

Abstract
Human glioblastoma multiforme (GBM) is an aggressive cancer with a very poor prognosis. Cripto‐1 (CR‐1) has a key regulatory role in embryogenesis, while in adult tissue re‐expression of CR‐1 has been correlated to malignant progression in solid cancers of non‐neuronal origin. As CR‐1 expression has yet to be described in cerebral cancer and CR‐1 is regulated by signaling pathways dysregulated in GBM, we aimed to investigate CR‐1 in the context of expression in GBM. The study was performed using enzyme‐linked immunosorbent assay (ELISA), Western blotting, polymerase chain reaction (PCR) and immunohistochemistry to analyze the blood and tissue from 28 GBM and 4 low‐grade glioma patients. Within the patient cohort, we found high CR‐1 protein levels in blood plasma to significantly correlate with a shorter overall survival. We identified CR‐1 in different areas of GBM tissue, including perivascular tumor cells, and in endothelial cells. Collectively, our data suggest that CR‐1 could be a prognostic biomarker for GBM with the potential of being a therapeutic target.
Keywords: CR‐1, endothelial proliferation, glioblastoma multiforme, microvasculature, plasma biomarker, tumor niche
Introduction
Glioblastoma multiforme (GBM) brain tumors expand rapidly by infiltrating the surrounding brain tissue and present themselves with extensive endothelial proliferation and multiple necrotic areas surrounded by palisading tumor cells 39. Despite new therapeutic strategies and better clinical diagnostics, GBM still remains a fatal disease 50. In addition to the generation of new vasculature, migrating tumor‐initiating cells are GBM trademarks 23, 39. These populations of tumor‐initiating cells have been identified using a number of stem cell markers and characterized by their ability to form neurospheres, exhibit unlimited proliferation potential with multi‐lineage differentiation and elicit resistance to cytostatic drugs 32, 46, 47. With an ever‐changing microenvironment, the growing GBM tumor is composed of necrotic, proliferative and highly vascularized areas. This suggests that in the search for tumor‐initiating cells within the heterogeneous population of GBM cells, a combination of characteristic markers for both the tumor niches and the contained tumor‐initiating cells are to be used.
Human Cripto‐1 (CR‐1), also known as teratocarcinoma‐derived growth factor 1 (TDGF1), regulates essential steps in early embryogenesis and has a key role in processes such as cell migration, angiogenesis and stem cell maintenance 7, 16, 21, 58, 60. In adult tissues, CR‐1 is expressed at low levels in several different tissue types and organs 51, 57, although a higher expression is detected in colon and cortex of adrenal gland 51, 57. Pathological re‐expression is seen in a number of solid cancers and inflammatory conditions where the protein has been shown to be upregulated 18, 25, 29, 40, 42. Numerous studies have demonstrated high expression levels of CR‐1 to correlate with malignant transformation, tumor invasiveness, metastatic spreading and, hence, poor prognosis 26, 29, 56, 59. As a glycosylphosphatidylinositol (GPI) anchored membrane protein, CR‐1 can be cleaved by GPI phospholipase (GPI‐PLD) and released as a soluble protein into the extracellular matrix 10. The cleaved‐free form of CR‐1 has been detected in the bloodstream of colon and breast cancer patients, and shown to be applicable as a serological marker of malignancy and disease progression 8. Currently, the cancer‐related re‐expression of CR‐1 is being mapped; however, it is not fully understood. Recent findings of Smad‐binding elements and hypoxia‐responsive elements (HRE) in the promotor region of the CR‐1 gene point to regulation by transforming growth factor β (TGFβ) family members and the oxygen‐dependent hypoxia‐inducible factor 1α (HIF1α) 4, 36. In addition, CR‐1 has been shown to be modulated by the canonical Wnt/b‐catenin signaling pathway 30. The modulation by TGFβ family members and the influence of the Wnt/b‐catenin signaling pathway are of high relevance in the context of brain tumors as both of them are dysregulated in GBM 36, 43.
Based on the known role of CR‐1 in fetal development and its modulation by pathways known to be dysregulated in GBM, CR‐1 could be a relevant marker in GBM. In this study, we investigated the possible association of CR‐1 with GBM in the context of brain tumors. This was performed by analyzing a cohort of GBM patient tissue and blood plasma samples for CR‐1 levels and supported by histologic analysis to identify the localization of CR‐1‐positive cells in tumor tissue. The results indicated that CR‐1 could be a potential prognostic marker and a target applicable for new GBM therapy.
Material and Methods
Patient samples
All protocols were reviewed and approved by the Regional Committee on Biomedical Research Ethics in Northern Jutland prior to the study. Patients provided informed consent for the use of samples from planed tumor resection surgery at the Department of Neurosurgery, Aalborg University Hospital, Denmark. The diagnoses were confirmed by immunohistochemistry at the Institute of Pathology, Aalborg University Hospital. Out of the 47 patients initially enrolled, only 31 patients were included for further analysis. These patients fulfilled the requirements of being diagnosed with GBM or lower grade glioma for the first time and not having received previous treatment. In total, 28 GBM and 4 grade III glioma patients were included (see Table 1). Mean age was 63 years (range 27–86) for GBM patients. Blood samples were drawn before surgery start, and the tumor tissue was resected alongside the diagnostic biopsy. Tumor tissue and blood samples were labeled with the letter T and a consecutive number for identification. Because of limited amounts of tissue, not all samples were used for downstream analysis.
Table 1.
