

Abstract
LGI1 mutations lead to an autosomal dominant form of epilepsy. Lgi1 mutant null mice develop seizures and show abnormal neuronal excitability. A fine structure analysis of the cortex in these mice demonstrated a subtle cortical dysplasia, preferentially affecting layers II–IV, associated with increased Foxp2 and Cux1‐expressing neurons leading to blurring of the cortical layers. The hypercellularity observed in the null cortex resulted from an admixture of highly branched mature pyramidal neurons with short and poorly aligned axons as revealed by Golgi staining and immature small neurons with branched disoriented dendrites with reduced spine density and undersized, morphologically altered and round‐headed spines. In vitro, hippocampal neurons revealed poor neurite outgrowth in null mice as well as reduced synapse formation. Electron microscopy demonstrated reduced spine‐localized asymmetric (axospinous) synapses with postsynaptic densities and vesicle‐loaded synapses in the mutant null cortex. The overall pathology in the null mice suggested cortical dyslamination most likely because of mislocalization of late‐born neurons, with an admixture of those carrying suboptimally developed axons and dendrites with reduced functional synapses with normal neurons. Our study suggests that LGI1 has a role in regulating cortical development, which is increasingly becoming recognized as one of the causes of idiopathic epilepsy.
Keywords: cortical dysplasia, LGI1, mouse model, neurite outgrowth
Introduction
The LGI1 gene was originally characterized at a 10q24 chromosome translocation breakpoint in a glioma cell line 11 and was shown to suppress cell motility and invasion in these cells 29. LGI1 is a secreted protein 22, 53, 55, carrying a leucine‐rich repeat (LRR) motif near the N‐terminus 56 and a repeat structure forming a β‐propeller structure at the C‐terminus 59. Both of these motifs are indicative of protein–protein binding functions and LRR proteins are frequently involved in phenotypes related to development of the nervous system 28. The locus for autosomal dominant lateral temporal lobe epilepsy (ADLTE), also known as autosomal dominant partial epilepsy with auditory features (ADPEAF) was also localized to 10q24 40 and mutation studies identified LGI1 as the gene that predisposed to ADLTE 26. This rare form of partial epilepsy is characterized by seizures accompanied by acoustic auras 37, 40. Onset of seizures in this disorder ranges from 4 from 59 years of age and shows ∼65% penetrance. Homozygous inactivation of LGI1 in mice leads to early onset of seizures and premature death 10, 19, 63. Electrophysiological studies in these mice identify abnormalities of synapse function in newborn mice, although whether these abnormalities affected the pre‐ or postsynaptic membrane remains controversial 19, 63. LGI1 binds to the ADAM22 and ADAM23 proteins 18, 30, 49, which are located on the post‐ and presynaptic membranes, respectively, and mice with homozygous deletions of ADAM22 and ADAM23 also show tremor and seizures 35, 41, 50, demonstrating the importance of their interaction with LGI1 in the pathophysiology of seizures. Analysis of isolated neurons from ADAM23 null mice demonstrates that addition of the LGI1 protein results in outgrowth of neurites, implicating it in the formation of synapses 41. Although LGI1 is one of only a few genes predisposing to epilepsy that does not encode a structural component of an ion channel, its association with abnormal synaptic transmission in the mature brain has suggested that this may be one potential underlying cause of ADLTE.
Although early immunohistochemical studies of the mature brain suggested that the LGI1 protein was largely expressed in mature neurons 26, 37, we recently demonstrated that LGI1 is also expressed extensively in the developing mouse brain during embryogenesis 54, especially in regions such as the subventricular zone (SVZ) and the ganglionic eminence (GE). The SVZ is rich in neuronal stem cells and is a major site of neurogenesis. The GE is a transitory structure that guides cell and axon migration during formation of the neocortex, particularly during tangential cell migration. Analysis of the developing fetal brain showed that LGI1 was expressed in cells expressing both nestin, a marker of neuronal stem cells, and doublecortin, a marker for migrating neuroblasts 54. These observations suggested a potential role for LGI1 in the early events in the development of the cortex. The suggestion that LGI1 could influence movement and invasion in glioma cells 29 was considered at variance with its lack of expression in glial cells and the lack of an increased incidence in glioma development in ADLTE patients 20. It has become clear, however, that gliomas are likely derived from a neuronal precursor cell rather than a cell committed to the glial lineage 25, 61 and emerging evidence suggests that this precursor cell may emanate from the subventricular zone 47. It is perhaps not surprising, therefore, that gene expression studies in glioma cells forced to express LGI1 show dysregulation of genes involved in molecular pathways that influence critical events associated with cell movement phenotypes such as axon guidance, neurite outgrowth and cone collapse, all of which are associated with actin cytoskeleton reorganization that facilitate cell movement 31. Taken together, these studies suggested another possible role for LGI1 in the developing brain that might affect corticogenesis. Abnormal corticogenesis is increasingly becoming recognized as a cause of epilepsy 27, 36. To investigate whether LGI1 affects corticogenesis, we performed a detailed analysis of the mutant null mouse brain in newborn mice with a special focus on the development of the cortex, which reveals subtle abnormalities in the outer layers II–IV consistent with mild cortical dysplasia (CD). In these animals, cortical layering is disorganized, blurred and shows abnormalities involving the formation and alignment of dendrites and axons as well as increased cellularity in the outer layers, which we show are derived from late‐born, CUX1‐expressing cells.
Materials and Methods
Histological, immunohistochemistry and Golgi‐cox staining
We previously reported the generation of the Lgi1 null mice through a chromosome engineering methodology 63 and animals used in this study were derived from this colony. All analyses were performed on experimental animals with littermate‐matched controls. Animal experiments were conducted in accordance with the institutional ethical guidelines for animal experiments and adhered to the safety guidelines for gene manipulation experiments issued by the Georgia Regents University (GRU) Committee on the use of Animal Care. Mice were anesthetized with isoflurane inhalation (Butler Animal Health Supply, Dublin, OH, USA), perfused intracardially with phosphate buffered saline (PBS) and fixed in a 4% paraformaldehyde solution in PBS. In the studies involving the expression of cortical cell layer markers (Cux1, Foxp2 and Tbr1), whole brain samples were analyzed from three different control and experimental mice at post‐natal day 11 (P11). However, more than 11 mice per group (mutant and control), age ranging from P7 to P20, underwent histopathological analysis of the brain. Except where mentioned, sections were prepared from the whole brain at bregma 1 mm, bregma −2.30 mm and bregma −4.30 mm and included all six layers of both hemispheres. Two sections from each of these three regions were prepared for each of the brains per group, which were closely matched using anatomical landmarks from the rat brain with stereotaxic coordinates as a reference resource 45. An average of 32 randomly selected areas throughout the cortex, including both hemispheres, were photographed. Cell counting was performed using a non‐stereological method, which is based on the notion that cells have a single nucleus. Thus, counting nuclei is equivalent to counting cells. The total number of nuclei with the specific cell phenotype under study (eg, NeuN, Tbr1, Foxp2, Cux1) was counted on each of the photographs (magnification 400×) using ImageJ software (NIH, Bethesda, MD). All nuclei on the field were counted. Sections were counterstained with hematoxylin and the relative proportion of cells expressing the marker of interest was expressed as a fraction of total cells in the image from control and mutant mice. Using this approach, >5000 cells were analyzed from each mouse brain. Counts were then averaged over the ∼32 images per brain and expressed as the number of cells per mm2 using ImageJ conversion software. Hematoxylin and Nissl staining were performed using standard protocols.
