

Abstract
Spinal cord injury (SCI) represents a devastating central nervous system disease that still lacks sufficient therapies. Here, dogs are increasingly recognized as a preclinical animal model for the development of future therapies. The aim of this study was a detailed characterization of axonopathy in canine intervertebral disc disease, which produces a mixed contusive and compressive injury and functions as a spontaneous translational animal model for human SCI. The results revealed an early occurrence of ultrastructurally distinct axonal swelling. Immunohistochemically, enhanced axonal expression of β‐amyloid precursor protein, non‐phosphorylated neurofilament (n‐NF) and growth‐associated protein‐43 was detected in the epicenter during acute canine SCI. Indicative of a progressive axonopathy, these changes showed a cranial and caudally accentuated spatial progression in the subacute disease phase. In canine spinal cord slice cultures, immunoreactivity of axons was confined to n‐NF. Real‐time quantitative polymerase chain reaction of naturally traumatized tissue and slice cultures revealed a temporally distinct dysregulation of the matrix metalloproteinases (MMP)‐2 and MMP‐9 with a dominating expression of the latter. Contrasting to early axonopathy, diminished myelin basic protein immunoreactivity and phagocytosis were delayed. The results present a basis for assessing new therapies in the canine animal model for translational research that might allow partial extrapolation to human SCI.
Keywords: amyloid precursor protein, dog, growth‐associated protein‐43, intervertebral disc disease, spinal cord slice culture, ultrastructure
Introduction
Spinal cord injury (SCI) is a devastating traumatic central nervous system (CNS) disease with an annual incidence rate ranging from 12.1 to 57.8 cases per million in humans [for a review, see 88]. Because of the lack of restitutional therapies, SCI significantly deteriorates the quality of life indicating the need for novel therapeutic strategies including pharmacological interventions and cell transplantation approaches 29, 44.
Following SCI, secondary injury processes include numerous pathological consequences such as axonal damage, vascular alterations, immune processes, necrosis and apoptosis 5, 67. The current concepts on these processes are broadly based on well established and highly standardized rodent models that are extremely suitable to mimic certain aspects of the disease. However, multiple lines of evidence indicate that there is a number of significant differences between experimental SCI in rodents and the clinical disease in humans 33, 38, 53, 76, 78, 79, 90. Besides discrepancies in anatomy and neurology, there is minimal inflammation, a protracted Wallerian degeneration, and reduced astroglial scar formation and demyelination in human SCI compared with rodents 33, 68, 72, 76. Moreover, SCI in humans is characterized by a mixture of contusive/compressive forces resulting from structures located ventral to the spinal cord, whereas most rodent models are usually based on mechanical forces applied to the dorsally exposed spinal cord and mimic different, however not combined, primary injuries such as contusion, compression, and hemisection, respectively 18, 33, 38. These important differences indicate the necessity of additional translational animal models more closely resembling the complexity of the heterogeneous human disease 33.
As companion animals, dogs have gained increasing notice as a bridging model between highly standardized experimental studies and heterogeneous clinical human CNS diseases such as multiple sclerosis (MS), epilepsy and SCI 6, 7, 11, 38, 51, 53, 75, 79. Intervertebral disc disease (IVDD) in dogs is one of the most common canine neurological diseases 28 and is commonly characterized by explosive spontaneous herniation of the degenerated intervertebral disc into the vertebral canal, causing a mixed contusive‐compressive injury to the spinal cord. Following the strong demand for large animal models in preclinical and clinical SCI research 49, canine IVDD is increasingly recognized as a valuable naturally occurring animal model mimicking the complexity of the human counterpart 10, 11, 25, 38, 53, 78, 79. Similar to most cases of human SCI, canine IVDD causes primary injury resulting from structures located ventral to the spinal cord 38, 53. However, the most evident strength of this model is its spontaneity, making it highly suitable to validate findings obtained in experimental studies and extrapolate them into the clinical human disease and therapeutic trials 38, 53, 78, 79. Consequently, dogs are nowadays additionally acknowledged as a bridging species for clinical trials of cell transplantation approaches as a therapy for SCI 37, 39, 53, 61, 90.
Based on recent observations in demyelinating diseases such as MS and its viral animal models 22, 36, 47, 75, 84, 85, the hypothesis arose that early axonal pathology during canine SCI might represent a key feature and a pivotal trigger for subsequent immune processes that are known to occur during subacute canine SCI 79.
Axonal damage is a central hallmark of naturally occurring SCI and one of the major pathological features leading to clinical disability because of conduction blockage 74. The morphology of degenerative and regenerative axonal changes in the traumatized CNS has been sophisticatedly characterized by the studies of Ramon y Cajal almost 100 years ago 16. In experimental SCI, axonopathy is associated with disturbances in fast axonal transport mechanisms and altered neurofilament phosphorylation 19, 45, 63, 82. Several mechanisms are involved in axonopathy, and here matrix metalloproteinases (MMPs), and especially the gelatinases MMP‐2 and MMP‐9, have shown to be directly and indirectly linked to axonal damage and regeneration in rodent models 88. Similarly, MMP‐9 in the cerebrospinal fluid of dogs with IVDD correlates with injury severity 52, 58; however, data on their expression in the local damaged tissue during spontaneous SCI are extremely rare.
In addition, several recent reports demonstrate the existence of significant axonal regeneration attempts following experimental traumatic CNS damage 3, 24, 26, 55, 74, 86, whereas knowledge on the expression of axonal growth‐promoting molecules such as growth‐associated protein (GAP)‐43 in naturally occurring SCI is strikingly sparse.
As recently stated in an opinion survey, the SCI community highly agrees upon an increasing demand of translational large animal models to validate findings obtained in experimental studies on rodents 49. As dogs are increasingly used as a preclinical model for the development of future therapeutic approaches such as cell transplantations 37, 39, 78, 90, a detailed understanding of the pathomechanisms involved in spontaneous canine SCI represents a prerequisite prior to the efficacy assessment of such strategies. Thus, the aim of the present study was a detailed characterization of the spatio‐temporal development of degenerative changes in the spinal cord white matter in a clinically relevant translational animal model for human SCI with a special focus on axonal pathology.
Materials and Methods
Sampling of spinal cord tissue of naturally traumatized dogs
The patients included in the present study are identical to those described in an earlier study 79. The dogs were examined by board‐certified veterinary neurologists (VMS and AT) at the Small Animal Clinic of the University of Veterinary Medicine, Hannover (Germany). Because of a poor prognosis, based on clinical signs such as para‐ or tetraplegia with subsequent severe gait deficiencies, partially with complete loss of pain perception, the dogs were sacrificed on the owner's request. With the owners' consent, the dogs were subsequently submitted to necropsy, which was performed within 2–4 hours after euthanasia. Spinal cord tissue of 17 dogs of different breeds with clinically diagnosed IVDD and a mean age of 7.0 years (minimum: 3.5 years; maximum: 12 years, dog no. 11–27) was included in the study [Table 1; 79]. The spinal cord was removed after manual dorsal disconnection of the vertebral laminae. The vertebral canal was evaluated for the presence of intervertebral disc protrusion and herniation, respectively. Precisely at the site of the intervertebral disc herniation, the spinal cord was transected using a razor blade and a tissue block measuring 1.5 cm in length was removed. Exactly at the site of the epicenter, this block was subsequently apportioned into two blocks resulting in one block measuring 0.5 cm [epicenter, localization (Loc.) 0] for paraffin embedding, and one block measuring 1 cm for molecular investigations. For real‐time quantitative polymerase chain reaction (qPCR) the native 1‐cm thick portion from the epicenter was immediately embedded in Tissue‐Tec® O.C.T.TM (OCT) compound (Sakura Finetech Europe BV, Zoetenwoude, the Netherlands), snap‐frozen in liquid nitrogen and stored at −80°C 57, 79. Frozen tissue was not available from dog no. 16 and 21. To evaluate the dimensions of degenerative white matter changes, additional tissue blocks, each measuring 0.5 cm in length, were collected at 1.5, 3, 4.5, 6, 7.5 and 9 cm cranial (Loc. 1.5, 3, 4.5, 6, 7.5, and 9) and caudal (Loc. −1.5, −3, −4.5, −6, −7.5, and −9) from the epicenter, respectively, resulting in a total number of 13 examined localizations per animal. Three dogs (No. 18, 19 and 27; Table 1) were not available for complete necropsy. However, with the owners' consent, a piece of spinal cord tissue measuring 1.5 cm in length was removed post‐mortally from the epicenter (Loc. 0). The 0.5‐cm thick block of each localization was immediately fixed in 10% neutral‐buffered formalin for 4 days followed by paraffin embedding. In addition, spinal cord tissue of neurologically healthy control dogs (n = 10, mean age: 6.9 years, dogs no. 1–10) was sampled as described earlier. Based on clinically relevant time points for future therapeutic approaches such as cell transplantations and pharmacological treatment strategies, the cases were grouped according to the temporal interval between the occurrence of the first neurologic deficits and euthanasia. While group 1 was assigned to control animals, group 2 included animals with a mean duration of 2.55 days of SCI (acute; n = 9; minimum: 1 day; maximum: 4 days), and group 3 consisted of animals with a mean duration of 7.58 days of SCI (subacute; n = 8; minimum: 5 days; maximum: 10 days). Dachshunds and dachshund mixed breeds were overrepresented (n = 9). After 1 day of formalin fixation, 1‐mm thin slices were taken from the 0.5 cm measuring formalin‐fixed block of the epicenter of selected animals [dogs no. 4, 8, 10 (controls); 11, 12, 14, 16 (acute SCI); 20, 21, 22, 23, 26 (subacute SCI) ] and post‐fixed in 5% glutaraldehyde and 4% paraformaldehyde buffered in 1% sodium‐cacodylate (pH 7.2) for 24 hours and subsequently embedded in epoxy resin for transmission electron and immunoelectron microscopy, as described previously 47, 87.
