

Abstract
Pituitary adenomas are common neuroendocrine neoplasms arising from adenohypophysial cells. Recent progress in our understanding of pituitary tumorigenesis as well as pathways involved in molecular cytodifferentiation of the adenohypophysis has impacted on the classification of pituitary adenomas. The detailed comprehensive classification of pituitary adenomas is now well recognized to reflect specific clinical features and genetic changes that predict targeted treatments, as well as prognostic information for patients with pituitary adenomas. Therefore, the clinical responsibility of pathologists is not only limited to the distinction of pituitary adenomas from other sellar lesions, but also to provide a comprehensive subtype classification using appropriate ancillary tools. In this article, we highlight an approach to clinical diagnosis and pitfalls in the classification of these common neoplasms.
Keywords: corticotroph adenoma, gonadotroph adenoma, immunohistochemistry, lactotroph adenoma, pituitary adenoma, somatotroph adenoma, thyrotroph adenoma, transcription factors
INTRODUCTION
Pituitary adenomas are benign clonal neuroendocrine proliferations arising from adenohypophysial cells 1, 7, 28, 29, 57. With modern methods of imaging and biochemical analysis of hormonal activity, the most recent data suggest that pituitary adenomas are common, occurring in almost 20% of the general population 2, 6, 7, 10, 21. The majority of these tumors are clinically nonfunctioning tumors that are now recognized to be of gonadotroph differentiation, but a significant proportion are prolactinomas that cause clinical symptoms and are usually treated medically 7, 17, 22. The detailed comprehensive subtyping of pituitary adenomas is now well recognized to reflect specific clinical features and genetic changes that predict targeted treatments for some patients with pituitary disorders 2, 6, 7, 10, 13, 15, 45. Therefore, the clinical responsibility of pathologists is not only limited to the distinction of pituitary adenomas from other sellar lesions, but also to provide a comprehensive subtype classification using appropriate ancillary tools. In this article, we highlight an approach to clinical diagnosis and pitfalls in the diagnosis and classification of these tumors.
WHY CLINICAL INFORMATION IS IMPORTANT
Despite the importance of clinicopathological correlation, many pathologists are still faced with diagnosing a pituitary lesion in the absence of clinical information. In most instances it is possible to determine a remarkable amount of information with careful morphologic and immunohistochemical evaluation. However, as in any field of medicine, it behooves the physician to obtain all relevant clinical information to provide an informed and valuable consultation.
Similar to other neuroendocrine tumors, the functional status of a pituitary adenoma is defined by the presence of clinical symptoms, not by immunohistochemistry (7). The use of the term “silent adenoma” should be restricted to lesions that have no evidence of clinical or biochemical abnormality. Inadequate clinical evaluation should not be a reason to make the diagnosis of a “silent” lesion 2, 6, 7, 10. Careful review of preoperative clinical and biochemical data is a necessary part of the consultation and should be requested if not readily available.
Clinically functioning pituitary adenomas produce hormones in excess and give rise to specific clinical syndromes. Among these syndromes, Cushing's disease and acromegaly can be lethal and require immediate treatment. While prolactin (PRL)‐producing adenomas are the most common type of functioning adenoma 2, 6, 7, 10, 18, 21, hyperprolactinemia does not necessarily indicate a PRL‐producing adenoma, as mild hyperprolactinemia (<200 ng/mL or <2000 mU/L) is a common finding associated with any sellar mass that causes stalk compression and interrupts blood flow that regulates tonic inhibition of PRL 6, 7, 18, 37, 39, 56. Moreover, some patients with Cushing's disease may exhibit hyperprolactinemia in the absence of stalk compression; this is attributable to the stimulatory action of several derivatives of proopiomelanocortin (POMC), especially β‐endorphin, on release of PRL (7). On the other hand, diabetes insipidus or cranial nerve deficits are uncommon in pituitary adenomas and may suggest the possibility of other inflammatory disorders, hematolymphoid neoplasms or metastatic carcinomas in the sellar region (7).
Clinically nonfunctioning pituitary adenomas are detected either as incidentalomas, radiologically or at autopsy, or when they become large enough to produce symptoms related to intracranial mass, such as a visual field defect, headache or hypopituitarism 2, 6, 7, 21.
Women often present at a younger age and have a higher incidence of PRL‐producing adenomas and adrenocorticotropic hormone (ACTH)‐producing adenomas, whereas men tend to present in middle or older age with clinically nonfunctioning adenomas 2, 6, 7, 10. Pediatric pituitary adenomas are infrequent and exhibit a female preponderance (35); only a small proportion of these lesions are clinically nonfunctioning adenomas 35, 36.
The administration of drugs can result in reversible morphological changes in the adenoma tissue 6, 7, 9, 44. One of the best examples is the morphologic effects of dopamine agonists (eg, bromocriptine) in PRL‐producing adenomas. The tumor becomes more cellular due to marked shrinkage in cell size, predominantly affecting the tumor cell cytoplasm and the nuclei become heterochromatic and irregular 6, 7, 9, 44. Perivascular and interstitial fibrosis with variable hemorrhage are also associated with these cellular changes 7, 9, 44. The interpretation of these findings can sometimes be challenging, as the morphological features resemble lymphoma or lymphocytic hypophysitis; therefore, immunohistochemical staining is required to address this differential diagnosis.
The response to bromocriptine in most patients with lactotroph adenomas is also of clinical importance, as sparsely granulated lactotroph adenomas have striking and rapid reduction of serum PRL levels when therapy is initiated, and within days to weeks there is symptomatic and radiologic evidence of tumor shrinkage 7, 9. Absence of this typical response in a patient with prolactinoma suggests the possibility of other aggressive rare PRL‐producing pituitary adenomas such as acidophil stem cell adenoma or densely granulated lactotroph adenoma. Similarly, acromegalic patients who fail surgical resection and do not show significant response to long‐acting somatostatin analogues usually harbor the more aggressive sparsely granulated somatotroph adenoma 7, 10, 13, 15.
Pituitary adenomas can sometimes undergo acute hemorrhagic necrosis, usually due to ischemia that results in a sudden onset of headache, visual symptoms, altered mental status and hormonal dysfunction 7, 50, 53, 54. The visual symptoms of pituitary apoplexy usually include both visual acuity impairment and visual field impairment from involvement of the optic nerve and/or chiasm, as well as ocular motility dysfunction from involvement of the cranial nerves in the cavernous sinus 50, 53, 54. Some studies show predominance of nonfunctional adenomas among apoplectic adenomas while other studies show a higher prevalence in functioning adenomas, with prolactinomas having the highest risk (53). A recent study highlighted that patients with classical pituitary apoplexy may exhibit recurrent pituitary tumor growth and therefore these patients need continued postoperative surveillance if they have not had postoperative radiotherapy (50). Moreover, pituitary apoplexy may mimic a variety of other lesions including subarachnoid hemorrhage because of ruptured intracranial aneurysm; therefore, the histological confirmation of apoplexy in a pituitary adenoma is of clinical importance.
THE IMPACT OF RADIOLOGICAL FINDINGS
Radiologic examination provides a method of classification of pituitary adenomas based on tumor size and degree of local invasion. These data are of critical importance to surgeons when planning a surgical resection. While microadenomas are lesions that measure less than 1 cm in diameter, macroadenomas are larger than 1 cm 2, 6, 7, 18.
Pituitary adenomas that are often associated with aggressive behavior include sparsely granulated somatotroph adenoma, densely granulated lactotroph adenoma, acidophil stem cell adenoma, thyrotroph adenoma, sparsely granulated corticotroph adenoma, Crooke cell adenoma and silent subtype III adenoma 2, 6, 7, 10, 13, 15, 27. These aggressive adenomas are usually invasive macroadenomas. Other less aggressive adenoma subtypes can sometimes be invasive; sparsely granulated lactotroph adenomas in men often present as large macroadenomas that exhibit sinonasal invasion. Some adenomas such as silent subtype III adenomas and acidophil stem cell adenomas reveal characteristic preferential downward invasive growth with significant bone invasion and parasellar extension rather than the more typical suprasellar expansion (7). An acromegalic patient with a hypointense pituitary mass on the T2 sequence of magnetic resonance imaging (MRI) usually harbors a densely granulated somatotroph adenoma.