Immunohistochemical staining was performed as described previously 54. Briefly, brains (three control and three mutant brains) were embedded in paraffin and sectioned at 5 μm. Coronal sections at bregma 1 mm, bregma −2.30 mm and bregma −4.30 mm were collected onto plus‐charged glass slides (Fisher Scientific, Pittsburgh, PA, USA). Paraffin sections were first de‐waxed in xylene, rehydrated in an alcohol gradient, permeabilized in 0.1% triton and reacted with specific primary and secondary antibodies. Primary antibodies were diluted 1:200 in blocking solution and incubated over night at 4°C. Secondary biotinylated antibodies were diluted 1:200 in blocking buffer and incubated for 30 minutes at 37°C. Images were obtained using an inverted Zeiss microscope (Carl Zeiss Microscopy, Gottingen, Germany) (Observer. D1) attached to an AxioCam HRc (Carl Zeiss Microscopy, Gottingen, Germany) with AxioVision LE software. The ultravision detection system (Thermo Scientific, Thermo Fisher Scientific Inc., Fremont, CA, USA) was used for development of 3,3'diaminobenzidine (DAB) plus substrate system ready‐to‐use horseradish peroxidase streptavidin (RTU) as the chromogen. Antibodies were obtained from the following sources: FOXP2 rabbit polyclonal (Abcam, Cambridge, MA, USA); mouse anti‐NeuN (Millipore, Darmstadt, Germany); Tbr1 rabbit polyclonal (Abcam); CDP (Cux1) rabbit polyclonal (Santa Cruz Biotechnology, Dallas, Texas, USA); anti‐GAD67 (Abcam). NeuN and anti‐GAD67 immunostaining was performed using the Vector M.O.M Immunodetection Kit according to the manufacturer's recommendations (Vector Laboratories, Burlingame, CA) to minimize background staining. Data analysis was performed using Excel software and statistical differences were determined using the unpaired (two samples) Student's t‐test.
Golgi‐Cox staining was performed as described by Anderson and Felten 1. Whole brains from six different mice (three control and three mutant brains) were placed in Golgi‐Cox solution at room temperature in the dark for 30 days. After the impregnation period, brains were placed in 30% sucrose for 3 to 5 days at room temperature in the dark, prior to vibratome (VT 100E, Leica, Wetzlar, Germany) sectioning in 6% sucrose. Sections (200 μm) were first incubated in the dark in ammonium hydroxide for 30 minutes and rinsed in distilled water for 1 minute. After rinsing, sections were fixed for 30 minutes in Kodak fixative solution, counterstained with 1% toluidine blue solution and mounted onto plus‐charged glass slides (Fisher Scientific). Images were obtained using an inverted Zeiss microscope (Observer. D1) attached to an AxioCam HRc with AxioVision LE software. Data were analyzed using Excel software and statistical differences were determined using the Student's t‐test.
Electron microscopy analysis of dendritic spines
For electron microscopy studies, coronal sections of the fixed brains were prepared and the tissue was postfixed in 2% osmium tetroxide in sodium cacodylate (NaCAC) buffer, stained en bloc with 2% uranyl acetate, dehydrated using a graded ethanol series and embedded in Epon‐Araldite resin (Electron Microscopy Sciences, Hartfield, PA). Thin sections were cut with a diamond knife using a Leica EM UC6 ultramicrotome (Leica Microsystems, Inc, Bannockburn, IL, USA), collected on copper grids and stained with 2% uranyl acetate and lead citrate. Cells were observed in a JEM 1230 transmission electron microscope (JEOL USA Inc., Peabody, MA, USA) at 110 kV and imaged with an UltraScan 4000 CCD camera and First Light digital camera controller (Gatan Inc., Pleasanton, CA, USA). Images were acquired from the primary somatosensory cortex which spanned all six layers. In these experiments, two control and two mutant brains were used. Four sections from each brain were generated and 20 microscope fields per brain were randomly selected at the same magnification to photograph. The total number of axospinous synapses in each of the 20 randomly selected images from each brain was counted using cell counter plug‐ins from ImageJ software. The counts were then normalized by calculating the number of synapses per μm2. Only asymmetric synapses were counted, which are defined as contacts with a clear presynaptic terminal domain (visible synaptic vesicles) and postsynaptic density (PSD) 2. Data were analyzed using Excel software and statistical differences were determined using the Student's t‐test.
Preparation and immunocytochemical staining of hippocampal neurons
Cell cultures were prepared from hippocampi of P1 mice using a modified method previously described 3, 12. Briefly, hippocampi were dissected and collected in Leibovitz's medium and dissociated by treatment with trypsin (0.125% for 15 minutes at 37°C). The tissue was triturated and passed through a 40 μm cell strainer. The supernatant was removed after centrifugation at 1000 rpm for 5 minutes. Cells were plated onto poly‐L‐lysine treated glass coverslips and cultured for 3 to 4 h in MEM‐F12 with 10% fetal bovine serum (FBS) and penicillin‐streptomycin. After allowing cells to adhere to the substrate, coverslips were transferred into dishes containing neurobasal supplemented with B‐27, L‐glutamine, penicillin‐streptomycin and 55 mM beta‐mercaptoethanol. Immunocytochemistry staining using a synaptophysin antibody (Abcam) was performed as described previously 55. Confocal images were obtained using a Zeiss microscope and LSM510 software (Carl Zeiss Microscopy, Gottingen, Deutschland).
Results
Loss of Lgi1 leads to cortical dysplasia
A detailed histopathological analysis of the hippocampus and cortex using NeuN staining was performed on brains obtained from Lgi1 null mice and their wild‐type littermates at P11. P11 was chosen because it was before the earliest day (P12) that seizures were observed in our colony of Lgi1 null mice, although the majority occurred later, between days 14 and 21. Thus, any abnormality observed at this age is unlikely to result from the effect of persistent, intermittent seizures. NeuN analysis of the hippocampus did not identify any differences between the wild‐type and mutant null mice (Supporting Information Figure S1). In contrast, analyzing the somatosensory cortex (Figure 1), we observed a subtle disruption of the ordered six‐layered tangential structure in the mutant null mice, with loss of organization of both horizontal as well as columnar arrangement of neurons, lack of size gradient and directional abnormalities. The major abnormality was dyslamination of layers II–IV, which was associated with an increase in the average number of neurons per mm2, compared with the cortex from wild‐type littermates (Figure 1, Supporting Information Figure S2). Because it was not possible to clearly distinguish layers II–IV, it was not possible to accurately count the total number of neurons in each of these individual layers in the mutant mice, but there were significantly more neurons in the mutant mice throughout layers II–IV compared with wild‐type litter mates (Figure 1). Cortical layers V and VI showed minimal dyslamination, where the large and small pyramidal and non‐pyramidal neurons were not well positioned or directionally oriented. Microcolumns, sometimes associated with cortical dysplasia 6 were not noted. Histopathological analysis of mice at P7 showed the same cortical dysplasia seen in the P11 mice (not shown) demonstrating that the abnormal phenotype is not an effect associated with later maturation. In the somatosensory cortex, there appears to be a more haphazard admixture of small pyramidal neurons with more rounded granular or immature neurons in the mutant mice which were concentrated in layers II and IV, consistent with the interpretation of superficial cortical dyslamination (Figure 1).