Table 1.
Identification, type of spinal cord injury, time elapsed after spinal cord injury, group, age, sex and breed of dogs used in the present study.
| Dog No. | Type of SCI | Time post SCI (D) | Groupa | Age (Y)/Sex | Breed |
|---|---|---|---|---|---|
| 1 | Control | — | 1 | 0.5/F | Beagle |
| 2 | Control | — | 1 | 0.5/F | Beagle |
| 3 | Control | — | 1 | 0.5/F | Beagle |
| 4 | Control | — | 1 | 0.9/M | Labrador retriever |
| 5 | Control | — | 1 | 8/MN | Small munsterlander |
| 6 | Control | — | 1 | 13/F | Dachshund |
| 7 | Control | — | 1 | 10/F | Dachshund |
| 8 | Control | — | 1 | 14/M | Mongrel |
| 9 | Control | — | 1 | 10/M | Pit bull terrier |
| 10 | Control | — | 1 | 11.5/M | Golden retriever |
| 11 | IVD herniation Th10/11 | 4 | 2 | 9/M | Dachshund |
| 12 | IVD herniation L2/3 | 3.5 | 2 | 4/M | Dachshund‐mix |
| 13 | IVD herniation Th12/13 | 3 | 2 | 7/F | Dachshund |
| 14 | IVD herniation Th12/13 | 4 | 2 | 5/M | Dachshund |
| 15 | IVD herniation C6/7 | 1.5 | 2 | 12/M | Fox terrier |
| 16 | IVD herniation Th13/L1 | 3 | 2 | 3.5/M | Pekingese |
| 17 | IVD herniation L2/3 | 1.5 | 2 | 9/M | Mongrel |
| 18 | IVD herniation L3/4 | 1.5 | 2 | 7/M | Bernese mountain dog |
| 19 | IVD herniation L5/6 | 1 | 2 | 5/M | Hanoverian scenthound |
| 20 | IVD herniation Th11/12 | 7 | 3 | 8/FN | Hanoverian scenthound |
| 21 | IVD herniation Th12/13 | 8 | 3 | 8/M | Dachshund |
| 22 | IVD herniation Th11/12 | 10 | 3 | 7/M | Dachshund |
| 23 | IVD herniation Th13/L1 | 7 | 3 | 4/M | Dachshund |
| 24 | IVD herniation C5/6 | >5 | 3 | 10/F | Dachshund |
| 25 | IVD protrusion Th12/13 | 10 | 3 | 7/M | Mongrel |
| 26 | IVD protrusion Th12/13 | 8 | 3 | 10/F | Mongrel |
| 27 | IVD herniation C2/3 | 5 | 3 | 5/M | Dachshund |
Group 1 (control group); group 2 (acute SCI for 1–4 days); group 3 (subacute SCI for 5 days and longer).
C = cervical vertebra; D = days; F = female; IVD = intervertebral disc; L = lumbar vertebra; M = male; N = neutered; SCI = spinal cord injury; Th = thoracic vertebra; Y = years.
Organotypic spinal cord slice cultures as an in vitro model for axonal pathology in canine SCI
To comparatively evaluate axonal changes in a simplified in vitro model devoid of peripheral blood supply and systemic environmental factors, an organotypic spinal cord slice culturing system was used as described 79. Briefly, spinal cord tissue of six 5‐month‐old, neurologically healthy beagle dogs that served as control dogs in an unrelated study was aseptically collected immediately after death and placed into medium [79, 80; pH 7.4, with slight modification] at 4°C. After removing the dura mater, spinal cord slices (350 μm) were produced using a McIllwain Tissue Chopper® (H. Saur Laborbedarf, Reutlingen, Germany) as described 36, 43, 79. Slices were then transferred onto Millicell membrane inserts (Millipore, Billerica, MA, USA) and placed into 6‐well culture plates, filled with 1 mL medium. Comparable with the mean duration of naturally occurring SCI in the investigated canine population of the present study, slices were cultivated for 0, 3 and 9 days, respectively. As the generation of the slices itself represents a massive mechanical injury, the time in culture is assumed to correlate to time post SCI 62, 79. In the context of the present study, the generation of slices is characterized by axonal transection and thus, this in vitro model was included to elucidate potential commonalities and differences to axonal pathology after naturally occurring mixed contusive‐compressive SCI. After removal from the culturing system, slices were fixed in 10% neutral‐buffered formalin for 48 hours. Slices immediately fixed after chopping (day 0) served as control tissue. Subsequently, spinal cord slices were routinely embedded in paraffin wax and sectioned for histology and immunohistochemistry. Additionally, for real‐time qPCR slices were snap‐frozen in OCT compound as described earlier.
Histology and immunohistochemistry of naturally traumatized spinal cord tissue
To evaluate white matter alterations in naturally traumatized canine spinal cords, paraffin‐embedded sections with a thickness of 4 μm were prepared from each of the 13 localizations per animal and from the epicenters of dog 18, 19 and 27, respectively, using hematoxylin and eosin (H&E) staining. Immunohistochemistry was performed on selected paraffin‐embedded serial sections (Loc. 0, 3, −3, 9) of traumatized and control animals.
After mounting on SuperFrost® Plus slides (Menzel–Gläser, Menzel GmbH & Co KG, Braunschweig, Germany), sections were incubated with mono‐ and polyclonal antibodies directed against phosphorylated neurofilament (p‐NF), non‐phosphorylated neurofilament (n‐NF), β‐amyloid precusor protein (β‐APP), GAP‐43, and myelin basic protein (MBP; Table 2). Additionally, immunohistochemistry for the detection of axonal changes was performed on cultured organotypic slices. Different pretreatments were applied if necessary (Table 2). The avidin–biotin–peroxidase complex method was performed as described previously 47, 75. Briefly, tissue was deparaffinized in Roticlear® (Carl Roth GmbH, Karlsruhe, Germany) and hydrated with graded alcohols. Endogenous peroxidase activity was blocked by 0.5% H2O2 in methanol and sections were incubated overnight at 4°C with primary antibodies. For controls, the specific monoclonal antibodies were replaced by ascitic fluid from non‐immunized Bagg albino/cJ (BALB/cJ) mice. Serum from non‐immunized rabbits in a dilution according to the concentration of the primary antibodies served as negative control for the used polyclonal antibodies. Incubation with primary antibodies was followed by biotinylated goat‐anti‐mouse or goat‐anti‐rabbit antibodies (secondary antibodies), respectively, for 30 minutes at room temperature, followed by the avidin–biotin–peroxidase complex (Vectastain Elite ABC Kit, PK6100, Vector Laboratories, Burlingame, CA, USA) for 30 minutes at room temperature, incubation with 3,3‐diaminobenzidine‐tetrahydrochloride–H2O2 in 0.1 M imidazole, pH 7.1 for 5 minutes and slight counterstaining with Mayer's hemalaun.
Table 2.
Antibodies used for immunohistochemistry.
| Antibody | Supplier | Catalog number | Source and clonality | Pretreatment and dilutiona |
|---|---|---|---|---|
| Phosphorylated neurofilament | Sternberger monoclonals | SMI312 | Mouse MC | Triton X (0,25% in PBS, 15 min, 37°C) 1:4000 |
| Non‐phosphorylated neurofilament | Sternberger monoclonals | SMI311 | Mouse MC | Citrate buffer (pH 6.0, 20 min, MW) 1:1000 |
| β‐amyloid precursor protein | Chemicon | M348 | Mouse MC | Citrate buffer (pH 6.0, 20 min, MW) 1:800 |
| Growth‐associated protein‐43 | Millipore | AB5220 | Rabbit PC | Citrate buffer (pH 6.0, 20 min, MW) 1:600 |
| Myelin basic protein | Chemicon | AB980 | Rabbit PC | Citrate buffer (pH 6.0, 20 min, MW) 1:800 |
Diluted in PBS with 1% bovine serum albumin.
MC = monoclonal; MW = microwave (800 W); PBS = phosphate‐buffered saline; PC = polyclonal.
Transmission electron microscopy and immunoelectron microscopy for GAP‐43
Ultrastructural analysis of white matter changes in naturally traumatized dogs was performed on epoxy resin‐embedded spinal cord tissue as described 47, 79, 87. Ultrathin sections were cut with a diamond knife and transferred to 200‐mesh nickel grids.