In patients with corticotroph adenomas, there tends to be an inverse correlation between tumor size and symptomatology that is because of the existence of two distinct tumor variants, hence the saying “Small tumor, big Cushing and Big tumor, small Cushing.” The classical florid Cushing's disease is usually associated with a small microadenoma that is a densely granulated corticotroph adenoma (7). In contrast, Crooke cell adenomas, sparsely granulated corticotroph adenomas and silent corticotroph adenomas are usually macroadenomas and are associated with cyclic Cushing's disease, less florid Cushing signs or no clinical features, respectively 7, 25, 27, 32, 40. Another clinical challenge associated with Cushing's disease is that MRI may fail to localize a microadenoma. Nontumorous corticotrophs populate the median wedge and some corticotroph adenomas are found in this location; however, others may be present in the lateral wings and may show lateralization of blood flow. Therefore, when MRI findings are not helpful, selective petrosal sinus sampling with corticotropin realising hormone (CRH) stimulation is used to localize lesions 23, 42, 48.
DIAGNOSTIC STEPS IN THE ASSESSMENT OF SELLAR LESIONS
The sellar region is the site of a large number of morphological entities arising from the pituitary gland and other adjacent anatomical structures, including meninges, blood vessels, brain and nerves 2, 6, 7, 10. Therefore, the initial evaluation of a sellar lesion starts on hematoxylin‐eosin stained slides, which allow the distinction of primary adenohypophysial pathologies from other sellar entities (Figure 1). The handling of tissue obtained at surgery should ensure adequate fixation in formalin for histology and immunohistochemistry. Once a pituitary lesion is determined to be composed of epithelial cells with neuroendocrine differentiation originating from adenohypophysial cells, several steps should be undertaken. First, the lesion must be identified as hyperplasia or adenoma. Pituitary adenomas reveal total breakdown of normal acinar architecture on Gordon‐Sweet silver stain, which distinguishes neoplasia from hyperplasia that retains an acinar reticulin pattern. The second step is to identify the cell subpopulation responsible for this proliferation. This is usually performed by using immunohistochemistry and/or electron microscopy. Finally, the behavior and potential therapy of choice must be determined. Ploidy analyses, proliferation markers including Ki‐67 (MIB‐1), the purine‐binding factor nm23 or topoisomerase II alpha have been proposed to be useful in this regard but the value of these markers remains controversial 2, 3, 4, 6, 7, 10, 24, 26, 34, 38, 59, 60, 64. The best prognosticator still remains accurate subtyping of the pituitary adenoma based on hormone content and cell structure 2, 3, 6, 7, 10.
Figure 1.
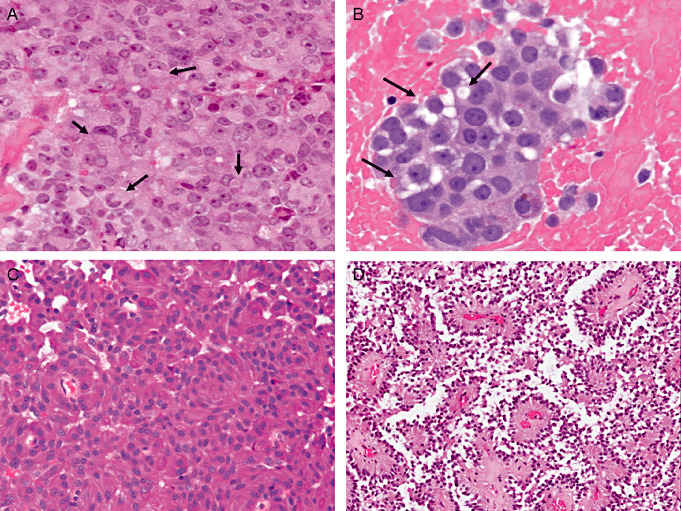
Light microscopic characteristics of pituitary adenomas. A. Sparsely granulated somatotroph adenomas are composed of chromophobic cells that contain eccentric nuclei, which are pushed to the cell periphery and indented by fibrous bodies (arrows). B. Acidophil stem cell adenomas are composed of eosinophilic “oncocytic” cells that exhibit cytoplasmic vacuolization because of mitochondrial dilatation and giant mitochondria. C. Densely granulated corticotroph adenomas and silent corticotroph type I adenomas are composed of tumor cells with basophilic cytoplasm due to the numerous adrenocorticotropic hormone‐containing secretory granules. D. Perivascular pseudorosette formation and a pseudopapillary appearance are characteristic features of gonadotroph adenomas.
Recent interest has focussed on MGMT (06‐methylguanine‐DNA methyltransferase), a DNA repair protein that is a biomarker used to predict response to temozolomide treatment in gliomas 16, 43. Aggressive PRL‐ and ACTH‐producing adenomas have shown response to temozolomide therapy, up to 73% and 60%, respectively (43). However, clinical response was not predicted by MGMT status in a recent large series that examined both MGMT promoter methylation and MGMT immunohistochemistry expression (16). Although temozolamide seems to be promising for an acute response in some aggressive pituitary adenomas, prospective large clinical trials are still needed to determine the long‐term efficacy of this treatment and the role of MGMT expression in predicting response.
DETAILED CLASSIFICATION OF PITUITARY ADENOMAS
The 2004 World Health Organization histological typing of endocrine tumors has illustrated tremendous progress in our understanding of pituitary tumors 3, 18. One of the major weaknesses of this classification is that ICD codes are only provided for typical pituitary adenoma (8272/0), atypical pituitary adenoma (8272/1) and pituitary carcinoma (8272/3) 3, 18. Despite this, the bulk of this classification provides the detailed classification of pituitary adenomas. As indicated previously, the most important clinical and prognostic features of pituitary adenomas remain the hormonal profile and subtype classification 2, 3, 6, 7, 10. In order to facilitate the reporting process, a synoptic checklist for pituitary lesions has been introduced recently to the literature and, like other cancer checklists, it should become a part of the routine examination of specimens from patients with primary pituitary tumors 7, 46 (Table 1). Many of the ultrastructural features that were recognized as characterizing specific tumour types are now easily identified by immunohistochemistry 2, 6, 7, 8, 10, 18. Therefore, the accurate classification of pituitary adenomas currently relies on the use of immunohistochemical characteristics of tumor cells.
Table 1.
Synoptic report: Pituitary gland. Abbreviations: Pit‐1 = pituitary specific transcription factor 1; ER‐α = estrogen receptor alpha; Tpit = T‐box transcription factor; α‐SU = alpha subunit; GH = growth hormone; PRL = prolactin; β‐TSH = thyroid stimulating hormone; ACTH = adrenocorticotropic hormone; β‐FSH = follicle stimulating hormone; β‐LH = luteinizing hormone.