Figure 1.
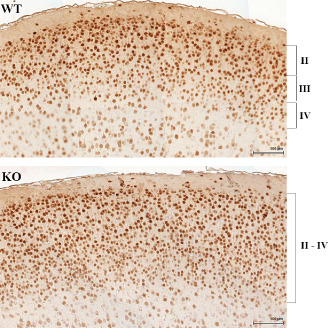
Cortical dysplasia in LGI1 null mice. Comparison of the NeuN‐stained somatosensory cortex shows the increased cellularity in the outer layers II–IV in the mutant null (KO) mouse compared with wild‐type (WT) littermates that leads to blurring of the outer cortical layers.
Deletion of the Lgi1 gene perturbs migration of late‐born cortical neurons
Neurocorticogenesis in mice begins around E10.5 and extends through E17.5–18.5. Normally, preplate cells are born earliest, during E10.5–E13.5, followed by cortical plate cells during E13.5–E16.5. The cortical layers are populated in order, from the deepest (layer VI) to the most superficial (layer II). Early‐born neurons that will populate the lower layers (V–VI) are generated at ∼E12.5, compared with late‐born neurons which will populate cortical cell layers II–IV 8, 16.
To determine the origin of the cells causing the increased cell numbers in the upper layers of the cerebral cortex in P11 mutant null mice, we analyzed cells using the Tbr1, Foxp2 and Cux1 layer‐specific cell markers.
The Tbr1 transcription factor is expressed shortly after cortical progenitor cells begin to differentiate and so it is typically expressed in cells in the lower cortical layers, which identifies early‐born neurons that will occupy the preplate and layer VI 23. No significant difference in the number of Tbr1‐expressing cells using anti‐Tbr1 antibodies was seen between the mutant and wild‐type mice (data not shown), suggesting that the differentiation and radial migration of early‐born neurons have not been significantly affected by loss of Lgi1.
Foxp2 is expressed in a subtype of Tbr1 neurons that populate layers VI and IV 24. Analysis of these cells using an anti‐Foxp2 antibody showed a 72% increase (control: 1.83 ± 0.2 cells/mm2; Lgi1 null: 3.1 ± 0.1 cells/mm2) of Foxp2‐positive neurons within layer IV of the cortex of Lgi1 null mice (Figure 2), including the motor and sensory cortices. There was also a significant increase in the average number of neurons per square area of Foxp2‐expressing cells in layer VI, although this was not as pronounced as in layer IV (control 1.85 ± 0.08 cells/mm2; Lgi1 null: 2.2 ± 0.1 cells/mm2).
Figure 2.
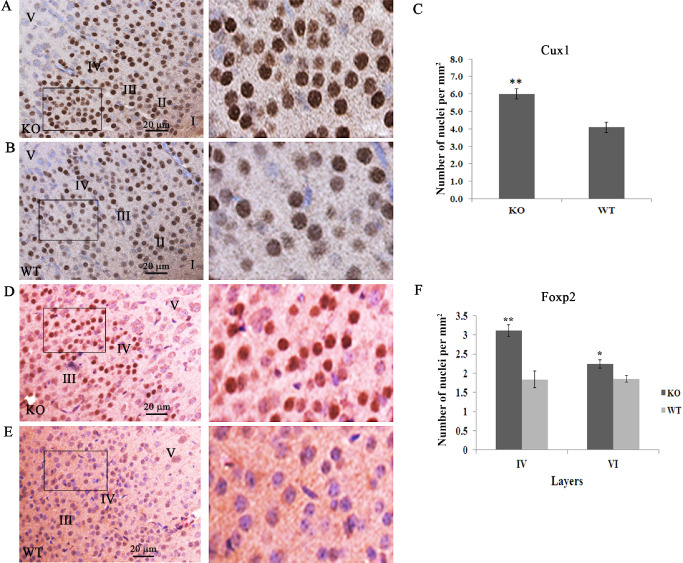
Increased cellularity in layers II–IV involves late‐born neurons. Increased cellularity of Cux1‐expressing cells in the outer cortical layers is demonstrated using Cux1‐specific antibodies in mutant null (A) compared with normal (B) mice. The boxed areas are shown at higher magnification on the right. Quantitation of Cux1‐positive cells in layers II–IV shows a ∼40% increase in the mutant mice (C). The same increase was seen for Foxp2‐positive cells in the mutant mice (D) compared with normal (E). The boxed areas are shown at higher magnification on the right. Quantitation in layers IV and VI (F) shows a significant difference in the number of Foxp2‐positive cells. (**P < 0.001; *P < 0.05).
Cux1 has been identified as a restricted marker for neurons in the upper cortical layers (II–IV) and is highly expressed in the somatosensory cortex 17, 21. Using an anti‐Cux1 antibody, we demonstrated that there is a 47% increase (control: 4.1 ± 0.3 cells/mm2; Lgi1 null: 6.0 ± 0.3 cells/mm2) in the number of neurons in layers II–IV in the P11 Lgi1 null cortex, compared with littermate controls (Figure 2 and Supporting Information Figure S3). We found no difference in the number of cells per square area using Nissl staining (control 193.3 ± 25.6 cells/mm2; Lgi1 null: 192.3 ± 31 cells/mm2). Thus, loss of Lgi1 appears to result in alteration of either neuronal migration or terminal translocation 38, 39 throughout layers II–VI of motor and sensory cortices, but preferentially affects late developing neurons of layers II to IV. There also appears to be relatively haphazard admixture of pyramidal neurons and the rounded granular neurons in layer III in the mutant mice, so that layers III and IV appear to be merging imperceptibly in these mice compared with wild‐type mice. Similar changes were seen in the auditory cortex (Supporting Information Figure S4).
Using an anti‐GAD67 antibody to identify GABAergic interneurons throughout the cortex (Figure 3), we observed no significant difference between the number of cells in the wild‐type mice (30.63 ± 1.32 cells/mm2) compared with the mutant null (27.36 ± 1.59 cells/mm2) mice (P = 0.125).
Figure 3.
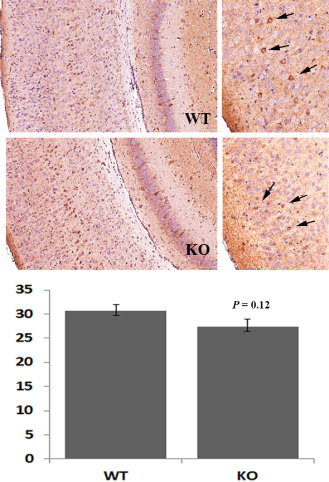
Distribution of interneurons is unaffected by loss of LGI1. GAD67 staining of the somatosensory cortex was used to determine the relative proportions of GABAergic interneurons between the wild‐type (WT) and mutant null (KO) mice. In the right hand panels, the relative distribution of the GAD67 interneurons (arrows) can be seen. Quantification using three different fields from five different areas of the cortex of five mice from each subgroup shows no significant different in interneurons between the mutant and WT mice.
