

Abstract
Pathological processing of tau protein during the formation and maturation of neurofibrillary tangles (NFTs) includes abnormal phosphorylation, conformational changes and truncation of the C‐terminus at aspartic‐acid421 (apoptotic product) and glutamic‐acid391 residues. Abnormal phosphorylation and misfolding may serve as recognition signals for ubiquitin‐targeting and proteosomal processing. For this reason, we sought to determine whether ubiquitin‐targeting of tau is associated with particular tau modifications that herald specific stages of NFTs maturation in the hippocampus of Alzheimer's disease cases. Using multiple tau antibodies, we found that 30% of the total load of NFTs is ubiquitin‐associated. As reported previously ubiquitin immunoreactivity was associated with markers of phosphorylated tau in certain NFTs; however, a strong association was also found between ubiquitin and the earliest known truncation event at aspartic‐acid421. These findings indicate that tau protein in the NFTs may be dually subjected to both apoptotic and proteosomal processing. By contrast ubiquitin immunoreactivity was poorly associated with truncation of tau at glutamic‐acid391, suggesting that this proteolytic event may be independent of proteosomal activity. It would appear, therefore, that ubiquitin targeting of tau protein occurs at NFTs in the early and intermediate stages of the maturation.
Keywords: Alzheimer's disease, neurofibrillary tangles, phosphorylated tau, proteasome, truncation, ubiquitin
INTRODUCTION
Alzheimer's disease (AD) is a neuropathological condition characterized by loss of memory and progressive decline of cognitive functions. In AD, these alterations are the result of neuronal loss, mainly produced by parenchymal accumulation of the β‐amyloid peptide (Aβ) into plaques and the intracellular development of neurofibrillary tangles (NFTs) in the hippocampus and cerebral cortex 5, 18, 76. Abnormally processed tau protein becomes assembled into paired helical filaments (PHFs) that accumulate in large amount within the neuronal cytoplasm to coalesce into NFTs 40, 41, 67. Abnormal phosphorylation of tau protein has been considered one of the earliest events in AD neurodegeneration mainly because it has been found in the form of early amorphous nonfibrillar cytoplasmic aggregates preceding the formation of NFTs 7, 10, 73, 84. Abnormal properties, such as the reduced ability to bind and preserve microtubule structure in neuronal cells, have been attributed to the hyperphosphorylated state of tau protein 34, 47, 52. This alteration may affect axonal transport indirectly and later lead to neuronal death in AD 13, 51.
Additionally, abnormally phosphorylated tau protein purified from the brains of AD patients was able to self‐polymerize into abnormal filaments and also to nucleate the assembly of normal nonphosphorylated tau 1, 38. These and other findings led some groups to propose that abnormal phosphorylation of tau protein is a key event that triggers the neurodegeneration process reported in AD 1, 2, 3, 38.
In addition to abnormal phosphorylation, conformational changes 37, 48, 49 as well as cleavage of tau protein have been described as two accompanying pathological events that are also involved in the self‐aggregation of tau and microtubule disruption 24, 25, 31, 83. Although tau protein under physiological conditions has a random coil structure 17, 44, it has been proposed that in AD, conformational changes could actually be driving its nucleation and aggregation into insoluble structures that contain a pathological β‐sheet conformation 8, 70.
Recently, in a report in which we analyzed the time course of the early modifications of tau protein accumulating in the brain of AD patients, we found that abnormal phosphorylation of tau precedes the conformational change recognized by the Alz‐50 antibody and that recognized by Tau‐C3 antibody, which recognizes a C‐terminal truncation site at aspartic acid421 (Asp421) (57).
In vitro, this truncated variant of tau aggregated at a higher rate than that reported for the wild‐type full‐length tau; overexpressed in cultured neuronal and non‐neuronal cells, it induced apoptotic changes 16, 26, 69. In the brains of AD patients, Asp421‐truncated tau localized with NFTs and other manifestations of the fibrillar pathology 28, 33. This truncation is associated with Alz‐50 conformation of tau and the formation of NFTs, appearing as the tau filaments coalesce into the NFTs. Tau‐C3 staining persists in the NFTs as it matures, losing its Alz‐50 signature, being replaced by a new conformation involving the folding of the proline‐rich region over the third microtubule binding repeat (32). In late‐stage NFTs this conformation is preserved; however, the C‐terminus of tau is further truncated at the glutamic acid391 (Glu391) position by an, as yet, unidentified proteolytic process. Another antibody, MN423, that specifically recognizes this truncated tau variant replaces the Tau‐C3 staining. This appears to be the terminal event in NFTs maturation 9, 31.
In the disease, alteration of the normal process of protein turnover, proteolysis and elimination of misfolded products may be necessitated. Hence, abnormally phosphorylated and misfolded tau proteins may induce proteosomal, lysosomal and apoptotic activities to metabolize the same substrate. At present, only a few instances suggesting multiple routes of degradation acting coordinately during tau processing and aggregation in AD have been reported 4, 23.
In AD, the role of the proteolytic processing of abnormal proteins via the ubiquitin proteasome system (UPS) has long been investigated, and several components of this system have been found to be increased in the brain of AD patients 21, 46. Some studies indicated that tau protein, in the form of fibrillar lesions, is labeled with ubiquitin in AD brains 53, 59, 65, while others demonstrated ubiquitin labeling of insoluble tau pools isolated from fresh AD brain tissue 19, 78. Generally, however, these findings have been interpreted as unsuccessful ubiquitin targeting of tau to be processed by the proteasomal system. We have placed ubiquitin labeling of tau in the context of NFTs maturation and determined that this attempt to target tau to the proteasome occurs predominantly during the early to intermediate stages of NFTs evolution. Ubiquitin labeling was associated with the phosphorylation of tau protein, but also coincided with the early truncation of tau at Asp421 by caspase. This result is indicative of an attempt at dual processing of tau protein by apoptotic and proteosomal pathways associated with its aggregation in AD. Because later truncation of tau protein at Glu391 was seldom associated with ubiquitin in the NFTs, we believe that different proteolytic mechanisms other than apoptosis and proteosomal degradation could be involved in the generation of this cleavage site.
MATERIALS AND METHODS
Brain tissue
Brain tissue sampling
All experiments are in accordance with the Declaration of Helsinki. The research was formally approved by the local ethics commission of the Prague Psychiatric Center, Prague, Czech Republic, and is in agreement with Laws 129‐2003 and 130‐2003. In total, 26 human brains recovered by autopsy of demented people (all patients of the Psychiatric Hospital Bohnice and were in advanced stages of dementia) were obtained from Patol s.r.o. laboratory (Prague, Czech Republic). The post‐mortem intervals were less than 24 h. All brain regions analyzed were obtained from the left hemisphere. After dissection, the samples were fixed in 10% paraformaldehyde until assayed.
Basic histological analysis
Five brain areas (from the neocortex: gyrus frontalis medius, gyrus temporalis superior et medius, and lobulus parietalis inferior; from the hippocampus: gyrus parahippocampalis; and from the cerebellum: lobulus semilunaris inferior) were histologically evaluated using a silver‐staining technique. The samples were divided into two groups; AD [nine patients with clinically diagnosed dementia, number of senile plaques in given areas of the cortex and hippocampus greater than would be expected for their age, with the criteria used consistent with the work of Mirra et al (56)] and mixed dementia cases (17 patients with the AD changes but also developing vascular alterations, eg, numbered minute focal impairments or lacunae, attenuated brain weight 50–100 × g). Forty‐five‐micrometer thick sucrose‐protected sections from the hippocampus were obtained by cryotomy, postfixed in 4% paraformaldehyde dissolved in phosphate‐buffered saline (PBS), and kept at 4°C. Additional post‐mortem, fresh brain samples from the temporal cortex of two AD patients were supplied by the Brain Bank of the Center of Research and Advanced Studies of the National Polytechnic Institute (CINVESTAV‐IPN), Mexico City. These procedures were also in accordance with the Declaration of Helsinki and formally approved by the local ethics commission of the CINVESTAV‐IPN, Mexico City.