Patient overview
| Patient ID | Gender | Age at diagnosis | Resection area | Cripto‐1 protein in blood (ng/mL) | Overall survival after resection (weeks) |
|---|---|---|---|---|---|
| Glioblastoma multiforme | |||||
| T1 | F | 53 | Left frontal and temporal lobe | NA | 142* |
| T2 | M | 57 | Right occipital lobe | 3.3 | 133* |
| T3 | M | 56 | Right temporal and occipital lobe | 2.0 | 122 |
| T4 | M | 74 | Right occipital and temporal lobe | 4.3 | 119 |
| T5 | F | 65 | Right occipital lobe | 1.7 | 87 |
| T6 | F | 60 | Right frontal lobe, premotor cortex | 8.5 | 73 |
| T7 | F | 54 | Right occipital lobe | 1.5 | 76 |
| T8 | M | 43 | Right frontal lobe | 0.7 | 48 |
| T9 | F | 79 | Left occipital lobe | 0.3 | 14 |
| T10 | F | 75 | Left frontal lobe | 0.3 | 16 |
| T11 | F | 61 | Right frontal lobe | 0.4 | 81* |
| T12 | M | 40 | Right occipital and parietal lobe | 0.7 | 74 |
| T13 | F | 27 | Right temporal and occipital lobe | 0.5 | 75* |
| T14 | M | 75 | Left temporal lobe | 0.2 | 11 |
| T15 | F | 85 | Left temporal lobe | 1.9 | 5 |
| T16 | F | 63 | Left frontal lobe | 1.3 | 37 |
| T17 | M | 65 | Right frontal lobe, premotor cortex | 0.8 | 59* |
| T18 | M | 78 | Left frontal lobe | 1.7 | 4 |
| T19 | F | 32 | Left frontal lobe | 0.6 | 52* |
| T20 | M | 73 | Right hemisphere | 0.8 | 22 |
| T21 | F | 56 | Right frontal lobe | 1.2 | 37* |
| T22 | M | 82 | Left temporal lobe | 1.6 | 18 |
| T23 | M | 68 | Right parietal lobe | 1.8 | 7 |
| T24 | M | 78 | Right temporal lobe | 29.6 | 7 |
| T25 | M | 67 | Left parietal lobe | 2.4 | 24* |
| T26 | M | 36 | Frontal lobe | 0.5 | 24* |
| T27 | F | 72 | Left parietal lobe | 0.4 | 20* |
| T28 | M | 86 | Left temporal lobe | 0.6 | 14* |
| Mean | 63 ± 3 | 2.6 ± 1.1 | |||
| Low‐grade gliomas | |||||
| T29 | M | 48 | Right frontal lobe | 0.5 | 496* |
| T30 | M | 49 | Right frontal lobe | 0.4 | 601* |
| T31 | M | 48 | Left frontal lobe | 0.4 | 111* |
| T32 | M | 42 | Left temporal lobe | 0.9 | 95* |
| Mean | 47 ± 2 | 0.54 ± 0.1 | |||
*Alive at analysis.
F = female; M = male; NA = not available.
Cell culture
The human GBM cell line, U87 (American Type Culture Collection, Cat. No. HTB‐14), was cultured in Dulbecco's modified Eagle's medium‐F12 (DMEM‐F12) (Lonza, Copenhagen, Denmark) supplemented with 10% fetal calf serum (FCS), 1% penicillin/streptomycin (Invitrogen Life Technologies, Naerum, Denmark) and 0.5% gentamicin (Invitrogen Life Technologies). Cultures were kept in a humidified atmosphere containing 5% CO2 buffered with ambient air at 37°C. Medium was changed twice per week.
Xenograft model
Prior to xenografting, U87 cells were cultured as previously described. For the investigation of U87 intracerebral tumorigenicity, nuclear magnetic resonance imaging (NMRI) mice (n = 13) were inoculated with 50 000 cells in the striatum. Animals were anesthetized by subcutaneous injection of 0.1 mL/10 g body weight of Hypnorm (VetaPharma, Leeds, UK), Dormicum (B. Braun Medical AS, Fredriksberg, Denmark) and sterile water in a ratio of 1:1:1. According to previous studies, and to ensure tumor development, the total duration of the experiments was set to 21 days. Animals were euthanized by intraperitoneal injection of a high dose of anesthesia. Subsequently, mice were sacrificed by transcardial perfusion fixation with formalin.
All animal experiments and surgical procedures were approved by the Danish Veterinary and Food Administration.
Reverse transcription polymerase chain reaction (RT‐PCR)
Total RNA from tissue and cells was isolated using the Aurum total RNA mini kit (Bio‐Rad, Copenhagen, Denmark) according to manufacturer's instructions. Complementary DNA (cDNA) was synthesized using the iScript cDNA synthesis kit (Bio‐Rad) according to the standard protocol with 800 ng/μL input total RNA. The cDNA synthesis was performed using the Perkin Elmer GeneAmp PCR system (Waltham, MA, USA).
For RT‐PCR, samples were mixed with DreamTaq (Fermentas, VWR, Herlev, Denmark) according to the manufacturer's protocol, with 0.188 μM of forward and reverse primers as listed below. PCR amplification was performed with the thermal cycle program: 3 minutes at 95°C, 35 cycles of 30 s at 95°C, 30 s at 60°C, 60 s at 72°C and one final extension at 72°C for 7.5 minutes.
| Primers: | ||
| CR‐1 | Fw: | 5′‐ATGCTGGGGTCCTTTTGTGCCT‐3′ |
| Rv: | 5′‐GGGCACAGACCCACAGTTCTCTTT‐3′ | |
| GAPDH | Fw: | 5′‐AGATCCCTCCAAAATCAAGTGG‐3′ |
| Rv: | 5′‐GGCAGAGATGATGACCCTTTT‐3′ | |
A blank “no template” control was included as a negative control. Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) served as loading control and reference gene 54. The RNA extract from the U87 GBM cell line was used as a positive control 33. PCR products were run on a 2% agarose gel (ONBIO Inc., Ontario, Canada) 50 mV for 30 minutes. Analysis was performed using a Kodak Image Station 4000 mm Pro (Carestream Health Denmark, Brøndby, Denmark).
Real‐time quantitative RT‐PCR was performed using the Bio‐Rad IQ SYBR® Green Supermix (Bio‐Rad) according to the standard protocol. PCR amplification was performed using 25 μL 1:100 dilution of cDNA as template with primers for CR‐1 and the housekeeping gene HPRT1 54, using SYBR Green PCR supermix (Bio‐Rad, Hercules, CA, USA). The amplification was performed with the thermal cycle program: 3 minutes at 95°C, 40 cycles of 15 s at 95°C, 30 s at 60°C. Product specificity was tested using a melting curve. A fourfold serial dilution was used to generate a standard curve from a pool of cDNA samples to calculate the relative starting quantity. Hypoxanthine‐guanine phosphoribosyltransferase (HPRT) was used as the internal control for normalization. A blank “no template” control was included as a negative control.
