated microglial activation. J Neurosci 29:11982–11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Riedl SJ, Salvesen GS (2007) The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol 8:405–413. [DOI] [PubMed] [Google Scholar]
- 133. Rivera‐Escalera F, Matousek SB, Ghosh S, Olschowka JA, O'Banion MK (2014) Interleukin‐1beta mediated amyloid plaque clearance is independent of CCR2 signaling in the APP/PS1 mouse model of Alzheimer's disease. Neurobiol Dis 69:124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Rivers‐Auty J, Brough D (2015) Potassium efflux fires the canon: potassium efflux as a common trigger for canonical and noncanonical NLRP3 pathways. Eur J Immunol 45:2758–2761. [DOI] [PubMed] [Google Scholar]
- 135. Roher AE, Weiss N, Kokjohn TA, Kuo Y‐M, Kalback W, Anthony J et al (2002) Increased A beta peptides and reduced cholesterol and myelin proteins characterize white matter degeneration in Alzheimer's disease. Biochemistry 41:11080–11090. [DOI] [PubMed] [Google Scholar]
- 136. Ross FM, Allan SM, Rothwell NJ, Verkhratsky A (2003) A dual role for interleukin‐1 in LTP in mouse hippocampal slices. J Neuroimmunol 144:61–67. [DOI] [PubMed] [Google Scholar]
- 137. Russo BC, Brown MJ, Nau GJ (2013) MyD88‐dependent signaling prolongs survival and reduces bacterial burden during pulmonary infection with virulent Francisella tularensis. Am J Pathol 183:1223–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Sanz JM, Chiozzi P, Ferrari D, Colaianna M, Idzko M, Falzoni S et al (2009) Activation of microglia by amyloid β requires P2X7 receptor expression. J Immunol 182:4378–4385. [DOI] [PubMed] [Google Scholar]
- 139. Saresella M, La Rosa F, Piancone F, Zoppis M, Marventano I, Calabrese E et al (2016) The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer's disease. Mol Neurodegener 11:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Sborgi L, Ravotti F, Dandey VP, Dick MS, Mazur A, Reckel S et al (2015) Structure and assembly of the mouse ASC inflammasome by combined NMR spectroscopy and cryo‐electron microscopy. Proc Natl Acad Sci U S A 112:13237–13242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Schmid‐Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E et al (2016) A genome‐wide crispr (clustered regularly interspaced short palindromic repeats) screen identifies nek7 as an essential component of nlrp3 inflammasome activation. J Biol Chem 291:103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Schroder K, Tschopp J (2010) The inflammasomes. Cell 140:821–832. [DOI] [PubMed] [Google Scholar]
- 143. Sheng WS, Hu S, Feng A, Rock RB (2013) Reactive oxygen species from human astrocytes induced functional impairment and oxidative damage. Neurochem Res 38:2148–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M et al (2016) NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol 17:250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H et al (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526:660–665. [DOI] [PubMed] [Google Scholar]
- 146. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P et al (2014) Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514:187–192. [DOI] [PubMed] [Google Scholar]
- 147. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S et al (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36:401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Simard AR, Soulet D, Gowing G, Julien J‐P, Rivest S (2006) Bone marrow‐derived microglia play a critical role in restricting senile plaque formation in alzheimer's disease. Neuron 49:489–502. [DOI] [PubMed] [Google Scholar]
- 149. Sofroniew MV (2015) Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci 16:249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Sperlagh B, Illes P (2014) P2X7 receptor: an emerging target in central nervous system diseases. Trends Pharmacol Sci 35:537–547. [DOI] [PubMed] [Google Scholar]
- 151. Stutz A, Horvath GL, Stahl R, Franklin BS, Kolbe C‐CL, Geyer M et al (2014) 177: The NLRP3 inflammasome is regulated by phosphorylation and ubiquitinylation. Cytokine 70:70–71. [Google Scholar]
- 152. Subaiea GM, Adwan LI, Ahmed AH, Stevens KE, Zawia NH (2013) Short‐term treatment with tolfenamic acid improves cognitive functions in Alzheimer's disease mice. Neurobiol Aging 34:2421–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Subaiea GM, Ahmed AH, Adwan LI, Zawia NH (2015) Reduction of amyloid‐beta deposition and attenuation of memory deficits by tolfenamic acid. J Alzheimers Dis 43:425–433. [DOI] [PubMed] [Google Scholar]