Immunohistochemistry
Hippocampal brain sections were processed for horseradish peroxidase (HRP) immunohistochemistry, as described previously (9). In the present study, brain sections were incubated by free floating in a solution of 0.01% Triton (Sigma Chemical Co., St. Louis, MO, USA) in tris buffered saline (TBS‐t) containing a rabbit polyclonal antibody to ubiquitin (Millipore Corp., Temecula, CA, USA) at a dilution of 1:500. Sections were incubated overnight at 4°C and washed the next day with TBS‐t. Then a HRP‐conjugated secondary antibody to rabbit immunoglobulins (Ig) (DakoCytomation, Glostrup, Denmark) was incubated in TBS‐t for 2 h at room temperature. After developing the HRP enzymatic reaction, the sections were dehydrated and preserved in a mixture of distyrene, tricresyl phosphate and xylene (DPX) (BDH Laboratory Supplies, Poole, UK).
Stained samples were visualized with both 20×[numerial aperture (NA): 0.5] and 40× (NA: 0.75) Plan‐Fluor Lens in a Nikon Eclipse‐80i Microscope (Nikon Corp., Tokyo, Japan). Images were obtained and recorded by using a Nikon digital sight‐DG‐Ri1 camera controlled with the Nikon NIS‐Elements AR‐3.0‐ SP7 software included in the system (Nikon).
Double‐ and triple‐labeling immunofluorescence
Sections of the hippocampus of demented individuals were processed for double and triple immunolabeling as we reported previously 9, 30. In separated incubations, the rabbit polyclonal antibody to ubiquitin (Millipore) was combined with one of different mouse monoclonal antibodies directed to specific epitopes in the molecule of tau (Table 1). From frozen stocks prepared at 1 µg/µL, the tau antibodies were diluted as follows: PHF1 (1:5000), Tau‐C3 (1:2000), MN423 (1:500) and Tau‐46.1 (1:8000). Primary antibodies were incubated at 4°C overnight, and the next day, sections were incubated with the corresponding secondary antibodies: fluorescein‐isothiocyanate (FITC)‐tagged anti‐rabbit (Jackson Immuno‐Research Laboratories, Inc. West Grove, PA, USA) and tetramethylrhodamine‐isothiocyanate (TRITC)‐tagged anti‐mouse “γ”‐specific chain (Jackson Immuno‐Research Laboratories). Samples were washed in TBS‐t and then incubated in 2% Sudan black‐B in 70% ethanol (ICN Biomedicals Inc., Aurora, OH, USA) (36).
Table 1.
Characteristics of the antibodies used in this study.
| Antibody | Epitope | Host/class | Source of reference |
|---|---|---|---|
| Anti‐ubiquitin | Ubiquitin‐protein conjugates | Rabbit polyclonal/IgG | Millipore Corp. |
| PHF‐1 | Phospho‐tau (Ser396,404) | Mouse/IgG | Otvos et al 1994 (64) |
| Tau‐C3 | Truncated tau at Asp421 | Mouse/IgG | Gamblin et al (28) |
| Tau‐46.1 | C‐terminus of tau (428–441) | Mouse/IgG | Kosik et al, 1988 (42); Carmel et al (14) |
| MN423 | Truncated tau at Glu391 | Mouse/IgG | Novak et al, 1991 (62) |
| Alz50 | Tau conformational change (5–15, 312–322) | Mouse/IgM | Carmel et al (14) |
In some experiments sections were incubated with the primary rabbit polyclonal antibody to ubiquitin and counterstained with either 0.001% thiazin red (TR) (Sigma) or 0.1% thioflavin‐S (ThS) (ICN Biomedicals) to visualize fibrillary aggregates showing a β‐pleated sheet structure (55).
For triple labeling, Alz‐50 antibody was also included in some experiments (1:5000) in combination with the polyclonal antiubiquitin and monoclonal antibodies to truncated tau (Tau‐C3 or MN423) (Table 1).
In this case, the secondary antibodies were selected as follows: FITC‐tagged anti‐rabbit IgG, Cy5‐tagged anti‐mouse “γ”‐specific and TRITC‐tagged anti‐mouse “µ”‐specific (Jackson Immuno‐Research Laboratories). At the end of all the incubations, the brain sections were mounted in the antiquenching media Vecta‐Shield (Vector Laboratories, Inc., Burlingame, CA, USA).
Confocal microscopy
The immunolabeled sections from the hippocampus were viewed and analyzed in a TCP‐SP2 confocal laser scanning microscope (Leica Microsytems, Heidelberg, Germany). Samples were observed through a 63× Plan‐apochromat oil immersion lens (NA: 1.32) to evaluate colocalization in single optical sections. The double‐ and triple‐labeled images were obtained by scanning from the top of the sample to 15‐µm depth through the z‐axis. Horizontal z‐sections were collected and analyzed individually for colocalization patterns and projected as superimposed stacks of two‐dimensional images. The resulting images were obtained and analyzed by using the Leica Confocal Software‐Lite V. 2.61 included in the confocal system.
Quantitative analysis
By following our previous criteria to quantify single‐labeled and double‐labeled immunoattached NFTs under epifluorescence 33, 58, we evaluated the number of labeled NFTs in a sampling area covering 1.46 mm2 in the hippocampus. The number of labeled NFTs was expressed as the average of at least eight randomly selected sampling‐areas.
Fluorescence images were captured by using a digital camera AxioCam MRc5 coupled to a Zeiss Axio Imager.z1 upright microscope (Carl Zeiss Imaging Solution, GmgH, Jena, Thuringen, Germany). A 20× Plan‐Neofluar lens (NA: 0.5) was used to record representative fields and a 40× Plan‐Neofluar lens (NA: 0.75) was used for more critical observations. Thus, single‐labeled fluorescent images were sequentially collected by using the AxioVision 40 V.4.5 software included with the system (Carl Zeiss Imaging Solution) and the colocalization patterns were later obtained and analyzed by using the merging program STG Picture Merge V.1.0 (downloaded from http://www.stgsys.com).
For statistical analysis, a Student's t‐test was used to compare two groups, whereas for multiple‐group comparisons a one‐way analysis of variance (ANOVA) was used, followed by Tukey's multiple comparison test. The statistical analysis was done with GraphPad Prism statistics software, version 3.0 (GraphPad software, Inc., San Diego, CA, USA).
Purification of insoluble tau from the brains of AD patients
Insoluble tau protein (A68) fraction was isolated from the brains of AD patients in accordance to previous protocols 11, 45, 82 and subjected to dot‐blot analysis with the polyclonal antibody to ubiquitin and the monoclonal antibodies to tau, Tau‐46.1 (Table 1) and Tau‐5, an IgG monoclonal antibody that recognizes amino acids 210–230 in all tau variants (14).
Samples of the A68 fraction were processed for dot‐blot analysis by using a Bio‐Dot SF microfiltration apparatus (Bio‐Rad Laboratories Inc., Hercules, CA, USA). The samples were absorbed on nitrocellulose membranes after vacuum pumping and then blocked in a solution of 5% nonfat dry milk in TBS‐1% Tween (Sigma) for 1 h at room temperature and gentle shaking. After washing in TBS‐1% Tween, membranes were incubated with either the rabbit polyclonal antibody to ubiquitin (1:1000), Tau‐46.1(1:1500) or Tau‐5 (1:2000) antibodies to tau protein for 1 h at room temperature.
