

Abstract
Changes in microglia function are involved in Alzheimer's disease (AD) for which ageing is the major risk factor. We evaluated microglial cell process morphologies and their gray matter coverage (arborized area) during ageing and in the presence and absence of AD pathology in autopsied human neocortex. Microglial cell processes were reduced in length, showed less branching and reduced arborized area with aging (case range 52–98 years). This occurred during normal ageing and without microglia dystrophy or changes in cell density. There was a larger reduction in process length and arborized area in AD compared to aged‐matched control microglia. In AD cases, on average, 49%–64% of microglia had discontinuous and/or punctate Iba1 labeled processes instead of continuous Iba1 distribution. Up to 16% of aged‐matched control microglia displayed discontinuous or punctate features. There was no change in the density of microglial cell bodies in gray matter during ageing or AD. This demonstrates that human microglia show progressive cell process retraction without cell loss during ageing. Additional changes in microglia occur with AD including Iba1 protein puncta and discontinuity. We suggest that reduced microglial arborized area may be an aging‐related correlate of AD in humans. These variations in microglial cells during ageing and in AD could reflect changes in neural‐glial interactions which are emerging as key to mechanisms involved in ageing and neurodegenerative disease.
Keywords: microglia, aging, Alzheimer's disease, tau, morphology, confocal microscopy, image analysis, dystrophic microglia, immunofluorescence, post‐mortem human brain tissue, Iba1 protein, tau protein, confocal microscopy, imaging
Introduction
Loss of cortical synapses and progression of cognitive deficits in Alzheimer's disease (AD) coincides with hyperphosphorylation, impaired sorting and aggregation of microtubule‐associated protein tau into filaments within neural cell bodies (neurofibrillary tangles: NFTs) and their processes (neuropil threads and neuritic plaques) 44. In the AD brain, microglia cluster at a subset of neuritic plaque lesions, however the role played by these cells in disease progression is controversial 31, 37. Microglial cell activation has been hypothesised to give rise to chronic inflammation and neurotoxicity which may play roles in disease pathogenesis 14. Alternatively, non‐inflammatory activation pathways in microglia have recently been shown to mediate synapse loss in AD mouse models 15. Coupled with the capacity to express receptors for a wide range of neurotransmitters and immune molecules, microglia can interact with neurons and respond to changes in homeostasis 25, 30, 42. Ligands and receptors expressed by microglia play dual roles in synaptic consolidation and plasticity in the normal non‐inflamed developing and adult brain 33, 35. It stands to reason that aberrant regulation of these functions could be factors in neurological disease. Polymorphisms in genes encoding microglia‐specific proteins related to phagocytic and protein degradation pathways increase the risk of AD 8, 10, 19, 24.
Microglia exist in a dynamic surveillant state, which involves mainly unmoving cell bodies and primary cell process branches but active higher order long thin branches in constant motion to monitor the surrounding microenvironment including direct interactions with astrocytes, neurons and blood vessels 25. The cells are arranged in the gray matter with a grid‐like non‐overlapping tiled pattern which maximises coverage of the parenchyma. Microglial cell bodies can also be motile, demonstrating cellular migration along chemokine gradients such as ATP 7. The resultant migration involves a transient shift in morphology, from the ramified phenotype with numerous long, thin processes and small cell body, to an amoeboid shape featuring fewer processes and a larger cell body that moves through the tissue 7, 17. In mouse models of AD, impairment of microglial cell body and process motility has been observed 16. Evidence for microglial dystrophy in the human AD brain has also been suggested by changes in morphological structure, offering a new perspective for a role of microglial dysfunction in AD 40. Aging related changes in microglia have been measured in rodent brain 5, 13.
We were interested in how microglial cells are organized within the normal ageing human brain and in AD. A number of markers of microglial cells have been described that include mostly membrane proteins or markers associated with phagocytic functions 1, 6, 28, 31. Most of these antigens are expressed to varying context‐driven extents in resident microglia as well as in some circulating macrophages and they show variable preservation in autopsied human brain. Iba1 is a cytoskeletal‐associated Ca binding protein that, in brain, is predominantly expressed in resident microglial cells. Because of its distribution throughout the entire cell body and processes of the majority, if not all microglia, and its preservation in autopsied brain, Iba1 antibody labeling is readily used to evaluate human microglial cell numbers and morphologies 38. Using Iba1 labeling, we confirm the presence of morphologically altered microglia in AD. We further demonstrate shortening of microglial cell processes and their reduced coverage of brain parenchyma with ageing, independent of AD. The observed shortening of microglial processes could reflect age‐related changes in the neurons, vessels and/or astroglia that microglial cells dynamically interact with. Mechanisms to mediate the activity of microglia particularly in their synaptic maintenance and neural support functions are a potential strategy to reduce the age‐related element of AD risk.
Results
No significant difference in microglial cell density in AD cortical gray matter compared to aged‐matched controls
Using Iba1 antibodies, microglial cells were labeled and compared in autopsied AD (n = 7, mean age 84 ± 11 years) and control brains (n = 5, mean age 82 ± 10 years). Two brain regions—layers II‐III of the inferior temporal and anterior cingulate cortical gray matter were sampled using 360 µm2 regions of interests (ROIs) separated along the section (10 ROIs per region) (Supporting Information Figure 1). The density of microglial cells and their morphological characteristics were measured in each ROI and averaged across all ROIs per brain region and case. Microglial cells in inferior temporal and anterior cingulate cortical gray matter were also sampled in the same way in three younger pathology‐free cases (n = 3, mean age 55 ± 4 years) (these younger cases were not pooled in the control group for comparison against AD cases).
The density of microglial cell bodies differed from case‐to‐case ranging from 69 to 156 cells per mm2. To investigate the relationship with AD pathology, neuritic plaques, neuropil threads and neurofibrillary tangles, were labeled with total tau antibodies (Figure 1). Although we observed some clustering of microglia at neuritic plaques in AD cases (Figure 1), the mean number of cells per mm2 in each brain region showed no significant differences between the controls and the AD cases (Figure 2).
Figure 1.