| Procedure (select all that apply) |
| ____ Transsphenoidal resection |
| ____ Transcranial resection |
| ____ Other (specify): __________ |
| ____ Not specified |
| Clinical features |
| ____ Functional |
| Hormone excess (specify): __________ |
| ____ Clinically nonfunctioning |
| Tumor size (from imaging) |
| Greatest dimension: ____ cm |
| *Additional dimensions: ____×____ cm |
| ____ Cannot be determined |
| *Received: |
| *____ Fresh |
| *____ In formalin |
| *____ Other |
| *Specimen integrity |
| *____ Intact |
| *____ Fragmented |
| Specimen size |
| ____×____×____cm |
| *Specimen weight |
| *____ grams |
| Histologic features |
| Reticulin |
| ____ Intact |
| ____ Expanded |
| ____ Disrupted |
| PAS |
| ____ Positive |
| ____ Negative |
| Infiltrating tumor |
| ____ Positive |
| (specify tissue): __________ |
| ____ Negative |
| ____ Cannot be determined |
| Immunohistochemistry (select all positive) |
| ____ Pit‐1 |
| ____ ER‐α |
| ____ Tpit |
| ____ SF‐1 |
| ____α‐SU |
| ____ GH |
| ____ PRL |
| ____β‐TSH |
| ____ ACTH |
| ____β‐FSH |
| ____β‐LH |
| ____ LMWK (CAM5.2) Diffuse: ____ |
| Fibrous bodies: ____ |
| Perinuclear: ____ |
| Mixed: ____ |
| ____ %MIB‐1 LI |
| ____ p53 |
| ____ Others (specify):__________ |
| Tumor type |
| Pituitary adenoma |
| Subtype |
| ____ Densely granulated somatotroph adenoma |
| ____ Sparsely granulated somatotroph adenoma |
| ____ Mammosomatotroph adenoma |
| ____ Mixed somatotroph‐lactotroph adenoma |
| ____ Sparsely granulated lactotroph adenonoma |
| ____ Densely granulated lactotroph adenonoma |
| ____ Acidophil stem cell adenoma |
| ____ Thyrotroph adenoma |
| ____ Densely granulated corticotroph adenoma |
| ____ Sparsely granulated corticotroph adenoma |
| ____ Gonadotroph adenoma |
| ____ Unusual plurihormonal adenoma |
| ____ Null cell adenoma |
| ____ Oncocytoma |
| ____ Other (specify):__________ |
| ____ Typical |
| ____ Atypical |
| Hyperplasia |
| ____ Cell type (specify): __________ |
| Pituitary carcinoma |
| ____ Cell type (specify): __________ |
| ____ Location of metastases __________ |
| Craniopharyngioma |
| ____ Papillary |
| ____ Adamantinomatous |
| Other |
| ____ Gangliocytoma |
| ____ Paraganglioma |
| ____ Meningioma |
| ____ Spindle cell oncocytoma |
| ____ Pituicytoma |
| ____ Granular cell tumor |
| ____ Other (specify): __________ |
| Additional pathologic findings |
| Nontumorous adenohyphysis: |
| ____ Present |
| *____ Crooke hyaline change |
| ____ Not identified |
| Neurohypohysis |
| ____ Present |
| ____ Not identified |
| *Comment(s) |
The immunopanel includes pituitary transcription factors [pituitary specific transcription factor 1 (Pit‐1), t‐box transcription factor (Tpit), steroidogenic factor 1 (SF‐1), estrogen receptor alpha (ER‐α), GATA binding protein 2 (GATA‐2)] (Figure 2), monoclonal antibodies against pituitary hormones [growth hormone (GH), PRL, β‐thyroid stimulating hormone (TSH), ACTH, β‐follicle stimulating hormone (FSH), β‐luteinizing hormone (LH), alpha subunit], and low molecular weight keratin (LMWK) (CAM5.2) and MIB‐1 (Ki67) 2, 3, 6, 7, 8, 10, 46 (Table 2, Figure 3). In some institutions, p53 staining forms a part of the routine panel (46). The cytoplasmic appearance of pituitary adenomas usually correlates with the content of hormone‐containing secretory granules. The terminology “densely” vs. “sparsely” granulated correlates with the extent of immunohistochemical positivity for the adenohypophysial hormone and/or with the density of electron‐dense secretory granules observed on electron microscopy. The periodic acid schiff (PAS) stain is useful to highlight secretory granules of corticotrophs, thyrotrophs and gonadotrophs 2, 6, 7, 10, 18. While densely granulated corticotrophs are intensely positive for PAS, thyrotrophs and gonadotrophs are focally positive 2, 6, 7, 10.
Figure 2.
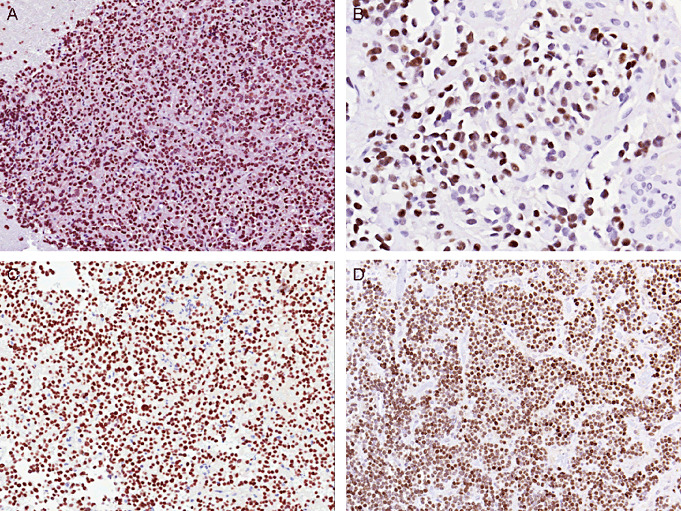
Immunolocalization of pituitary transcription factors. A. Pituitary specific transcription factor 1 is a nuclear transcription factor that is expressed in all GH/PRL/thyroid stimulating hormone family tumors. In this picture, a densely granulated somatotroph adenoma is illustrated. B. Estrogen receptor alpha (ER‐α) is expressed in PRL‐producing adenomas and gonadotroph adenomas. Of note, the levels of expression tend to be low and if the tissue is poorly fixed, they can be negative. In this picture, a sparsely granulated lactotroph adenoma is illustrated. C. T‐box transcription factor is the corticotroph lineage‐specific pituitary transcription factor. In this picture, a densely granulated corticotroph adenoma is illustrated. D. Depending on fixation time, gonadotroph adenomas may be positive for ER‐α. Among gonadotroph cytodifferentiation markers, steroidogenic factor 1 (SF‐1) is the only gonadotroph lineage‐specific transcription factor in the pituitary gland. This picture illustrates positivity for SF‐1 in a hormone inactive gonadotroph adenoma.
Table 2.
Classification of pituitary adenomas. Abbreviations: Pit‐1 = pituitary specific transcription factor 1; GH = growth hormone; PRL = prolactin; TSH = thyroid stimulating hormone; ACTH = adrenocorticotropic hormone; SF‐1 = steroidogenic factor 1; α‐SU = alpha subunit; ER‐α = estrogen receptor alpha; GATA‐2 = GATA binding protein 2; Tpit = T‐box transcription factor; β‐FSH=follicle stimulating hormone; β‐LH = luteinizing hormone.
| Adenoma subtypes | Immunoreactivities | CAM 5.2 |
|---|---|---|
| Pit‐1 (GH/PRL/TSH) family tumors | ||
| GH‐producing adenomas | ||
| Densely granulated somatotroph adenoma | Pit‐1, GH (diffuse), α‐SU | Perinuclear |
| Intermediate type somatotroph adenoma* | Pit‐1, GH (diffuse), α‐SU | Few fibrous bodies |
| Sparsely granulated somatotroph adenoma | Pit‐1, GH (weak) | Fibrous bodies (>90%) |
| Mammosomatotroph adenoma | Pit‐1, ER‐α†, α‐SU | |
| Mixed somatotroph and lactotroph adenomas | Pit‐1, ER‐α†, α‐SU | |
| GH‐producing plurihormonal adenoma | Pit‐1, (ER‐α†), (GATA‐2) | |
| PRL‐producing adenomas | ||
| Sparsely granulated lactotroph adenoma | Pit‐1, ER‐α†, PRL (Golgi) | |
| Densely granulated lactotroph adenoma | Pit‐1, ER‐α†, PRL (Diffuse) | |
| Acidophil stem cell adenomas | Pit‐1, ER‐α†, PRL (Diffuse), GH (variable) | Few fibrous bodies |
| TSH‐producing adenomas | ||
| Thyrotroph adenoma | Pit‐1, GATA‐2 | |
| Monomorphous Pit‐1 lineage plurihormonal adenoma | ||
| Silent subtype 3 adenoma | Pit‐1, (ER‐α†, α‐SU), GH/PRL/TSH (variable) | |
| T pit (ACTH) family tumors | ||
| Densely granulated corticotroph adenoma | Tpit, ACTH (strong, diffuse) | Strong diffuse |
| Sparsely granulated corticotroph adenoma | Tpit, ACTH (weak, variable) | Strong diffuse |
| Crooke cell adenoma | Tpit, ACTH (juxtanuclear and peripheral) | Ring‐like pattern |
| SF‐1 (Gonadotroph) family tumors | ||
| Hormone active gonadotroph adenoma | SF‐1, ER‐α†, GATA‐2, α‐SU, β‐FSH, β‐LH | Usually negative |
| Hormone‐inactive gonadotroph adenoma | SF‐1, ER‐α†, GATA‐2, α‐SU (variable) | Usually negative |
| Transcription factor and hormone negative adenoma | ||
| Null cell adenoma | Negative for all transcription factors and hormones | Variable positive |
| Polymorphous plurihormonal adenoma | ||
| Plurihormonal adenoma, NOS | Multiple |
This tumor is usually classified as densely granulated somatotroph adenoma as their biology is similar to densely granulated somatotroph adenomas.
ER‐α is sensitive to fixation and can be very focally and weakly positive.
Figure 3.