The corresponding HRP‐tagged secondary antibodies to rabbit and mouse IgG (DakoCytomation) were incubated in TBS‐1% Tween for 1 h. After washing with TBS‐1% Tween, membranes were incubated with ECL‐Plus Western Lightning detection reagent 1 (Perkin Elmer Inc., San Jose, CA, USA) for 30 s. The membranes were then exposed to an X‐ray film (Kodak Medical X‐ray, general‐purpose‐blue, Eastman Kodak Company, Rochester, NY, USA) for 10 s in the dark room.
RESULTS
Ubiquitin is associated with the neurofibrillary pathology in the brain of demented patients
The brains of a group of demented patients, composed of AD and cases with mixed dementia, were analyzed to determine the presence of ubiquitin in the neurofibrillary pathology. As previously described (39) in the hippocampus of AD subjects, ubiquitin is usually associated with NFTs (Figure 1). These structures, immunoreactive to a polyclonal antibody to ubiquitin, displayed both the intracellular and extracellular profiles, with the former having the most common presentation of ubiquitin‐targeted NFTs (arrows in Figure 1A). The fibrillary feature of these structures was confirmed by using immunofluorescence in combination with either TR or ThS 48, 50. Some amounts of NFTs were stained with TR of which many examples were also immunoreactive to the ubiquitin antibody (arrows in Figure 1B). However, many examples of NFTs only stained by TR were also seen (asterisk in Figure 1B).
Figure 1.
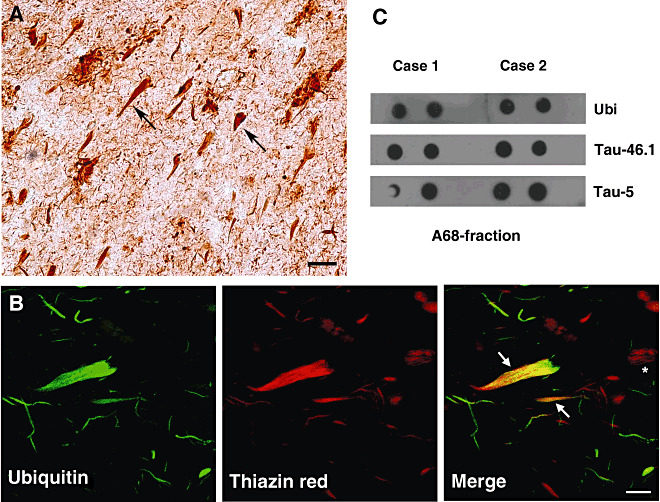
Ubiquitin is associated with tau protein in neurofibrillary tangles (NFTs). Ubiquitin‐positive NFTs (arrows) and clusters of neuropil threads are observed by immunoperoxidase in the hippocampus of Alzheimer's disease (AD) patients (A). Confocal microscopy images of ubiquitin immunoreactivity and thiazin red (TR) staining (B). Note in the merge channel, the colocalization displayed in some NFTs (arrows). However ubiquitin (Ubi) is not present in all the NFTs detected by TR (asterisk). Dot‐blot analysis of the A68 fraction, purified from the brain of two AD patients, also confirms that ubiquitin is a constituent of fibrillary aggregates of tau protein (C). Positive spots correspond to the use of a polyclonal antibody to ubiquitin, and two monoclonal antibodies to tau protein (Tau‐46.1 and Tau‐5). Scale bars 35 µm in (A), and 6 µm in panel (B).
To confirm that ubiquitin is in close association with tau fibrillary structures, we used the A68 fraction (composed of aggregated tau protein) purified from the brains of some AD patients 11, 43. By using dot‐blot analysis with the same polyclonal antibody to ubiquitin, it was confirmed that ubiquitin is a constituent of the A68 fraction (Figure 1C). Some samples were also processed for tau antibodies, such as Tau‐46.1 and Tau‐5, to confirm the presence of tau in this insoluble fraction (Figure 1C).
Quantitative analysis of the load of NFTs composed of ubiquitin
To obtain clear evidence of the contribution of ubiquitin in the development of NFTs in the hippocampus, we quantified the number of ubiquitin‐targeted NFTs and compared with the total load of NFTs. In the whole population of demented individuals, the number of NFTs (mean ± standard error) detected with ThS (14.7 ± 2.8) was significantly greater than the number of ubiquitin‐targeted NFTs (4.8 ± 0.9) (P = 0.0017). No significant differences were found for both the total load of NFTs (ThS positive) and the number of ubiquitin‐targeted NFTs when they were compared between AD and mixed dementia cases.
In the whole group of cases, the proportion of NFTs detected with ThS that were composed of ubiquitin (colocalization between both markers) was approximately 30% (28.1 ± 2.9). This proportion was not significantly modified when the samples were subdivided into AD (33.2 ± 5.2) and mixed dementia (26.9 ± 3.3) subgroups (P = 0.5632, by one‐way ANOVA).
Ubiquitin is associated with Asp421‐truncated tau protein in NFTs
To evaluate the possibility that ubiquitin‐targeting may contribute to the development of NFTs, we analyzed the association of ubiquitin with tau protein undergoing several post‐translational modifications, such as phosphorylation at Ser396,404 and two major truncations at the C‐terminus of tau protein occurring at Asp421 and Glu391 (57).
As shown in Figure 2, there is a close association between ubiquitin and tau protein in the NFTs. Colocalization was found between ubiquitin and Asp421‐truncated tau (Figure 2A), Ser396,404‐phosphorylated tau (Figure 2B), and the C‐terminus intact tau protein (Figure 2C). However, remarkably almost no colocalization was found between ubiquitin and Glu391‐truncated tau in the NFTs (Figure 2D). Quantitative analysis of distinctive tau‐NFTs colocalizing with ubiquitin is shown in Figure 3. When the populations of distinct tau NFTs were compared with each other, the only significant difference was found between the number of PHF1 and Tau‐46.1‐positive NFTs (P < 0.05, in Figure 3A). When colocalization was analyzed for all the combinations between tau and ubiquitin in the NFTs, the most common association was found between ubiquitin and phosphorylated tau at Ser396,404 (Figure 3B). Ubiquitin and Asp421‐truncated tau were also associated in a population of NFTs showing a significantly higher rate of colocalization than that observed for NFTs composed of ubiquitin and C‐terminus intact tau protein (Figure 3B). By contrast, significantly less number of NFTs displayed colocalization between ubiquitin and Glu391‐truncated tau (Figure 3B).
Figure 2.