| Primers: | ||
| CR‐1 | Fw: | 5′‐ACCTGGCCTTCAGAGATGACAGCA‐3′ |
| Rv: | 5′‐ATGCCTGAGGAAAGCAGCGGAGCT‐3′ | |
| HPRT | Fw: | 5′‐TGAGGATTTGGAAAGGGTGT‐3′ |
| Rv: | 5′‐GAGCACACAGAGGGCTACAA‐3′ | |
Western blotting
Tissue samples were lysed on ice using radioimmunoprecipitation assay (RIPA) buffer [0.1% (v/v) sodium dodecyl sulfate (SDS), 50 mM Tris‐HCl (pH 7.4; Sigma‐Aldrich, Broenby, Denmark), 1% (v/v) Igepal (Bie and Berntsen, Aabyhoej, Denmark), 0.25% (w/v) sodium deoxycholate, 1 mM ethylenediaminetetraacetic acid (EDTA), 150 mM NaCl, 1× Mini Protease Inhibitor Cocktail (Roche Diagnostics, Indianapolis, IN, USA)]. Protein concentrations were determined using the Pierce BCA Protein Assay Kit (Thermo Scientific, Rockford, IL, USA). A total amount of 50 μg protein extract was heat denaturated, electrophoresed through a 12% (w/v) polyacrylamide‐SDS gel and transferred onto a nitrocellulose membrane using the iBlot transfer apparatus (Invitrogen Life Technologies). Membranes were blocked using phosphate‐buffered saline with Tween 20 (PBST) (0.5% Tween‐20, Sigma‐Aldrich, St. Louis, MO, USA) containing 5% skimmed milk (Fluka, Sigma‐Aldrich, St. Louis, MO, USA) and incubated at 4°C. Goat antihuman CR‐1 polyclonal antibody (R&D Systems, Oxon, UK) was used at a dilution of 1:1000, and mouse anti‐β‐actin monoclonal antibody (clone AC‐15, Sigma‐Aldrich, St. Louis, MO, USA) was used as standard reference (1/5000) and loading control. Samples were incubated overnight at 4°C on a rocking shaker. After sequential washing three times in PBST, CR‐1 and β‐actin secondary antibodies conjugated to horseradish peroxidase were incubated at room temperature for 1 h. After further washing of the membrane, the antibody–antigen complex was visualized using enhanced chemiluminescence (Amersham ECL Plus, GE Healthcare, Chalfont St. Giles, UK). Signal acquisition was accomplished using a Kodak Image Station 4000 mm Pro. Protein extracts from U87 were used as a positive control.
Enzyme‐linked immunosorbent assay (ELISA)
CR‐1 protein concentrations in tumor tissue lysate and blood plasma were determined using the human CR‐1 DuoSet ELISA kit (R&D Systems) according to the manufacturer's protocol. Blood plasma was isolated by differential sedimentation centrifugation 10 minutes at 3000 g with no break at room temperature. Tumor tissue lysate was prepared as described for Western blotting. CR‐1 protein levels were normalized to the general protein content measured by the Pierce BCA Protein Assay Kit. CR‐1 quantification was performed in triplicate measures for blood plasma and in duplicate for GBM tissue.
Immunofluorescence immunohistochemistry
Formalin‐fixed tissue was embedded in TissueTek (Sakura, Finetek Europe B.V., The Netherlands) and sectioned at 40‐μm thickness. Tissue sectioning was carried out using a cryostat (microtome cryostat, HM 505 N, Microm, Germany). Primary antibodies used were mouse anti‐CD31 (clone JC70A, Dako, Glostrup, Denmark) and polyclonal rabbit antihuman CR‐1 (Millipore Corporation, Billerica, MA, USA) both at dilution 1:200. Isotype control, rabbit IgG (Dako) was included for evaluation of the specificity. Prior to staining all sections were blocked for 1 h in blocking buffer [0.3% Triton‐X (Sigma‐Aldrich) and 5% swine serum in PBS]. All sections were incubated overnight with primary antibodies at 4°C and subsequently washed 3 × 10 minutes in washing buffer (blocking buffer diluted 1:50 in PBS). Secondary antibodies used were Alexa Fluor 488 mouse anti‐rabbit (Invitrogen Life Technologies) and Alexa Fluor 594 goat anti‐mouse (Invitrogen Life Technologies) at dilution 1:50. Secondary antibodies were incubated for 90 minutes at room temperature. Additionally, cells were stained with 1:1000 diluted 4′,6‐diamidino‐2‐2‐phenylindole (DAPI) (Sigma‐Aldrich) for 10 minutes at room temperature. Mounting medium (Dako) was used as an antifade reagent. The Zeiss Axio Observer Z1 microscope (Brock and Mickelsen, Birkeroed, Denmark) was used to produce images that were processed using ZEN 2012 (blue edition) software (Zeiss, Jena, Germany).
Bright‐field immunohistochemistry
The histological material consisted of archival tumor tissue material from three patients (T6, T7 and T11), which had been routinely formalin fixed and paraffin embedded. The three patients were selected to represent the full range of CR‐1 concentrations found in the blood plasma including a low, an average and a high level. Tissue sections with a thickness of 3 μm were cut and mounted on Superfrost+ slides, dried overnight at room temperature, baked for 1 h at 60°C and deparaffinized. Protocol optimization, that is, identification of the protocol giving the best signal‐to‐noise ratio, was performed by testing different heat‐induced epitope retrieval (HIER) settings (time and buffer pH) and primary antibody concentrations. A control panel of >100 normal and neoplastic tissues, routinely used in pathology as well as the U87 xenograft formalin fixed and paraffin embedded, was run to evaluate the sensitivity and specificity of the antibody.
The staining reactions were carried out in a BenchMark Ultra Slide Staining System (Ventana, Tucson, AZ, USA) with the optimized protocols as follows: HIER was performed using Ventana Cell Conditioning 1 (CC1, pH 8.5) at 99°C for 48 minutes. Endogenous peroxidase was blocked using 3% hydrogen peroxide inhibitor (OptiView DAB kit, Ventana). The primary antibody, CR‐1 (rabbit polyclonal, Millipore Corporation, Billerica, MA, USA), was diluted 1:1500 and incubated with the slides for 32 minutes at 36°C. After washing in buffer, a multimer‐based detection system (OptiView DAB IHC Detection Kit, Ventana) was applied for visualization. Finally, the slides were counterstained in hematoxylin, submitted to bluing reagent, washed in water, dehydrated in graded alcohol and coverslipped.
Statistics
SPSS 21 (IBM, Kgs. Lyngby, Denmark) was used for statistical analysis. Data are presented as mean. For survival analysis based on blood CR‐1 levels, Kaplan–Meier plots and nonparametric statistics (Mantel–Cox test) were used. The patient cohort was divided into CR‐1 above and CR‐1 below the mean control level to compare the overall survival in the two groups. The cutoff value was set based on the average control level. Statistical significance was assigned to differences with P < 0.05.
Results
CR‐1 expression in glioblastoma patient samples
To evaluate the expression of CR‐1 in GBM tumor tissue, RT‐PCR, quantitative RT‐PCR, Western blot analysis and ELISA were performed. CR‐1 transcript was identified in nine representative GBM tumor samples, with amplicon size comparable with that found in the positive control, U87 37 (Figure 1A). CR‐1 transcript was differentially expressed in a group of representative GBM tumor samples with an average relative expression level of 2.9, and expression levels in some instances were twofold higher than that found in the positive control, U87 37 (Figure 1B). The analysis of CR‐1 protein using Western blot analysis and ELISA showed expected bands between 25 and 37 kDa (Figure 1C), with protein concentrations ranging from 0.6 to 14.3 ng/mL (Figure 1D).
Figure 1.