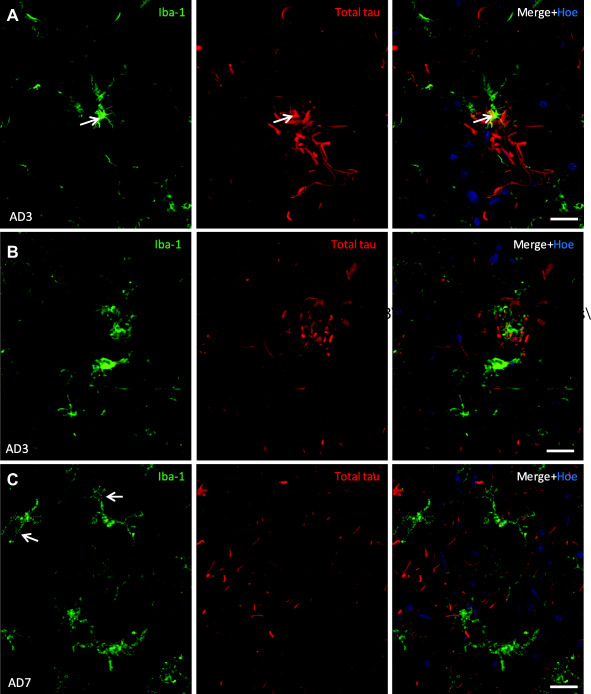
Microglial cells cluster at a subset of neuritic plaques in human AD cortical gray matter. A. A de‐ramified microglial cell with intense Iba1 labeling (arrow) localizes to a neuritic plaque containing tau positive dystrophic neurites in AD inferior temporal cortex. B. Another example of a neuritic plaque intertwined with multiple microglial cell processes in AD cingulate cortex. C. AD cingulate cortex, gray matter contains microglial cells with altered morphology (arrows: punctate Iba1 labeling) and neuropil threads. Data are represented here as maximum intensity projections from confocal z‐stacks, of 17–18 μm deep. Scale bars = 20μm.
Figure 2.
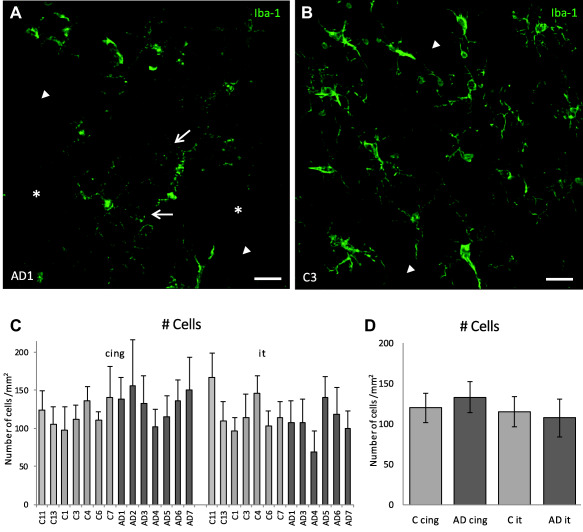
Altered morphology of cells but no significant difference in microglial cell numbers in AD compared to aged‐matched control human gray matter. Images are of inferior temporal cortical gray matter shown as maximum intensity projections of 20 µm deep confocal z‐stacks. A. Microglia in AD tissue show altered morphologies including punctate Iba1 in processes (arrows) and some spaces devoid of cells (asterisks). B. Microglia within control tissue display extensive branching of their processes with uniform, continuous Iba‐1 labeling along the length of the processes. C. The mean number of microglial cell bodies per mm2 of gray matter for each individual case. Error bars represent standard deviation for n = 10 ROIs. D. There was no significant difference in microglial cell density in AD compared to aged matched control gray matter in neither the cingulate (cing) (P = 0.249) nor the inferior temporal (it) cortex (P = 0.566). Note: AD2 tissue was not available from the inferior temporal region so this case was only available for analysis of the cingulate region. Scale bars 20 µm.
Microglial cell processes are morphologically altered in AD
In the AD cases, Iba1 protein was found within small puncta in microglial cell bodies and along their processes (Figure 2a arrows; higher magnification images in Figures 3 and 4a). Control microglia showed Iba1 labeling that was continuously distributed along cell processes (Figures 2b and 3a). The microglia with punctate Iba1 distribution in their processes contained intact nuclei as imaged with Hoechst (Figure 3b,c). Although punctate, the cells retained their branching processes (Figure 3b). To test whether other proteins also located to Iba1 puncta, cells were double labeled with actin‐associated protein cofilin (which is known to label microglia in AD brain) 29. This showed that some puncta contained both Iba1 and cofilin (Figure 3c, arrow). Cofilin negative, Iba1 protein positive puncta were also present (Figure 3c, arrowhead) and cofilin protein was present in some areas of the cell that were not labeled with Iba1. In addition to punctate Iba‐1 labeling, in AD cases we also observed microglia with discontinuous processes, exhibiting breaks in Iba‐1 labeling (Figure 4a, bottom left) similar to previous reports of dystrophic microglia 18, 40. These cells with discontinuous Iba1 labeling in cell processes still contained intact nuclei (Supporting Information Figure 2).
Figure 3.
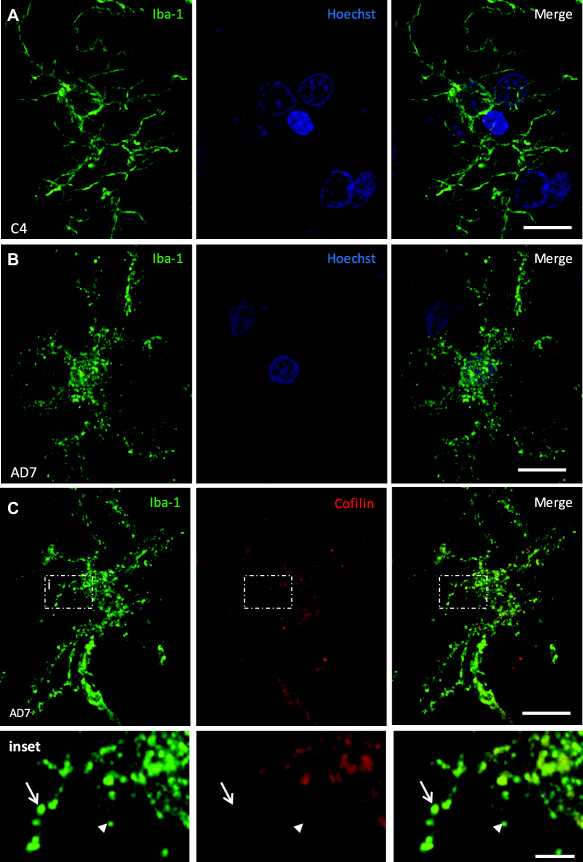
Microglia morphology is altered in AD compared to age‐matched controls. Maximum intensity projections are displayed of 20 µm deep confocal z‐stacks. A. Ramified cells in aged‐matched control cases have abundant, long, thin, branched processes with smooth continuous labeling of Iba1 and intact nuclei. B. Many microglia in AD cases display punctate Iba1 labeling along their processes but the nuclei (Hoechst labeled) remain intact. C. Cofilin co‐localized with Iba‐1 in some but not all of the Iba1 staining in AD microglial cell processes. (Inset: arrow and arrowhead, respectively). Co‐localization analysis revealed a Pearson's Coefficient of 0.434 and Mander's Coefficients of 0.611 (Cofilin over Iba‐1) and 0.422 (Iba‐1 over Cofilin). Scale bars a‐c= 10 µm; inset = 2 µm.
Figure 4.