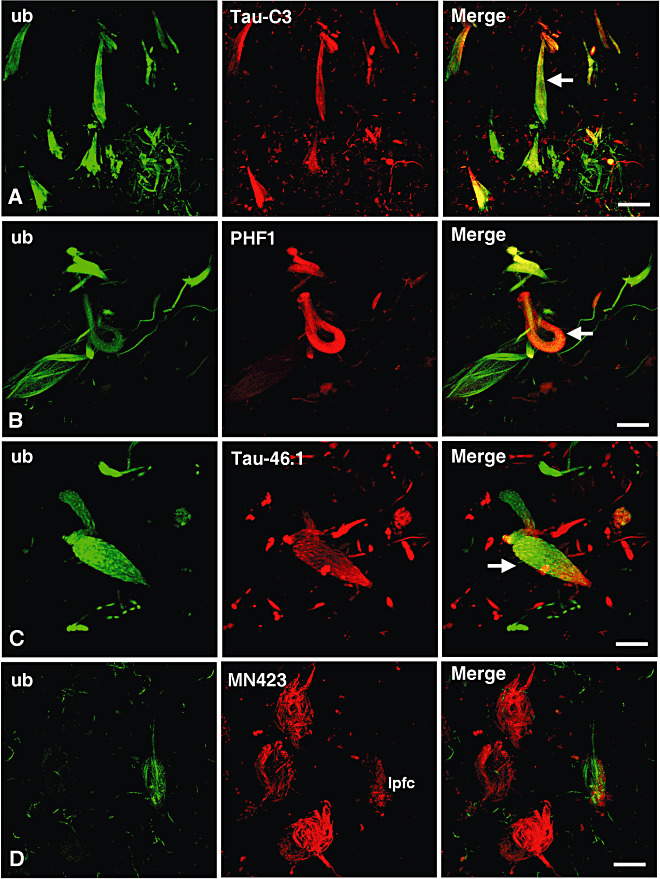
Colocalization between ubiquitin and tau protein in the neurofibrillary tangles (NFTs) of demented patients. Double‐labeling immunofluorescence and confocal microscopy illustrate the patterns of colocalization between ubiquitin (ub: polyclonal antibody to ubiquitin) and different presentations of tau protein (red channel) in the NFTs. Those structures composed of Asp421‐truncated tau (Tau‐C3 antibody), Ser396,404‐phosphorylated tau [paired helical filaments (PHF)1 antibody] and C‐terminus intact tau protein (Tau‐46.1 antibody) were constantly attached by ubiquitin [see arrows in the merge channels of (A–C)]. However, almost no colocalization was found between ubiquitin and the Glu391‐truncated tau protein (MN423 antibody) in most of the NFTs analyzed (D). Note in (D) that the NFTs recognized by either ubiquitin or MN423 have a different morphology. Moreover ubiquitin was commonly detected in neuropil threads and dystrophic neurites in addition to the NFTs. In (D), lpfc: lipofucsin. Scale bars 27 µm in (A), 10 µm in (B), 6 µm in (C) and 14 µm in (D).
Figure 3.
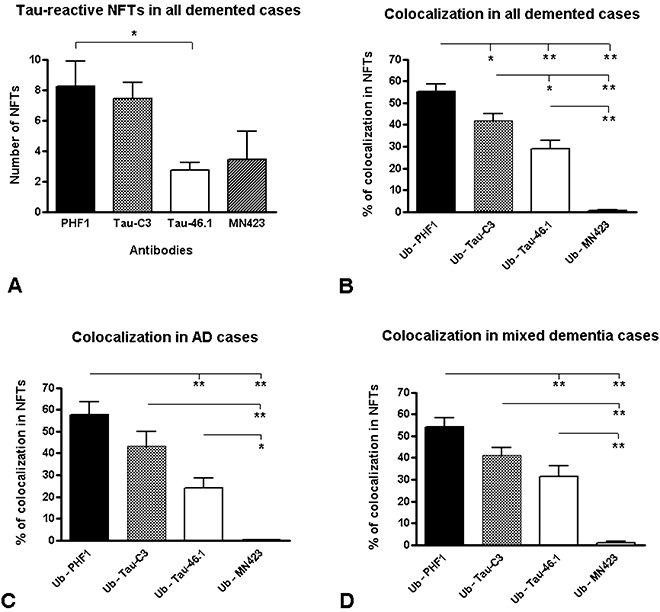
Quantitative colocalization of ubiquitin and tau proteins in neurofibrillary tangles (NFTs). A. NFTs immunoreactive to distinct antibodies to tau protein. A significant difference was found by using a one‐way analysis of variance (ANOVA) (P = 0.009) followed by Tukey's multiple comparison test to determine specific differences between all the tau antibodies. Only the number of NFTs immunoreactive to paired helical filaments (PHF)1 was significantly greater than that of the NFTs recognized by the Tau‐46.1 antibody. B. In the whole group of demented patients, colocalization was expressed as the percentage of NFTs composed of ubiquitin (Ub: polyclonal antibody to ubiquitin) and tau protein (recognized independently by Tau‐C3, MN423, PHF1 and Tau‐46.1). Colocalization was found in most of the combinations; however, almost undetected numbers of NFTs were composed of ubiquitin and tau truncated at the Glu391 (Ub ‐MN423), which was also consistent when the group was separated as AD (C) and mixed dementia patients (D). The same statistical analysis as in (A) was done in (B–D) (see the text for details). *P < 0.05; **P < 0.001.
The whole group of demented patients was subdivided into AD and mixed dementia cases, and the samples then analyzed independently for ubiquitin‐tau colocalization as above (Figure 3C,D). The same pattern of colocalization was measured for both groups, with the ubiquitin‐Ser396,404‐phosphorylated tau (Ub–PHF1) and ubiquitin–Asp421‐truncated tau (Ub–Tau‐C3) having the higher rate of colocalization in the NFTs (Figure 3C,D). In both groups of cases, again colocalization between ubiquitin and Glu391‐truncated tau in NFTs (Ub–MN423) was significantly reduced (Figure 3C,D). All these results indicate that ubiquitin targeting of tau in the NFTs is more associated with both phosphoryation and the early truncation of the C‐terminus (Asp421).
Because truncation of the C‐terminus of tau protein has been considered a key mechanism that predicts the state of maturation of the NFTs, and because in this study ubiquitin targeting seems to be more associated with the minimum truncation of tau protein, we sought more detail of the proteolytic state of the C‐terminus of tau in ubiquitin‐attached NFTs.
For this purpose, we performed triple‐labeling experiments by including the Alz‐50 antibody, which has been demonstrated to recognize early and intermediate NFTs (30). As shown in Figure 4A,B, triple colocalization was found in Alz‐50‐positive NFTs also composed of ubiquitin and Asp421‐truncated tau (positive to Tau‐C3). In contrast, in Alz‐50‐negative NFTs (more advanced stage of maturation), which are commonly composed of truncated tau at the Glu391 (recognized with the MN423 antibody), no attachment of ubiquitin was found (Figure 4C,D). These results indicate that ubiquitin‐targeting of tau protein could be more associated with the early stages of tau degradation (Asp421) than the proteolytic processes involved in the generation of the Glu391‐cleavage at the C‐terminus of tau.
Figure 4.
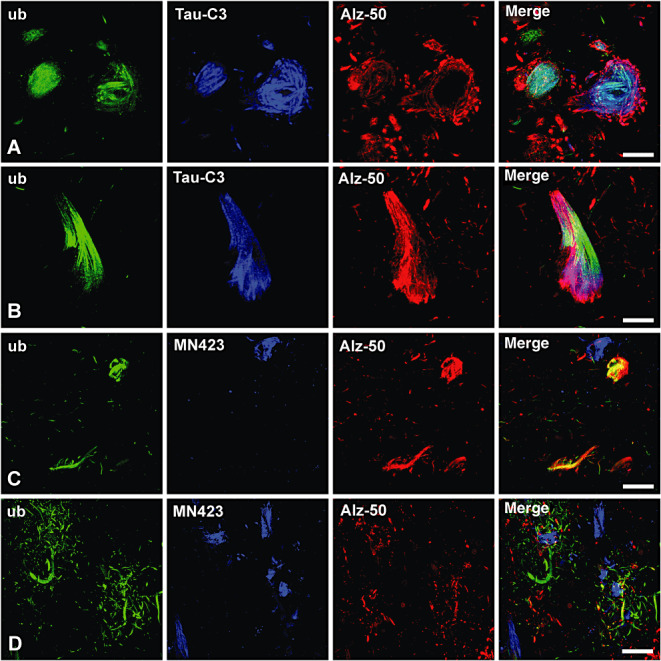
In neurofibrillary tangles (NFTs), ubiquitin colocalizes with Asp421‐truncated tau but not with tau cleaved at the Glu391. Triple labeling by including the Alz‐50 antibody confirms that early NFTs detected by the Tau‐C3 antibody (for Asp421‐truncated tau) are also composed of ubiquitin (ub) (A) and (B). In contrast, mature NFTs immunoreactive to MN423 (to tau cleaved at the Glu391) were little composed of ubiquitin (C–D). Note in (B) the coexistence for the three markers in the same NFT, whereas in (C) only the double‐labeled NFTs are composed of ubiquitin and stained with Alz‐50 antibody. In panels (C–D), the NFTs recognized by MN423 remain unassociated with other markers and have an unmerged blue color (see the merge channel). These MN423‐positive NFTs sometimes were surrounded by a crown of neuropil treads composed of ubiquitin [see merge channel in (D)]. Scale bars 9 µm in (A), 10 µm in (B), 22 µm in (C) and 24 µm in (D).