Although Golgi‐Cox staining is not quantitative because of the random staining of neurons by this procedure, counting neurons in the various layers from the Golgi‐stained sections of somatosensory cortex (Figure 4) supports the observation of increased number of neurons in the outer cortical layers. Six different fields were selected randomly from sections of cortex from three mutant null mice and three wild‐type mice. Because of the blurring of the margins within layers II–IV, we could not accurately define the individual layers in the mutant mouse. When the total number of stained neurons in layer II–IV was compared, there was a significant increase in the mutant mice (Figure 4). When we compared layers V and VI in the same fields, there was no difference between the mutant null (0.3258 ± 0.06 cells/per mm2) and the wild‐type (0.3648 ± 0.02 cells/mm2) mice. Similarly, in layer VI, there were 0.3772 ± 0.04 cells/mm2 and 0.3823 ± 0.06 cells/mm2 in the mutant and wild‐type mice, respectively. These data support the primary observation that there are increased numbers of neurons restricted to the outer cortical layers.
Figure 4.
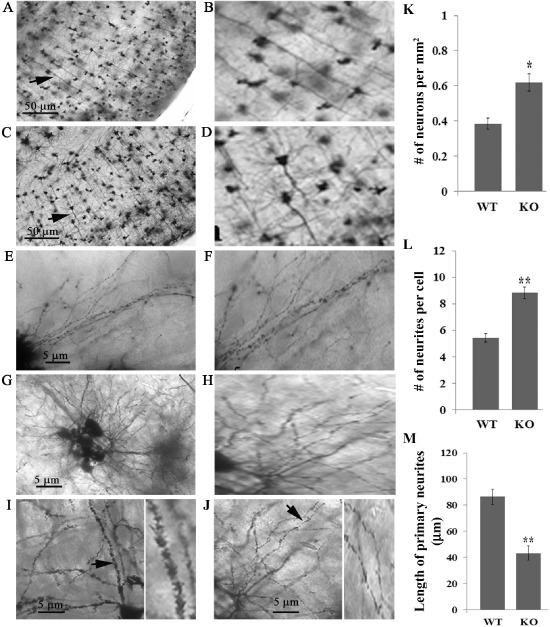
Lgi1 mutant null mice exhibit abnormalities in axon structure. Representative image of Golgi‐Cox staining from normal (A) and mutant null (C) mice at P11 shows misalignment of axons in the mutant mice compared with the normal mice (arrows). Right panels (B and D) show the arrowed neurons from A and C at higher magnification. The dendritic tree complexity is also increased in mutant mice (G) compared with normal mice (E). Right panels (F and H) show a higher magnification segment of the axon from the mutant neurons (G) compared with control neurons (E). When dentrite morphology is compared, the normal mice (I) show long well‐directed dendrites with smooth contours and pointed ends (arrow). A higher magnification image is shown to the right. In the mutant mice (J), shorter and irregular dendrites are seen with fuzzy contours and round‐headed ends (arrow). A higher magnification is shown to the right. Quantitation of the images shows increased neurons in the outer cortical layers of the mutant mice (K) as well as increased number of neurites (L) and decreased neurite length (M).
Lgi1 null mice have reduced numbers of axospinous synapses in the cerebral cortex
NeuN staining defines total neurons in the cortex but does not allow examination of axon and neurite structure from these neurons. To determine whether loss of Lgi1 affects these structures, we performed Golgi‐Cox staining of the Lgi1 null cortex at P11 and showed that the neurons in layers II to IV (and to a lesser extent layers V and VI) showed highly branched pyramidal neurons with short and poorly aligned axons, admixed with small oval‐to‐round cells with branched disoriented dendrites, suggesting immature neurons (Figure 4). To investigate the abnormal neuronal fine structure, we studied higher magnifications of Golgi‐Cox staining as well as higher resolution electron microscopic analysis. Golgi‐Cox staining revealed altered spine morphology as well as a decrease in the number of spines along the dendrites of the mutant null neurons. The Lgi1 null spines were round headed, short in length and undersized (Figure 4). This phenotype is characterized of increased neuronal activity that results from increased calcium influx, which may lead to seizures 52. In contrast, spines in neurons from wild‐type mice were longer and the heads were sharp (Figure 4). Analysis of the neurons in cortical layers II–IV showed that there were increased numbers of primary neurites in the cortical neurons from the mutant mouse. To quantify this observation, we compared the number of neurites in the mutant and wild‐type mice. Morphologically distinct neurons within the sections were identified at random and, as shown in Figure 4, after counting from only 12 neurons in mutant and wild type, the difference was highly significantly increased (P = 1.16E‐06) in the mutant mice. In addition, the length of the neurites in the mutant mice was significantly smaller (P = 1.06E‐05) than those in the wild‐type littermates (Figure 4).
To determine if a reduced number of spines in the mutant null neurons correlated with a similar reduction in the distribution of functional synapses, we analyzed the incidence of PSD using electron microscopy (Figure 5). The PSD identifies the electron dense postsynaptic region juxtaposed to the (active) presynaptic vesicle release site. The incidence of spine‐localized, asymmetric (axospinous) synapses in the mutant null cortex was reduced by 37.5% (control: 0.14 ± 0.007 spines/μm2; Lgi1 null: 0.088 ± 0.007 spines/μm2). In addition, the number of vesicle‐loaded synapses was decreased in the mutant mice and, although there appears to be a larger number of synapses, many of them contained few, if any, vesicles suggesting they are non‐functional (Figure 5). These findings suggest that Lgi1 may be one genetic determinant of neuronal plasticity and provide insights into its role in regulating cortical lamination and synapse formation.
Figure 5.
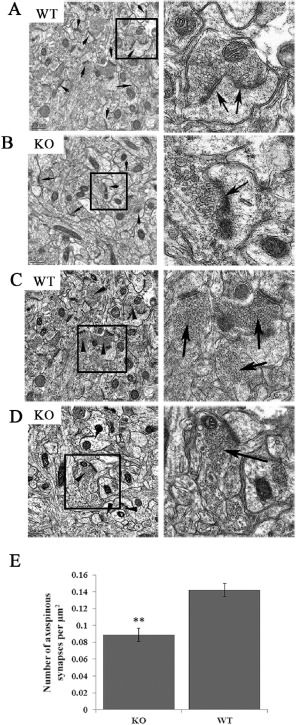
Reduced spine distribution in the cortex from Lgi1 mutant mice. Electron micrographs from the cortex of P17 wild‐type (A) and mutant (KO) null mice (B) show a reduced PSD frequency (arrows) in the mutant mice. Right panels show spines in higher magnification. Analysis of vesicle‐loaded synapses (D) shows increased smaller, irregular and attenuated axonal boutons in the mutant null mice with fewer synaptic vesicles admixed with a few well‐formed vesicle‐rich asymmetric synapses (right) compared with mostly well‐formed axonal boutons in P17 normal mice (C, shown at higher magnification on the right). Quantification of functional synapses (see text) demonstrates a ∼35% reduction in the mutant mice (E) (**P < 0.001).