Additionally, for immunoelectron microscopical demonstration of GAP‐43 representative sections from a dog with subacute SCI were stained with immunogold as described with slight modification 48. Ultrathin sections were pretreated with 10% sodium‐metaperiodate for 10 minutes, followed by rinsing in distilled water three times for 5 minutes 9. Sections were incubated in phosphate‐buffered saline (PBS) containing 0.05 M glycine for 20 minutes and washed in PBS twice for 5 minutes, followed by a pretreatment with 5% goat serum, 0.5% bovine serum albumin (BSA) and 0.1% cold water fish skin (CWFS) gelatine diluted in PBS for 30 minutes. The primary antibody (GAP‐43, Table 2) diluted in PBS with 0.5% BSA and 0.1% CWFS gelatine was applied overnight at 4°C. After rinsing in PBS with 0.5% BSA and 0.1% CWFS gelatine, sections were incubated with goat‐anti‐rabbit antibodies conjugated with 10 nm gold particles (Plano, Wetzlar, Germany; diluted 1:40 in PBS) for 60 minutes, followed by rinsing in PBS with 0.5% BSA and 0.1% CWFS gelatine and in PBS alone.
After fixation with 2% glutaraldehyde in PBS, samples were washed in distilled water. All ultrathin sections were stained with 2% aqueous uranyl acetate with lead citrate and examined with a transmission electron microscope (EM 10C, Zeiss, Oberkochen, Germany) 47, 79, 87.
Evaluation of transversal spinal cord sections
The white matter of H&E‐stained spinal cord transverse sections was divided into six areas, followed by semiquantitative evaluation of axonal swellings and phagocytic responses (0 = no changes; 1 = 1–7 swollen axons/1–3 phagocytes; 2 = 8–15 swollen axons/4–6 phagocytes; and 3 = more than 15 swollen axons/more than 6 phagocytes per spinal cord area). Subsequently, the overall mean value for the white matter of each transverse spinal cord section was calculated (sum of scores of six white matter areas divided by 6) and used for statistical analysis 79. Additionally, necrotic and hemorrhagic changes in the white and gray matter were evaluated semiquantitatively (0 = no changes; 1 = mild; 2 = moderate; and 3 = severe necrotic/hemorrhagic changes per spinal cord area) resulting in a combined overall mean value per spinal cord transverse section for necrosis/hemorrhage.
Analysis of immunohistochemistry for n‐NF, β‐APP and GAP‐43 was performed by counting immuno‐positive and ‐negative axons per 0.063 mm2 in 30 randomly chosen high‐power fields of the spinal cord white matter in vivo and of five randomly chosen high‐power fields in organotypic spinal cord slices using a counting grid. Subsequently, the percentage of immuno‐positive axons per spinal cord cross section was calculated 79.
To determine the p‐NF/n‐NF ratio, immunoreactivity for p‐NF and n‐NF was additionally analyzed morphometrically by measuring the respective immunopositive area after manually outlining the total white matter plane, employing the analySIS® 3.1 software (SOFT Imaging System, Münster, Germany). Subsequently, the immunopositive area was calculated as percentage of the total white matter. The same method was used to analyze the MBP‐positive area per spinal cord white matter 47.
qPCR
MMP‐2 and MMP‐9 mRNA were quantified using real‐time qPCR as described 69. Briefly, RNA was extracted from OCT‐embedded frozen spinal cord tissue of naturally traumatized dogs and organotypic slice cultures using RNeasy Lipid Mini Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. As positive controls, RNA was further isolated from confluent cell layers of a canine macrophage cell line [DH82; 69, 81] using TRIzol (Invitrogen, Life Technologies GmbH, Darmstadt, Germany), digested with RNase‐Free DNAse (Qiagen) and purified using a silica gel‐based membrane (RNeasy Mini, Qiagen) according to the manufacturer's protocols. Total RNA was reversely transcribed to complementary DNA (cDNA) using the Omniscript kit (Qiagen) with RNase Out (Invitrogen) and Random Primers (Promega, Mannheim, Germany) following the manufacturer's instructions. Reverse transcription was performed using 1 μg RNA in 1 × reverse transcriptase reaction (RT) buffer with 0.5 mM of each deoxynucleotide triphosphate (dNTP), 10 μM random hexamers (Promega), 0.5 U/μL RNAse inhibitor (RNAse Out, Invitrogen) and 0.2 U/μL Omniscript Reverse Transcriptase (Qiagen) in a total volume of 20 μL at 37°C for 1 hour. For the generation of standard dilution series qualitative PCR was performed with cDNA from DH82 cells using primers specific for the housekeeping genes glyceraldehyde 3‐phosphate dehydrogenase (GAPDH), hypoxanthine‐guanine phosphoribosyltransferase (HPRT), and elongation factor (EF)‐1α, and for MMP‐2 and MMP‐9, respectively (Table 3; 69, 79, 81). Qualitative PCR was performed using a PTC200 thermocycler (Biozym Diagnostic, Hessisch–Oldendorf, Germany) under the conditions as described 69, 79, 81. The annealing temperature was adjusted to 56°C (MMP‐2, ‐9, HPRT), 59°C (GAPDH), and 60°C (EF‐1α). PCR products were analyzed by agarose gel electrophoresis.
Table 3.
Target gene, sequence, amplicon length and GenBank accession number of primers used for qualitative polymerase chain reaction analysis for the generation of standards.
| Target gene | Primer sequence | Amplicon length [BP] | GenBank accession |
|---|---|---|---|
| MMP‐2 | Forward: 5′‐CCAAGAACTTCCGTCTGTCC‐3′ | 606 | AF177217 |
| Reverse: 5′‐AGCTATGACCACTGCCTTGC‐3′ | |||
| MMP‐9 | Forward: 5′‐GAGGTTCGACGTGAAGGCGCAGAT‐3′ | 200 | NM_004994 |
| Reverse: 5′‐AGGTCACGTAGCCCACTTCGTCCAC‐3′ | |||
| HPRT | Forward: 5′‐TAAAAGTAATTGGTGGAGAT‐3′ | 133 | CFU16661 |
| Reverse: 5′‐ATTATACTGCGCGACCAAG‐3′ | |||
| EF‐1α | Forward: 5′‐AGCCCTTGCGCCTGCCTCTC‐3′ | 219 | X03558 |
| Reverse: 5′‐CAGACACATTCTTGACATTGAAGC‐3′ | |||
| GAPDH | Forward: 5′‐AAGGTCGGAGTCAACGGATT‐3′ | 365 | AB038240 |
| Reverse: 5′‐GCAGAAGAAGCAGAGATGATG‐3′ |
BP = base pairs; EF‐1α = elongation factor 1α; GAPDH = glyceraldehyde‐3‐phosphate‐dehydrogenase; HPRT = hypoxanthine‐guanine phosphoribosyltransferase; MMP = matrix metalloproteinase.
Real‐time qPCR and data analysis were performed using the Mx3005P QPCR System (Agilent Technologies, Waldbronn, Germany) as described 69, 81. In addition to the cDNA samples, 10‐fold serial dilutions of purified, agarose gel extracted (NucleoSpin Extract II Kit, Macherey‐Nagel, Düren, Germany) qualitative PCR products from 102 to 108 copies per sample were used as templates to generate standard curves for estimation of copy numbers on each plate. Standards, samples, and no‐template controls were run in duplicates. Primers used for real‐time qPCR are listed in Table 4. The annealing temperature was adjusted to 60°C (HPRT, EF‐1α) and 64°C (MMP‐2, GAPDH). Specificity of the reaction was determined by melting curve analysis. In the case of MMP‐9, real‐time qPCR was performed using 0.025 U/μL SureStart Taq DNA Polymerase in 1 × Core PCR buffer with 5.0 mM MgCl2, 300 nM of each primer, 200 nM TaqMan probe, 76 nM Rox as reference dye and 200 μM dNTP mix 69. Consecutively, gene expression values were normalized to 100 ng transcribed RNA. Normalization of the genes of interest was performed against the three housekeeping genes, GAPDH, EF‐1α and HPRT, using the software geNorm® (Ghent University Hospital Center for Medical Genetics; available at http://medgen.ugent.be/~jvdesomp/genorm/), as described 79, 89. Briefly, the software detects the most stable reference genes of which the geometric means are used to calculate a normalization factor for the genes of interest.
Table 4.
Target gene, sequence, amplicon length and GenBank accession number of primers used for real–time quantitative polymerase chain reaction analysis.
| Target gene | Primer sequence | Amplicon length (BP) | GenBank accession |
|---|---|---|---|
| MMP‐2 | Forward: 5′‐GGAGATCTTCTTCTTCAAGGACCG‐3′ | 89 | AF177217 |
| Reverse: 5′‐AGAATGTGGCTACCAGCAGGG‐3′ | |||
| MMP‐9 | Forward: 5′‐CATGACATCTTCCAGTACCAAG‐3′ | 85 | AB006421 |
| Reverse: 5′‐GGTTCACCTCATTCCGAGAA‐3′ | |||
| Probe: 5′‐FAMCTACTTCTGCCAGGACCGCTTCTACTTAMRA‐3′ | |||
| HPRT | Forward: 5′‐GAGATGACCTCTCAACTTTAACTGAAAA‐3′ | 89 | CFU16661 |
| Reverse: 5′‐GGGAAGCAAGGTTTGCATTG‐3′ | |||
| EF‐1α | Forward: 5′‐CAAAAACGACCCACCAATGG‐3′ | 68 | AY195837 |
| Reverse: 5′‐GGCCTGGATGGTTCAGGATA‐3′ | |||
| GAPDH | Forward: 5′‐GTCATCAACGGGAAGTCCATCTC‐3′ | 84 | AB038240 |
| Reverse: 5′‐AACATACTCAGCACCAGCATCAC‐3′ |
BP = base pairs; EF‐1α = elongation factor 1α; GAPDH = glyceraldehyde‐3‐phosphate‐dehydrogenase; HPRT = hypoxanthine‐guanine phosphoribosyltransferase; MMP = matrix metalloproteinase.