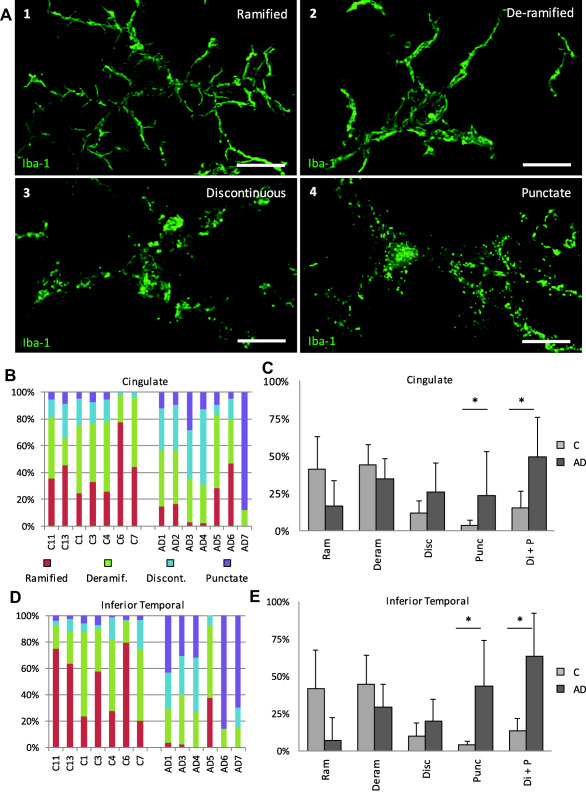
Characterization and quantification of morphological features of microglia in cingulate and interior temporal gray matter. A. Microglia have highly polymorphic processes which were categorized into four main groups for quantification: (1) ramified: microglia with abundant, long, thin, branched processes and continuous Iba1 distribution; (2) deramified: microglia with fewer processes that were shortened and thickened whilst retaining continuous distribution of Iba1; (3) discontinuous: microglial processes with discontinuities in Iba1 labeling in multiple places and (4) punctate: microglia that had processes with punctate Iba‐1 labeling. Maximum intensity confocal projections of an Iba‐1 labeled microglia in each category are shown (A 1–4). A reduction in the % of cells with continuous Iba1 labeling (ramified or deramified processes) and corresponding increase in the discontinuous and punctate features was seen in AD compared to age matched controls both in the cingulate (B, C) and inferior temporal (D, E) regions. Discontinuous and punctate categories were pooled to illustrate in control compared to AD cases the change from continuous Iba1 labeling to discontinuous/punctate processes (Di + P) (C, E). Statistical significance is marked by * when P > 0.05. For the cingulate region: n1 = 5, n2 = 7; Di + P P = 0.013; punctate P < 0.05. For the inferior temporal region: n1 = 5, n2 = 6; punctate P = 0.025; Di + P P < 0.05. Scale bars = 10 µm. Error bars represent standard deviation.
Quantification of morphology types in control and AD gray matter reveals a transition in AD microglia to punctate and/or discontinuous Iba1 labeling
In this cohort of AD cases, we confirm that the whole spectrum of microglial cell morphologies can be present within the same brain section. Given this range of morphologies, quantification of the relative contribution of each of the morphologies in AD and in controls was needed. We grouped cells into one of four categories according to morphology—ramified (multiple long, thin highly branched processes and continuously distributed Iba1), deramified (fewer, shorter, thickened processes and continuous Iba1), discontinuous (distinct dis‐continuities in Iba1 labeling in multiple places) or punctate (numerous small Iba1 puncta along the processes) (see materials and methods). Images of an example of a cell in each category are illustrated in Figure 4a. Examples of punctate cells compared to ramified cells with uniform Iba1 distribution are illustrated in Figure 3. These categories separate the cells into distinct continuous or discontinuous Iba1 labeling groups and give a further estimation of the degree of ramification in cells with continuous labeling (the degree of ramification is further assessed below with skeletal analysis).
The results showed variation between cases in the % of cells in each category (Figure 4b,d), but overall, continuous Iba1 labeling (ramified or deramified processes) was found in the controls while the AD cases had higher proportions of microglia with discontinuous and/or punctate processes (Figure 4c,e). The inferior temporal cortex showed a more pronounced transition from continuous labeling to discontinuous and/or punctate processes compared to the cingulate cortex (Figure 4c,e). In AD inferior temporal cortex, 7% of cells were ramified, 29% deramified, 20% discontinuous and 44% punctate. In control inferior temporal cortex, on average 42% of cells were ramified, 45% deramified, 10% discontinuous and 4% punctate. In AD cingulate cortex, 16% of cells were ramified, 35% deramified, 26% discontinuous and 23% punctate. In control cingulate cortex on average 41% of cells were ramified, 44% deramified, 12% discontinuous and 4% punctate. Hence in AD inferior temporal and cingulate cortices, 49%–63% of cells showed non‐continuous Iba1 labeling in their processes (discontinuous and punctate categories combined). This contrasted with control cases where 14%–15% of cells had non‐continuous distribution of Iba1 along processes. We note that some cells in the deramified groups from all cases may have been perivascular macrophages which represent a minor component of Iba1 expressing cells in brain and would be indistinguishable from fully deramified amoeboid microglia in this analysis.
Microglial cell process length and arborization area is reduced in AD cortical gray matter compared to age‐matched controls
Next we wanted to determine whether the broad morphological transitions in AD brain reflected quantifiable changes in parameters related to microglial cell function such as cell process length and expanse of branching. Microglial cells and processes were defined by polygon overlays and the total length of branches and number of junctions (points at which microglial cell processes branch) were measured within the polygons (Supporting Information Figure 2c). This was done in three of the ROIs per brain region per case. The mean branch length per mm2 was significantly smaller in the inferior temporal cortical gray matter of AD cases compared to the controls (Figure 5a,b). The branch length parameter was not significantly different in AD compared to control cases in the cingulate region.
Figure 5.
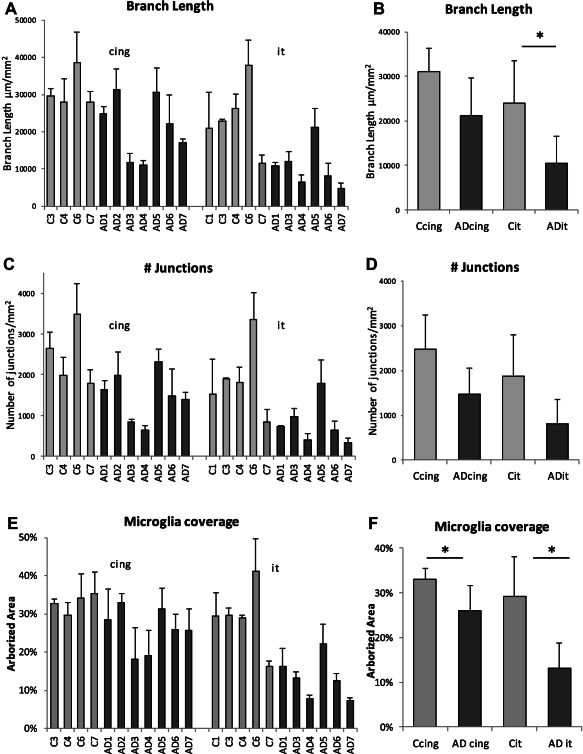
Microglial cell process length and arborization area is reduced in AD compared to aged‐matched control human gray matter (A,B,C,D). Microglial cell process lengths (mean branch length) showed a statistically significant reduction in AD inferior temporal (P = 0.035) but not cingulate (P = 0.060) gray matter compared to aged matched controls (A,B). The number of branch points (junctions) in microglial cell processes in the AD inferior temporal gray matter and the cingulate region were not significantly different in AD compared to age matched controls (P = 0.131 and 0.072, respectively) (C,D). There was a significant reduction in the arborized area of microglial cell processes in AD cases compared to age matched controls in both the cingulate (P = 0.019) and inferior temporal (P = 0.013) gray matter. Error bars represent standard deviation.