- 154. Summersgill H, England H, Lopez‐Castejon G, Lawrence CB, Luheshi NM, Pahle J et al (2014) Zinc depletion regulates the processing and secretion of IL‐1β. Cell Death Dis 5:1:e1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Sutinen EM, Pirttilä T, Anderson G, Salminen A, Ojala JO, Carpentier M et al (2012) Pro‐inflammatory interleukin‐18 increases Alzheimer's disease‐associated amyloid‐β production in human neuron‐like cells. J Neuroinflammation 9:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Sy M, Kitazawa M, Medeiros R, Whitman L, Cheng D, Lane TE et al (2011) Inflammation induced by infection potentiates tau pathological features in transgenic mice. Am J Pathol 178:2811–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140:805–820. [DOI] [PubMed] [Google Scholar]
- 158. Talreja D, Singh PK, Kumar A (2015) In vivo role of tlr2 and myd88 signaling in eliciting innate immune responses in staphylococcal endophthalmitis. Invest Ophthalmol Vis Sci 56:1719–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Tan M‐S, Tan L, Jiang T, Zhu X‐C, Wang H‐F, Jia C‐D et al (2014) Amyloid‐β induces NLRP1‐dependent neuronal pyroptosis in models of Alzheimer's disease. Cell Death Dis 5:e1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Tosto G, Reitz C (2013) Genome‐wide association studies in Alzheimer's disease: a review. Curr Neurol Neurosci Rep 13:381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Tsay H‐J, Huang Y‐C, Huang F‐L, Chen C‐P, Tsai Y‐C, Wang Y‐H et al (2013) Amyloid β peptide‐mediated neurotoxicity is attenuated by the proliferating microglia more potently than by the quiescent phenotype. J Biomed Sci 20:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Vermeer SE, Prins ND, den Heijer T, Hofman A, Koudstaal PJ, Breteler MMB (2003) Silent brain infarcts and the risk of dementia and cognitive decline. N Engl J Med 348:1215–1222. [DOI] [PubMed] [Google Scholar]
- 163. Walsh JG, Muruve DA, Power C (2014) Inflammasomes in the CNS. Nat Rev Neurosci 15:84–97. [DOI] [PubMed] [Google Scholar]
- 164. Wang C, Sun B, Zhou Y, Grubb A, Gan L (2012) Cathepsin B degrades amyloid‐beta in mice expressing wild‐type human amyloid precursor protein. J Biol Chem 287:39834–39841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Wang X, Feuerstein GZ, Gu J‐L, Lysko PG, Yue T‐L (1995) Interleukin‐1β induces expression of adhesion molecules in human vascular smooth muscle cells and enhances adhesion of leukocytes to smooth muscle cells. Atherosclerosis 115:89–98. [DOI] [PubMed] [Google Scholar]
- 166. Wattmo C, Londos E, Minthon L (2014) Risk factors that affect life expectancy in Alzheimer's disease: a 15‐year follow‐up. Dement Geriatr Cogn Disord 38:286–299. [DOI] [PubMed] [Google Scholar]
- 167. Weldon DT, Rogers SD, Ghilardi JR, Finke MP, Cleary JP, O'Hare E et al (1998) Fibrillar beta‐amyloid induces microglial phagocytosis, expression of inducible nitric oxide synthase, and loss of a select population of neurons in the rat CNS in vivo. J Neurosci 18:2161–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Whitmer RA, Gunderson EP, Barrett‐Connor E, Quesenberry CP, Yaffe K (2005) Obesity in middle age and future risk of dementia: a 27 year longitudinal population based study. bmj 330:1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Wilkinson BL, Landreth GE (2006) The microglial NADPH oxidase complex as a source of oxidative stress in Alzheimer's disease. J Neuroinflammation 3:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Wu Y, Tibrewal N, Birge RB (2006) Phosphatidylserine recognition by phagocytes: a view to a kill. Trends Cell Biol 16:189–197. [DOI] [PubMed] [Google Scholar]
- 171. Yasuno F, Kosaka J, Ota M, Higuchi M, Ito H, Fujimura Y et al (2012) Increased binding of peripheral benzodiazepine receptor in mild cognitive impairment‐dementia converters measured by positron emission tomography with [(1)(1)C]DAA1106. Psychiatry Res 203:67–74. [DOI] [PubMed] [Google Scholar]
- 172. Zenaro E, Pietronigro E, Bianca V, Della Piacentino G, Marongiu L, Budui S et al (2015) Neutrophils promote Alzheimer's disease‐like pathology and cognitive decline via LFA‐1 integrin. Nat Med 21:880–886. [DOI] [PubMed] [Google Scholar]
- 173. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J (2010) Thioredoxin‐interacting protein links oxidative stress to inflammasome activation. Nat Immunol 11:136–140. [DOI] [PubMed] [Google Scholar]
- 174. Zimmer ER, Leuzy A, Bened‐Breitner J, Gauthier S, Rosa‐Neto P (2014) Tracking neuroinflammation in Alzheimer's disease: the role of positron emission tomography imaging. J Neuroinflammation 11:120. [DOI] [PMC free article] [PubMed] [Google Scholar]