Statistical analysis and illustrations
For all statistical analyses nonparametric tests were applied. Comparison of data between the three different groups was conducted using the Kruskal–Wallis test. If significant P‐values were obtained, subsequent groupwise comparisons were done using the Mann–Whitney U‐test followed by alpha adjustment according to the method of Bonferroni (alpha value divided by 3) to minimize type I errors in multiple hypothesis testing. Thus, for Mann–Whitney U‐test, P‐values lower than 0.017 were considered as significant.
For related samples such as different spinal cord localizations per group and different time points of the slice culturing system, the Friedman test was applied. Subsequent pairwise comparisons were conducted using the signed rank test. For the correlation of MMP copynumbers and immunohistochemical data of the axonal markers, Spearman's rank correlation analysis was performed for both naturally traumatized groups. The software used for statistical analyses was Statistical Analysis System (SAS) for Windows, version 9.1 (SAS Institute, Cary, NC, USA) and the Statistical Package for the Social Sciences (SPSS version 18.0; SPSS Inc., Chicago, IL, USA), respectively. For the creation of box‐and‐whisker plots, GraphPad Prism version 5.0 for Windows (GraphPad Software Inc., La Jolla, CA, USA) was used.
Immunohistochemical images were created using an Olympus BX‐51 digital camera microscope [Olympus Optical Co. (Europe) GmbH, Hamburg, Germany] and stored as tagged image file format (tiff) files using cell‐D imaging software (Olympus Soft Imaging Solutions GmbH, Münster). Further processing, including adjustment of contrast and brightness if necessary, was performed with Adobe® Photoshop® 7.0 (Adobe Systems, Inc., San Jose, CA).
Results
Alterations and spatio‐temporal extension of changes in naturally traumatized spinal cord tissue
The spinal cord tissues of control animals showed no morphologic changes apart from single swollen axons in a few animals. The most prominent findings were in dogs with acute SCI axonal changes (see later; Figures 1A and 2A). In dogs with subacute SCI (group 3) prominent myelin changes occurred in addition to axonal changes (see later; Figures 1 and 2B,D). In dogs with acute SCI, occasional mild to moderate, multifocal hemorrhages, and/or necrosis were seen, mainly restricted to the epicenter and localization 1.5 (Figure 2E). These changes were accompanied by minimal numbers of neutrophilic granulocytes in the epicenter of animal no. 13. During subacute SCI, mild necrotic and/or hemorrhagic changes, which were mainly restricted to the epicenter, could be noted in some animals of group 3 (Figure 2F).
Figure 1.
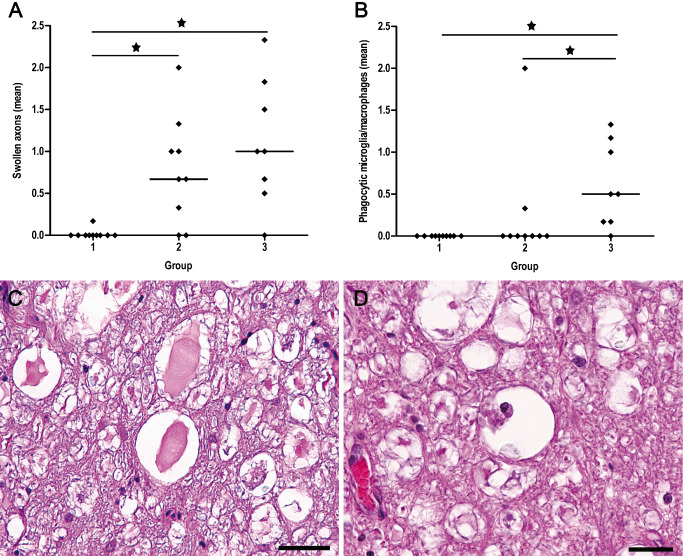
Semiquantitative analysis and H&E staining of swollen axons (A,C) and phagocytic microglia/macrophages (B,D) in the lesion epicenter. (A) Scatter plots with median illustrate the mean values of semiquantitative evaluation of swollen axons in H&E‐stained sections that are significantly increased in dogs with SCI compared with controls. (B) Increased numbers of phagocytic cells are exclusively noted in dogs with subacute SCI compared with controls and dogs with acute SCI (group 2). Group 1 = control animals. Group 2 = dogs with acute SCI. Group 3 = dogs with subacute SCI. Statistically significant differences are marked with an asterisk (Kruskal–Wallis test with subsequent pairwise Mann–Whitney U‐test, alpha adjusted according to the method of Bonferroni). (C) Swollen axons are a prominent finding in animals with SCI and are mainly located in the ventral funiculi of the white matter (epicenter of animal no. 20). (D) Phagocytic cells, mainly located in virtually empty and dilated myelin sheaths, are nearly completely restricted to animals with subacute SCI (group 3; epicenter of animal no. 23). Scale bar (C) = 50 μm (D) = 25 μm. H&E = hematoxylin and eosin; SCI = spinal cord injury.
Figure 2.
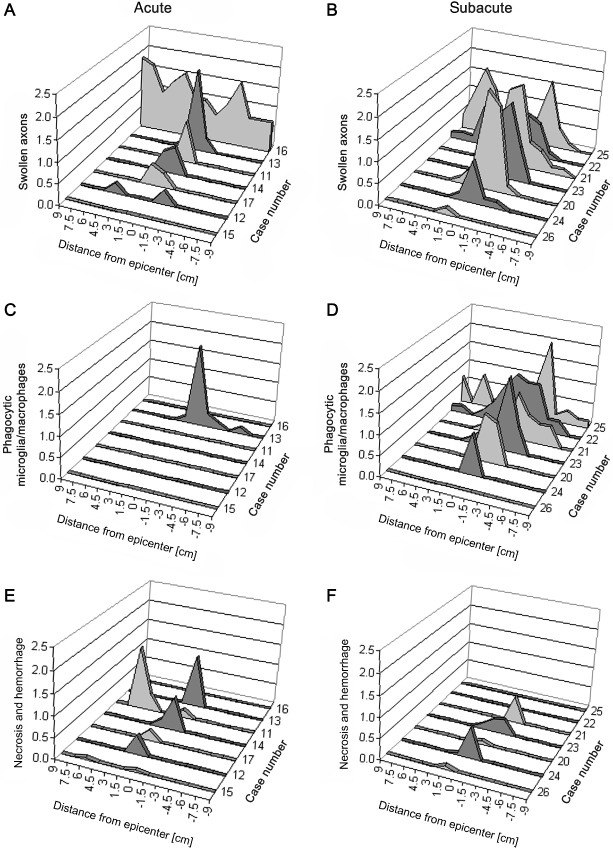
Extension of axonal changes (A,B), phagocytic microglia/macrophages (C,D) and necrotic/haemorrhagic changes (E,F) in dogs with acute (group 2) and subacute (group 3) SCI. Minus signs on the X‐axis symbolize caudal (‐) and numbers without signs cranial direction, respectively. (A) Semiquantitatively evaluated swollen axons in dogs with acute SCI (group 2) are mainly located in the epicenter (0). One animal shows a significant spread of swollen axons over the entire investigated length of the spinal cord. (B) Axonal damage in dogs with subacute SCI (group 3) spreads over long distances with an accentuation to the caudal direction. (C) Phagocytic microglia/macrophages are restricted to a single animal with acute SCI. (D) In contrast, most dogs with subacute SCI show significant numbers of phagocytic microglia/macrophages extending over long distances from the epicenter (0). (E) Mild to moderate, multifocal hemorrhages and/or necrosis are seen in animals with acute SCI, mainly restricted to the epicenter and localization 1.5. (F) Some animals with subacute SCI showed mild, multifocal hemorrhages and/or necrosis, mainly restricted to the epicenter. SCI = spinal cord injury.