The area under the polygon overlays was measured to provide an estimate of the % area of parenchyma covered by microglia in AD compared to aged‐matched control brain. The arborized area coverage by microglial cells and processes was significantly reduced in AD compared to control gray matter for both cingulate and inferior temporal cortices (Figure 5e,f). The effect was greatest in the inferior temporal cortex where there was a twofold reduction in arborized area. This measurement reflects a “snapshot” of the brain area covered by microglia at autopsy with the inferior temporal cortex particularly affected by reduced coverage in AD.
Confocal imaging and 3D analysis shows reduced process lengths and branching in AD microglia compared to control cells
AD and control microglial processes were further evaluated at the single cell level by morphometric analysis of high resolution confocal z‐stacks of individual cells. Animations of AD and control microglial cells from the inferior temporal cortical gray matter reconstructed in 3D are available in Supporting Information figures 3a and 3b. It can be seen both visually (Figure 6a,b; Supporting Information Figure 3) and numerically (Figure 6c–g) that there is an alteration in these parameters in the AD microglia compared to control cells. The reduction in cell volume (Figure 6c), number of branches (Figure 6d) and length of branches (Figure 6e) were significant in AD microglia compared to the age matched control microglia. This is consistent with the results above and demonstrates a loss of process length and branching complexity in individual cells. A significant reduction in the straightness of the processes was also observed in AD microglia compared to the control cells (straightness represents the direct distance between two branch points (h) per length (l) of the filament: straightness = h/l) (Figure 6f). There was no significant difference in the diameter of the processes in AD compared to control cells (Figure 6g).
Figure 6.
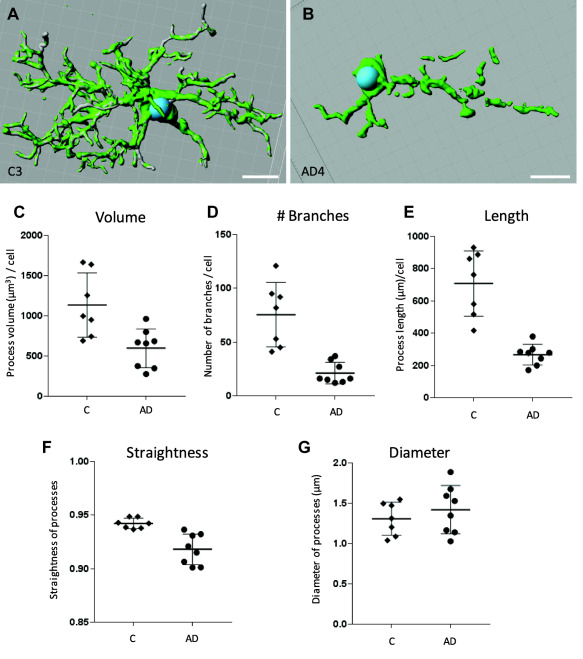
Three‐dimensional analysis of high resolution confocal z‐stacks of representative individual microglial cells from four control cases and four AD cases in inferior temporal human gray matter demonstrates reduced process length and branching in AD microglia. Three‐dimensional surface rendered images of control (A) and AD (B) cells. Volume data produced from the constructed surface render images of control and AD cells showed a significant decrease in the total volume of the processes in AD compared to control microglial cells (P = 0.028) (C) filament tracing analysis showed a reduction in the total number of branches (P = 0.042) (D) and total length of the processes per cell (P = 0.003) (E) in AD microglia compared to control cells. A reduction in straightness of the processes (P = 0.002) was also observed in AD microglia compared to control (F). There was no significant difference (P = 0.460) in the diameter of the processes in AD compared to control microglial cells (G) Scale bars = 10 µm. Error bars represent standard deviation.
Ageing related reduction in microglial cell process length, branching and arborized area in humans
We were interested in whether ageing related changes occur in microglial cells in the human brain independent of the presence of AD pathology. The aged‐matched controls from the experiments above, as well as the additional three younger cases (mean age 55 ± 4 years) were used in this analysis of microglial cells in inferior temporal cortical gray matter. The numbers of cells, the length of processes, number of branching junctions and % arborized area were plotted against age (Figure 7). There was no correlation between age and microglial cell numbers per mm2 in human inferior temporal cortical gray matter (R 2 =0.011 for non‐AD and 0.005 for the AD cases, respectively) (Figure 7a). Significant reductions in microglial cell process length (R 2 = 0.658) (Figure 6b), degree of process branching (number of junctions) (R 2 = 0.647) (Figure 6c) and % gray matter parenchyma coverage by microglia cell processes (% arborized area) (R 2 = 0.600) (Figure 7d) were seen with ageing in the non‐AD cases. Among AD cases, there was no correlation with age and these parameters (Figure 7d).
Figure 7.
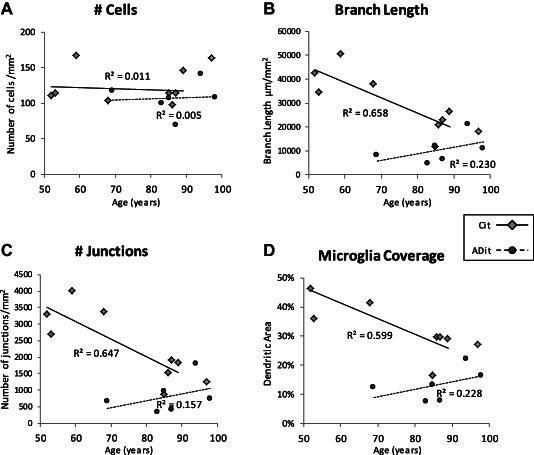
Age‐related reduction in microglial cell process length, branching and area of arborization in inferior temporal gray matter. There was no significant correlation of microglial cell density with age in inferior temporal gray matter in control or in AD brain (A). In pathology free young and age‐matched controls, microglial cell process branch length (B); degree of branching (number of junctions) (C) and % gray matter coverage (arborized area) reduced with age (R 2 = 0.658, 0.647, 0.600, respectively; n = 8 non‐AD cases) (diamonds). Among AD cases (n = 6) (circles) there was no correlation of these parameters with age (branch length R 2 = 0.230; number of junctions R 2 = 0.157; arborized area R 2 = 0.228). Cit – aged‐matched and young control inferior temporal gray matter. ADit – AD inferior temporal gray matter.