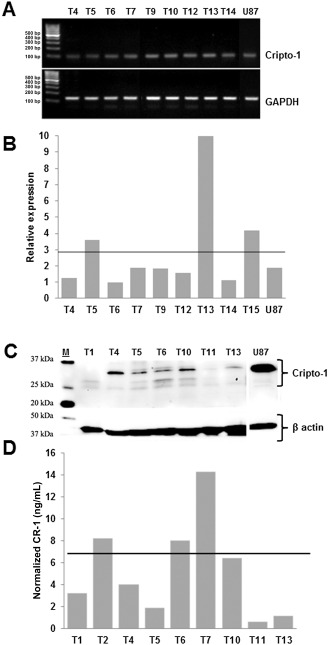
Presence of Cripto‐1 ( CR ‐1) in glioblastoma multiforme ( GBM ) tumor tissue. A. CR‐1 messenger RNA (mRNA) by reverse transcription polymerase chain reaction (RT‐PCR) in GBM tumor samples. B. CR‐1 mRNA by quantitative RT‐PCR in GBM tumor samples. Normalization was performed relative to hypoxanthine‐guanine phosphoribosyltransferase (HPRT). Horizontal line depicts the mean relative expression for GBM samples of 2.9. C. CR‐1 protein in GBM tumor samples by Western blotting. D. CR‐1 protein in GBM tumor samples quantified by enzyme‐linked immunosorbent assay (ELISA). Horizontal line represents the mean normalized concentration of 5.3 ng/mL. Normalization is performed relative to the total protein contents of each sample. Tx denotes the tumor samples of individual patients.
Elevated CR‐1 protein in the blood of glioblastoma patients
In GBM patient blood samples, CR‐1 protein concentrations were detected with an average of 2.51 ng/mL. This was more than fourfold higher compared with the average level in healthy control plasma and plasma from patients diagnosed with lower grade gliomas (Figure 2). The mean CR‐1 concentration (2.57 ± 1.1 ng/mL) found in GBM patients was higher than normal (0.59 ± 0.1 ng/mL). Low‐grade glioma patients had CR‐1 levels (0.54 ± 0.1 ng/mL) comparable with the control group. Looking at individual patients, more than 70% of the newly diagnosed GBM patients presented with a higher CR‐1 concentration in the plasma compared with the control group. An overview of CR‐1 levels found in plasma is depicted in Table 1. The results showed a considerable patient variability that mirrored the variable CR‐1 concentrations in tissue (Figure 1C).
Figure 2.
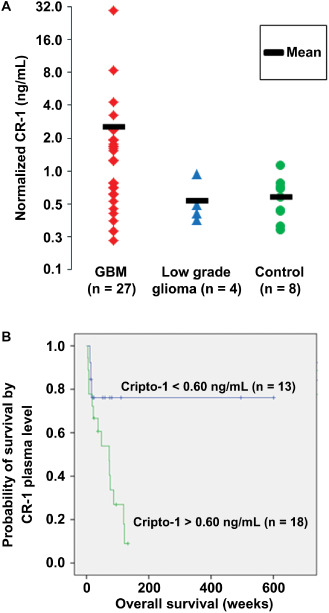
Cripto‐1 ( CR ‐1) in glioma patient blood and its association with overall survival of the patients. A. CR‐1 protein in glioblastoma multiforme (GBM) plasma samples, normal control plasma and plasma of lower grade glioma patient. Normalization is performed relative to the total protein contents of each sample. B. Kaplan–Meier plot showing overall survival in glioblastoma and low‐grade glioma patients by plasma levels of CR‐1 above or below the average control level set at 0.60 ng/mL. Symbols on the line indicate patients alive at the time of analysis.
In order to depict the CR‐1 level as a function of patient overall survival, a Kaplan–Meier plot was made (Figure 2B). The patient cohort including both GBM and low‐grade glioma patients was divided according to their CR‐1 blood levels as being above or below the normal levels found in the control group (0.6 ng/mL). Kaplan–Meier analysis revealed longer survival to significantly correlate with CR‐1 levels equal to or below the control concentrations.
CR‐1‐positive tumor cells have dual phenotypes
To investigate the location of the CR‐1‐positive cells in GBM tissue, immunofluorescence analysis was carried out on tumor tissue sections using the endothelial marker, CD31 together with CR‐1. The results of the staining for CR‐1 and CD31 showed CR‐1‐positive cells in part residing in different areas (see Figure 3). One population of CR‐1‐positive cells, presumably tumor cells, was located around glomeruloid blood vessels separate from endothelial cells. Here, around the vasculature, CR‐1 expression was observed in areas of higher cell densities (Figure 3A). However, in other areas endothelial cells in the tumor microvasculature were found to co‐express CR‐1 (Figure 3B). In these areas, the vasculature was bordering areas of low cell density depicted by the sparse blue nuclear staining analogous to areas of typical necrosis.
Figure 3.
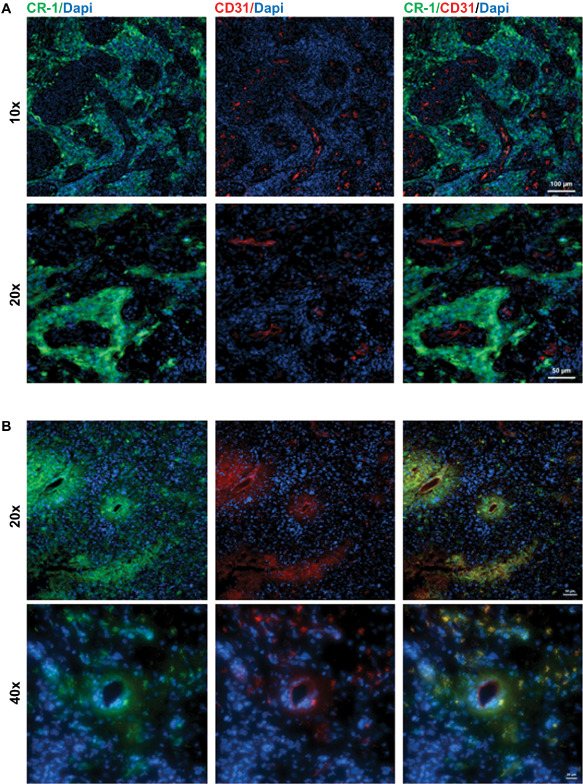
Co‐localization of Cripto‐1 ( CR ‐1) with CD 31 in patients T 24 and T 25. A. CR‐1‐positive cells present in the niche surrounding glomeruloid vasculature but not co‐expressed with the endothelial marker CD31 in patient T24. Scale bar indicates 100 μm (top panel) and 50 μm (lower panel). B. Areas containing microvasculature with CR‐1‐positive cells co‐localizing with CD31 expressing endothelial cells lining capillaries in patient T25. Scale bar indicates 50 μm (top panel) and 20 μm (lower panel).