Swollen axons
Axonal swelling was a prominent finding in traumatized dogs and mainly localized in ventral and ventro‐lateral tracts of the spinal cord white matter (Figure 1C). With regard to the epicenters, significantly higher mean scores for swollen axons were detected in animals with acute (group 2) and subacute (group 3) SCI compared with controls (Kruskal–Wallis test: P = 0.001; groupwise comparison by Mann–Whitney U‐test: controls vs. acute SCI: P = 0.002; controls vs. subacute SCI P = 0.001; Figure 1A). Although generally more pronounced in dogs with subacute SCI there was no statistical difference in the number of swollen axons in the epicenter between both traumatized groups. However, there was a difference between animals with acute and subacute SCI regarding the cranio‐caudal extension of axonal swellings. In animals with acute SCI (group 2), swollen axons were nearly restricted to the epicenter with an extension of up to 1.5 cm in cranial and caudal direction, respectively (Figure 2A). In one animal (no. 16), an extension of up to 9.0 cm in both directions could be noted. In contrast, a remarkable overall extension of axonal swelling was noted in animals with subacute SCI (group 3), becoming less pronounced distant from the epicenter (Figure 2B). In these dogs, the numbers of swollen axons were similar in both, the epicenter and at Loc. −3 (P > 0.05), compared with Locs. 3 and 9 (P = 0.001). The most widespread axonal changes were seen in the spinal cord of dog no. 25, where swollen axons could be detected up to a distance of 9 cm from the epicenter in cranial and 6 cm in caudal direction (Figure 2B). Extension of morphological axonal changes in other animals of group 3 varied from 13.5 cm in dog no. 22 to 1.5 cm in dog no. 26 (Figure 2B).
Phagocytic microglia/macrophages
In dogs with acute SCI (group 2), phagocytic microglia/macrophages were restricted to a single animal (no. 13) located within the epicenter and extending 1.5 cm in cranial and 6 cm in caudal direction (Figure 2C). A delayed occurrence of phagocytic cells, mainly located in virtually empty and dilated myelin sheaths, was nearly completely restricted to animals with subacute SCI (Kruskal–Wallis test: P < 0.001; groupwise comparison by Mann–Whitney U‐test: controls vs. subacute SCI: P < 0.001; acute SCI vs. subacute SCI: P = 0.007; Figure 1B,D). In these animals, phagocytes were most prominent in the epicenter, but parallel to axonal swelling markedly extended to cranial and/or caudal localizations, where the changes became less pronounced (Figure 2D). The most widespread changes were seen within the spinal cord of dog no. 25 (Figure 2D). In this individual, phagocytic cells were detectable up to a distance of 9 cm from the epicenter.
Immunohistochemical detection of axonal changes and MBP expression in naturally traumatized spinal cord tissue
To further characterize axonal changes, immunhistochemistry for the detection of neurofilament phosphorylation, axonal transport mechanisms and potential axonal regeneration attempts was performed. The expression of p‐NF, n‐NF, β‐APP and GAP‐43 was investigated in the white matter of the four earlier‐mentioned spinal cord localizations per animal (Loc. 0, 3, −3, 9). Additionally, myelin pathology was determined by MBP‐specific immunohistochemistry.
p‐NF expression was found in normally sized axons of all groups as well as in swollen axons of dogs with SCI (Figure 3A–C).
Figure 3.
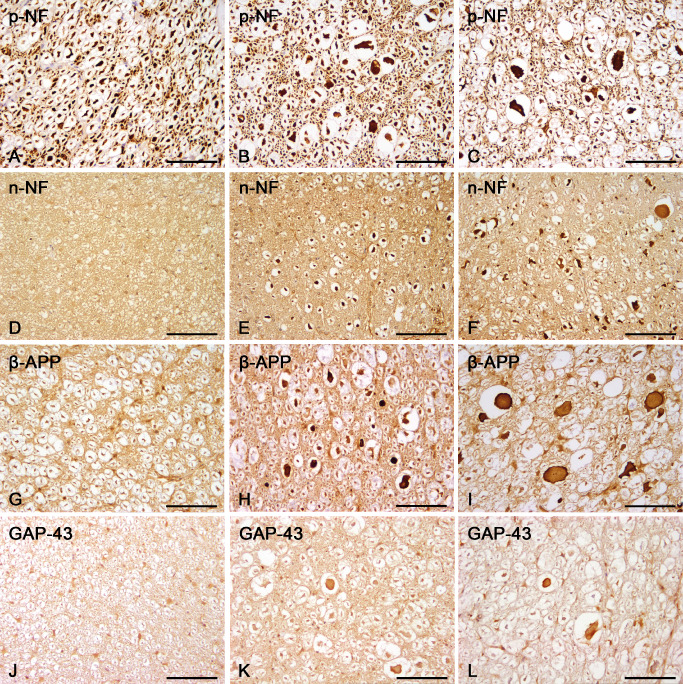
Immunohistochemistry of axonal changes in controls and the lesion epicenter of canine spinal cord injury (SCI). (A–C) Axonal expression of p‐NF. p‐NF is expressed by healthy axons in control animals (A) and additionally by swollen axons in dogs with acute (B) and subacute (C) SCI. (D–F) Axonal expression of n‐NF. n‐NF is not expressed in axons of control animals (D) whereas there is a significant n‐NF expression in swollen and normal‐diameter axons of dogs with acute (E) and subacute (F) trauma. (G–I) Axonal expression of β‐APP. β‐APP is confined to some glial cells and not expressed by healthy axons in control animals (G). In dogs with acute (H) and subacute (I) SCI β‐APP is mainly detected in swollen axons. (J–L) Axonal expression of GAP‐43. GAP‐43 is not expressed by healthy axons in control animals (J) whereas in dogs with acute (K) and subacute (L) trauma swollen axons expressing GAP‐43 are found within dilated myelin sheaths. Scale bar (A–L) = 100 μm. Avidin–biotin–complex method and staining dye 3,3′‐diaminobenzidine. β‐APP = β‐amyloid precursor protein; GAP = growth‐associated protein; n‐NF = non‐phosphorylated neurofilament; p‐NF = phosphorylated neurofilament.
n‐NF expression was restricted to neuronal perikarya in control animals, whereas in both groups of traumatized dogs, n‐NF was additionally expressed by a subpopulation of swollen axons and in several axons with a regularly appearing diameter (Figure 3D–F). The number of n‐NF‐positive axons was thus significantly increased in the epicenter of dogs with acute and subacute SCI compared with controls (Kruskal–Wallis test: P = 0.003; groupwise comparison by Mann–Whitney U‐test: controls vs. acute SCI: P = 0.001; controls vs. subacute SCI: P = 0.016; Figure 4A). Comparing spinal cord localizations within traumatized groups, there were no significant differences between the epicenter and distant sites, indicating a widespread expression of n‐NF within the entire investigated spinal cord segments.
Figure 4.
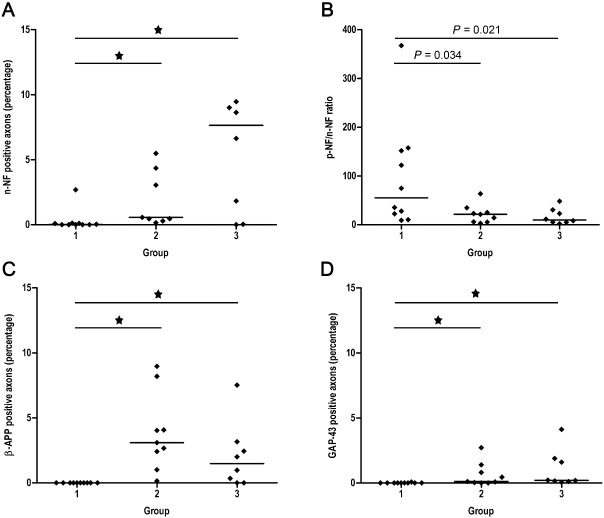
Scatter plots with median illustrating the results of the quantitative immunohistochemical analysis of axonal changes in the lesion epicenter of canine SCI. Group 1 = controls, group 2 = dogs with acute SCI, group 3 = dogs with subacute SCI. Statistically significant differences are marked with an asterisk (Kruskal–Wallis test with subsequent pairwise Mann–Whitney U ‐test, alpha adjusted according to the method of Bonferroni). (A) The percentage of axons expressing n‐NF is increased in dogs with acute and subacute SCI compared with controls. (B) Depiction of the p‐NF/n‐NF ratio as determined by morphometrical assessment of the immunopositive total white matter. Because of the relative increase in n‐NF expression there is a decrease of the p‐NF/n‐NF ratio in dogs with acute and subacute SCI compared with controls. (C) The percentage of axons expressing β‐amyloid precursor protein is increased in dogs with SCI compared with controls. (D) Similarly, the percentage of axons expressing growth‐associated protein‐43 is increased in traumatized dogs compared with healthy controls. n‐NF = non‐phosphorylated neurofilament; p‐NF = phosphorylated neurofilament; SCI = spinal cord injury.
To evaluate the relation between p‐NF and n‐NF expression, additional morphometrical analyses were performed, and the p‐NF/n‐NF‐ratio was subsequently calculated. Morphometrically, because of a relative increase of the n‐NF immunoreactive total white matter area in groups 2 and 3, there was a significant decrease of the p‐NF/n‐NF‐ratio in the epicenter of both traumatized groups compared with controls (Kruskal–Wallis test: P = 0.03). However, P‐values of groupwise comparisons did not reach the level of significance when alpha was adjusted according to the method of Bonferroni (adjusted P‐value for significance level = 0.017; controls vs. acute SCI: P = 0.034; controls vs. subacute SCI: P = 0.021; Figure 4B).