Neurite development and synapse formation is impaired in Lgi1 null neurons
The EM studies demonstrated that synapse development and distribution are impaired in the Lgi1 null cortex. Hippocampal neurons from wild‐type and mutant null cells were introduced into cell culture and the neurite networks that built up were revealed using anti‐synaptophysin antibodies. Axons and dendrites that emerge in these cultures form synapses with one another and exhibit a normal synaptic polarity where axons are typically presynaptic and dendrites are postsynaptic 4. After ∼15 days in culture, when both axons and dendrites are well developed, immunostaining for synaptophysin was concentrated in brightly fluorescence puncta around cell bodies and along thinning radially oriented processes that are identified as dendrites (Figure 6). In both of these cultures, synaptophysin accumulation could be seen along the length of the neurons (Figure 6). Synaptophysin immunoreactivity could also be detected in the perinuclear region of the cell body, where the Golgi complex is located. The observed distribution of synaptophysin in these cells is consistent with the distribution of presynaptic boutons in hippocampal neurons as reported by Bartlett and Banker 4. The number of synapses in cultured neurons correlates with the focal accumulation of synaptic vesicle proteins such as synaptophysin. To compare neuronal cultures prepared from the wild‐type and Lgi1 mutant null mice, we counted the number of synaptophysin‐positive boutons per 100 μm length of the neurites. From confocal images, three different fields were randomly selected from nine different images (n = 27). In this analysis, the mean number of boutons per unit length was 6.3 ± 1.46 in the wild‐type mice compared with 1.85 ± 1.44 in the Lgi1 null mice (P = 8.54E‐06). In the neurons from the mutant null mice, therefore, there were fewer neurites extending from the cell bodies and reduced synapse formation, compared with those seen in cells from wild‐type littermate (Figure 6A).
Figure 6.
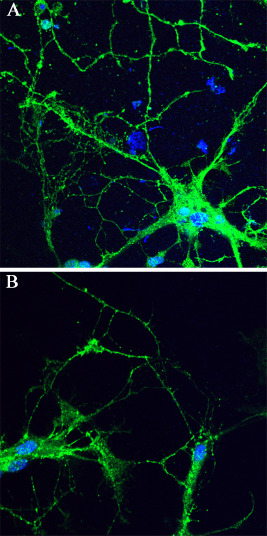
Analysis of neurite network formation in vitro. Cultures of hippocampal neurons from wild‐type (A) and Lgi1 null (B) mice stained with anti‐synaptophysin antibodies shows vastly reduced neurites and synapse formation in hippocampal neurons after 14–15 days in culture.
Discussion
The development of the cerebral cortex involves a complex cascade of events involving precursor cell proliferation, well‐regulated cell migration and eventual mature cortical organization, including acquisition of differentiated neuronal morphology, synaptogenesis and functionality. During early embryogenesis, neural progenitor cells in the ventricular and subventricular zones proliferate to generate the neurons that will form the cortical plate. After exiting the cell cycle, neurons then migrate into the cortical plate, successively bypassing each other to form the normal laminar structure of the cerebral cortex, where the deep layers (V–VI) include the early‐born neurons and the outer layers (II–IV) contain the late‐born neurons. Once these neurons have reached their position in the cortex, they extend neurites and establish synaptic connections that perform specific functions 2.
Cortical dysplasias are congenital malformations of cortical development 5, 9, where cortical neurons fail to migrate appropriately, leading to a disorganization of the normal structure of the cortex, which is increasingly being diagnosed as a source of symptomatic focal epilepsy in pediatric and adult patients 7, 46, 57. Local interactions of dysmature neurons with normal neurons are thought to promote seizures 9. The congenital nature of these focal cortical dysplasias (FCDs) suggests that they may result, at least in some cases, from inherited genetic abnormalities. Recently, a revised classification of FCDs placed this defect in the neuronal migration disorders 6, 42. FCD in younger patients 21, in addition to showing an abnormal laminar architecture in the cortex and hypercellularity in layer II, showed distinct populations of immature cells at the interface of layers I and II 58. In this system, FCDs are classified into types I, II and III based on histopathological appearance, where type 1a shows abnormal radial cortical lamination, type 1b shows abnormal tangential cortical lamination and type 1c shows a combination of abnormal radial and tangential cortical lamination 6. FCD type 1b is further characterized by the presence of immature and small pyramidal neurons in layers II–IV with disoriented dendrites and abnormal development of the six‐layer tangential arrangement of the cortex. Other pathologies within this category are restricted to abnormal layering of layers II or IV or both. While it is acknowledged that mouse and human brain development differ in many respects, the cortical dysplasia associated with Lgi1 deficiency results from subtle abnormalities in layers II–IV of the cerebral cortex, characterized by increased numbers of cells in these layers leading to dyslamination. Although distinct, the pathology associated with the loss of Lgi1 most closely resembles the International League Against Epilepsy (ILEA) FCD type 1b.
Inactivation of Lgi1 leads to a complex series of structural changes in the cortex in mutant null mice leading to cortical dysplasia manifesting as subtle abnormalities in layers II–IV of the somatosensory and auditory cortex, characterized by increased numbers of cells in these layers leading to dyslamination. Cortical dysplasia involving increased numbers in Cux1‐ and Foxp2‐expressing neurons in superficial cortical cell layers represents a novel observation in cortical laminar malformations which have been more extensively studied for layers V and VI, where overlapping dispersion of layer V cells beyond their usual cortical layer localization have been demonstrated, indicating misplaced neurons 48.
Cortical dysplasia has been suggested as one underlying cause of seizures 7 and the observation of cortical dysplasia in Lgi1 null mice provides one potential explanation for the observed spontaneous seizures seen in these animals. It has also been shown, however, that Lgi1 null mice exhibit abnormal synaptic transmission in neurons in mature cortex and hippocampus associated with abnormalities at the pre‐ and postsynaptic membranes 19, 63. Lgi1 also binds to the ADAM23 and ADAM22 proteins located on the pre‐ and postsynaptic membranes, respectively 18, 30, 49. One suggestion has been that Lgi1 forms a bridge across these membranes to facilitate synaptic transmission 18, although this mechanism has been modified based on ultrastructural predictions of binding sites for the ADAM proteins within Lgi1 32. The demonstration, however, that Lgi1 co‐immunoprecipitates with the Kv1 channel, by binding to the Kvβ subunit, implicated Lgi1 in synaptic function 51. LGI1 is a secreted protein, however, how it binds to an intracellular component of a potassium channel is not clear. Demonstrations that Lgi1 also binds to the Kv1.4 and Kvβ proteins in the axonal terminal 51 and that it removes the rapid inactivation of the channel mediated by Kvβ suggested that this tight association with presynaptic channels may also be involved in the seizure phenotype. Indeed, we noticed that the excitatory synaptic transmission was increased in the absence of Lgi1 in mutant mice, although the effect was weak 63.
Relating genotype–phenotype observations in mutant null mice with those seen in human genetic disease, however, has its limitations. The Lgi1 null mouse, for example, represents the extreme genetic form of this disease with both alleles inactivated which leads to a seizure phenotype that is completely penetrant. In ADLTE, however, the penetrance of Lgi1 mutations is lower, as might be expected as the heterozygous mutations in these patients affects only one allele which gives rise to a milder phenotype through haploin sufficiency. Heterozygous mutations in mice do not give rise to overt seizures, but it has been clearly shown that they are more susceptible to pharmacologically induced seizures than wild‐type littermates 10. Unfortunately, there have been no reports of histological or ultrastructural examination of the cortex from ADLTE patients to determine whether a similar dyslamination is seen in this disorder. Although ADLTE patients typically do not show prominent abnormalities using magnetic resonance imaging (MRI) 14, 34, 44, isolated reports have described various possible malformations in the brains of these patients. Kobayashi et al 27, for example, suggested the possibility of mild temporal lobe abnormalities in members of a large family with ADTE and Pisano et al 43 suggested that LGI1 mutations could determine subtle structural changes in the lateral temporal cortex underlying focal epilepsy. Using voxel‐based analysis, Tessa et al 60 suggested that structural abnormalities occurred in the left lateral temporal lobe in patients with LGI1 mutations.