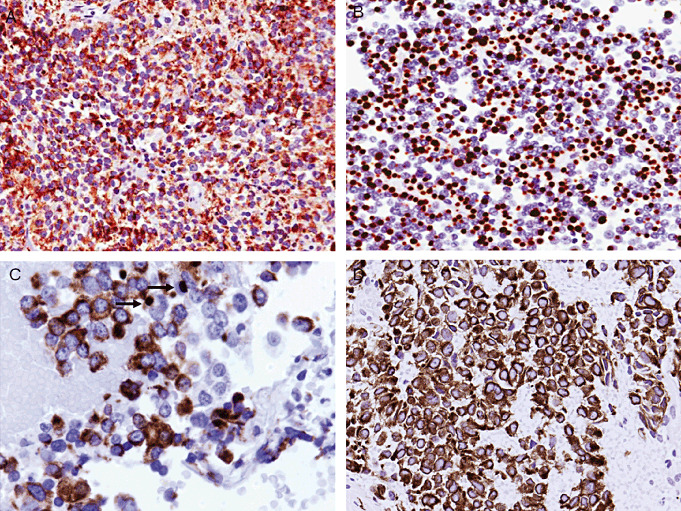
Keratin staining patterns in pituitary adenomas using CAM5.2. A. Perinuclear staining is a feature of densely granulated somatotroph adenomas. B. Paranuclear “fibrous bodies” are the most conspicuous feature of a sparsely granulated somatotroph adenoma. C. Occasional fibrous bodies (arrows) can be seen in acidophil stem cell adenomas. D. Diffuse and strong cytoplasmic staining is characteristic of corticotroph adenomas.
Adenomas that cause GH excess constitute 10%–15% of all adenomas and are classified into five clinically relevant histological subtypes 2, 6, 7, 10. Regardless of the subtype, all GH‐producing adenomas are positive for Pit‐1 2, 6, 7, 10, 18.
-
(i)
Densely granulated somatotroph adenoma (DGSA): These tumors are composed of eosinophilic cells that are positive for Pit‐1 2, 6, 7, 10, 18 (Figure 2A). GH is diffusely positive and α‐subunit (α‐SU) is often expressed 7, 62. LMWK reveals a characteristic perinuclear pattern 2, 6, 7, 10, 18 (Figure 3A). DGSAs are usually responsive to somatostatin analogues as they are usually associated with somatic activation of Gsα proteins that result in high cAMP levels 2, 6, 7, 10. Somatostatin analogues activate the inhibitory subunit of G proteins and reduce cAMP levels 7, 10.
-
(ii)
Sparsely granulated somatotroph adenoma (SGSA): These tumors are composed of chromophobic to pale eosinophilic cells that are positive for Pit‐1 2, 6, 7, 10, 18. The pathognomonic feature of this tumor is the presence of keratin aggresomes, known as “fibrous bodies”2, 6, 7, 10, 18, 30, 47, 49. The nuclei exhibit variable pleomorphism and tend to be pushed to cell periphery and indented by fibrous bodies (Figure 1A). These tumors exhibit variable positivity for GH and are usually negative for α‐SU 2, 6, 7, 10, 62. Of note, LMWK (CAM5.2) reveals characteristic paranuclear globular fibrous bodies in the majority of the tumor cells 2, 6, 7, 10, 18, 47, 49 (Figure 3B). Unlike DGSAs, SGSAs do not respond well to somatostatin analogues. These lesions are thought to lack the high levels of cAMP that predict a response to these agents; instead, they have altered STAT signaling that in some cases has been shown to be because of somatic GH receptor mutations (histidine to leucine mutation in the extracellular domain of the GH receptor of the somatotroph that impairs glycoyslation‐mediated receptor processing) and signaling in GH autoregulation 2, 6, 7, 10, 13. The discrimination of this entity is of clinical interest as Pegvisomant, a GH receptor antagonist, is used in patients with SGSAs that do not respond significantly to somatostatin analogues 2, 6, 7, 10, 13, 15, 45, 49. Some DGSAs may exhibit occasional fibrous bodies and these are classified as “intermediate‐type somatotroph adenomas”(47). As their biology does not differ from DGSAs, they are classified in the spectrum of DGSAs 7, 47.
-
(iii)
Mammosomatotroph adenoma: These tumors resemble DGSAs, are composed of eosinophilic cells and are diffusely positive for Pit‐1 and GH, but also express ER‐α, PRL and α‐SU. LMWK reveals a perinuclear staining pattern 2, 6, 7, 10.
-
(iv)
Mixed somatotroph and lactotroph adenoma: The most common combination is DGSA and sparsely granulated lactotroph adenoma 2, 6, 7, 10. GH and α‐SU are positive in DGSA component, and positivity for ER‐α and PRL with a Golgi pattern is seen in the lactotroph adenoma component 2, 6, 7, 10. The immunoprofile of this tumor resembles that of mammosomatotroph adenomas and the distinction is usually an academic challenge rather than prognostic. This distinction usually requires ultrastructural examination 2, 6, 7, 10, 30; a monomorphous proliferation that exhibits pleomorphic and heterogeneous secretory granules and misplaced exocytosis distinguishes mammosomatotroph adenomas from mixed somatotroph and lactotroph adenomas that have two distinct cell populations 7, 30.
-
(v)
Plurihormonal GH‐producing adenoma: These tumors are predominantly composed of densely granulated somatotrophs with variable mammosomatotroph and thyrotroph differentiation 2, 6, 7, 10. They are diffusely positive for Pit‐1, ER‐α, GH, PRL, β‐TSH and α‐SU, and usually exhibit perinuclear LMWK positivity 2, 6, 7, 10.
Adenomas that cause PRL excess constitute 30% of all adenomas and are classified into three clinically relevant histological subtypes 2, 6, 7, 10. Regardless of the subtype, all PRL‐producing adenomas are positive for Pit‐1 and ER‐α2, 6, 7, 10, 18.
-
(i)
Sparsely granulated lactotroph adenoma (SGLA): This tumor is the most common pituitary adenoma that responds dramatically to dopamine agonists 2, 6, 7, 10. SGLAs are composed of chromophobic cells that exhibit solid, trabecular or papillary growth. They are positive for Pit‐1 and ER‐α (Figure 2B), and are usually negative for α‐SU 2, 6, 7, 10. PRL reveals a characteristic juxtanuclear pattern of staining reflecting Golgi localization rather than diffuse cytoplasmic accumulation of secretory granules 2, 6, 7, 10. ER‐α is sensitive to fixation and may be artifactually negative.
-
(ii)
Densely granulated lactotroph adenoma (DGLA): These tumors are composed of eosinophilic cells that are positive for Pit‐1 and ER‐α. PRL reveals diffuse cytoplasmic positivity 2, 6, 7, 10.
-
(iii)
Acidophil stem cell adenoma (ASCA): These lesions are composed of cells that exhibit the cytoplasmic eosinophilia of oncocytic change in addition to clear cytoplasmic vacuoles corresponding to megamitochondria 2, 6, 7, 10, 31 (Figure 1B). They are strongly positive for Pit‐1 and ER‐α. PRL reveals variable intensity and distribution, and rarely Golgi‐type staining can be observed 2, 6, 7, 10. Focal GH positivity is common. LMWK reveals scattered fibrous bodies 2, 6, 7, 10 (Figure 3C). Even in the absence of GH‐production by a PRL‐producing adenoma, oncocytic change with occasional fibrous bodies should suggest the diagnosis of ASCA. Ultrastructural examination reveals numerous enlarged mitochondria, forming giant mitochondria with loss of cristae and harboring electron dense tubular structures 2, 6, 7, 10, 31.
Adenomas that cause TSH excess constitute less than 1% of all pituitary adenomas 2, 6, 7, 10, 14. These thyrotroph adenomas are aggressive tumors, composed of polygonal, angulated or spindle‐shaped chromophobic cells that exhibit indistinct cell borders and solid growth 2, 6, 7, 10. They exhibit nuclear pleomorphism and fibrosis similar to silent subtype III adenomas 2, 6, 7, 10, 33. They are positive for Pit‐1, GATA‐2, α‐SU and β‐TSH 2, 6, 7, 10, 63, 65. Immunoreactivity for β‐TSH varies from cell to cell; the staining pattern usually allows recognition of angular cellular morphology of tumor cells 2, 6, 7, 10.
Adenomas that cause ACTH excess constitute 10%–15% of all adenomas and are classified into three clinically relevant histological subtypes 2, 6, 7, 10, 18:
-
(i)
Densely granulated corticotroph adenoma (DGCA): DGCAs are composed of basophilic cells (Figure 1C) that are strongly positive for PAS. Tpit (Figure 2C) and LMWK (Figure 3D) are strongly expressed and ACTH is usually strong and diffuse 2, 6, 7, 10, 18. These tumors are usually microadenomas and require surgical resection.
-
(ii)
Sparsely granulated corticotroph adenoma (SGCA): SGCAs are aggressive tumors composed of chromophobic cells. Tpit and LMWK are strongly expressed. Positivity for ACTH is variable and usually focal and weak 2, 6, 7, 10, 18. PAS is faintly positive.