β‐APP expression was confined to neuronal perikarya and some glial cells of the white matter in control animals. In traumatized dogs β‐APP was additionally found in a remarkable number of swollen axons resulting in a significant increase of axonal β‐APP expression in animals with acute and subacute SCI (Kruskal–Wallis test: P < 0.001; groupwise comparison by Mann–Whitney U‐test: controls vs. acute SCI: P < 0.001; controls vs. subacute SCI: P = 0.006; Figures 3G–I and 4C). In dogs with acute trauma (group 2), there was no significant cranio‐caudal extension of axonal β‐APP expression, as β‐APP positive axons were confined to the epicenter compared with other localizations (P < 0.001). In contrast, in dogs with subacute SCI (group 3), the number of β‐APP positive axons was increased in both the epicenter and at localization −3, indicating a significant progressive caudal extension (P = 0.001).
Although confined to a comparatively low absolute number of axons, GAP‐43, a marker for axonal regeneration, was expressed in a subpopulation of axons only in traumatized animals (Figure 3J–L), resulting in a significant upregulation of GAP‐43 immunoreactivity in the epicenter of both traumatized groups (Kruskal–Wallis test: P = 0.001; groupwise comparison by Mann–Whitney U‐test: controls vs. acute SCI: P = 0.003; controls vs. subacute SCI: P < 0.001; Figure 4D). In dogs with acute SCI (group 2), GAP‐43 positive axons were exclusively detected in the epicenter (P < 0.001), whereas in dogs with subacute SCI (group 3), comparable with β‐APP, the number of GAP‐43 positive axons was additionally significantly increased at localization −3 (P < 0.001).
In contrast to axonal changes, MBP immunoreactivity of the white matter in the lesion epicenter was significantly decreased exclusively in dogs with subacute SCI (Kruskal–Wallis test: P = 0.024; groupwise comparison by Mann–Whitney U‐test: controls vs. subacute SCI: P = 0.016; acute SCI vs. subacute SCI: P = 0.021; data not shown). Regarding the extension of MBP‐immunoreactive area in dogs with subacute SCI (group 3) a tendency for decreased MBP‐expression was observed in the epicenter compared with localization 9, although not reaching the level of significance (P = 0.058).
Axonal changes in spinal cord slice cultures
In order to comparatively evaluate phenotypical axonal changes in a simplified in vitro model, devoid of influences from the periphery, canine organotypic spinal cord slice cultures were investigated. Culturing of spinal cord slices was accompanied by a notable swelling of transected axons beginning at day 3 in culture, whereas slices of the 0‐day control exhibited a regular and organotypic morphology without remarkable axonal alterations. Comparison of immunohistochemical axonal markers in spinal cord slice cultures of days 0, 3 and 9 revealed a significantly higher number of n‐NF–positive axons in slices cultivated for 3 and 9 days, respectively, compared with 0‐day controls (P = 0.046 and 0.025, respectively). In contrast to spinal cord tissue of naturally traumatized dogs, β‐APP and GAP‐43 were not present in axons of spinal cord slices.
Ultrastructural changes in naturally traumatized spinal cord tissue
In spinal cord tissue of control animals, axons occupied most of the intramyelinic space and were characterized by an axoplasm consisting of well‐organized neurofilaments and a moderate number of normal‐sized organelles such as the mitochondria (Figure 5A). Mild lamellar separations of the surrounding myelin sheaths were observed in control and traumatized animals and are thus interpreted as changes attributed to fixation artifacts (Figure 5A–D). Degenerative axonal alterations were not noted in controls. In contrast, axons of traumatized animals showed various degenerative changes such as an increase in the periaxonal space, excessive myelin fragmentation, vesicular myelin formation and accumulation of vesicular axoplasmic structures and electron‐dense bodies, mostly accompanied by a marked axonal swelling (Figure 5B,C). Further, axoplasmic neurofilaments appeared nonparallel and disintegrated (Figure 5D, inset). Such changes were most prominent in dogs with subacute SCI (group 3). In animals with acute SCI (group 2), similar degenerative axonal changes were seen, but there was still a significant number of axons without obvious alterations. Interestingly, in one animal with subacute trauma (group 3; no. 20), several swollen axons lacking dense body accumulation, but massively filled with impressive numbers of mitochondria were seen (Figure 5D). To further characterize these mitochondria‐rich swollen axons, immunoelectron microscopy for the detection of GAP‐43 was performed on representative ultrathin sections of this animal, revealing axoplasmic clusters of immunogold particles labeling GAP‐43 in close proximity to mitochondria, potentially associated to microtubules (Figure 5E, inset).
Figure 5.
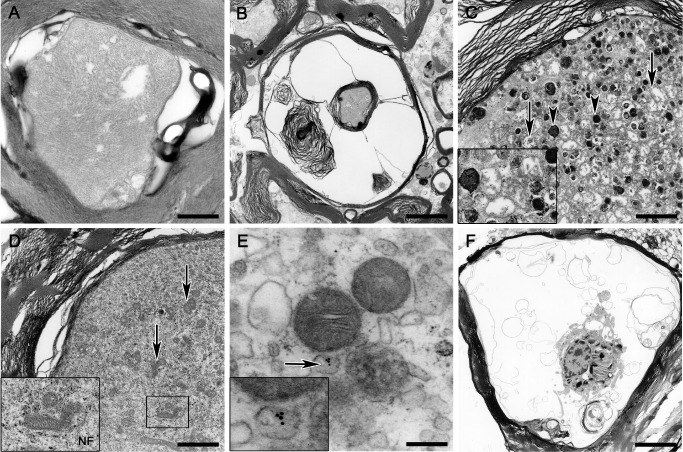
Transmission electron microscopy. (A) Intact axon of a control animal exhibiting mild myelin changes interpreted as fixation artifacts. (B) Myelin sheath dilatation and myelin fragmentation in an animal with acute SCI. (C) Axon swelling and axoplasmic degeneration with accumulation of numerous electron‐dense bodies (arrowheads; inset) and swollen mitochondria (arrows; inset) in an animal with subacute SCI. (D) Marked axoplasmic accumulation of mitochondria (arrows; inset) intermingled with numerous neurofilaments (inset) without dense body formation in a swollen axon of an animal with subacute SCI. (E) Immunoelectron microscopic illustration of potentially microtubule‐associated gold particles specifically labeling growth‐associated protein‐43 (arrow, inset) in a mitochondria‐rich swollen axon of the same animal as in (D). (F) Large phagocytic cell located within an empty myelin sheath showing phagocytosed myelin and axonal remnants in an animal with subacute SCI. A scale bar = 0.5 μm; B scale bar = 2.7 μm; C,D scale bar = 1.7 μm; E scale bar = 0.2 μm; F scale bar = 5.4 μm. SCI = spinal cord injury.
Solely in animals with subacute SCI, large phagocytic cells were detected, mainly located within empty myelin sheaths, showing phagocytosed myelin and axonal remnants consistent with axono‐ and myelinophagia (Figure 5F). However, naked axons indicating obvious demyelination were only rarely observed. The vast majority of degenerated axons, even in dogs with subacute SCI, were still ensheathed by myelin lamellae. Consequently, there were no hints for remyelination events neither by Schwann cells nor by oligodendrocytes.
Quantification of matrix MMPs‐2 and 9 in spinal cord tissue of naturally traumatized dogs and in organotypic slice cultures
To evaluate potential molecular alterations in the orchestration of the gelatinases MMP‐2 and MMP‐9, both representing crucial modulators involved in tissue damage and regeneration during secondary injury processes following SCI, transcripts were quantified using real‐time qPCR. Compared with controls, MMP‐2 was significantly downregulated in dogs with acute SCI (Kruskal–Wallis test: P = 0.015; groupwise comparison by Mann–Whitney U‐test: controls vs. acute SCI: P = 0.003), whereas there was no difference between controls and dogs with subacute SCI (Figure 6A). In contrast, MMP‐9 exhibited a statistically significant upregulation during acute SCI (Kruskal–Wallis test: P = 0.023; groupwise comparison by Mann–Whitney U‐test: controls vs. acute SCI: P = 0.006), while there were no changes in expression levels during subacute SCI (Figure 6B). Moreover, MMP‐9 mRNA levels exhibited a moderate positive correlation to β‐APP expression (Spearman's rank correlation; r = 0.54; P = 0.04). In organotypic slice cultures, MMP‐2 expression was not changed on days 3 and 9 in culture compared with control slices of day 0. However, pairwise comparison revealed a significantly higher expression of MMP‐2 on day 9 compared with day 3 (P = 0.031; Figure 6C). MMP‐9 was significantly upregulated on days 3 and 9 in culture compared with the control slices of day 0 (P = 0.013; Figure 6D).
Figure 6.