Immunohistochemical localization of CR‐1 in paraffin‐embedded GBM tissue
A selection of tumors, T6, T7 and T11 with classical GBM features and CR‐1 plasma levels spanning the observed range, were chosen. Typically, as identified by the Institute of Pathology, these tumors contained necrotic areas (low cell density), atypical endothelial hyperplasia and proliferation indices of 20%–40% indicated by Ki67‐positive staining (data not shown). Bright‐field immunohistochemical analysis of GBM tissue for CR‐1 expression resulted in staining that was somewhat varied between the individual tumor samples (Figure 4). In the CR‐1 low patient T11, no significant staining or areas of necrotic was observed (Figure 4A), whereas areas were seen to have a more intense stain compared with the background in T6 and T7. Here, necrotic areas were surrounded by CR‐1 immune reactive pseudopalisading cells (Figure 4B). Similar to the immunofluorescence staining, there was evidence for CR‐1 staining adjacent to glomeruloid blood vessels in patient T7 (Figure 4C,D) and newly formed microvasculature in patient T6 (Figure 4E,F). The antibody specificity was confirmed as CR‐1 staining in U87 xenografts in mouse brains revealed two cell populations with strong and moderate cytoplasmic staining, while the mouse brain tissue was negative (Figure 4G). Furthermore, distinct CR‐1 expression was found in endocrine tumors of the small intestine (Figure 4H) and neuroendocrine cells of the colon (Figure 4I), but not in normal brain (data not shown).
Figure 4.
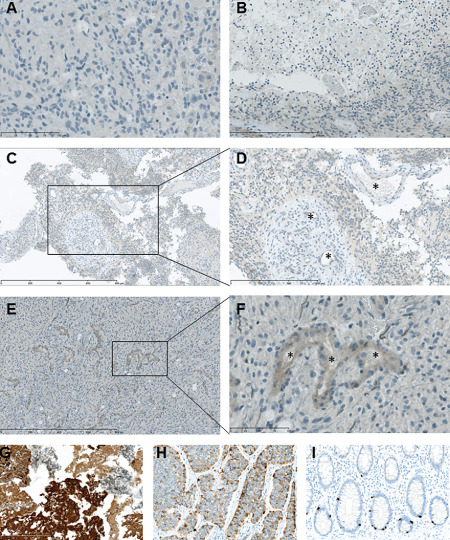
Immunohistochemical staining for Cripto‐1 ( CR ‐1) in glioblastoma tissue from patients presenting CR ‐1 low and high plasma levels, respectively. A. CR‐1 staining in CR‐1 low patient T11. Barely perceptible staining in endothelial cells (left). B. Faint CR‐1 staining in pseudopalisading cells around necrotic area of low cell density in patient T6. C,D. Faint CR‐1 staining in the vicinity of glomeruloid vasculature in CR‐1 high patient T7. E,F. Moderate CR‐1 staining in endothelial cells in CR‐1 high patient T6. G. CR‐1 staining of formalin‐fixed, paraffin‐embedded U87 cerebral xenograft showing strong and medium mainly cytoplasmic reaction in tumor cells and no staining in murine neural cells. H. CR‐1 staining of small intestine neuroendocrine tumor (carcinoid) showing a distinct, heterogenous cytoplasmic staining. I. CR‐1 staining of colon mucosa showing strong cytoplasmic staining of neuroendocrine crypt cells. *Vessel lumen. Scale bar indicates 800 μm (C and E), 300 μm (B and D), 100 μm (A and F) and 200 μm (G, H and I).
Discussion
Here, the expression of CR‐1 in GBM tissue and patient‐matched blood samples with regard to protein concentration and a vascular niche‐dependent expression pattern was investigated. Previously, CR‐1 expression has been observed in the context of Nodal regulation in cultured glioma stem cells. In the study by Lee et al 33, Nodal expression linked CR‐1 to increased proliferation and invasiveness brought about by the upregulation of specific proteases in a GBM cell line. However, CR‐1 expression has yet to be described in situ in cerebral tumors.
In GBM tissue, CR‐1‐positive areas were found within the cell dense palisading regions and around or within the vasculature. The regulatory mechanisms that drives CR‐1 expression are not well defined, but it has been shown that the promoter region contains a number of specific DNA‐binding domains and response elements. One of which is the HRE that is important in the regulation of cell proliferation 4, 12, 28. Hypoxia, through HIF‐1α, directly regulates CR‐1 expression implying that CR‐1 is a target gene in the hypoxic niche and hence an expected finding within regions of vascular genesis caused by hypoxia 4, 5. Among the best known hallmarks of GBM, gliomas initially grow along existing blood vessels and progress by co‐option of normal vasculature 41. As the tumor growth exceeds the capacity of the existing blood supply, necrosis and hypoxia sets in and eventually initiates angiogenesis 53. During this process, tumor tissue expands under variable oxygen tensions resulting in niche formation with areas of necrosis, endothelial proliferation and palisading cells 11. GBM cancer‐initiating cells have been shown to reside in hypoxic areas of GBM tumors 35, 44. Hence, finding CR‐1‐positive areas in or near microvasculature and in areas void of cells could indicate that CR‐1 reflects the presence of cancer‐initiating cells of variable hypoxia‐adapted phenotypes similar to those previously described for the niche‐dependent phenotypes of CD133‐positive tumor‐initiating cells 15. The expression of prominin‐1, (CD133) identified by Singh et al, is preferentially used in the identification of cancer‐initiating cells in GBM 46, 47. For the current work, we performed immunohistochemical analysis to investigate the co‐localization of CR‐1 and CD133 expressing cells. However, this was unsuccessful because of the specificity of the CD133 antibody 31. One could speculate if CR‐1 along with a pallet other markers for stemness, such as Sox2 and Oct4, could be better markers of cancer‐initiating cells in GBM. Because a number of studies have demonstrated that the CD133‐negative populations of cells also harbor tumor‐forming properties, this marker needs to be supported by other validated markers of cancer‐initiating cells 2, 17, 45. Based on the heterogenic nature of individual GBM tumors, it seems plausible that a combination of several markers should therefore be used. Based on the findings presented in this study, CR‐1 could be an interesting candidate to further investigate.