-
(iii)
Crooke cell adenoma: These unusual and aggressive adenomas are composed of large chromophobic or eosinophilic cells with prominent Crooke's hyaline change 2, 6, 7, 10, 18. Crooke's hyaline change is a morphologic alteration characteristic of nontumorous corticotrophs which are subject to feedback suppression by elevated circulating levels of cortisol 2, 6, 7, 10. Accumulation of keratin filaments gives the cell cytoplasm a glassy hyaline appearance (Figure 4A). This phenomenon results in relocation of PAS‐ and ACTH‐positive secretory granules to the cell periphery and a juxtanuclear location (Figure 4B,C). Granular ACTH reactivity is restricted at the cell periphery and adjacent to the nucleus (Figure 4C). Therefore, these cells are identified by their typical “ring‐like” abundant LMWK positivity 2, 6, 7, 10, 18 (Figure 4D). Atypical large pleomorphic cells of a Crooke cell adenoma may mimic mature neurons of gangliocytomas or metastatic carcinomas (7). The demonstration of Tpit confirms corticotroph lineage 7, 8.
Figure 4.
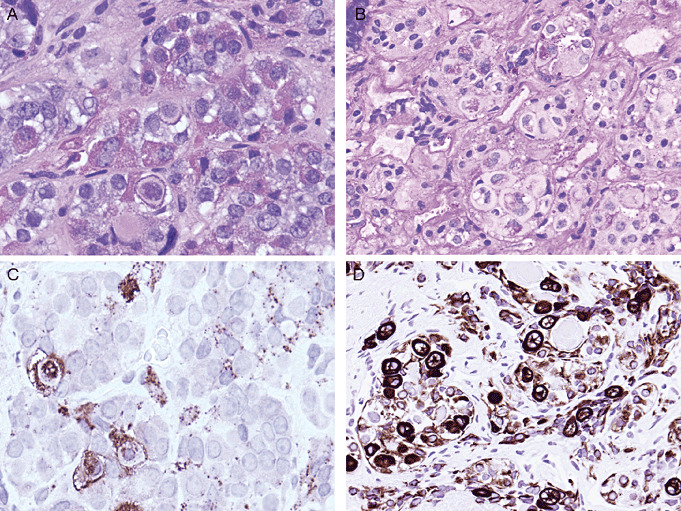
Crooke's hyaline change. (A) This morphologic alteration is characteristic of nontumorous corticotrophs (except those of the intermediate lobe) which are subject to feedback suppression by elevated circulating levels of cortisol. Accumulation of keratin filaments gives the cell cytoplasm a glassy hyaline appearance. This phenomenon results in relocation of (B) Periodic acid schiff‐ and (C) Adrenocorticotropic hormone‐staining that is localized to secretory granules that are pushed to the cell periphery and a juxtanuclear location. (D) These cells are identified by their typical strong “ring‐like” LMWK positivity.
Gonadotroph adenomas constitute 30% of pituitary adenomas and are the most common clinically non‐functioning pituitary adenomas 2, 6, 7, 10, 22. By definition, regardless of the expression of gonadotropins (β‐LH, β‐FSH), positivity for SF‐1 (Figure 2D) and/or ER‐α in a hormone‐negative pituitary adenoma is diagnostic of a gonadotroph adenoma 7, 10. Similar to thyrotroph adenomas, which are negative for SF‐1 and ER‐α, GATA‐2 is also expressed in gonadotroph adenomas 7, 8, 10. Depending on fixation time, gonadotroph adenomas may be positive for ER‐α, but the levels tend to be low and if the tissue is poorly fixed, they can be negative. Among gonadotroph cytodifferentiation markers, SF‐1 is the only gonadotroph lineage specific transcription factor in the pituitary gland. These tumors are usually composed of chromophobic cells that exhibit solid, trabecular, sinusoidal or papillary architecture and prominent pseudorosette formations around blood vessels 2, 6, 7, 10 (Figure 1D). Differentiated forms exhibit marked polarization of cells with elongated cell process and eccentric nuclei (7). These forms are more frequently positive for α‐SU, β‐FSH or β‐LH (7). However, as mentioned before, these tumors can be negative for hormones and α‐SU.
Silent subtype III adenomas are aggressive monomorphous plurihormonal adenomas of Pit‐1 lineage; they stain diffusely for Pit‐1 2, 6, 7, 10, 20. They may be associated with hyperprolactinemia or acromegaly or they may have no clinical evidence of hormone excess. Focal immunoreactivity for one or more Pit‐1 lineage adenohypophysial hormones (GH, PRL, β‐TSH) is usually observed 2, 6, 7, 10, 20. They exhibit variable nuclear atypia and stromal fibrosis similar to thyrotroph adenomas 2, 6, 7, 10. Focal positivity for ER‐α, GH and/or PRL favors silent subtype III adenoma in a sparsely granulated pituitary adenoma that reveals positivity for Pit‐1 and β‐TSH. Radiologically they can mimic ASCAs; they are characterized by downward invasive growth. Ultrastructural examination reveals nuclear inclusions, so‐called “spheridia,” which are typical of silent subtype III adenomas, and they do not exhibit the mitochondrial alterations of ASCAs 2, 6, 7, 10, 18, 20, 33. Our experience suggests that ASCAs and silent subtype III adenomas represent poorly differentiated Pit‐1 lineage adenomas.
Plurimorphous plurihormonal adenomas are extremely rare 2, 6, 7, 10. Several reports of various combinations of hormones in a single tumor are found in the literature (7). In some instances, trapped nontumorous cells can be mistaken for adenoma cells and an adenoma may be misclassified as plurihormonal based on this misinterpretation. The application of highly specific monoclonal antibodies and the understanding of cell differentiation pathways have clarified many of the controversies. It is essential to use monoclonal antisera especially for glycoprotein hormones in order to prevent nonspecific cross‐reactivities that can lead to misclassification of an adenoma as plurihormonal. The use of antibodies to identify cell‐specific pituitary transcription factors is equally important in order to highlight if the tumor has a polymorphous phenotype. The presence of a few scattered cells that are positive for other hormones is not unequivocal evidence of a plurihormonal adenoma.
Null cell adenomas are composed of adenohypophysial cells that do not exhibit any evidence of cell‐type specific differentiation using pituitary transcription factors, hormones and ultrastructural features 2, 6, 7, 10. Use of appropriate pituitary transcription factors (SF1, ER‐α) prevents hormone‐immunonegative gonadotroph adenomas from being mistakenly diagnosed as null cell adenoma. Null cell adenomas can exhibit oncocytic change, and they are classified as oncocytomas (7).
Patients with clinically nonfunctioning adenomas do not usually have evidence of hormone hypersecretion in vivo 2, 6, 7, 10, 18. Gonadotroph adenomas represent the vast majority of clinically nonfunctioning pituitary adenomas. Null cell adenomas are also by definition clinically nonfunctioning adenomas 2, 6, 7, 10, 18. However, each cell lineage may exhibit a clinically non‐functioning variant. The pattern of immunohistochemical staining and ultrastructural appearance of silent pituitary adenomas resemble those of functioning counterparts as described previously. Among silent corticotroph adenomas, two morphologic variants have been described 2, 6, 7. While the features of type I silent corticotroph adenomas exhibit morphological and immunohistochemical features of clinically functioning densely granulated basophilic corticotroph adenomas, type II silent corticotroph adenomas resemble the uncommon chromophobic sparsely granulated corticotroph adenomas. Of note, it is important to distinguish silent corticotroph adenomas from other clinically nonfunctioning pituitary adenomas as silent corticotroph adenomas have an aggressive behavior characterized by high recurrence rate, invasion and propensity to undergo hemorrhagic infarction (7). Rare Crooke cell adenomas can sometimes be clinically silent (7).
WHAT DOES ATYPICAL ADENOMA MEAN?
Invasive pituitary adenomas that exhibit increased mitotic activity, MIB‐1 labeling index >3% or extensive p53 expression are considered “atypical adenomas”3, 7, 18. However, this terminology does not seem to reflect any biological superiority to the aggressive histological subtypes determined by the accurate classification of pituitary adenomas 2, 3, 6, 7, 10, 18. The diagnosis of pituitary carcinoma is restricted to adenohypophysial proliferations that exhibit cerebrospinal and/or systemic metastasis 3, 7, 18. There are no morphological criteria to distinguish locally aggressive or atypical adenomas from carcinomas when the tumor is confined to the sella. In this context, it is important to remember that pituitary adenomas can originate from ectopic pituitary tissues located in the nasal cavity, nasopharynx, temporal bone, clivus and third ventricle 5, 7, 19, 66; therefore, they should not be mistaken for metastatic pituitary carcinoma or invasive pituitary adenomas.