In this study, the majority of the analysis of the cortex was performed in mice at P11, even though the majority of mice showed onset of grossly observable seizures between days 14 and 21, because we noted seizures in rare animals at P12. The same latency was reported by Fukata et al 19. In the colony described by Charbrol et al 10, however, seizures were reported at P10, which were accompanied by epileptiform electroencephalography (EEG) recordings. Although we did not perform EEG recordings in our colony, some of the Lgi1 null mice might possibly have experienced seizure activity that went unnoticed and that the pathology of cortex in these mice could be in part due to these seizures. We do not, however, think that this is the case as (i) we evaluated the CD in six different mice and all showed the same abnormalities; (ii) we also performed histological analysis of P7 mice which showed the same CD as the P11 mice and there are no reports of Lgi1 null mice experiencing seizures at this age; and (iii) no abnormalities were observed in our analysis of the hippocampus in the Lgi1 null mice, which might have been expected if they had experienced seizures as the hippocampus is typically more severely affected than the cortex as a result of seizures.
Developmental abnormalities identified in the mutant null mice point to a much more complex etiology than the cortical dysplasia. EM ultrastructural analysis of functional synapses in the Lgi1 null mice showed an increased number of non‐functional synapses as determined by the morphology of synaptic vesicles in the termini. In many cases, although connections could be seen, there were no PSD and, in many of these cases, there were few vesicles. Decreased PSD indicates decreased asymmetric (presumably glutaminergic) excitatory synapses, which seems paradoxical to the occurrence of seizures. This observation, in the context of increased smaller, irregular and attenuated axonal boutons, with fewer synaptic vesicles, however, is consistent with the idea that many highly branched axons may not be making functional connections. Moreover, the admixture of well‐formed, vesicle‐rich asymmetric synapses, forming PSD with attenuated synaptic boutons of highly branched axons, is consistent with reports that local interactions of dysmature neurons with normal neurons promotes seizures 9. Of note, no significant loss of neuropil, multisynaptic giant axonal spines or astrogliosis was identified. The ultrastructural observations described here, therefore, are inconsistent with reported post‐seizure changes 2, 62 and favor a developmental malformation. The demonstration of increased Cux1‐expressing cells together with abnormalities in dendrite formation in Lgi1 null mice are consistent with the observations of altered numbers and orientation of dendritic branches leading to neuronal dysfunction seen in cognitive disorders including epilepsy 13 and are often the only morphologic defects detectable in post‐mortem studies of non‐syndromic mental retardation 15.
In summary, we have shown that Lgi1, a non‐ion channel gene 11, 33, plays an important role in cortical development and synapse plasticity. The dysplasia phenotype seen in mice null for Lgi1 is hypercellularity in the outer layers leading to a blurring of the cortical layers. Molecular studies implicate Lgi1 in a variety of signaling cascades that impact on actin cytoskeleton reorganization that is required for cell movement 31. While it is possible that loss of Lgi1‐regulated signaling cascades promotes excessive glial‐guided migration of neurons during development, we cannot exclude the possibility that Lgi1 may also impact mechanisms that lead to somatic translocation 38, 39. The Lgi1 null mouse model, however, provides a unique opportunity to examine the relationship between cortical dysplasia and epilepsy.
Supporting information
Figure S1. Representative areas of NeuN stained regions of the hippocampus in wild‐type (WT) and mutant null (KO) mice shows no difference in cell positioning or organization between the two groups.
Figure S2. Abnormal cortical lamination in Lgi1 null mice. Representative analysis of NeuN stained cortex from P11 wild‐type (A) and Lgi1 null (D) mice demonstrates hypercellularity in the superficial cortex with a subtle blurring of the individual cortical layers in the mutant null mice. At higher resolution (E and F respectively), the increased cellularity in the outer layers in the null mice is clearly seen compared with normal littermates (B and C). A comparison between P17 wild‐type mice, shown in panel G (H/I at higher magnification) and mutant null mice (J–L), which had not experienced seizures shows the same phenotype (shown at higher resolution in K and L, respectively). Scale bars in G/J and H/K represent the same region of layers I–IV for comparison.
Figure S3. Comparison of the CUX1‐stained somatosensory cortex shows the increased cellularity in the outer layers II–IV in the mutant null (KO) mouse compared with wild‐type (WT) littermates. In addition, there is a relatively haphazard admixture of pyramidal neurons and the rounded granular neurons in the region for layer III in the mutant mice, so that layers III and IV appear to be merging imperceptibly in these mice compared with WT mice.
Figure S4. Comparison of the NeuN‐stained auditory cortex shows the increased cellularity in the outer layers II–IV in the mutant null (KO) mouse compared with wild‐type (WT) littermates.
References
- 1. Anderson WJ, Felten DL (1982) A modified Golgi‐Cox technique for morphological characterization of serotonergic neurons. J Histochem Cytochem 30:750–755. [DOI] [PubMed] [Google Scholar]
- 2. Barkovich AJ, Kuzniecky RI, Jackson GD, Guerrini R, Dobyns WB (2005) A developmental and genetic classification for malformations of cortical development. Neurology 65:1873–1887. [DOI] [PubMed] [Google Scholar]
- 3. Bartlett WP, Banker GA (1984) An electron microscopic study of the development of axons and dendrites by hippocampal neurons in culture. I. Cells which develop without intracellular contacts. J Neuroscience 4:1944–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bartlett WP, Banker GA (1984) An electron microscopic study of the development of axons and dendrites by hippocampal neurons in culture. II. Synaptic relationships. J Neuroscience 4:1954–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Battaglia G, Becker AJ, Loturco J, Represa A, Baraban SC, Roper SN, Vezzani A (2009) Basic mechanisms of MCD in animal models. Epileptic Disord 11:206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blümcke I, Thom M, Aronica E, Armstrong DD, Vinters HV, Palmini A et al (2011) The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc task force of the ILAE diagnostic methods commission. Epilepsia 52:158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buoni S, Zannolli R, Miracco C, Macucci F, Hayek J, Burroni L et al (2008) Focal cortical dysplasia type 1b as a cause of severe epilepsy with multiple independent spike foci. Brain Dev 30:53–58. [DOI] [PubMed] [Google Scholar]