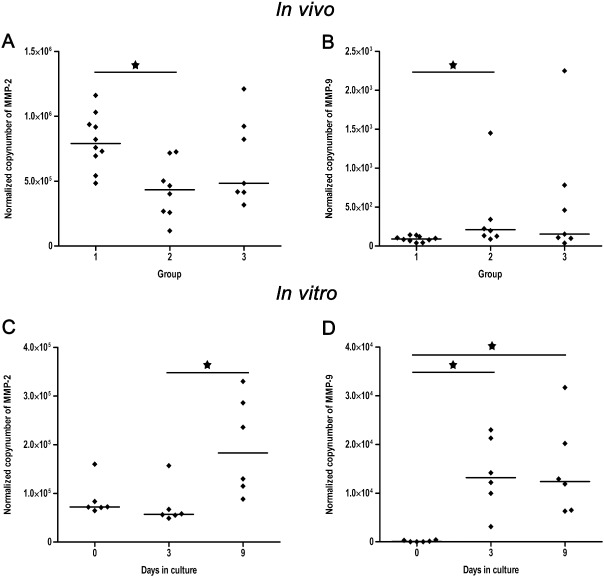
Scatter plots with median illustrating the results of the real‐time quantitative polymerase chain reaction for the analysis of normalised mRNA levels per 100 ng transcribed RNA of the gelatinases MMP‐2 and MMP‐9 in naturally traumatized dogs (A,B) and organotypic spinal cord slice cultures (C,D). Group 1 = controls, group 2 = dogs with acute SCI, group 3 = dogs with subacute SCI. 0, 3, 9 = days in culture. Statistically significant differences are marked with an asterisk [Kruskal–Wallis test with subsequent pairwise Mann–Whitney U ‐test, alpha adjusted according to the method of Bonferroni (A,B); Friedman test and subsequent pairwise signed rank test (C,D)]. (A) MMP‐2 mRNA is transiently downregulated in dogs with acute SCI compared with controls. (B) MMP‐9 shows a significant upregulation in dogs with acute SCI compared with controls. (C) In organotypic spinal cord slices MMP‐2 shows a delayed upregulation on day 9 compared with day 3 in culture. (D) MMP‐9 is upregulated on day 3 and 9 in cultivated spinal cord slices compared with the control tissue of 0 days. MMP = matrix metalloproteinases; SCI = spinal cord injury.
Discussion
The aim of the present study was to detail the spatio‐temporal development of white matter changes in a translational large animal model for preclinical SCI research. The present report represents the first multidirectional approach to the spatio‐temporal development of axonal changes in canine SCI, applying different methods such as histology, immunohistochemistry, transmission electron microscopy and real‐time qPCR. Given the fact that dogs are increasingly recognized as a valuable large animal model for clinical studies upon treatment strategies for SCI 37, 39, 78, 90, these data provide a basis for the development of future clinical applications such as stem cell transplantations in the canine animal model. The present data may additionally help to validate findings obtained in experimental studies and might be comparatively evaluated in the light of reported data on the neuropathology of human SCI 21, 41.
The investigated population of dogs doubtlessly exhibits a relatively high inter‐individual variability, which increases the risk of statistical outliers and limits the explanatory and statistical power of the provided results. However, the present data clearly indicate that axonopathy is a major feature of naturally occurring canine SCI, implying that this might represent a valuable target for future therapies. Axonopathy during canine naturally occurring IVDD‐related SCI is characterized by prominent and early occurring axonal swellings in the white matter accentuated in the ventral parts of the lesion epicenter as demonstrated by H&E staining. However, immunohistochemistry revealed that such axonal swellings (i.e. spheroids) represent considerably heterogeneous changes.
β‐APP expression in axons of the present study is in accordance to studies on experimental rodent and canine SCI as well as reports on naturally occurring SCI in humans 1, 20, 54, 70. In humans, β‐APP expression in axons can be demonstrated as early as 30 minutes after primary injury 20, and is still detectable after several years 1. Similar to the present results, a significant expansion of β‐APP immunopositivity of axons in segments far distant from the epicenter was demonstrated following human SCI 20. This underlines the possibility that axons do not necessarily need to be transected to exhibit β‐APP immunoreactivity, but single continuous axons might rather exhibit multiple β‐APP swellings 8, 19. Contrasting to experimental SCI, in most cases of naturally occurring SCI the primary injury is caused by structures located ventral to the spinal cord leading to generally more pronounced axonal damage in the ventral parts of the white matter 33, 38, 53. This might have consequences for clinical therapies 78. In combination with the neuroanatomical situation this fact might also explain that Wallerian‐like degeneration of the distal axon subsequently proceeds mainly to the caudal direction as it was detected in subacutely injured dogs of the present study.
Morphometrical assessment of the total p‐NF‐ and n‐NF–positive white matter area revealed a trend toward a decrease of the p‐NF/n‐NF ratio in both dogs with acute and subacute SCI because of the relative increase of n‐NF in damaged axons. Comparable with β‐APP, there was a significant expansion of axonal n‐NF immunoreactivity remote from the lesion site indicating that altered neurofilament phosphorylation might affect axons on their entire length. However, contrasting to β‐APP, n‐NF expression was also noted in a remarkable number of axons with a normal appearing diameter, suggesting that n‐NF is a suitable marker for early axonal damage. Comparably, in traumatic brain injury in rats, neurofilament compaction is known to occur independently from β‐APP immunoreactivity and is often found in thin and elongate axons whereas β‐APP expression in combination with neurofilament alteration or alone is predominantly seen in axonal swellings 82.
The present study is the first report of axonal GAP‐43 expression in a clinically relevant model for naturally occurring SCI. Moreover, a significant progressive extension of axonal GAP‐43 immunopositivity into segments caudal to the lesion site in dogs with subacute SCI was demonstrated indicative of a progressive neuroplastic response apart from the epicenter. Although GAP‐43 is an extensively used marker for axonal regeneration in experimental SCI 2, 23, 26, 30, 34, 55, 83, data on its expression during naturally occurring diseases are strikingly sparse 73. As early as 1928, Ramon y Cajal has recognized and outstandingly characterized abortive regenerative axonal changes in the traumatized CNS 16. Indeed, many axons start regeneration attempts in spinal injured mice within 6–24 hours demonstrating significant spontaneous regeneration in SCI, which might be a much more common process during SCI than previously thought 33, 43. However, these attempts seem to fail, probably because of the inability of axons to navigate in a proper direction 43. Abortive axonal regeneration is additionally believed to be attributed to the expression of inhibitory factors such as the myelin‐derived molecules Nogo and myelin‐associated glycoprotein or astrocyte‐derived chondroitin sulfate proteoglycanes 61, 93. Although the functional consequence remains unclear so far, the discovery of spontaneous axonal GAP‐43 expression in naturally occurring canine SCI might be relevant for future therapies. In fact, enhanced GAP‐43 expression in axons is inducible by the transplantation of regeneration‐promoting stem cells and glial cells in rodent models 3, 42.
Organotypic slice cultures of the CNS have been used as an in vitro model for SCI previously 17, 32, 40, 46, 62, 77, 79. While most authors have used tissue from rodents, recent reports have demonstrated that even culturing spinal cord slices from human post‐mortem tissue is possible and represents a valuable alternative for the assessment of therapeutic strategies for SCI 40. Organotypic slice cultures offer the opportunity to study endogenous changes in the local CNS environment without interference of systemic factors. We have previously reported that culturing of canine organotypic spinal cord slices is associated with a pro‐inflammatory microenvironment and an increase of MHC class II expressing microglial cells, highly similar to the in vivo situation. Thus, the hypothesis arose that culturing of spinal cord slices might additionally induce axonopathy comparable with naturally traumatized spinal cord tissue. Interestingly, the results of the present study revealed an increased expression of n‐NF after 3 and 9 days in cultured slices, whereas GAP‐43 and β‐APP expression were not noted in vitro. Artificial bilateral axotomy might explain that there is no accumulation of proteins that need to undergo axonal transport mechanisms. However, the fact that n‐NF expression is—comparable with the in vivo situation—increased in axons after 3 and 9 days in culture, demonstrates that cytoskeletal alterations might initially represent a local, but not immediately occurring phenomenon. Thus, the present results clearly demonstrate that anti–β‐APP and anti–n‐NF antibodies—both extensively used markers for axonal injury in general—are markers that react with distinct axonal changes and should not be considered as uniform and interchangeable. Thus, only combined immunohistochemical approaches might be sufficient to characterize the complexity of traumatic axonal damage 82.
Axonal damage in canine SCI was characterized by marked ultrastructural axoplasmic alterations highly comparable with reported changes in severed axons from rodents, monkeys and dogs 4, 12, 31, 50, 71, 78. In addition, in the present study, immunoelectron microscopy identified a subclass of organelle‐rich axonal swellings as being immunoreactive for GAP‐43. In fact, mitochondria are the key player in axonal energy supply and play a significant role in axonal development and regeneration 56. Axonal swellings with excessive organelle accumulations are commonly observed following SCI and have previously been believed to represent the terminals of the proximal stump of the interrupted axon 12.
Although phagocytic cells consistent with myelinophages have been observed, neither impressive widespread demyelination nor remyelinating changes were noted in the present study. However, from a detailed study on naturally occurring canine SCI, it is known that demyelination is followed by delayed remyelination by both, oligodendrocytes and Schwann cells after longer survival periods 78. The relatively short time period after the initial injury in the recent study population might be the explanation for the lacking detection of such events.
In contrast to the early occurrence of axonal changes, degenerative changes such as myelin loss and phagocytosis were restricted to dogs with subacute SCI hence highly reflecting processes in humans. Buss et al report a gradual loss of MBP immunoreactivity in spinal cord tissue of humans with SCI lasting for years after the primary injury 14. In the present study, axonal damage precedes myelin loss and immune responses. However, axonal damage is increasingly progressing over time and thus reflects secondary injury processes 64, 65, 66, 76. This is highly similar to the inside‐out theory in neurodegenerative diseases, which is based on the assumption that primary axonopathy functions as an early trigger for subsequent demyelination and responses of microglia/macrophages 8, 22, 75, 84, 85. Indeed, focal axonal swellings occur prior to demyelination in MS and experimental autoimmune encephalomyelitis [EAE; 59] revealing high similarities between SCI and neurodegenerative diseases in terms of Wallerian‐like degeneration processes.