THE ROLE OF ELECTRON MICROSCOPY
Many of the ultrastructural features that were recognized as characterizing specific tumor types are now identified by immunohistochemistry. For example, the hallmark of SGSAs, fibrous bodies are now easily identified with LMWK (CAM5.2) 2, 6, 7, 10. However, there still remain occasional situations that require electron microscopy for accurate classification 2, 6, 7, 10. These include characterization of unusual pituitary adenomas, identification of giant mitochondria or dilated mitochondria in ASCAs (Figure 5A), identification of spheridia in silent subtype III adenomas (Figure 5B), the academic distinction of mammosomatotroph adenomas from mixed somatotroph and lactotroph adenomas, and sometimes confirmation of adenohypophysial neuroendocrine differentiation in null cell adenomas 2, 6, 7, 10, 18, 20, 30, 31, 33.
Figure 5.
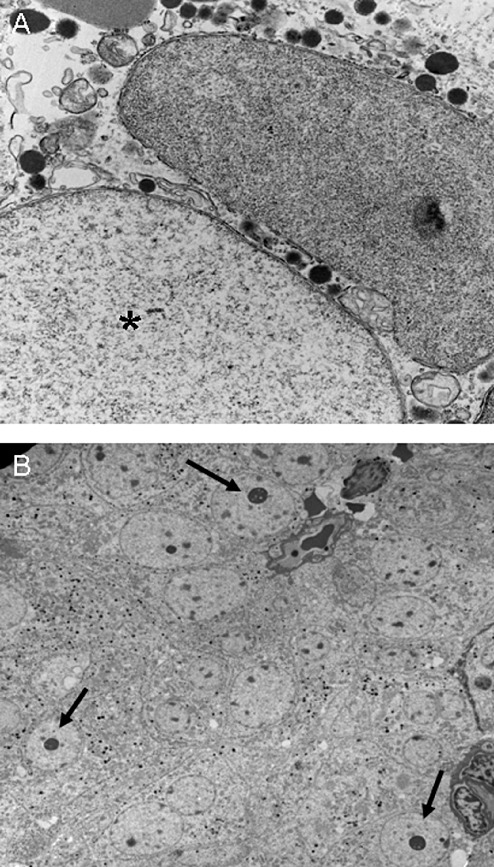
Ultrastructural features of some rare pituitary adenomas. A. Giant (*) and dilated mitochondria are features of the acidophil stem cell adenoma. B. Spheridia (arrows) are characteristic of silent subtype III adenomas.
THE IMPORTANCE OF THE NONTUMOROUS HYPOPHYSIS
The assessment of nontumorous adenohypophysis can sometimes illuminate the underlying pathogenesis of a pituitary adenoma. Patients with long‐standing primary hypothyroidism may develop thyrotroph hyperplasia and thyrotroph adenoma arising in background of hyperplasia. Similarly, Addison's disease results in corticotroph hyperplasia that can mimic adenoma (7). In addition, mammosomatotroph hyperplasia unassociated with a known growth hormone releasing hormone (GHRH)‐producing tumor has been reported in association with McCune–Albright syndrome where GNAS1 mutations can result in constitutive activation of the G proteins mediating GHRH stimulation and formation of a pituitary adenoma 7, 10. Similarly, other genetic conditions such as MEN1 syndrome (MEN1) and Carney complex (PRAKR1A) can be associated with pituitary hyperplasia (eg, somatotroph hyperplasia) surrounding a dominant pituitary adenoma 7, 10, 18. Of note, CRH‐ and/or GnRH‐producing sellar gangliocytomas can be associated with corticotroph hyperplasia/adenoma, and somatotroph hyperplasia/adenoma, respectively 7, 11, 12, 18, 51. Sellar gangliocytomas can be seen intermingled within pituitary adenomas and in such situations the large ganglion cells may be scarce 7, 52, 55, 58.
Careful examination of nontumorous adenohypophyseal parenchyma can also provide some additional important data. The presence or absence of Crooke's hyaline change in the nontumorous adenohypophysis should be carefully assessed in patients with hypercortisolism 2, 6, 7, 10 (Figure 4). Of note, intermediate lobe corticotrophs are not sensitive to suppression of hypercortisolemia and are unlikely to exhibit Crooke's hyaline change (7). Hypercortisolism may have other causes including alcoholism, obesity, several drugs (estrogens, diphenylhydantoin) and pregnancy (7). Obesity also can result in a state of relative hypercortisolemia and the distinction of these various forms of “pseudo‐Cushing's” syndrome from Cushing's syndrome can be challenging (7). Given the difficulty to localize microadenomas by radiology, as well as challenges in the clinical workup, the initial surgery may only reveal small fragments of nontumorous adenohypophysis. In this situation, the identification of Crooke's hyaline change is of clinical significance, as patients with corticotroph hyperplasia and pseudo‐Cushing's syndrome do not usually exhibit Crooke's hyaline change. It is important to remember that Crooke's hyaline change is seen in the adenohypophysis of patients with primary adrenal hypercortisolism, iatrogenic hypercortisolism or ectopic ACTH syndrome; in Cushing's disease, however, it is usually confined to the suppressed nontumorous corticotroph population. Moreover, Crooke's hyaline change can sometimes be focal in the nontumorous corticotrophs or can be seen focally in a corticotroph adenoma; these findings may suggest an abnormality in the feedback autoregulation of corticotrophs and therefore is an indication for careful clinical surveillance 2, 6, 7, 10.
NEUROENDOCRINE MIMICS OF PITUITARY ADENOMAS
Sometimes pituitary adenomas can be negative for keratins, pituitary transcription factors and hormones, but positive for neuroendocrine markers 2, 6, 7, 10. In this situation, the differential diagnosis includes sellar paraganglioma and null cell adenoma. To complicate this quandry, metastatic neuroendocrine carcinomas can sometimes also be negative for keratins (7). In this setting, positivity for tyrosine hydroxylase is diagnostic of paraganglioma and positivity for other transcription factors [thyroid transcription factor 1 (TTF‐1), caudal type homeobox transcription factor 2] favors metastatic neuroendocrine carcinomas 7, 61. It is of note that TTF‐1 is expressed in high‐grade neuroendocrine carcinomas, regardless of their site of origin, as well as in medullary thyroid carcinomas and those originating from lung. However, normal pituicytes, as well as lesions arising from ventral neuroectoderm such as granular cell tumors, spindle cell oncocytomas and pituicytomas, are also positive for TTF‐1 7, 41. As these lesions are negative for neuroendocrine markers, their distinction is relatively easy when TTF‐1 is used in a panel. Moreover, it is important to remember that pituitary adenomas do not often express peptides of thyroid, gut and pancreas. Metastatic neuroendocrine carcinomas can express ACTH, CRH, GHRH and α‐SU, but not GH, PRL, β‐TSH, β‐FSH or β‐LH (7). Furthermore, MIB‐1 is usually high in metastatic carcinomas and usually low in pituitary adenomas.
CONCLUSIONS
There has been significant progress in our understanding of pituitary tumorigenesis including markers involved in the molecular cytodifferentiation of adenohypophysis. Detailed classification of pituitary adenomas based on immunohistochemical characteristic of tumor cells reflects specific clinical features and genetic changes that predict targeted treatments, as well as prognosis. Therefore, the application of markers of molecular cytodifferentiation of adenohypohysis, including pituitary transcription factors (Pit‐1, Tpit, SF‐1, ER‐α, GATA‐2), along with monoclonal antibodies against pituitary hormones, LMWK and MIB‐1, are crucial in the diagnostic assessment and accurate classification of pituitary adenomas.