DISCUSSION
Ubiquitin‐targeting of tau protein in NFTs
In this study we investigated the possible association of ubiquitin with particular tau modifications that herald specific stages of NFTs maturation within the brains of AD patients. Using immunological probes that recognize different post‐translational modifications of tau has allowed us to determine the timeline of NFTs formation and its association with ubiquitin.
In previous reports, we proposed a chronological model of the pathological processing of tau during the evolution of AD using antibodies that recognize specific modifications within the tau molecule 9, 30, 31, 33. The sequence of events that promotes the assembly of soluble tau into PHFs and NFTs has been characterized by sequential phosphorylation events at different amino acids with the consequential development of conformational changes 25, 49, 57. Conformational changes within a protein may be subjected to degradation by different proteolytic systems, including proteosomal, lysosomal and apoptotic components in an attempt to eliminate these abnormal or toxic molecules from the cytoplasm 35, 54, 71, 79. We proposed in our model that truncation of tau at Asp421 occurs after the conformational changes of the tau molecule induced by abnormal phosphorylation events (57). As mentioned above, truncation of tau at Asp421 appears to be an early event during the disease process and attributed to caspase activity. However the protease responsible for sequential cleavage of tau at Glu391 has yet to be elucidated. The contribution of the UPS in neurodegenerative diseases, including AD, has been extensively studied 15, 63, 75, 77. For instance, one of the earliest evidence implicating a link between the UPS and tau pathology arose when ubiquitin was localized in PHFs 59, 60, 66 and NFTs 6, 65, 74 within the brains of AD patients. More recently, ubiquitin was found to be associated with a fraction of purified PHF‐tau isolated from the brains of AD patients by immunoaffinity with the MC1 antibody (19). In this study, by using tandem mass spectrometry, Cripps and colleagues elegantly showed that PHF‐tau is a target for ubiquitin and that it selectively binds to residues Lys254, Lys311 and Lys353 located within the microtubule binding domain (19).
Within this body of work, we corroborate the presence of ubiquitin within the NFTs (Figure 1) and found that the density of ubiquitin‐attached NFTs is significantly less than the total load of NFTs. Only 30% of these structures contained ubiquitin as a component. However, controversy has risen regarding the timing of ubiquitin targeting during NFTs maturation. For instance, some evidence indicates that late NFTs are highly targeted with ubiquitin 6, 20, whereas other groups reported that ubiquitin is not associated with extracellular NFTs (59).
All together, these results indicate that ubiquitin is a conspicuous component of the neurofibrillary pathology in AD, and this, therefore, suggests a more significant role for ubiquitin during the maturation of the NFTs in AD.
Phosphorylation, truncation and ubiquitin‐targeting of tau protein during the maturation of the NFTs
Phosphorylation of tau is thought to be an early event that, besides promoting a misfolding state of the molecule, serves as recognition signal to be targeted with ubiquitin and degraded by the UPS through the action of the carboxy terminus of the hsc70‐interacting protein (CHIP hsc70) complex 68, 72. Because some studies reported that tau is not a proteasome substrate 22, 27, the central role of tau degradation by proteosomal activity is still under debate. It is not clear if partial proteolysis of tau protein eliciting some domains may accelerate its self‐aggregation and cause more toxicity. If this is the case, ubiquitin targeting of tau and proteasome degradation may contribute to the formation of tau aggregates in the brain of AD cases. In this regard, it has been proposed that polyubiquitin‐targeting of tau by CHIP may facilitate the formation of insoluble tau filaments. This event coincidentally, was preceded by phosphorylation and caspase‐3‐cleavage of tau (23).
The regulatory activity of tau processing by several of the UPS complexes has been investigated in cell and animal models 4, 23, but information about the final contribution of this system in AD patients is based on the analysis of lesions composed of tau and other associated proteins.
Our results obtained from AD brain tissue corroborate that ubiquitin is closely associated with phosphorylated tau in the early neurofibrillary pathology (2, 3), but more intriguing is that in a considerable number of the NFTs, ubiquitin was also associated with truncated tau at the Asp421 (2, 3). This result may provide evidence to suggest that a single substrate can be subjected to dual processing involving apoptotic and proteosomal degradation. In a recent report, sequential proteosomal, apoptotic and lysosomal processing of tau was produced in prostaglandin‐J2‐treated neuroblastoma SK‐N‐SH cells 12, 22, 27. Because of the specific truncation of tau by caspase at Asp421 (4), it is difficult to predict that this cleavage can also be generated by proteosomal activity. As shown in Figure 3, the number of NFTs composed of C‐terminus intact‐tau protein and ubiquitin is reduced, which suggests that ubiquitin targeting of tau occurs in a molecule that is truncated early as was occurring at the Asp421 (Figure 3B–D). These observations also reinforce the concept of two synchronized proteolytic activities within tau protein, namely apoptosis and proteosome activity. These results are also in agreement with those studies that proposed that polyubiquitin targeting follows phosphorylation and truncation (28).
By timing the event of ubiquitin targeting of tau protein according to the patterns of colocalization within this study, we further demonstrated that this event is more related to the early and intermediate stages of the maturation of the NFTs. As shown in Figure 4A,B, close association at a different degree was found between ubiquitin, truncation of tau at Asp421 and N‐terminus folding of tau recognized by Alz‐50 antibody (23). This is the first finding in situ that demonstrates that ubiquitin targeting of tau protein is associated with a specific truncation site (Asp421) at the C‐terminus, but also preserving intact the N‐terminus in a folded conformation. All these events taking place at the early and intermediate stages of the maturation of the NFTs depicts a temporal course of incidence at any single NFT, which is also in agreement with the concept of chimeric NFTs having different degrees of colocalization 14, 30.
In previous studies, we also identified an advanced stage of tau processing during the maturation of the NFTs, namely truncation of tau at Glu391 (9). In these NFTs, distinct tau molecules were also associated with Glu391‐truncated tau; however, they were no longer than the Ser396 residue 9, 30, 33. The origin of this truncation is still debated and a putative intracellular enzyme responsible for the cleavage at Glu391 is still unknown (30). Hence a potential role for proteosomal degradation emerges as a possible explanation. Nevertheless, in the present study we found a scant colocalization between ubiquitin and the Glu391‐truncated tau (Figure 3B–D) with almost total segregation of these proteins in two different populations of the NFTs. These structures composed mostly of Glu391‐truncated tau were also negative to Alz‐50 and ubiquitin, confirming that all these events are mutually exclusive when they occur within NFTs in AD (Figure 4C,D).
The lack of this association may indicate that truncation of this protein at Glu391 is not related to ubiquitin targeting and therefore, to the consecutive degradation through proteosomal components.
One explanation may be that previous to the Glu391‐truncation, ubiquitin is removed from the C‐ terminus of tau protein, though if this were true, the transitional stages of partial colocalization would be identified in the NFTs. In addition, it was reported that several ubiquitin‐targeting sites of tau protein lay over the flanking region of the repeated domains 9, 29, which are known to be conserved in the minimum PHF‐core composed of Glu391‐truncated tau (72).