Discussion
Microglia play important roles in AD aetiology: this is known first through histological evidence showing that microglia interact with pathogenic lesions and undergo changes in their morphology in AD brain 36, 40. Second, polymorphisms in genes encoding microglial proteins are linked to increased risk of AD 10, 14. Third, alterations in the levels of proteins related to microglial cell phagocytic function and autophagy have been found in AD and in mouse models of AD demonstrating an altered disease‐related microglial cell gene expression profile 19, 45. Animal models have demonstrated that microglial cell processes are dynamic: they constantly monitor the normal brain, participating in the removal of dysfunctional synapses and providing trophic factors to neurons thereby supporting plasticity of neuronal connectivity25, 34. Recent studies in AD mouse models suggest that microglial processes mediate early Aβ‐dependent synapse loss, acting through complement protein receptors and that they are also involved in the spread of tau pathology throughout interconnected brain regions 2, 15. We aimed to evaluate the length and extent of microglial cell process arborization in human neocortical gray matter brain and to determine whether there are changes in these properties associated with ageing and/or with AD pathology.
While there are other markers of microglial cells, for our purposes, we found that Iba1 was most optimal as Iba1 antibodies labeled all microglial cells and revealed Iba1 distribution along the extent of all processes allowing their lengths to be tracked. We observed a change in distribution (in up to 44% of cells) of Iba1 protein into small puncta within AD microglial cell processes. The Iba1 protein puncta seen in these AD microglia could represent a response to the changing activity of or loss of synapses that occurs in AD and the corresponding neural‐microglia interactions that occur as the disease progresses 15. In this context, the puncta could reflect the localization of Iba1 in phagosomes 26 but further investigation is needed to evaluate this. In addition to cells with numerous small Iba1 puncta, we also found other AD microglial cells with discontinuous processes giving them a fragmented appearance (up to 26% of cells). Discontinuous Iba1 labeling along processes has previously been characterized as representing dystrophic microglia (described by cytoplasmic beading or spheroids, cytoplasmic fragmentation and decreased process arborization) 3, 40. These authors interpreted the morphology of AD microglia as reflecting a dystrophy or senescence and loss of function. However, there has been no molecular evidence for microglial cell degeneration or senescence in human brain and the morphology of microglial cells may also reflect a response to degenerating neurons or synapses rather than a cell‐autonomous functional decline in the microglia themselves. Aged‐matched control inferior temporal and cingulate cortices had an average of just 4% of microglia with punctate Iba1 and up to 12% discontinuous cells. The extent of discontinuous and punctate microglia in AD was higher in the inferior temporal compared to the cingulate cortex of AD cases. This may reflect an increased level of cortical atrophy in the AD inferior temporal cortex 12. The extent of discontinuous Iba1 labeling of microglial processes measured here is consistent with a recent report of ∼25% dystrophic microglia in AD hippocampus 3. Furthermore, the reduced straightness of AD compared with control microglial processes measured in 3D analysis of confocal z‐stacks, is consistent with descriptions of tortuosity often seen in “dystrophic” cells 39. The consistency of the appearance of Iba1‐discontinuous/dystrophic cells within these independent cohorts of autopsied AD brains and across affected brain regions strongly suggests that this is a robust marker for AD and reflects a hallmark feature of this disease aetiology 3, 40.
We further show significant decreases in cell process length and reduced arborized area of gray matter coverage by the microglial cell processes in AD cases compared to age‐matched controls. These features are consistent with a loss in AD of the distal thin motile cell processes that monitor the parenchyma. Recent publications have presented evidence from mouse models and human genetics for a mechanism whereby age‐accelerated microglial‐mediated synaptic pruning is a mediator of synapse loss and neurodegeneration 15, 20, 35. In this context, the reduction in the length and arborization of microglial processes that we observe in the AD cases, may in part, reflect functions in response to signalling from neurons during aberrant tau phosphorylation and mis‐localization and stripping of dysfunctional synapses. Further investigations should examine the relationship between synaptic markers and microglial cells in human brain. Furthermore, microglial morphology changes would be likely to occur as a response to later stage neurodegeneration and brain atrophy. Analysis demonstrated that the microglial cell morphology parameters plotted against post‐mortem interval showed no correlation for any of the parameters. We therefore do not believe that the discontinuities in Iba1 labeling are related to post‐mortem interval. We note that the discontinuity of Iba1 staining in some microglia may have reduced the ability of Iba1 immunostaining to fully represent the length of the processes (ie, processes between puncta exist but not observed). This may have led to an underestimation of the process lengths in AD in the 2D image analysis. Conversely, Iba1 discontinuous labeling is not necessarily an under‐representation of the cell processes: we cannot completely exclude that this labeling represents a true image of the processes and that the cell processes are breaking down in AD.
Ageing is the strongest correlate to incidence of AD. Age‐related changes in microglial cell morphology and motility have been described in numerous animal models 5, 7, 13, 41, 43. In rodents, normal ageing is reported to bring about changes in the resident microglial cell population, with differences noted in terms of number of cells, their distribution, morphology and responses to injury 5, 7, 13. By contrast, in humans, microglial cell morphological changes associated with normal ageing outside the context of disease have not been extensively studied. We demonstrate that independent of AD and without the widespread appearance of punctate or discontinuous Iba1, human microglial cell processes shorten with age. This is associated with a reduced arborization area. A corresponding reduced % of gray matter surveillance capacity could be a consequence of the reduced arborization of processes. Although our study is limited to just eight control cases of middle to late age, the data are consistent with results from animal models. It would be interesting to build on this analysis with the addition of more cases and brain regions including younger controls < 50 years, although these cases are rarer in tissue banks. In mice, ageing is also associated with reduced branching and shortening of microglial processes 5. Therefore, as in rodents 5, 7, 13, human microglia may exhibit diminished functionality with age including reduced capacity for outgrowth of processes and homeostatic behavior. Functional changes in aged rodent microglia also included delayed recovery following immune response, reduced phagocytic ability and reduced capacity for migration 7. In humans, we and others show that morphological changes are not necessarily accompanied by changes in overall microglial cell numbers in gray matter during disease or with ageing 4, 27, 36. Although a significant increase in Iba1 positive microglial cell numbers in AD white matter within the temporal cortex has recently been reported in one study 27. Local migration and clustering, for example at amyloid plaques, may qualitatively give the appearance of increased density of cells in some regions 11, 28, 31, 32, 36, 37. At present, causes of microglial cell changes with ageing are unknown. Known age‐related changes in neurons and synapses may cause the microglia to reduce the area of arborization. Alternatively or in addition, sources of functional decline have been hypothesized such as oxidative damage that accumulates in microglia with aging, toxicity of an increased demand for clearance of protein aggregates, changes in gene expression profiles, altered activity of telomerase or altered local proliferative capacity 9, 21, 24, 27. Microglial cell functional change during ageing may undermine neuroprotection in AD and contribute to a compromised response to the neurodegenerative cascade. Hence, rather than suppression of microglial function (eg, with anti‐inflammatory strategies), perhaps strategies to maintain normal microglial function may help limit neurodegeneration and slow cognitive decline.