- 8. Caviness VS, Bhide PG, Nowakowski RS (2008) Histogenetic process leading to the laminated neocortex: migration is only a part of the story. Dev Neurosci 230:82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cepeda C, Andre VM, Levine MS, Salanion N, Miyata H, Vinters HV, Mathern GW (2006) Epileptogenesis in pediatric cortical dysplasia: the dysmature cerebral developmental hypothesis. Epilepsy Behav 9:219–235. [DOI] [PubMed] [Google Scholar]
- 10. Chabrol E, Navarro V, Provenzano G, Cohen I, Dinocourt C, Rivaud‐Péchoux S et al (2010) Electroclinical characterization of epileptic seizures in leucine‐rich, glioma‐inactivated 1‐deficient mice. Brain 133:2749–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chernova O, Somerville RPT, Cowell JK (1998) A novel gene, LGI1, from region 10q24, is rearranged and downregulated in malignant brain tumors. Oncogene 17:2873–2881. [DOI] [PubMed] [Google Scholar]
- 12. Coras R, Siebzehnrubl FA, Pauli E, Huttner HB, Njunting M, Kobow K et al (2010) Low proliferation and differentiation capacities of adult hippocampal stem cells correlate with memory dysfunction in humans. Brain 133:3359–3372. [DOI] [PubMed] [Google Scholar]
- 13. Cubelos B, Nieto M (2010) Intrinsic programs regulating dendrites and synapses in the upper layer neurons of the cortex. Commun Integr Biol 3:483–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Di Bonaventura C, Carni M, Diani E, Fattouch J, Vaudano EA, Egeo G et al (2009) Drug resistant ADLTE and recurrent partial status epilepticus with dysphasic features in a family with a novel LGI1mutation: electroclinical, genetic, and EEG/fMRI findings. Epilepsia 50:2481–2486. [DOI] [PubMed] [Google Scholar]
- 15. Dierssen M, Ramakers GJ (2006) Dendritic pathology in mental retardation: from molecular genetics to neurobiology. Genes Brain Behav 5:48–60. [DOI] [PubMed] [Google Scholar]
- 16. Fairen A, Cobas A, Fonseca M (1986) Times of generation of glutamic acid decarboxylase immunoreactive neurons in mouse somatosensory cortex. J Comp Neurol 251:67–83. [DOI] [PubMed] [Google Scholar]
- 17. Ferrere A, Vitalis T, Gingras H, Gaspar P, Cases O (2006) Expression of Cux‐1 and Cux‐2 in the developing somatosensory cortex of normal and barrel‐defective mice. Anat Rec A Discov Mol Cell Evol Biol 288:158–165. [DOI] [PubMed] [Google Scholar]
- 18. Fukata Y, Adesnik H, Iwanaga T, Bredt DS, Nicoll RA, Fukata M (2006) Epilepsy‐related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science 313:1792–1795. [DOI] [PubMed] [Google Scholar]
- 19. Fukata Y, Lovero KL, Iwanaga T, Watanabe A, Yokoi N, Tabuchi K et al (2010) Disruption of Lgi1‐linked synaptic complex causes abnormal synaptic transmission and epilepsy. Proc Natl Acad Sci U S A 107:3799–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gu W, Brodtkorb E, Piepoli T, Finocchiaro G, Steinlein OK (2005) LGI1: a gene involved in epileptogenesis and glioma progression? Neurogenetics 6:59–66. [DOI] [PubMed] [Google Scholar]
- 21. Hadjivassiliou G, Martinian L, Squier W, Blumcke I, Aronica E, Sisodiya SM, Thom M (2010) The application of cortical layer markers in the evaluation of cortical dysplasias in epilepsy. Acta Neuropathol 120:517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Head K, Gong S, Joseph S, Wang C, Burkhardt T, Rossi MR et al (2007) Defining the expression pattern of the Lgi1 gene in BAC transgenic mice. Mamm Genome 18:328–337. [DOI] [PubMed] [Google Scholar]
- 23. Hevner RF, Shi L, Justice N, Hsueh Y‐P, Sheng M, Smiga S et al (2001) Tbr1 regulates differentiation of the preplate and layer 6. Neuron 29:353–366. [DOI] [PubMed] [Google Scholar]
- 24. Hisaoka T, Nakamura Y, Senba E, Morikawa Y (2010) The forkhead transcription factors, Foxp1 and Foxp2, identify different subpopulations of projection neurons in the mouse cerebral cortex. Neuroscience 166:551–563. [DOI] [PubMed] [Google Scholar]
- 25. Ilkanizadeh S, Lau J, Huang M, Foster DJ, Wong R, Frantz A et al (2014) Glial progenitors as targets for transformation in glioma. Adv Cancer Res 121:1–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kalachikov S, Evgrafov O, Ross B, Winawer M, Barker‐Cummings C, Martinelli Boneschi F et al (2002) Mutations in LGI1 cause autosomal‐dominant partial epilepsy with auditory features. Nat Genet 30:335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kobayashi E, Santos NF, Torres FR, Secolin R, Sardinha LA, Lopez‐Cendes I, Cendes F (2003) Magnetic resonance imaging abnormalities in familial temporal lob e epilepsy with auditory auras. Arch Neurol 60:1546–1551. [DOI] [PubMed] [Google Scholar]
- 28. Kobe B, Kajava AV (2001) The leucine‐rich repeat as a protein recognition motif. Curr Opin Struct Biol 11:725–732. [DOI] [PubMed] [Google Scholar]
- 29. Kunapuli P, Chitta K, Cowell JK (2003) Suppression of the cell proliferation and invasion phenotypes in glioma cells by the LGI1 gene. Oncogene 22:3985–3991. [DOI] [PubMed] [Google Scholar]
- 30. Kunapuli P, Jang G, Kazim L, Cowell JK (2009) Mass spectrometry identifies LGI1‐interacting proteins that are involved in synaptic vesicle function in the human brain. J Molec Neurosci 39:137–143. [DOI] [PubMed] [Google Scholar]
- 31. Kunapuli P, Lo K, Hawthorn L, Cowell JK (2010) Reexpression of LGI1 in glioma cells results in dysregulation of genes implicated in the canonical axon guidance pathway. Genomics 95:93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leonardi E, Andressa S, Vanin S, Busolin G, Nobile C, Tosatto SCE (2011) A computational model of the LGI1 protein suggests a common binding site for ADAM proteins. PLoS ONE 6:E1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lerche H, Jurkat‐Rott K, Lehmann‐Horn F (2001) Ion channels and epilepsy. Am J Med Genet 106:146–159. [DOI] [PubMed] [Google Scholar]
- 34. Michelucci R, Poza JJ, Sofia V, de Feo MR, Binelli S, Bisulli F et al (2003) Autosomal dominant lateral temporal epilepsy: clinical spectrum, new epitempin mutations, and genetic heterogeneity in seven European families. Epilepsy 44:1289–1297. [DOI] [PubMed] [Google Scholar]
- 35. Mitchell KJ, Pinsonm KI, Kelly OG, Brennan J, Zupicich J, Scherz P et al (2001) Functional analysis of secreted and transmembrane proteins critical to mouse development. Nat Genet 28:241–249. [DOI] [PubMed] [Google Scholar]
- 36. Mochida GH, Walsh CA (2004) Genetic basis of developmental malformations of the cerebral cortex. Arch Neurol 61:637–640. [DOI] [PubMed] [Google Scholar]
- 37. Morante‐Redolate JM, Gorostidi‐Pagola A, Piquer‐Sirerol S, Saenz A, Poza JR, Galan J et al (2002) Mutations in the LGI1/Epitempin gene on 10q24 cause autosomal dominant lateral temporal epilepsy. Hum Mol Genet 11:1119–1128. [DOI] [PubMed] [Google Scholar]