At present, the generation of the Glu391‐cleavage of tau is not clear, and more cytoplasmic enzymes need to be analyzed. The lysosomal system emerges as another possibility, and previous reports indicate that tau can be fragmented by this complex 61, 80, 81. However, whether this proteolysis eliminates pathologic phosphorylated tau or that this fragmentation produces more toxic entities with aggregation properties is still unknown and needs to be further investigated. In this study we showed evidence that ubiquitin targeting of tau occurs during the early and mostly at intermediate stages of the maturation of the NFTs (Figure 5), following the sequence: phosphorylation →Asp421‐truncation →, = ubiquitin targeting. At this stage, proteosomal degradation of tau may eliminate several domains of the molecule, mainly at the extreme N‐ and C‐ termini.
Figure 5.
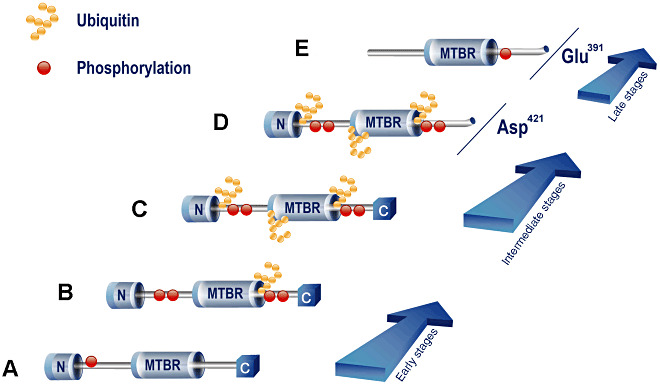
Ubiquitin targeting is associated with Asp421‐truncation but not with Glu391‐cleavage of tau during the maturation of the NFTs in AD. At early stages of the maturation of the NFTs, the major modification in tau is the abnormal phosphorylation of several residues (A–B). Truncation of tau at Asp421 may also occur at early stages, but it is more predominant at intermediate stages (C–D). In late‐stage NFTs (E), Glu391‐truncated tau remains as the major component. Ubiquitin targeting of tau seems to occur at early and mostly at intermediate stages of the maturation of the NFTs (B–D), in close association with abnormal phosphorylation and Asp421‐truncation (B–D). Scant association is found between ubiquitin and Glu391‐truncated tau (E).
The advanced stages of the maturation of the NFTs are still characterized by the occurrence of truncation of tau at Glu391, but not associated with ubiquitin‐targeting (Asp421‐truncation →, = ubiquitin targeting → . . . ? . . . →Glu391‐truncation). Thus, we can conclude that ubiquitin targeting and proteosomal activity may contribute to the proteolytic processing of tau protein during the formation and maturation of NFTs in AD.
ACKNOWLEDGMENTS
The authors thank Dr. Virginia Lee for the use of Tau‐46.1 and PHF1 antibodies, Dr. Peter Davis for the use of Alz‐50 and PHF1 antibodies, Michael Novak for the use of MN423 antibody. We thank Maria de Jesús Garcia‐Sierra for artwork design. We also want to thank to Dr. Ellis Glazier who edited this English‐language text. This study was financially supported by CONACyT‐ México to FG‐S grants 59651, 113772, and sabbatical fellowship 93628. JJJ‐B was supported by an international scholarship from CONACyT‐Mexico. Supports from the Ministry of Health of the Czech Republic for ZK and DR, project MZ0PCP2005, are also acknowledged.
REFERENCES
- 1. Alonso AC, Grundke‐Iqbal I, Iqbal K (1996) Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med 2:783–787. [DOI] [PubMed] [Google Scholar]
- 2. Alonso AD, Grundke‐Iqbal I, Barra HS, Iqbal K (1997) Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule‐associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci U S A 94:298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alonso AD, Di Clerico J, Li B, Corbo CP, Alaniz ME, Grundke‐Iqbal I, Iqbal K (2010) Phosphorylation of tau at Thr212, Thr231, and Ser262 combined causes neurodegeneration. J Biol Chem 285:30851–30860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arnaud LT, Myeku N, Figueiredo‐Pereira ME (2009) Proteasome‐caspase‐cathepsin sequence leading to tau pathology induced by prostaglandin J2 in neuronal cells. J Neurochem 110:328–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ball MJ (1977) Neuronal loss, neurofibrillary tangles and granulovacuolar degeneration in the hippocampus with ageing and dementia. A quantitative study. Acta Neuropathol 37:111–118. [DOI] [PubMed] [Google Scholar]
- 6. Bancher C, Brunner C, Lassmann H, Budka H, Jellinger K, Seitelberger F et al (1989) Tau and ubiquitin immunoreactivity at different stages of formation of Alzheimer neurofibrillary tangles. Prog Clin Biol Res 317:837–848. [PubMed] [Google Scholar]
- 7. Bancher C, Brunner C, Lassmann H, Budka H, Jellinger K, Wiche G et al (1989) Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer's disease. Brain Res 477:90–99. [DOI] [PubMed] [Google Scholar]
- 8. Barghorn S, Davies P, Mandelkow E (2004) Tau paired helical filaments from Alzheimer's disease brain and assembled in vitro are based on beta‐structure in the core domain. Biochemistry 43:1694–1703. [DOI] [PubMed] [Google Scholar]
- 9. Basurto‐Islas G, Luna‐Munoz J, Guillozet‐Bongaarts AL, Binder LI, Mena R, Garcia‐Sierra F (2008) Accumulation of aspartic acid421‐ and glutamic acid391‐cleaved tau in neurofibrillary tangles correlates with progression in Alzheimer disease. J Neuropathol Exp Neurol 67:470–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Braak H, Del Tredici K (2004) Alzheimer's disease: intraneuronal alterations precede insoluble amyloid‐beta formation. Neurobiol Aging 25:713–718. [DOI] [PubMed] [Google Scholar]
- 11. Brion JP, Hanger DP, Couck AM, Anderton BH (1991) A68 proteins in Alzheimer's disease are composed of several tau isoforms in a phosphorylated state which affects their electrophoretic mobilities. Biochem J 279(Pt 3):831–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brown MR, Bondada V, Keller JN, Thorpe J, Geddes JW (2005) Proteasome or calpain inhibition does not alter cellular tau levels in neuroblastoma cells or primary neurons. J Alzheimers Dis 7:15–24. [DOI] [PubMed] [Google Scholar]
- 13. Buee L, Troquier L, Burnouf S, Belarbi K, Van der Jeugd A, Ahmed T et al (2010) From tau phosphorylation to tau aggregation: what about neuronal death? Biochem Soc Trans 38:967–972. [DOI] [PubMed] [Google Scholar]
- 14. Carmel G, Mager EM, Binder LI, Kuret J (1996) The structural basis of monoclonal antibody Alz50's selectivity for Alzheimer's disease pathology. J Biol Chem 271:32789–32795. [DOI] [PubMed] [Google Scholar]
- 15. Chu CT, Caruso JL, Cummings TJ, Ervin J, Rosenberg C, Hulette CM (2000) Ubiquitin immunochemistry as a diagnostic aid for community pathologists evaluating patients who have dementia. Mod Pathol 13:420–426. [DOI] [PubMed] [Google Scholar]
- 16. Chung CW, Song YH, Kim IK, Yoon WJ, Ryu BR, Jo DG et al (2001) Proapoptotic effects of tau cleavage product generated by caspase‐3. Neurobiol Dis 8:162–172. [DOI] [PubMed] [Google Scholar]
- 17. Cleveland DW, Hwo SY, Kirschner MW (1977) Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Biol 116:227–247. [DOI] [PubMed] [Google Scholar]
- 18. Cras P, Kawai M, Lowery D, Gonzalez‐DeWhitt P, Greenberg B, Perry G (1991) Senile plaque neurites in Alzheimer disease accumulate amyloid precursor protein. Proc Natl Acad Sci U S A 88:7552–7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cripps D, Thomas SN, Jeng Y, Yang F, Davies P, Yang AJ (2006) Alzheimer disease‐specific conformation of hyperphosphorylated paired helical filament‐Tau is polyubiquitinated through Lys‐48, Lys‐11, and Lys‐6 ubiquitin conjugation. J Biol Chem 281:10825–10838. [DOI] [PubMed] [Google Scholar]