In summary, we show that the arborized area of microglial cell processes is reduced in AD accompanied by a striking change in microglial morphology manifesting in an altered distribution of Iba1 protein into numerous small puncta within cell bodies and processes and/or discontunous Iba1 labeling along processes. We further demonstrate an independent age‐related decrease in microglial arborization in human brain in the absence of any AD pathology or gross changes in Iba1 protein distribution. This suggests possible changes in the monitoring of the parenchyma with age. The resulting proportion of gray matter parenchyma surveyed by microglial cells and their processes may be reduced with age which could lead to compromised neural support and homeostasis. It is also likely that some of the observed reduced arborization of microglial processes is related to ageing related changes in neurons or their synapses. Indeed, interactions between neurons and glia during ageing and in diseases associated with ageing may prove key to the biological mechanisms of these processes.
Experimental Procedures
Human tissue
Post‐mortem human brain tissue was acquired from the New South Wales Brain Bank (NSWBB) network with approval by the University of Sydney Human Ethics committee. Formalin‐fixed free‐floating 50 µm sections from the inferior temporal cortex and anterior cingulate cortex of AD (n = 7, mean age 84 ± 11 years), age‐matched control (n = 5, mean age 82 ± 10 years) and younger control cases (n = 3, mean age 55 ± 4 years) were used in the study (Table 1). Cases were classified as control or cognitively impaired based on psychiatric dementia diagnosis prior to death. AD was confirmed by histopathological examination for the presence, location and extent of amyloid plaques, neuritic plaques and tau NFTs according to the National Institute on Aging guidelines for the neuropathological assessment of AD 22. Screening for synuclein and TDP pathologies was performed on all cases. Cases with co‐existing pathology were excluded from the study.
Table 1.
Case details. Cases are numbered in line with previous publication where these cases are reported (Rahman, Davies et al 2014). Cases were classified as control or cognitively impaired based on psychiatric dementia diagnosis prior to death. AD was then confirmed by histopathological examination for the presence, location and extent of amyloid plaques, neuritic plaques and tau NFTs according to the National Institute on Aging guidelines for the neuropathological assessment of AD (Montine, Phelps et al 2012). Screening for synuclein and TDP pathologies was performed on all cases. None of the listed cases had co‐existing pathology. Braak staging information for NFTs and CERAD criteria results of scoring neuritic plaques are provided in this table.
| Case | Classification | Age (years) | Sex | Post‐mortem interval (h) | Braak stage | CERAD | Clinical cause of death |
|---|---|---|---|---|---|---|---|
| C1 | Control | 86 | M | 15 | I | 0 | Cancer |
| C3 | Control | 87 | F | 5 | 0 | 0 | Cancer |
| C4 | Control | 89 | F | 23 | 0 | 0 | Metastatic adenocarcinoma |
| C6 | Control | 68 | M | 11 | 0 | 0 | Renal failure |
| C7 | Control | 85 | F | 23 | 0 | 0 | Pneumonia |
| C11 | Younger control | 59 | F | 24 | 0 | 0 | Respiratory |
| C12 | Younger control | 53 | M | 26 | 0 | 0 | Cardiac |
| C13 | Younger control | 52 | M | 28 | 0 | 0 | Cardiac |
| AD1 | AD | 98 | F | 11 | V | 2 | Cerebrovascular occlusion |
| AD2 | AD | 69 | M | 3 | VI | 3 | Carcinoma of colon |
| AD3 | AD | 85 | F | 10 | VI | 3 | Cardiorespiratory failure |
| AD4 | AD | 87 | F | 19 | VI | 2 | Cardiorespiratory failure |
| AD5 | AD | 94 | M | 22 | V | 2 | Renal failure |
| AD6 | AD | 69 | M | 18 | VI | 3 | Cardiorespiratory failure |
| AD7 | AD | 83 | F | 3 | V | 2 | Uraemia |
Immunolabeling
Immunolabeling was performed as previously described with minor modifications 29. Sections were incubated in sodium citrate buffer pH6.0 at 100°C for 10 minutes for antigen retrieval, before permeablising, blocking and then incubating in primary antibodies (Cofilin 1:500 (Sigma, cat # C8736) or total tau 1:500 (Dako) and Iba‐1 1:50 (Millipore, cat # MABN92) for 3 hours. Secondary antibodies were anti‐mouse‐Alexa 555 and anti‐rabbit‐Alexa 647 (Invitrogen). Hoechst was added at 10 µg/mL for the final 30 minutes of the secondary antibody incubation. Sections were mounted on slides, coverslipped with Prolong Gold (Invitrogen) and allowed to dry at room temperature for 24 hours before sealing with nail varnish.
Microglial cell density and morphology analysis
Sections from the seven AD cases (AD1, AD2, AD3, AD4, AD5, AD6 and AD7) and five age‐matched controls (C1, C3, C4, C6 and C7) were used to compare microglial cell density and morphology in AD vs. control inferior temporal cortex (Table 1). The same cases were used to compare cell density and morphology in cingulate cortex with the exception of AD2 where tissue from this region was not available. Data from the younger control cases (C11, C12 and C13) were not pooled with the age‐matched controls in comparison of AD vs. control cell counts and morphology.
Slides were given a number and imaged blindly. Widefield images of each slide were captured using an Olympus VS120 slide scanner. Overviews were taken of each section before mapping out 10 ROI of 360 µm2 (a total area of approximately 1.3 mm2 per section) within cortical layers II‐III distributed along the gray matter (Supporting Information Figure 1). A z‐stack (7μm) of each ROI was obtained using a 40×/0.95 oil immersion objective lens.
Olympus slide scanner image (.vsi) files were opened in Fiji using the BioFormats Importer plugin and a maximum intensity projection was produced and saved as a tiff file for subsequent analysis. The Iba1 labeled microglial cells were counted manually within the ten ROI's per case. Counting (and morphological analysis) was performed blindly without knowledge of the case number or whether AD or control. Morphological categories were established as follows; (1) ramified (multiple long, thin highly branched processes and continuous Iba1 distribution in the processes) (2) deramified (fewer, shorter, thickened processes whilst retaining a continuous distribution of Iba1) (3) discontinuous (dis‐continuities in Iba1 labeling in multiple places) and (4) punctate (numerous small Iba1 puncta along the processes which may have a ramified or deramified appearance). In the 10 ROIs per case, each cell was given one category only. So for example, punctate cells with ramified processes were categorized as “punctate”.
Two‐dimensional analysis of microglial cell process arborization
For comparison of AD vs. control microglial cell process arborization parameters (length, number of junctions, arborized area of processes), the same slide scanner images of AD and aged‐matched control cases as above were used except that control case C1, cingulate region had to be excluded from this part of the analysis. This was because unusually high autofluorescence of neurons in the cingulate tissue from this case made the subsequent skeletoniseation analysis inaccurate (microglial cell process lengths and junctions were highly over estimated). The arborization parameters were additionally measured in younger control cases C11, 12 and C13, but these data were not included in the aged‐matched control group bar graph used to compare against the AD group.