The presence of CR‐1‐positive cells both bordering glomeruloid blood vessels and located within the microvasculature was an interesting finding. Rapid cell proliferation demanding new blood supply and the formation of glomeruloid bodies are typical hallmarks of GBM 9, 20, 39. In fact, the magnitude of these unusual vascular formations is directly correlated to a more dismal prognosis 9. Therefore, finding CR‐1‐expressing cells residing in areas of rapid tumor growth characterized by greater cell proliferation suggests a link between CR‐1 and a role in GBM progression. Previous studies have shown that CR‐1 is known to have an essential role in angiogenesis‐linked tumor growth 1, 7, 19. As demonstrated by Bianco et al, CR‐1 overexpression has a promoting effect on tumor growth and formation of microvessels 7. Bianco et al also found that neovascularization and tumor growth could be inhibited using anti‐CR‐1 monoclonal antibodies, further emphasizing the role of CR‐1 in tumor progression by means of angiogenesis. Supportive of this observation, CR‐1 has been documented to be a direct interaction partner to angiogenin, a known inducer of vascularization 27, 49. Accordingly, the finding of CR‐1 in the microvasculature indicates an essential role of this protein in facilitating tumor growth. GBM tumor cells have been shown to be able to facilitate endothelial proliferation by endocrine signaling and, consequently, the sprouting of existing blood vessels to accommodate the rapid cell proliferation 53. Alternatively, neoangiogenesis could be driven by GBM tumor cells transdifferentiating into tumor endothelium 48, 55. These two scenarios may be used as an explanation for the finding of CR‐1‐positive cells co‐expressing the endothelial marker CD31 in one niche and CR‐1‐positive cells distinct from the tumor vasculature in other areas.
The bright‐field immunohistological staining was hoped to recapitulate the immunofluorescence staining. Weak‐level staining of CR‐1 protein was apparent in endothelial cells and perivascular tumor cells; however, crisply defined regions comparable with the positive controls were not seen. Especially the U87 xenografts stained strongly that could be explained by an upregulation of CR‐1 in response to the change of environment going from ambient in vitro cultures to the lower oxygen tension in vivo setting 13, 34. Similar results ranging from no to low levels of expression have been found previously in samples of breast ductal carcinoma, where CR‐1 expression has been reported 29. Nevertheless, despite similar findings, further optimization is required to determine the precise location of CR‐1 within the tumor microenvironment in paraffin‐embedded tissue. When looking at clinical pathological parameters in conjunction with CR‐1 expression, it was previously found to be significantly correlated with tumor size or volume 3, 29. So, despite the weak CR‐1 staining, the histological morphology of the GBM sections in the current work depicted the principal regions of interest such as palisading cells surrounding areas of necrosis and extensive vasculature. With respect to tumor heterogeneity, the location of tumor resection combined with tumor volume could be a key correlative factor here when looking for CR‐1 expression.
In the current work, we describe CR‐1 expression in GBM tissue and patient‐matched blood samples. From gene expression profiling, normal tissue of the temporal and parietal lobe and especially tissue from the cerebellar peduncles have been shown to have higher expression levels of CR‐1 compared with, for example, the occipital lobe tissue 51, 57. Therefore, tumor location, tumor volume and site of biopsy could all influence the detected protein and expression levels observed in tissue. This should be taken into account when evaluating CR‐1 levels in tissue. For the present study, the tumor location varied between patients and the site of biopsy or tumor resection was not controlled. Therefore, in this case, the inter‐patient variation of CR‐1 tissue protein levels cannot be expected to uniquely reflect the disease status. This was very evident when looking at the differential expression obtained with quantitative RT‐PCR and Western blotting. As for the CR‐1 protein detected in plasma, we investigated the correlation to overall patient survival. Here, the degree of tumor vascularization, a compromised blood–brain barrier and the tumor volume was expected to be major factors facilitating the CR‐1 release from the tumor to the general blood system. These three factors are present at an advanced disease stage. Hence, we expected CR‐1 plasma levels to be an indication of malignancy and a shorter overall survival from the time of diagnosis. Plasma levels of CR‐1 were variable between patients, however, in line with previously seen concentrations in blood from colon and breast cancer patients 8. Bianco et al 8 showed that CR‐1 plasma levels in the blood were correlated to malignant progression and poor prognosis. Similarly, the correlation between high CR‐1 concentrations in plasma and shorter overall survival was seen in our study. This way, CR‐1 could serve as a prognostic marker for malignant progression. In the context of glioma, a progression from lower to higher grade tumors or a sign of GBM relapse. On the other hand, in the case of GBM and glioma, CR‐1 plasma levels will not serve as a relevant candidate for diagnostic purposes as CR‐1 re‐expression is associated with several malignancies 18, 25, 29, 40, 42. Further studies are needed in order to evaluate CR‐1 in plasma as a prognostic marker for disease progression and tumor relapse.
The blood concentrations of CR‐1 correlated with overall survival suggestive of a GBM patient subdivision based on CR‐1 manifestation. A subdivision of the GBM diagnosis has been attempted on the basis of genetic tumor profiles. Recent progress has identified potential molecular signatures that are involved in stem cell self‐renewal and delineate pathways contributing to malignant progression in GBM 24, 38. Three to four subclasses have been identified based on global gene expression data. Of these, the two subclasses, characterized by genes involved in proliferation and a mesenchymal phenotype, show a shorter overall survival time 24, 38. Especially patients with a more mesenchymal expression profile pertaining to that of the epithelia to mesenchymal transition (EMT) have been linked to poor prognosis as a result of a higher degree of tumor invasiveness 14, 22, 37, 52. As CR‐1 has been linked to EMT and has an essential role in invasion and migration, this marker could be inferred as a factor of poor prognosis 5, 60. However, data presented in the current work also show that not all patients with a GBM diagnosis present with a high CR‐1 level in blood and tumor tissue. Therefore, we expect that elevated CR‐1 levels in the blood could be developed as a serologic marker of tumor progression in conjunction with gene expression profiling that reveals the distinct molecular subtypes of GBM. CR‐1‐targeted therapies are already being developed for other solid cancers with promising clinical outcomes 1, 6. In the case of GBM, these may be applicable to the subset of patients with CR‐1‐positive profiles.
Conclusion
Here, we describe CR‐1 expression in GBM tissue and blood. CR‐1 protein levels in plasma samples from GBM patients were found to be elevated compared with normal. Most interestingly, CR‐1 protein concentrations higher than normal were significantly correlated to shorter overall survival in the study cohort. In addition, three areas of CR‐1‐staining cells were seen in GBM tissue. Collectively, our data support that CR‐1 has the potential of being a new prognostic biomarker in GBM and might play a role in the tumor pathogenesis and progression.
Acknowledgments
We greatly appreciate the generous support from The Obel Family Foundation that made this experimental work possible. Bachelor medical students Line Frandsen, Julie Vedel, Sille Jensen, Ditte Hansen and Spogmai Zadran at Department of Health Science and Technology, Aalborg University are greatly acknowledged for their assistance in gathering the clinical data.