- 20. Dammer EB, Na CH, Xu P, Seyfried NT, Duong DM, Cheng D et al (2011) Polyubiquitin linkage profiles in three models of proteolytic stress suggest the etiology of Alzheimer disease. J Biol Chem 286:10457–10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de Vrij FM, Fischer DF, van Leeuwen FW, Hol EM (2004) Protein quality control in Alzheimer's disease by the ubiquitin proteasome system. Prog Neurobiol 74:249–270. [DOI] [PubMed] [Google Scholar]
- 22. Delobel P, Leroy O, Hamdane M, Sambo AV, Delacourte A, Buee L (2005) Proteasome inhibition and Tau proteolysis: an unexpected regulation. FEBS Lett 579:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dickey CA, Yue M, Lin WL, Dickson DW, Dunmore JH, Lee WC et al (2006) Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho‐ and caspase‐3‐cleaved tau species. J Neurosci 26:6985–6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ding H, Matthews TA, Johnson GV (2006) Site‐specific phosphorylation and caspase cleavage differentially impact tau‐microtubule interactions and tau aggregation. J Biol Chem 281:19107–19114. [DOI] [PubMed] [Google Scholar]
- 25. Du JT, Yu CH, Zhou LX, Wu WH, Lei P, Li Y et al (2007) Phosphorylation modulates the local conformation and self‐aggregation ability of a peptide from the fourth tau microtubule‐binding repeat. FEBS J 274:5012–5020. [DOI] [PubMed] [Google Scholar]
- 26. Fasulo L, Ugolini G, Visintin M, Bradbury A, Brancolini C, Verzillo V et al (2000) The neuronal microtubule‐associated protein tau is a substrate for caspase‐3 and an effector of apoptosis. J Neurochem 75:624–633. [DOI] [PubMed] [Google Scholar]
- 27. Feuillette S, Blard O, Lecourtois M, Frebourg T, Campion D, Dumanchin C (2005) Tau is not normally degraded by the proteasome. J Neurosci Res 80:400–405. [DOI] [PubMed] [Google Scholar]
- 28. Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL et al (2003) Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci U S A 100:10032–10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Garcia‐Sierra F, Wischik CM, Harrington CR, Luna‐Munoz J, Mena R (2001) Accumulation of C‐terminally truncated tau protein associated with vulnerability of the perforant pathway in early stages of neurofibrillary pathology in Alzheimer's disease. J Chem Neuroanat 22:65–77. [DOI] [PubMed] [Google Scholar]
- 30. Garcia‐Sierra F, Ghoshal N, Quinn B, Berry RW, Binder LI (2003) Conformational changes and truncation of tau protein during tangle evolution in Alzheimer's disease. J Alzheimers Dis 5:65–77. [DOI] [PubMed] [Google Scholar]
- 31. Garcia‐Sierra F, Mondragon‐Rodriguez S, Basurto‐Islas G (2008) Truncation of tau protein and its pathological significance in Alzheimer's disease. J Alzheimers Dis 14:401–409. [DOI] [PubMed] [Google Scholar]
- 32. Ghoshal N, Garcia‐Sierra F, Fu Y, Beckett LA, Mufson EJ, Kuret J et al (2001) Tau‐66: evidence for a novel tau conformation in Alzheimer's disease. J Neurochem 77:1372–1385. [DOI] [PubMed] [Google Scholar]
- 33. Guillozet‐Bongaarts AL, Garcia‐Sierra F, Reynolds MR, Horowitz PM, Fu Y, Wang T et al (2005) Tau truncation during neurofibrillary tangle evolution in Alzheimer's disease. Neurobiol Aging 26:1015–1022. [DOI] [PubMed] [Google Scholar]
- 34. Gustke N, Steiner B, Mandelkow EM, Biernat J, Meyer HE, Goedert M, Mandelkow E (1992) The Alzheimer‐like phosphorylation of tau protein reduces microtubule binding and involves Ser‐Pro and Thr‐Pro motifs. FEBS Lett 307:199–205. [DOI] [PubMed] [Google Scholar]
- 35. Haas AL, Warms JV, Hershko A, Rose IA (1982) Ubiquitin‐activating enzyme. Mechanism and role in protein‐ubiquitin conjugation. J Biol Chem 257:2543–2548. [PubMed] [Google Scholar]
- 36. Harman D (1989) Lipofuscin and ceroid formation: the cellular recycling system. Adv Exp Med Biol 266:3–15. [DOI] [PubMed] [Google Scholar]
- 37. Hyman BT, Augustinack JC, Ingelsson M (2005) Transcriptional and conformational changes of the tau molecule in Alzheimer's disease. Biochim Biophys Acta 1739:150–157. [DOI] [PubMed] [Google Scholar]
- 38. Iqbal K, Grundke‐Iqbal I (2008) Alzheimer neurofibrillary degeneration: significance, etiopathogenesis, therapeutics and prevention. J Cell Mol Med 12:38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Iwatsubo T, Hasegawa M, Esaki Y, Ihara Y (1992) Lack of ubiquitin immunoreactivities at both ends of neuropil threads. Possible bidirectional growth of neuropil threads. Am J Pathol 140:277–282. [PMC free article] [PubMed] [Google Scholar]
- 40. Kidd M (2006) The history of the paired helical filaments. J Alzheimers Dis 9:71–75. [DOI] [PubMed] [Google Scholar]
- 41. Kosik KS, Joachim CL, Selkoe DJ (1986) Microtubule‐associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A 83:4044–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kosik KS, Orecchio LD, Binder L, Trojanowski JQ, Lee VM, Lee G (1988) Epitopes that span the tau molecule are shared with paired helical filaments. Neuron 1:817–825. [DOI] [PubMed] [Google Scholar]
- 43. Ksiezak‐Reding H, Binder LI, Yen SH (1990) Alzheimer disease proteins (A68) share epitopes with tau but show distinct biochemical properties. J Neurosci Res 25:420–430. [DOI] [PubMed] [Google Scholar]
- 44. Lee G, Cowan N, Kirschner M (1988) The primary structure and heterogeneity of tau protein from mouse brain. Science 239:285–288. [DOI] [PubMed] [Google Scholar]
- 45. Lee VM, Balin BJ, Otvos L, Jr , Trojanowski JQ (1991) A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science 251:675–678. [DOI] [PubMed] [Google Scholar]
- 46. Lehman NL (2009) The ubiquitin proteasome system in neuropathology. Acta Neuropathol 118:329–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lichtenberg‐Kraag B, Mandelkow EM, Biernat J, Steiner B, Schroter C, Gustke N et al (1992) Phosphorylation‐dependent epitopes of neurofilament antibodies on tau protein and relationship with Alzheimer tau. Proc Natl Acad Sci U S A 89:5384–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Luna‐Munoz J, Garcia‐Sierra F, Falcon V, Menendez I, Chavez‐Macias L, Mena R (2005) Regional conformational change involving phosphorylation of tau protein at the Thr231, precedes the structural change detected by Alz‐50 antibody in Alzheimer's disease. J Alzheimers Dis 8:29–41. [DOI] [PubMed] [Google Scholar]
- 49. Luna‐Munoz J, Chavez‐Macias L, Garcia‐Sierra F, Mena R (2007) Earliest stages of tau conformational changes are related to the appearance of a sequence of specific phospho‐dependent tau epitopes in Alzheimer's disease. J Alzheimers Dis 12:365–375. [DOI] [PubMed] [Google Scholar]
- 50. Luna‐Munoz J, Peralta‐Ramirez J, Chavez‐Macias L, Harrington CR, Wischik CM, Mena R (2008) Thiazin red as a neuropathological tool for the rapid diagnosis of Alzheimer's disease in tissue imprints. Acta Neuropathol 116:507–515. [DOI] [PubMed] [Google Scholar]
- 51. Mandelkow E, Song YH, Schweers O, Marx A, Mandelkow EM (1995) On the structure of microtubules, tau, and paired helical filaments. Neurobiol Aging 16:347–354. [DOI] [PubMed] [Google Scholar]