Arborization parameters were analyzed in three ROIs per case. This was done using the Fiji polygon tool to mark out the area around the microglial processes including whole or partial microglia cells and processes in focus (Supporting Information Figure 1). After each polygon was drawn, it was saved, resulting in a polygon overlay for the entire ROI. The arborized area = the sum area of all of the polygons in the ROI divided by the entire ROI area. Skeletal analysis within the polygons was based on that previously described 7, 23 (Supporting Information Figure 1b, c). Pre‐processing of images included modifying the brightness and contrast of individual images to ensure that all microglia processes were included. All images were processed identically: sharpened before being converted to binary, despeckled and skeletonized using the Skeletonize3D plugin in Fiji. The polygon overlay was then used to screen the resulting skeleton image of each ROI. Analysis of the skeletons was completed using the AnalyzeSkeleton feature (Supporting Information Figure 1).
Three‐dimensional analysis of microglial cell processes
For high resolution analysis of microglial cell processes, representative cells were imaged from inferior temporal cortex of four AD cases (AD1, AD3, AD4 and AD6) and four control cases (C1, C3, C4 and C6). Highest resolution confocal z‐stacks of whole individual Iba1 labeled microglial cells were taken using a Zeiss LSM710 confocal microscope with a 100× NA1.4 Plan‐APOchromat oil immersion objective (0.22–0.44 µm intervals, pinhole maintained at 56 μm; laser excitation line 405 nm was used for Hoechst (nuclear staining) and 561nm for Alexa 555 (Iba‐1)). A total of seven control and eight AD microglial cells were imaged and analyzed.
Three‐dimensional surface rendering of each confocal imaged whole microglia cell was carried out using the automatic surface creation tool within the surpass function of Bitplane Imaris, using a constant threshold of 18.8 for measurement of the features of the processes. A mask was created to exclude the cell body and surrounding area in order to restrict subsequent analysis to the microglia processes. Filament tracing was completed using the manual autodepth filament tracing function with “cone” visualization. Measurements of this filament structure were automatically generated after application of smooth processes function to the structure and centre processes function for alignment of the cell body. The data acquired included total volume of processes per cell (µm3), number of branches per cell, total length of branches per cell, mean diameter of processes per cell and straightness of processes (the direct distance between two branch points (h) per length (l) of the filament (straightness = h/l)).
Statistical analysis
Graphical representation of the data and subsequent statistical analysis was carried out using Microsoft excel and GraphPad Prism. Group differences were determined using the Mann Whitney U test and P values are stated in the figure legends. Results were considered statistically significant for P < 0.050 (*).
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Figure 1. Quantification of the morphological features of the microglia processes was carried out by taking z‐stacks of 10 ROI per case using an Olympus slide scanner which were then converted to maximum intensity projection tiff files. (a) The 10 ROIs were from cortical laminae II‐III evenly distributed along the section (boxes). (b) An example of a maximum projection image from one ROI. Polygons outlining the microglial cell process extents are overlayed (middle panel). The images were thresholded and skeletonized in Fiji to enable branch length and number of junctions to be analyzed. (c) An example of a single cell cropped from a larger ROI, outlined (1), skeletonized (2) and junction pixels tagged (3,4). Scale bars = 1 mm (a), 50 µm (b), 10 µm (c).
Figure 2. Iba1 and cofilin labeled AD microglial cell (shown as a maximum intensity projection of a 20 µm deep confocal z‐stack). The cell displays completely discontinuous processes but an intact nucleus. Scale bar = 10 µm.
Figure 3. Three‐dimensional visualization of Iba1 labeled microglia cells in control (a) and AD (b) inferior temporal cortex showing original confocal images (left) and subsequent surface rendering in Bitplane Imaris (right). Punctate and fragmented processes are evident in the AD cell while the control cell is more ramified and contains continuous labeling of Iba1. Three‐dimensional animations available online, allow a more complete visualization of the cells. Scale bars = 10 µm.
SuppFig3A movie
SuppFig3B movie
Acknowledgments
We are grateful to the brain donors and their families who provided tissue via the Sydney Brain Bank at Neuroscience Research Australia and the NHMRC for funding the project. We would like to acknowledge the assistance of Twishi Gulati with technical advice, Greg Sutherland and Manuel Graeber for insightful comments on the manuscript.
Author Contributions
DSD designed the study, performed the experiments, analyzed data, produced the figures and co‐wrote the paper. JM analyzed data and co‐wrote the paper. TJ analyzed data. CG designed the study, analyzed data and co‐wrote the paper.
References
- 1. Ahmed Z, Shaw G, Sharma VP, Yang C, McGowan E, Dickson DW (2007) Actin‐binding proteins coronin‐1a and IBA‐1 are effective microglial markers for immunohistochemistry. J Histochem Cytochem 55:687–700. [DOI] [PubMed] [Google Scholar]
- 2. Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B (2015) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18:1584–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bachstetter AD, Van Eldik LJ, Schmitt FA, Neltner JH, Ighodaro ET, Webster SJ et al (2015) Disease‐related microglia heterogeneity in the hippocampus of Alzheimer's disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol Commun 3:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baig S, Wilcock GK, Love S (2005) Loss of perineuronal net N‐acetylgalactosamine in Alzheimer's disease. Acta Neuropathol 110:393–401. [DOI] [PubMed] [Google Scholar]
- 5. Baron R, Babcock AA, Nemirovsky A, Finsen B, Monsonego A (2014) Accelerated microglial pathology is associated with Abeta plaques in mouse models of Alzheimer's disease. Aging Cell 13:584–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB et al (2016) New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci USA 113:E1738–E1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Damani MR, Zhao L, Fontainhas AM, Amaral J, Fariss RN, Wong WT (2011) Age‐related alterations in the dynamic behavior of microglia. Aging Cell 10:263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De Strooper B, Karran E (2016) The cellular phase of Alzheimer's disease. Cell 164:603–615. [DOI] [PubMed] [Google Scholar]
- 9. Flanary BE, Streit WJ (2004) Progressive telomere shortening occurs in cultured rat microglia, but not astrocytes. Glia 45:75–88. [DOI] [PubMed] [Google Scholar]
- 10. Griciuc A, Serrano‐Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K et al (2013) Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78:631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Griffin WS, Sheng JG, Roberts GW, Mrak RE (1995) Interleukin‐1 expression in different plaque types in Alzheimer's disease: significance in plaque evolution. J Neuropathol Exp Neurol 54:276–281. [DOI] [PubMed] [Google Scholar]
- 12. Halliday GM, Double KL, Macdonald V, Kril JJ (2003) Identifying severely atrophic cortical subregions in Alzheimer's disease. Neurobiol Aging 24:797–806. [DOI] [PubMed] [Google Scholar]