References
- 1. Adkins HB, Bianco C, Schiffer SG, Rayhorn P, Zafari M, Cheung AE et al (2003) Antibody blockade of the Cripto CFC domain suppresses tumor cell growth in vivo . J Clin Invest 112:575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ et al (2007) CD133(+) and CD133(−) glioblastoma‐derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res 67:4010–4015. [DOI] [PubMed] [Google Scholar]
- 3. Bianco C, Castro NP, Baraty C, Rollman K, Held N, Rangel MC et al (2013) Regulation of human Cripto‐1 expression by nuclear receptors and DNA promoter methylation in human embryonal and breast cancer cells. J Cell Physiol 228:1174–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bianco C, Cotten C, Lonardo E, Strizzi L, Baraty C, Mancino M et al (2009) Cripto‐1 is required for hypoxia to induce cardiac differentiation of mouse embryonic stem cells. Am J Pathol 175:2146–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bianco C, Rangel MC, Castro NP, Nagaoka T, Rollman K, Gonzales M, Salomon DS (2010) Role of Cripto‐1 in stem cell maintenance and malignant progression. Am J Pathol 177:532–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bianco C, Salomon DS (2009) Human Cripto‐1 as a target for a cancer vaccine: WO2008040759. Expert Opin Ther Pat 19:141–144. [DOI] [PubMed] [Google Scholar]
- 7. Bianco C, Strizzi L, Ebert A, Chang C, Rehman A, Normanno N et al (2005) Role of human cripto‐1 in tumor angiogenesis. J Natl Cancer Inst 97:132–141. [DOI] [PubMed] [Google Scholar]
- 8. Bianco C, Strizzi L, Mancino M, Rehman A, Hamada S, Watanabe K et al (2006) Identification of cripto‐1 as a novel serologic marker for breast and colon cancer. Clin Cancer Res 12:5158–5164. [DOI] [PubMed] [Google Scholar]
- 9. Birner P, Piribauer M, Fischer I, Gatterbauer B, Marosi C, Ambros PF et al (2003) Vascular patterns in glioblastoma influence clinical outcome and associate with variable expression of angiogenic proteins: evidence for distinct angiogenic subtypes. Brain Pathol 13:133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brandt R, Normanno N, Gullick WJ, Lin JH, Harkins R, Schneider D et al (1994) Identification and biological characterization of an epidermal growth factor‐related protein: cripto‐1. J Biol Chem 269:17320–17328. [PubMed] [Google Scholar]
- 11. Brognaro E (2011) A theory and a model to understand glioblastoma development both in the bulk and in the microinfiltrated brain parenchyma. Neurochem Res 36:2145–2154. [DOI] [PubMed] [Google Scholar]
- 12. Bruick RK (2003) Oxygen sensing in the hypoxic response pathway: regulation of the hypoxia‐inducible transcription factor. Genes Dev 17:2614–2623. [DOI] [PubMed] [Google Scholar]
- 13. Camphausen K, Purow B, Sproull M, Scott T, Ozawa T, Deen DF, Tofilon PJ (2005) Influence of in vivo growth on human glioma cell line gene expression: convergent profiles under orthotopic conditions. Proc Natl Acad Sci U S A 102:8287–8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chou CH, Lieu AS, Wu CH, Chang LK, Loh JK, Lin RC et al (2010) Differential expression of hedgehog signaling components and Snail/E‐cadherin in human brain tumors. Oncol Rep 24:1225–1232. [DOI] [PubMed] [Google Scholar]
- 15. Christensen K, Schrøder HD, Kristensen BW (2011) CD133(+) niches and single cells in glioblastoma have different phenotypes. J Neurooncol 104:129–143. [DOI] [PubMed] [Google Scholar]
- 16. Ciccodicola A, Dono R, Obici S, Simeone A, Zollo M, Persico MG (1989) Molecular characterization of a gene of the “EGF family” expressed in undifferentiated human NTERA2 teratocarcinoma cells. EMBO J 8:1987–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clement V, Dutoit V, Marino D, Dietrich PY, Radovanovic I (2009) Limits of CD133 as a marker of glioma self‐renewing cells. Int J Cancer 125:244–248. [DOI] [PubMed] [Google Scholar]
- 18. D'Antonio A, Losito S, Pignata S, Grassi M, Perrone F, De Luca A et al (2002) Transforming growth factor alpha, amphiregulin and cripto‐1 are frequently expressed in advanced human ovarian carcinomas. Int J Oncol 21:941–948. [PubMed] [Google Scholar]
- 19. De Luca A, Arra C, D'Antonio A, Casamassimi A, Losito S, Ferraro P et al (2000) Simultaneous blockage of different EGF‐like growth factors results in efficient growth inhibition of human colon carcinoma xenografts. Oncogene 19:5863–5871. [DOI] [PubMed] [Google Scholar]
- 20. DeAngelis LM (2001) Brain tumors. N Engl J Med 344:114–123. [DOI] [PubMed] [Google Scholar]
- 21. Ding J, Yang L, Yan YT, Chen A, Desai N, Wynshaw‐Boris A, Shen MM (1998) Cripto is required for correct orientation of the anterior‐posterior axis in the mouse embryo. Nature 395:702–707. [DOI] [PubMed] [Google Scholar]
- 22. Elias MC, Tozer KR, Silber JR, Mikheeva S, Deng M, Morrison RS et al (2005) TWIST is expressed in human gliomas and promotes invasion. Neoplasia 7:824–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eramo A, Ricci‐Vitiani L, Zeuner A, Pallini R, Lotti F, Sette G et al (2006) Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ 13:1238–1241. [DOI] [PubMed] [Google Scholar]
- 24. Freije WA, Castro‐Vargas FE, Fang Z, Horvath S, Cloughesy T, Liau LM et al (2004) Gene expression profiling of gliomas strongly predicts survival. Cancer Res 64:6503–6510. [DOI] [PubMed] [Google Scholar]
- 25. Friess H, Yamanaka Y, Buchler M, Kobrin MS, Tahara E, Korc M (1994) Cripto, a member of the epidermal growth factor family, is over‐expressed in human pancreatic cancer and chronic pancreatitis. Int J Cancer 56:668–674. [DOI] [PubMed] [Google Scholar]
- 26. Fuchs IB, Lichtenegger W, Buehler H, Henrich W, Stein H, Kleine‐Tebbe A, Schaller G (2002) The prognostic significance of epithelial‐mesenchymal transition in breast cancer. Anticancer Res 22(6A):3415–3419. [PubMed] [Google Scholar]
- 27. Gao X, Xu Z (2008) Mechanisms of action of angiogenin. Acta Biochim Biophys Sin (Shanghai) 40:619–624. [DOI] [PubMed] [Google Scholar]
- 28. Goda N, Ryan HE, Khadivi B, McNulty W, Rickert RC, Johnson RS (2003) Hypoxia‐inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Mol Cell Biol 23:359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gong YP, Yarrow PM, Carmalt HL, Kwun SY, Kennedy CW, Lin BP et al (2007) Overexpression of Cripto and its prognostic significance in breast cancer: a study with long‐term survival. Eur J Surg Oncol 33:438–443. [DOI] [PubMed] [Google Scholar]