- 52. Mandelkow EM, Biernat J, Drewes G, Gustke N, Trinczek B, Mandelkow E (1995) Tau domains, phosphorylation, and interactions with microtubules. Neurobiol Aging 16:355–362. discussion 362–353. [DOI] [PubMed] [Google Scholar]
- 53. Manetto V, Perry G, Tabaton M, Mulvihill P, Fried VA, Smith HT et al (1988) Ubiquitin is associated with abnormal cytoplasmic filaments characteristic of neurodegenerative diseases. Proc Natl Acad Sci U S A 85:4501–4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Manetto V, Abdul‐Karim FW, Perry G, Tabaton M, Autilio‐Gambetti L, Gambetti P (1989) Selective presence of ubiquitin in intracellular inclusions. Am J Pathol 134:505–513. [PMC free article] [PubMed] [Google Scholar]
- 55. Mena R, Edwards P, Perez‐Olvera O, Wischik CM (1995) Monitoring pathological assembly of tau and beta‐amyloid proteins in Alzheimer's disease. Acta Neuropathol 89:50–56. [DOI] [PubMed] [Google Scholar]
- 56. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM et al (1991) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41:479–486. [DOI] [PubMed] [Google Scholar]
- 57. Mondragon‐Rodriguez S, Basurto‐Islas G, Santa‐Maria I, Mena R, Binder LI, Avila J et al (2008) Cleavage and conformational changes of tau protein follow phosphorylation during Alzheimer's disease. Int J Exp Pathol 89:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mondragon‐Rodriguez S, Mena R, Binder LI, Smith MA, Perry G, Garcia‐Sierra F (2008) Conformational changes and cleavage of tau in Pick bodies parallel the early processing of tau found in Alzheimer pathology. Neuropathol Appl Neurobiol 34:62–75. [DOI] [PubMed] [Google Scholar]
- 59. Mori H, Kondo J, Ihara Y (1987) Ubiquitin is a component of paired helical filaments in Alzheimer's disease. Science 235:1641–1644. [DOI] [PubMed] [Google Scholar]
- 60. Morishima‐Kawashima M, Hasegawa M, Takio K, Suzuki M, Titani K, Ihara Y (1993) Ubiquitin is conjugated with amino‐terminally processed tau in paired helical filaments. Neuron 10:1151–1160. [DOI] [PubMed] [Google Scholar]
- 61. Novak M, Wischik CM, Edwards P, Pannell R, Milstein C (1989) Characterisation of the first monoclonal antibody against the pronase resistant core of the Alzheimer PHF. Prog Clin Biol Res 317:755–761. [PubMed] [Google Scholar]
- 62. Novak M, Jakes R, Edwards PC, Milstein C, Wischik CM (1991) Difference between the tau protein of Alzheimer paired helical filament core and normal tau revealed by epitope analysis of monoclonal antibodies 423 and 7.51. Proc Natl Acad Sci USA 88:5837–5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Oddo S (2008) The ubiquitin‐proteasome system in Alzheimer's disease. J Cell Mol Med 12:363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Otvos L, Jr. , Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM (1994) Monoclonal antibody PHF‐1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res 39:669–673. [DOI] [PubMed] [Google Scholar]
- 65. Perry G, Friedman R, Shaw G, Chau V (1987) Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains. Proc Natl Acad Sci U S A 84:3033–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Perry G, Mulvihill P, Fried VA, Smith HT, Grundke‐Iqbal I, Iqbal K (1989) Immunochemical properties of ubiquitin conjugates in the paired helical filaments of Alzheimer disease. J Neurochem 52:1523–1528. [DOI] [PubMed] [Google Scholar]
- 67. Perry G, Kawai M, Tabaton M, Onorato M, Mulvihill P, Richey P et al (1991) Neuropil threads of Alzheimer's disease show a marked alteration of the normal cytoskeleton. J Neurosci 11:1748–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A et al (2004) CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet 13:703–714. [DOI] [PubMed] [Google Scholar]
- 69. Quintanilla RA, Matthews‐Roberson TA, Dolan PJ, Johnson GV (2009) Caspase‐cleaved tau expression induces mitochondrial dysfunction in immortalized cortical neurons: implications for the pathogenesis of Alzheimer disease. J Biol Chem 284:18754–18766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sadqi M, Hernandez F, Pan U, Perez M, Schaeberle MD, Avila J, Munoz V (2002) Alpha‐helix structure in Alzheimer's disease aggregates of tau‐protein. Biochemistry 41:7150–7155. [DOI] [PubMed] [Google Scholar]
- 71. Schwartz AL, Ciechanover A (2009) Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu Rev Pharmacol Toxicol 49:73–96. [DOI] [PubMed] [Google Scholar]
- 72. Shimura H, Schwartz D, Gygi SP, Kosik KS (2004) CHIP‐Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem 279:4869–4876. [DOI] [PubMed] [Google Scholar]
- 73. Sun Q, Gamblin TC (2009) Pseudohyperphosphorylation causing AD‐like changes in tau has significant effects on its polymerization. Biochemistry 48:6002–6011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tabaton M, Cammarata S, Mancardi G, Manetto V, Autilio‐Gambetti L, Perry G, Gambetti P (1991) Ultrastructural localization of beta‐amyloid, tau, and ubiquitin epitopes in extracellular neurofibrillary tangles. Proc Natl Acad Sci U S A 88:2098–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tai HC, Schuman EM (2008) Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci 9:826–838. [DOI] [PubMed] [Google Scholar]
- 76. Trojanowski JQ, Shin RW, Schmidt ML, Lee VM (1995) Relationship between plaques, tangles, and dystrophic processes in Alzheimer's disease. Neurobiol Aging 16:335–340. discussion 341–335. [DOI] [PubMed] [Google Scholar]
- 77. Upadhya SC, Hegde AN (2007) Role of the ubiquitin proteasome system in Alzheimer's disease. BMC Biochem 8(Suppl. 1):S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Vincent IJ, Davies P (1990) ATP‐induced loss of Alz‐50 immunoreactivity with the A68 proteins from Alzheimer brain is mediated by ubiquitin. Proc Natl Acad Sci U S A 87:4840–4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang Y, Martinez‐Vicente M, Kruger U, Kaushik S, Wong E, Mandelkow EM et al (2009) Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet 18:4153–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wischik CM, Novak M, Edwards PC, Klug A, Tichelaar W, Crowther RA (1988) Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A 85:4884–4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wischik CM, Novak M, Thogersen HC, Edwards PC, Runswick MJ, Jakes R et al (1988) Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A 85:4506–4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wischik CM, Edwards PC, Lai RY, Gertz HN, Xuereb JH, Paykel ES et al (1995) Quantitative analysis of tau protein in paired helical filament preparations: implications for the role of tau protein phosphorylation in PHF assembly in Alzheimer's disease. Neurobiol Aging 16:409–417. [DOI] [PubMed] [Google Scholar]
- 83. Yin H, Kuret J (2006) C‐terminal truncation modulates both nucleation and extension phases of tau fibrillization. FEBS Lett 580:211–215. [DOI] [PubMed] [Google Scholar]
- 84. Zhang Y, Tian Q, Zhang Q, Zhou X, Liu S, Wang JZ (2009) Hyperphosphorylation of microtubule‐associated tau protein plays dual role in neurodegeneration and neuroprotection. Pathophysiology 16:311–316. [DOI] [PubMed] [Google Scholar]