- 13. Hefendehl JK, Neher JJ, Suhs RB, Kohsaka S, Skodras A, Jucker M (2014) Homeostatic and injury‐induced microglia behavior in the aging brain. Aging Cell 13:60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL et al (2015) Neuroinflammation in Alzheimer's disease. Lancet Neurol 14:388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hong S, Beja‐Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S et al (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science (New York, NY) 352:712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krabbe G, Halle A, Matyash V, Rinnenthal JL, Eom GD, Bernhardt U et al (2013) Functional impairment of microglia coincides with Beta‐amyloid deposition in mice with Alzheimer‐like pathology. PLoS One 8:e60921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee JE, Liang KJ, Fariss RN, Wong WT (2008) Ex vivo dynamic imaging of retinal microglia using time‐lapse confocal microscopy. Invest Ophthalmol Vis Sci 49:4169–4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lopes KO, Sparks DL, Streit WJ (2008) Microglial dystrophy in the aged and Alzheimer's disease brain is associated with ferritin immunoreactivity. Glia 56:1048–1060. [DOI] [PubMed] [Google Scholar]
- 19. Lucin KM, O'Brien CE, Bieri G, Czirr E, Mosher KI, Abbey RJ et al (2013) Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer's disease. Neuron 79:873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang HY et al (2016) Progranulin deficiency promotes circuit‐specific synaptic pruning by microglia via complement activation. Cell 165:921–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ma W, Cojocaru R, Gotoh N, Gieser L, Villasmil R, Cogliati T et al (2013) Gene expression changes in aging retinal microglia: relationship to microglial support functions and regulation of activation. Neurobiol Aging 34:2310–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW et al (2012) National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morrison HW, Filosa JA (2013) A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J Neuroinflammation 10:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mosher KI, Wyss‐Coray T (2014) Microglial dysfunction in brain aging and Alzheimer's disease. Biochem Pharmacol 88:594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science (New York, NY) 308:1314–1318. [DOI] [PubMed] [Google Scholar]
- 26. Ohsawa K, Imai Y, Kanazawa H, Sasaki Y, Kohsaka S (2000) Involvement of Iba1 in membrane ruffling and phagocytosis of macrophages/microglia. J Cell Sci 113:3073–3084. [DOI] [PubMed] [Google Scholar]
- 27. Olmos‐Alonso A, Schetters ST, Sri S, Askew K, Mancuso R, Vargas‐Caballero M et al (2016) Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer's‐like pathology. Brain 139:891–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Perlmutter LS, Scott SA, Barron E, Chui HC (1992) MHC class II‐positive microglia in human brain: association with Alzheimer lesions. J Neurosci Res 33:549–558. [DOI] [PubMed] [Google Scholar]
- 29. Rahman T, Davies DS, Tannenberg RK, Fok S, Shepherd C, Dodd PR et al (2014) Cofilin rods and aggregates concur with tau pathology and the development of Alzheimer's disease. J Alzheimer's Dis 42:1443–1460. [DOI] [PubMed] [Google Scholar]
- 30. Richter N, Wendt S, Georgieva PB, Hambardzumyan D, Nolte C, Kettenmann H (2014) Glioma‐associated microglia and macrophages/monocytes display distinct electrophysiological properties and do not communicate via gap junctions. Neurosci Lett 583:130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rogers J, Luber‐Narod J, Styren SD, Civin WH (1988) Expression of immune system‐associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer's disease. Neurobiol Aging 9:339–349. [DOI] [PubMed] [Google Scholar]
- 32. Sasaki A, Yamaguchi H, Ogawa A, Sugihara S, Nakazato Y (1997) Microglial activation in early stages of amyloid beta protein deposition. Acta Neuropathol 94:316–322. [DOI] [PubMed] [Google Scholar]
- 33. Schafer DP, Stevens B (2015) Microglia function in central nervous system development and plasticity. Cold Spring Harb Perspect Biol 7:a020545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schafer DP, Lehrman EK, Stevens B (2013) The “quad‐partite” synapse: microglia‐synapse interactions in the developing and mature CNS. Glia 61:24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N et al (2016) Schizophrenia risk from complex variation of complement component 4. Nature 530:177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Serrano‐Pozo A, Gomez‐Isla T, Growdon JH, Frosch MP, Hyman BT (2013) A phenotypic change but not proliferation underlies glial responses in Alzheimer disease. Am J Pathol 182:2332–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Serrano‐Pozo A, Muzikansky A, Gomez‐Isla T, Growdon JH, Betensky RA, Frosch MP, Hyman BT (2013) Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer disease. J Neuropathol Exp Neurol 72:462–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Streit WJ (2004) Microglia and Alzheimer's disease pathogenesis. J Neurosci Res 77:1–8. [DOI] [PubMed] [Google Scholar]
- 39. Streit WJ, Sammons NW, Kuhns AJ, Sparks DL (2004) Dystrophic microglia in the aging human brain. Glia 45:208–212. [DOI] [PubMed] [Google Scholar]
- 40. Streit WJ, Braak H, Xue QS, Bechmann I (2009) Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol 118:475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tremblay ME, Zettel ML, Ison JR, Allen PD, Majewska AK (2012) Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. Glia 60:541–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wake H, Moorhouse AJ, Miyamoto A, Nabekura J (2013) Microglia: actively surveying and shaping neuronal circuit structure and function. Trends Neurosci 36:209–217. [DOI] [PubMed] [Google Scholar]
- 43. Wong WT (2013) Microglial aging in the healthy CNS: phenotypes, drivers, and rejuvenation. Front Cell Neurosci 7:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zempel H, Mandelkow E (2014) Lost after translation: missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci 37:721–732. [DOI] [PubMed] [Google Scholar]
- 45. Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA et al (2013) Integrated systems approach identifies genetic nodes and networks in late‐onset Alzheimer's disease. Cell 153:707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Figure 1. Quantification of the morphological features of the microglia processes was carried out by taking z‐stacks of 10 ROI per case using an Olympus slide scanner which were then converted to maximum intensity projection tiff files. (a) The 10 ROIs were from cortical laminae II‐III evenly distributed along the section (boxes). (b) An example of a maximum projection image from one ROI. Polygons outlining the microglial cell process extents are overlayed (middle panel). The images were thresholded and skeletonized in Fiji to enable branch length and number of junctions to be analyzed. (c) An example of a single cell cropped from a larger ROI, outlined (1), skeletonized (2) and junction pixels tagged (3,4). Scale bars = 1 mm (a), 50 µm (b), 10 µm (c).
Figure 2. Iba1 and cofilin labeled AD microglial cell (shown as a maximum intensity projection of a 20 µm deep confocal z‐stack). The cell displays completely discontinuous processes but an intact nucleus. Scale bar = 10 µm.
Figure 3. Three‐dimensional visualization of Iba1 labeled microglia cells in control (a) and AD (b) inferior temporal cortex showing original confocal images (left) and subsequent surface rendering in Bitplane Imaris (right). Punctate and fragmented processes are evident in the AD cell while the control cell is more ramified and contains continuous labeling of Iba1. Three‐dimensional animations available online, allow a more complete visualization of the cells. Scale bars = 10 µm.
SuppFig3A movie
SuppFig3B movie