REFERENCES
- 1. Alexander JM, Biller BMK, Bikkal H, Zervas NT, Arnold A, Klibanski A (1990) Clinically non‐functioning pituitary tumors are monoclonal in origin. J Clin Invest 86:336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Al‐Brahim NY, Asa SL (2006) My approach to pathology of the pituitary gland. J Clin Pathol 59:1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Al‐Shraim M, Asa SL (2006) The 2004 World Health Organization classification of pituitary tumors: what is new? Acta Neuropathol 111:1–7. [DOI] [PubMed] [Google Scholar]
- 4. Anniko M, Tribukait B, Wersäll J (1984) DNA ploidy and cell phase in human pituitary tumors. Cancer 53:1708–1713. [DOI] [PubMed] [Google Scholar]
- 5. Appel JG, Bergsneider M, Vinters H, Salamon N, Wang MB, Heaney AP (2011) Acromegaly due to an ectopic pituitary adenoma in the clivus: case report and review of literature. Pituitary DOI: 10.1007/s11102‐011‐0345‐9. [DOI] [PubMed] [Google Scholar]
- 6. Asa SL (2008) Practical pituitary pathology: what does the pathologist need to know? Arch Pathol Lab Med 132:1231–1240. [DOI] [PubMed] [Google Scholar]
- 7. Asa SL (2011) Tumors of the Pituitary Gland. Fascicle 15, 4th Series. The Atlas of Tumor Pathology. Armed Forces Institute of Pathology: Washington DC. [Google Scholar]
- 8. Asa SL, Ezzat S (1999) Molecular determinants of pituitary cytodifferentiation. Pituitary 1:159–168. [DOI] [PubMed] [Google Scholar]
- 9. Asa SL, Ezzat S (2002) Medical management of pituitary adenomas: structural and ultrastructural changes. Pituitary 5:133–139. [DOI] [PubMed] [Google Scholar]
- 10. Asa SL, Ezzat S (2009) The pathogenesis of pituitary tumors. Annu Rev Pathol 4:97–126. [DOI] [PubMed] [Google Scholar]
- 11. Asa SL, Kovacs K, Tindall GT, Barrow DL, Horvath E, Vecsei P (1984) Cushing's disease associated with an intrasellar gangliocytoma producing corticotrophin‐releasing factor. Ann Intern Med 101:789–793. [DOI] [PubMed] [Google Scholar]
- 12. Asa SL, Scheithauer BW, Bilbao JM, Horvath E, Ryan N, Kovacs K et al (1984) A case for hypothalamic acromegaly: a clinicopathological study of six patients with hypothalamic gangliocytomas producing growth hormone‐releasing factor. J Clin Endocrinol Metab 58:796–803. [DOI] [PubMed] [Google Scholar]
- 13. Asa SL, Digiovanni R, Jiang J, Ward ML, Loesch K, Yamada S et al (2007) A growth hormone receptor mutation impairs growth hormone autofeedback signaling in pituitary tumors. Cancer Res 67:7505–7511. [DOI] [PubMed] [Google Scholar]
- 14. Beck‐Peccoz P, Persani L (2002) Medical management of thyrotropin‐secreting pituitary adenomas. Pituitary 5:83–88. [DOI] [PubMed] [Google Scholar]
- 15. Bhayana S, Booth GL, Asa SL, Kovacs K, Ezzat S (2005) The implication of somatotroph adenoma phenotype to somatostatin analog responsiveness in acromegaly. J Clin Endocrinol Metab 90:6290–6295. [DOI] [PubMed] [Google Scholar]
- 16. Bush ZM, Longtine JA, Cunningham T, Schiff D, Jane JA Jr, Vance ML et al (2010) Temozolomide treatment for aggressive pituitary tumors: correlation of clinical outcome with O(6)‐methylguanine methyltransferase (MGMT) promoter methylation and expression. J Clin Endocrinol Metab 95:E280–E290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Colao A, Savastano S (2011) Medical treatment of prolactinomas. Nat Rev Endocrinol 7:267–278. [DOI] [PubMed] [Google Scholar]
- 18. DeLellis RA, Lloyd RV, Heitz PU, Eng C (eds) (2004) World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Endocrine Organs. IARC Press: Lyon. [Google Scholar]
- 19. Erdogan N, Sarsilmaz A, Boyraz EI, Ozturkcan S (2012) Ectopic pituitary adenoma presenting as a nasopharyngeal mass: CT and MRI findings. Clin Neurol Neurosurg 114:414–416. [DOI] [PubMed] [Google Scholar]
- 20. Erickson D, Scheithauer B, Atkinson J, Horvath E, Kovacs K, Lloyd RV, Young WF Jr (2009) Silent subtype 3 pituitary adenoma: a clinicopathologic analysis of the Mayo Clinic experience. Clin Endocrinol (Oxf) 71:92–99. [DOI] [PubMed] [Google Scholar]
- 21. Ezzat S, Asa SL, Couldwell WT, Barr CE, Dodge WE, Vance ML, McCutcheon IE (2004) The prevalence of pituitary adenomas: a systematic review. Cancer 101:613–619. [DOI] [PubMed] [Google Scholar]
- 22. Fernandez A, Karavitaki N, Wass JA (2010) Prevalence of pituitary adenomas: a community‐based, cross‐sectional study in Banbury (Oxfordshire, UK). Clin Endocrinol (Oxf) 72:377–382. [DOI] [PubMed] [Google Scholar]
- 23. Findling JW, Aron DC, Tyrrell JB Shinsako JH, Fitzgerald PA, Norman D, Wilson CB, Forsham PH (1981) Selective venous sampling for ACTH in Cushing's syndrome. Differentiation between Cushing's disease and the ectopic ACTH syndrome. Ann Intern Med 94:647–652. [DOI] [PubMed] [Google Scholar]
- 24. Fitzgibbons PL, Appley AJ, Turner RR, Bishop PC, Parker JW, Breeze RE et al (1988) Flow cytometric analysis of pituitary tumors. Correlation of nuclear antigen p105 and DNA content with clinical behavior. Cancer 62:1556–1560. [DOI] [PubMed] [Google Scholar]
- 25. Franscella S, Favod‐Coune C‐A, Pizzolato G, Asa SL, Gaillard R, Bernay J, Philippe J (1991) Pituitary corticotroph adenoma with Crooke's hyalinization. Endocr Pathol 2:111–116. [DOI] [PubMed] [Google Scholar]
- 26. Gandour‐Edwards R, Kapadia SB, Janecka IP, Martinez AJ, Barnes L (1995) Biologic markers of invasive pituitary adenomas involving the sphenoid sinus. Mod Pathol 8:160–164. [PubMed] [Google Scholar]
- 27. George DH, Scheithauer BW, Kovacs K, Horvath E, Young WF Jr, Lloyd RV, Meyer FB (2003) Crooke's cell adenoma of the pituitary: an aggressive variant of corticotroph adenoma. Am J Surg Pathol 27:1330–1336. [DOI] [PubMed] [Google Scholar]
- 28. Gicquel C, LeBouc Y, Luton J‐P, Girad F, Bertagna X (1992) Monoclonality of corticotroph macroadenomas in Cushing's disease. J Clin Endocrinol Metab 75:472–475. [DOI] [PubMed] [Google Scholar]
- 29. Herman V, Fagin J, Gonsky R, Kovacs K, Melmed S (1990) Clonal origin of pituitary adenomas. J Clin Endocrinol Metab 71:1427–1433. [DOI] [PubMed] [Google Scholar]
- 30. Horvath E (1994) Ultrastructural markers in the pathologic diagnosis of pituitary adenomas. Ultrastruct Pathol 18:171–179. [DOI] [PubMed] [Google Scholar]
- 31. Horvath E, Kovacs K, Singer W, Ezrin C, Kerenyi NA (1977) Acidophil stem cell adenoma of the human pituitary. Arch Pathol Lab Med 101:594–599. [PubMed] [Google Scholar]
- 32. Horvath E, Kovacs K, Killinger DW, Smyth HS, Platts ME, Singer W (1980) Silent corticotropic adenomas of the human pituitary gland. A histologic, immunocytologic, and ultrastructural study. Am J Pathol 98:617–638. [PMC free article] [PubMed] [Google Scholar]
- 33. Horvath E, Kovacs K, Smyth HS, Cusimano M, Singer W (2005) Silent adenoma subtype 3 of the pituitary‐immunohistochemical and ultrastructural classification: a review of 29 cases. Ultrastruct Pathol 29:511–524. [DOI] [PubMed] [Google Scholar]
- 34. Hsu DW, Hakim F, Biller BMK, de la Monte S, Zervas NT, Klibanski A, Hedley‐Whyte ET (1993) Significance of proliferating cell nuclear antigen index in predicting pituitary adenoma recurrence. J Neurosurg 78:753–761. [DOI] [PubMed] [Google Scholar]
- 35. Jagannathan J, Dumont AS, Jane JA Jr, Laws ER Jr (2005) Pediatric sellar tumors: diagnostic procedures and management. Neurosurg Focus 18:E6. [PubMed] [Google Scholar]