- 38. Nadarajah B, Parnavelas JG (2003) Radial glia and somal translocation of radial neurons in the developing cerebral cortex. Glia 43:33–36. [DOI] [PubMed] [Google Scholar]
- 39. Nadarajah B, Alifragis P, Wong RO, Parnavelas JG (2003) Neuronal migration in the developing cerebral cortex: observations based on real‐time imaging. Cereb Cortex 13:607–611. [DOI] [PubMed] [Google Scholar]
- 40. Ottman R, Risch N, Hauser WA, Pedley TA, Lee JH, Barker‐Cummings C et al (1995) Localization of a gene for partial epilepsy to chromosome 10q. Nat Genet 10:56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Owuor K, Harel NY, Englot DJ, Hisama F, Blumenfeld H, Strittmatter SM (2009) LGI1‐associated epilepsy through altered ADAM23‐dependent neuronal morphology. Mol Cell Neurosci 42:448–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Palmini A (2011) Revising the classification of focal cortical dysplasias. Epilepsia 52:188–189. [DOI] [PubMed] [Google Scholar]
- 43. Pisano T, Marini C, Brovedani P, Brizzolara D, Pruna D, Mei D et al (2005) Abnormal phonologic processing in familial lateral temporal lobe epilepsy due to a new LGI1 mutation. Epilepsia 4:118–123. [DOI] [PubMed] [Google Scholar]
- 44. Poza JJ, Sáenz A, Martínez‐Gil A, Cheron N, Cobo AM, Urtasun M et al (1999) Autosomal dominant lateral temporal epilepsy: clinical and genetic study of a large Basque pedigree linked to chromosome 10q. Ann Neurol 45:182–188. [DOI] [PubMed] [Google Scholar]
- 45. Praxinos G, Watson C (1998) The Rat Brain in Stereotaxic Coordinates. Academic Press. [Google Scholar]
- 46. Prayson RA, Melinda LE (1995) Cortical dysplasias: a histopathologic study of 52 cases of partial lobectomy in patients with epilepsy. Hum Pathol 26:493–500. [DOI] [PubMed] [Google Scholar]
- 47. Quiñones‐Hinojosa A, Chaichana K (2007) The human subventricular zone: a source of new cells and a potential source of brain tumors. Exp Neurology 205:313–324. [DOI] [PubMed] [Google Scholar]
- 48. Rossini L, Moroni RF, Tassi L, Watakabe A, Yamamori T, Spreafico R, Garbelli R (2011) Altered layer‐specific gene expression in cortical samples from patients with temporal lobe epilepsy. Epilepsia 52:1928–1937. [DOI] [PubMed] [Google Scholar]
- 49. Sagane K, Ishihama Y, Sugimoto H (2008) LGI1 and LGI4 bind to ADAM22, ADAM23 and ADAM11. Int J Biol Sci 4:387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sagane K, Hayakawa K, Kai J, Hirohashi T, Takahashi E, Miyamoto N et al (2005) Ataxia and peripheral nerve hypomyelination in ADAM22‐deficient mice. BMC Neurosci 6:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schulte U, Thumfaart JO, Klöcker N, Sailer CA, Bildl W, Biniossek M et al (2006) The epilepsy‐linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvβ1. Neuron 49:697–706. [DOI] [PubMed] [Google Scholar]
- 52. Segal M, Korkotian E, Murphy DD (2000) Dendritic spine formation and pruning: common cellular mechanisms? Trends Neurosci 23:53–57. [DOI] [PubMed] [Google Scholar]
- 53. Senechal KR, Thaller C, Noebels JL (2005) ADPEAF mutations reduce levels of secreted LGI1, a putative tumor suppressor protein linked to epilepsy. Hum Mol Genet 14:1613–1620. [DOI] [PubMed] [Google Scholar]
- 54. Silva J, Wang G, Cowell JK (2011) Temporal and spatial expression of the Lgi1 gene during mouse development using a BAC transgenic reporter system. BMC Neurosci 12:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sirerol‐Piquer MS, Ayerdi‐Izquierdo A, Morante‐Redolat JM, Herranz‐Pérez V, Favell K, Barker PA, Pérez‐Tur J (2006) The epilepsy gene LGI1 encodes a secreted glycoprotein that binds to the cell surface. Hum Mol Genet 15:3436–3445. [DOI] [PubMed] [Google Scholar]
- 56. Somerville RP, Chernova O, Liu S, Shoshan Y, Cowell JK (2000) Identification of the promoter, genomic structure, and mouse ortholog of LGI1. Mamm Genome 11:622–627. [DOI] [PubMed] [Google Scholar]
- 57. Spreafico R, Blumcke I (2010) Focal cortical dysplasias: clinical implication of neuropathological classification systems. Acta Neuropathol 120:359–367. [DOI] [PubMed] [Google Scholar]
- 58. Srikandarajah N, Martinian L, Sisodiya SM, Squier W, Blumcke I, Aronica E, Thom M (2009) Doublecortin expression in focal cortical dysplasia in epilepsy. Epilepsia 50:2619–2628. [DOI] [PubMed] [Google Scholar]
- 59. Staub E, Perez‐Tur J, Siebert R, Nobile C, Moschonas NK, Deloukas P, Hinzmann B (2002) The novel EPTP repeat defines a superfamily of proteins with implications in epileptic disorders. Trends Biochem Sci 27:441–444. [DOI] [PubMed] [Google Scholar]
- 60. Tessa C, Michelucci R, Nobile C, Giannelli M, Della Nave R, Testoni S et al (2007) Structural anomaly of left lateral temporal lobe in epilepsy due to mutated LGI1. Neurology 69:1298–1300. [DOI] [PubMed] [Google Scholar]
- 61. Westphal M1, Lamszus K (2011) The neurobiology of gliomas: from cell biology to the development of therapeutic approaches. Nat Rev Neurosci 12:495–508. [DOI] [PubMed] [Google Scholar]
- 62. Witcher MR, Park YD, Lee MR, Sharma S, Harris KM, Kirov SA (2010) Three‐dimensional relationships between perisynaptic astroglia and human hippocampal synapses. Glia 58:572–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yu EY, Wen L, Silva J, Li Z, Head K, Sossey‐Alaoui K et al (2010) Lgi1 null mutant mice exhibit myoclonic seizures and CA1 neuronal hyperexcitability. Hum Mol Genet 19:1702–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Representative areas of NeuN stained regions of the hippocampus in wild‐type (WT) and mutant null (KO) mice shows no difference in cell positioning or organization between the two groups.
Figure S2. Abnormal cortical lamination in Lgi1 null mice. Representative analysis of NeuN stained cortex from P11 wild‐type (A) and Lgi1 null (D) mice demonstrates hypercellularity in the superficial cortex with a subtle blurring of the individual cortical layers in the mutant null mice. At higher resolution (E and F respectively), the increased cellularity in the outer layers in the null mice is clearly seen compared with normal littermates (B and C). A comparison between P17 wild‐type mice, shown in panel G (H/I at higher magnification) and mutant null mice (J–L), which had not experienced seizures shows the same phenotype (shown at higher resolution in K and L, respectively). Scale bars in G/J and H/K represent the same region of layers I–IV for comparison.
Figure S3. Comparison of the CUX1‐stained somatosensory cortex shows the increased cellularity in the outer layers II–IV in the mutant null (KO) mouse compared with wild‐type (WT) littermates. In addition, there is a relatively haphazard admixture of pyramidal neurons and the rounded granular neurons in the region for layer III in the mutant mice, so that layers III and IV appear to be merging imperceptibly in these mice compared with WT mice.
Figure S4. Comparison of the NeuN‐stained auditory cortex shows the increased cellularity in the outer layers II–IV in the mutant null (KO) mouse compared with wild‐type (WT) littermates.