- 30. Hamada S, Watanabe K, Hirota M, Bianco C, Strizzi L, Mancino M et al (2007) beta‐Catenin/TCF/LEF regulate expression of the short form human Cripto‐1. Biochem Biophys Res Commun 355:240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hermansen SK, Christensen KG, Jensen SS, Kristensen BW (2011) Inconsistent immunohistochemical expression patterns of four different CD133 antibody clones in glioblastoma. J Histochem Cytochem 59:391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kang MK, Kang SK (2007) Tumorigenesis of chemotherapeutic drug‐resistant cancer stem‐like cells in brain glioma. Stem Cells Dev 16:837–847. [DOI] [PubMed] [Google Scholar]
- 33. Lee CC, Jan HJ, Lai JH, Ma HI, Hueng DY, Lee YC et al (2010) Nodal promotes growth and invasion in human gliomas. Oncogene 29:3110–3123. [DOI] [PubMed] [Google Scholar]
- 34. Li P, Zhou C, Xu L, Xiao H (2013) Hypoxia enhances stemness of cancer stem cells in glioblastoma: an in vitro study. Int J Med Sci 10:399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S et al (2009) Hypoxia‐inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15:501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mancino M, Strizzi L, Wechselberger C, Watanabe K, Gonzales M, Hamada S et al (2008) Regulation of human Cripto‐1 gene expression by TGF‐beta1 and BMP‐4 in embryonal and colon cancer cells. J Cell Physiol 215:192–203. [DOI] [PubMed] [Google Scholar]
- 37. Mikheeva SA, Mikheev AM, Petit A, Beyer R, Oxford RG, Khorasani L et al (2010) Rostomily RC TWIST1 promotes invasion through mesenchymal change in human glioblastoma. Mol Cancer 9:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD et al (2006) Molecular subclasses of high‐grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9:157–173. [DOI] [PubMed] [Google Scholar]
- 39. Plate KH, Risau W (1995) Angiogenesis in malignant gliomas. Glia 15:339–347. [DOI] [PubMed] [Google Scholar]
- 40. Saeki T, Stromberg K, Qi CF, Gullick WJ, Tahara E, Normanno N et al (1992) Differential immunohistochemical detection of amphiregulin and cripto in human normal colon and colorectal tumors. Cancer Res 52:3467–3473. [PubMed] [Google Scholar]
- 41. Sakariassen PO, Prestegarden L, Wang J, Skaftnesmo KO, Mahesparan R, Molthoff C et al (2006) Angiogenesis‐independent tumor growth mediated by stem‐like cancer cells. Proc Natl Acad Sci U S A 103:16466–16471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saloman DS, Bianco C, Ebert AD, Khan NI, De Santis M, Normanno N et al (2000) The EGF‐CFC family: novel epidermal growth factor‐related proteins in development and cancer. Endocr Relat Cancer 7:199–226. [DOI] [PubMed] [Google Scholar]
- 43. Sandberg CJ, Altschuler G, Jeong J, Stromme KK, Stangeland B, Murrell W et al (2013) Comparison of glioma stem cells to neural stem cells from the adult human brain identifies dysregulated Wnt‐signaling and a fingerprint associated with clinical outcome. Exp Cell Res 319:2230–2243. [DOI] [PubMed] [Google Scholar]
- 44. Seidel S, Garvalov BK, Wirta V, von Stechow L, Schanzer A, Meletis K et al (2010) A hypoxic niche regulates glioblastoma stem cells through hypoxia inducible factor 2 alpha. Brain 133(Pt 4):983–995. [DOI] [PubMed] [Google Scholar]
- 45. Shmelkov SV, Butler JM, Hooper AT, Hormigo A, Kushner J, Milde T et al (2008) CD133 expression is not restricted to stem cells, and both CD133+ and CD133− metastatic colon cancer cells initiate tumors. J Clin Invest 118:2111–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB (2003) Identification of a cancer stem cell in human brain tumors. Cancer Res 63:5821–5828. [PubMed] [Google Scholar]
- 47. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T et al (2004) Identification of human brain tumour initiating cells. Nature 432:396–401. [DOI] [PubMed] [Google Scholar]
- 48. Soda Y, Marumoto T, Friedmann‐Morvinski D, Soda M, Liu F, Michiue H et al (2011) From the cover: feature article: transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc Natl Acad Sci U S A 108:4274–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H et al (2005) A human protein–protein interaction network: a resource for annotating the proteome. Cell 122:957–968. [DOI] [PubMed] [Google Scholar]
- 50. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996. [DOI] [PubMed] [Google Scholar]
- 51. Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D et al (2004) A gene atlas of the mouse and human protein‐encoding transcriptomes. Proc Natl Acad Sci U S A 101:6062–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tso CL, Shintaku P, Chen J, Liu Q, Liu J, Chen Z et al (2006) Primary glioblastomas express mesenchymal stem‐like properties. Mol Cancer Res 4:607–619. [DOI] [PubMed] [Google Scholar]
- 53. Vajkoczy P, Farhadi M, Gaumann A, Heidenreich R, Erber R, Wunder A et al (2002) Microtumor growth initiates angiogenic sprouting with simultaneous expression of VEGF, VEGF receptor‐2, and angiopoietin‐2. J Clin Invest 109:777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Valente V, Teixeira SA, Neder L, Okamoto OK, Oba‐Shinjo SM, Marie SK et al (2009) Selection of suitable housekeeping genes for expression analysis in glioblastoma using quantitative RT‐PCR. BMC Mol Biol 10:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A et al (2010) Glioblastoma stem‐like cells give rise to tumour endothelium. Nature 468:829–833. [DOI] [PubMed] [Google Scholar]
- 56. Wechselberger C, Ebert AD, Bianco C, Khan NI, Sun Y, Wallace‐Jones B et al (2001) Cripto‐1 enhances migration and branching morphogenesis of mouse mammary epithelial cells. Exp Cell Res 266:95–105. [DOI] [PubMed] [Google Scholar]
- 57. Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S et al (2009) BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol 10:R130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Xu C, Liguori G, Persico MG, Adamson ED (1999) Abrogation of the Cripto gene in mouse leads to failure of postgastrulation morphogenesis and lack of differentiation of cardiomyocytes. Development 126:483–494. [DOI] [PubMed] [Google Scholar]
- 59. Xue C, Plieth D, Venkov C, Xu C, Neilson EG (2003) The gatekeeper effect of epithelial‐mesenchymal transition regulates the frequency of breast cancer metastasis. Cancer Res 63:3386–3394. [PubMed] [Google Scholar]
- 60. Zhong XY, Zhang LH, Jia SQ, Shi T, Niu ZJ, Du H et al (2008) Positive association of up‐regulated Cripto‐1 and down‐regulated E‐cadherin with tumour progression and poor prognosis in gastric cancer. Histopathology 52:560–568. [DOI] [PubMed] [Google Scholar]