- 36. Kane LA, Leinung MC, Scheithauer BW, Bergstralh EJ, Laws ER Jr, Groover RV et al (1994) Pituitary adenomas in childhood and adolescence. J Clin Endocrinol Metab 79:1135–1140. [DOI] [PubMed] [Google Scholar]
- 37. Karavitaki N, Thanabalasingham G, Shore HC, Trifanescu R, Ansorge O, Meston N et al (2006) Do the limits of serum prolactin in disconnection hyperprolactinaemia need re‐definition? A study of 226 patients with histologically verified non‐functioning pituitary macroadenoma. Clin Endocrinol 65:524–529. [DOI] [PubMed] [Google Scholar]
- 38. Knosp E, Kitz K, Perneczky A (1989) Proliferation activity in pituitary adenomas: measurement by monoclonal antibody Ki‐67. Neurosurgery 25:927–930. [PubMed] [Google Scholar]
- 39. Korevaar T, Wass J, Grossman A, Karavitaki N (2012) Disconnection hyperprolactinaemia in non‐adenomatous sellar/parasellar lesions practically never exceeds 2000 mU/L. Clin Endocrinol (Oxf) 76:602–603. [DOI] [PubMed] [Google Scholar]
- 40. Kovacs K, Horvath E, Bayley TA, Hassaram ST, Ezrin C (1978) Silent corticotroph cell adenoma with lysosomal accumulation and crinophagy. A distinct clinicopathologic entity. Am J Med 64:492–499. [DOI] [PubMed] [Google Scholar]
- 41. Lee EB, Tihan T, Scheithauer BW, Zhang PJ, Gonatas NK (2009) Thyroid transcription factor 1 expression in sellar tumors: a histogenetic marker? J Neuropathol Exp Neurol 68:482–488. [DOI] [PubMed] [Google Scholar]
- 42. Martines V, Mansueto G, Tosi F, Caruso B, Castello R, Procacci C (2003) Selective venous sampling in diagnosing ACTH‐dependent hypercortisolism. Radiol Med 105:356–361. [PubMed] [Google Scholar]
- 43. McCormack AI, Wass JA, Grossman AB (2011) Aggressive pituitary tumours: the role of temozolomide and the assessment of MGMT status. Eur J Clin Invest 41:1133–1148. [DOI] [PubMed] [Google Scholar]
- 44. Menucci M, Quiñones‐Hinojosa A, Burger P, Salvatori R (2011) Effect of dopaminergic drug treatment on surgical findings in prolactinomas. Pituitary 14:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Neggers SJ, van der Lely AJ (2011) Pegvisomant and improvement of quality of life in acromegalic patients. Horm Res Paediatr 76(Suppl. 1):102–105. [DOI] [PubMed] [Google Scholar]
- 46. Nosé V, Ezzat S, Horvath E, Kovacs K, Laws ER, Lloyd R et al (2011) Protocol for the examination of specimens from patients with primary pituitary tumors. Arch Pathol Lab Med 135:640–646. [DOI] [PubMed] [Google Scholar]
- 47. Obari A, Sano T, Ohyama K, Kudo E, Qian ZR, Yoneda A et al (2008) Clinicopathological features of growth hormone‐producing pituitary adenomas: difference among various types defined by cytokeratin distribution pattern including a transitional form. Endocr Pathol 19:82–91. [DOI] [PubMed] [Google Scholar]
- 48. Oldfield EH, Chrousos GP, Schulte HM, Schaaf M, McKeever PE, Krudy AG et al (1985) Preoperative lateralization of ACTH‐secreting pituitary microadenomas by bilateral and simultaneous inferior petrosal venous sinus sampling. N Engl J Med 312:100–103. [DOI] [PubMed] [Google Scholar]
- 49. Osamura RY, Egashira N, Kajiya H, Takei M, Tobita M, Miyakoshi T et al (2009) Pathology, pathogenesis and therapy of growth hormone (GH)‐producing pituitary adenomas: technical advances in histochemistry and their contribution. Acta Histochem Cytochem 29:95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pal A, Capatina C, Tenreiro AP, Guardiola PD, Byrne JV, Cudlip S et al (2011) Pituitary apoplexy in non‐functioning pituitary adenomas: long term follow up is important because of significant numbers of tumour recurrences. Clin Endocrinol (Oxf) 75:501–504. [DOI] [PubMed] [Google Scholar]
- 51. Puchner MJ, Lüdecke DK, Valdueza JM, Saeger W, Willig RP, Stalla GK, Odink RJ (1993) Cushing's disease in a child caused by a corticotropin‐releasing hormone‐secreting intrasellar gangliocytoma associated with an adrenocorticotropic hormone‐secreting pituitary adenoma. Neurosurgery 33:920–924; discussion 924–925. [DOI] [PubMed] [Google Scholar]
- 52. Puchner MJ, Lüdecke DK, Saeger W, Riedel M, Asa SL (1995) Gangliocytomas of the sellar region‐a review. Exp Clin Endocrinol Diabetes 103:129–149. [DOI] [PubMed] [Google Scholar]
- 53. Ranabir S, Baruah MP (2011) Pituitary apoplexy. Indian J Endocrinol Metab 15(Suppl. 3):S188–S196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Randeva HS, Schoebel J, Byrne J, Esiri M, Adams CB, Wass JA (1999) Classical pituitary apoplexy: clinical features, management and outcome. Clin Endocrinol (Oxf) 51:181–188. [DOI] [PubMed] [Google Scholar]
- 55. Saeger W, Puchner MJ, Lüdecke DK (1994) Combined sellar gangliocytoma and pituitary adenoma in acromegaly or Cushing's disease. A report of 3 cases. Virchows Arch 425:93–99. [DOI] [PubMed] [Google Scholar]
- 56. Salehi F, Doglietto F, Ridout R, Gentili F, Asa S, Zadeh G (2011) A case report and review of hyperprolactinemia that is not prolactinoma. Can J Neurol Sci 38:652–655. [DOI] [PubMed] [Google Scholar]
- 57. Schulte HM, Oldfield EH, Allolio B, Katz DA, Berkman RA, Ali IU (1991) Clonal composition of pituitary adenomas in patients with Cushing's disease: determination by X‐chromosome inactivation analysis. J Clin Endocrinol Metab 73:1302–1308. [DOI] [PubMed] [Google Scholar]
- 58. Serri O, Berthelet F, Bélair M, Vallette S, Asa SL (2008) An unusual association of a sellar gangliocytoma with a prolactinoma. Pituitary 11:85–87. [DOI] [PubMed] [Google Scholar]
- 59. Takino H, Herman V, Weiss M, Melmed S (1995) Purine‐binding factor (nm23) gene expression in pituitary tumors: marker of adenoma invasiveness. J Clin Endocrinol Metab 80:1733–1738. [DOI] [PubMed] [Google Scholar]
- 60. Thapar K, Kovacs K, Scheithauer BW, Stefaneanu L, Horvath E, Pernicone PJ et al (1996) Proliferative activity and invasiveness among pituitary adenomas and carcinomas: an analysis using the MIB‐1 antibody. Neurosurgery 38:99–107. [DOI] [PubMed] [Google Scholar]
- 61. Tischler AS (2008) Pheochromocytoma and extra‐adrenal paraganglioma: updates. Arch Pathol Lab Med 132:1272–1284. [DOI] [PubMed] [Google Scholar]
- 62. Trouillas J, Sassolas G, Guigard MP, Fonlupt P, Ansaneli‐Naves L, Perrin G (1996) Relationships between pathological diagnosis and clinical parameters in acromegaly. Metabolism 45:53–56. [DOI] [PubMed] [Google Scholar]
- 63. Umeoka K, Sanno N, Osamura RY, Teramoto A (2002) Expression of GATA‐2 in human pituitary adenomas. Mod Pathol 15:11–17. [DOI] [PubMed] [Google Scholar]
- 64. Vidal S, Kovacs K, Horvath E, Rotondo F, Kuroki T, Lloyd RV, Scheithauer BW (2002) Topoisomerase IIalpha expression in pituitary adenomas and carcinomas: relationship to tumor behavior. Mod Pathol 15:1205–1212. [DOI] [PubMed] [Google Scholar]
- 65. Wang EL, Qian ZR, Yamada S, Rahman MM, Inosita N, Kageji T et al (2009) Clinicopathological characterization of TSH‐producing adenomas: special reference to TSH‐immunoreactive but clinically non‐functioning adenomas. Endocr Pathol 20:209–220. [DOI] [PubMed] [Google Scholar]
- 66. Wang H, Yu W, Zhang Z, Xu W, Zhang F, Bao W (2010) Ectopic pituitary adenoma in the spheno‐orbital region. J Neuroophthalmol 30:135–137. [DOI] [PubMed] [Google Scholar]