In the present study, loss of MBP immunoreactivity coinciding with phagocytic responses might rather be attributed to myelin edema and lamellar splitting than true demyelination as totally demyelinated naked axons were rarely observed ultrastructurally. This is comparable with human SCI and experimental studies in monkeys where demyelinating processes seem to be minimal whereas there is robust demyelination in rodents 14, 33, 76.
Highly reflecting the immune response in humans 27, 72, an impressive upregulation of MHC class II on phagocytic cells in dogs with subacute SCI was shown in an initial study using the same population of dogs and in canine spinal cord slices incubated for 9 days in vitro 79. Strikingly, MBP‐positive areas in the present report and MHC class II immunoreactivity in the earlier investigation 79 show a negative correlation (r = −0.72; P = 0.001; Spearman's rank correlation test; data not shown). This demonstrates the pivotal role of microglia/macrophages during canine SCI and their possible relationship to progressing axonal damage and myelin pathology.
In order to investigate a potential temporal and local role of MMPs during this progressive degeneration, real‐time qPCR was performed. An upregulation of MMP‐9 during acute naturally occurring SCI and a prolonged upregulation in organotypic slice cultures was noticed. These findings are consistent with observations in experimental rodent models and human SCI 15, 27, 92. In rodent models, MMP‐9 activity peaks around 12–24 hours post injury and is believed to be a key mediator of the early pathogenesis of secondary injury 92. In fact, inhibition of MMP‐9 expression improves the locomotor outcome in spinal cord‐injured mice 60. MMP‐9 is known to be crucially involved in axonal dying‐back processes as inhibition of MMP‐9 was shown to reduce macrophage‐mediated axonal retraction 13. Interestingly, in dogs with IVDD, MMP‐9 activity in the cerebrospinal fluid correlates with injury severity, potentially suggestive of a deleterious role of this gelatinase in acute SCI 52, 58. The present communication is the first report of MMP transcripts in local spinal cord tissue of a large animal model for spontaneous SCI. The positive correlation of MMP‐9 with β‐APP expression certainly does not imply a causal relationship, and the upregulation of this gelatinase might represent an epiphenomenon. However, although the functional significance of this finding needs further investigation, the results indicate a crucial role of locally produced MMP‐9 during spontaneous canine SCI, potentially contributing to lesion progression. In contrast to MMP‐9, MMP‐2 exhibited a delayed upregulation in organotypic slice cultures, exclusively on day 9 in culture, and was even downregulated during acute naturally occurring SCI. Interestingly, MMP‐2 might play a temporally dependent beneficial role during SCI as it is involved in wound healing, recovery and inhibition of scar formation 91, 92. Indeed, multiple lines of evidence indicate that MMPs might also facilitate the growth and regeneration of axons 91. Locomotion is significantly impaired in spinal cord‐injured MMP‐2 null mice demonstrating that MMP‐2 promotes functional recovery after SCI in part by regulating axonal plasticity 35. The results of the present study indicate that an altered orchestration of gelatinases plays a crucial role during canine SCI. However future investigations are needed to clarify the explicit role of MMPs in canine SCI. These mediators may additionally be a relevant therapeutical target during canine SCI in the future. Furthermore, the fact that the investigated MMPs are upregulated in organotypic slice cultures is in concordance with a local tissue source of these molecules. Microglia/macrophages are well‐known as a main source of these gelatinases 27, 81, 92 underlining the crucial role of these cells during the pathogenesis of secondary injury. In addition, as MMPs are critically involved in matrix remodeling during SCI 92, elucidation of the temporal matrix development during canine SCI represents an interesting field for future studies.
In summary, the results of the present study demonstrate that (i) morphological alterations during canine SCI are dominated by early occurring primary axonopathy; (ii) axonal damage shows a significant long‐distance spatial and temporal progression during secondary injury; (iii) there is significant and early spontaneous axonal expression of growth‐related molecules following SCI; and (iv) axonal damage is followed by subsequent changes in myelin pathology and associated phagocytic responses. Moreover, although the functional relevance needs further investigation, morphological changes are accompanied by (v) a dominating and early upregulation of the potentially harmful gelatinase MMP‐9 with concurrent downregulation of MMP‐2 that is known to be involved in healing processes. These data are similar to findings in human SCI 21, 41 validating dogs as a sufficient intermediate model for preclinical research. Additionally, the provided results bear striking similarities to findings in models for demyelinating diseases suggesting common underlying pathomechanisms for axonopathy that trigger subsequent white matter degeneration. These findings present a basis for the discovery of novel therapeutic strategies in this large animal model for preclinical SCI research and substantiate that data obtained on canine IVDD might additionally in part be extrapolated to the human counterpart.
Acknowledgments
The authors are thankful to Kerstin Rohn, Petra Grünig, Caroline Schütz and Bettina Buck for excellent technical assistance. The authors also thank Dr. Karl Rohn, Department of Biometry, Epidemiology and Information Processing, University of Veterinary Medicine, Hannover, Germany, for statistical assistance. This work was supported by the German Research Foundation (FOR 1103; BA 815/10‐2). Ingo Spitzbarth was supported by a Georg‐Christoph‐Lichtenberg scholarship from the state of Lower Saxony.
Both authors contributed equally to the study
References
- 1. Ahlgren S, Li GL, Olsson Y (1996) Accumulation of beta‐amyloid precursor protein and ubiquitin in axons after spinal cord trauma in humans: immunohistochemical observations on autopsy material. Acta Neuropathol 92:49–55. [DOI] [PubMed] [Google Scholar]
- 2. Andrade MS, Mendonça LM, Chadi G (2010) Treadmill running protects spinal cord contusion from secondary degeneration. Brain Res 1346:266–278. [DOI] [PubMed] [Google Scholar]
- 3. Andrews MR, Stelzner DJ (2007) Evaluation of olfactory ensheathing and Schwann cells after implantation into a dorsal injury of adult rat spinal cord. J Neurotrauma 24:1773–1792. [DOI] [PubMed] [Google Scholar]
- 4. Anthes DL, Theriault E, Tator CH (1995) Characterization of axonal ultrastructural pathology following experimental spinal cord compression injury. Brain Res 702:1–16. [DOI] [PubMed] [Google Scholar]
- 5. Bareyre FM, Schwab ME (2003) Inflammation, degeneration and regeneration in the injured spinal cord: insights from DNA microarrays. Trends Neurosci 26:555–563. [DOI] [PubMed] [Google Scholar]
- 6. Beineke A, Markus S, Borlak J, Thum T, Baumgärtner W (2008) Increase of pro‐inflammatory cytokine expression in non‐demyelinating early cerebral lesions in nervous canine distemper. Viral Immunol 21:401–410. [DOI] [PubMed] [Google Scholar]
- 7. Beineke A, Puff C, Seehusen F, Baumgärtner W (2009) Pathogenesis and immunopathology of systemic and nervous canine distemper. Vet Immunol Immunopathol 127:1–18. [DOI] [PubMed] [Google Scholar]
- 8. Beirowski B, Nógrádi A, Babetto E, Garcia‐Alias G, Coleman MP (2010) Mechanisms of axonal spheroid formation in central nervous system Wallerian degeneration. J Neuropathol Exp Neurol 69:455–472. [DOI] [PubMed] [Google Scholar]
- 9. Bendayan M, Zollinger M (1983) Ultrastructural localization of antigenic sites on osmium‐fixed tissues applying the protein A‐gold technique. J Histochem Cytochem 31:101–109. [DOI] [PubMed] [Google Scholar]
- 10. Boekhoff TM, Flieshardt C, Ensinger EM, Fork M, Kramer S, Tipold A (2011) Quantitative magnetic resonance imaging characteristics: evaluation of prognostic value in the dog as a translational model for spinal cord injury. J Spinal Disord Tech 25:E81–E87. [DOI] [PubMed] [Google Scholar]
- 11. Boekhoff TM, Ensinger EM, Carlson R, Bock P, Baumgärtner W, Rohn K et al (2011) Microglial contribution to secondary injury evaluated in a large animal model of human spinal cord trauma. J Neurotrauma 29:1000–1011. [DOI] [PubMed] [Google Scholar]
- 12. Bresnahan JC (1978) An electron‐microscopic analysis of axonal alterations following blunt contusion of the spinal cord of the rhesus monkey (Macaca mulatta). J Neurol Sci 37:59–82. [DOI] [PubMed] [Google Scholar]
- 13. Busch SA, Horn KP, Silver DJ, Silver J (2009) Overcoming macrophage‐mediated axonal dieback following CNS injury. J Neurosci 29:9967–9976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buss A, Brook GA, Kakulas B, Martin D, Franzen R, Schoenen J et al (2003) Gradual loss of myelin and formation of an astrocytic scar during Wallerian degeneration in the human spinal cord. Brain 127:34–44. [DOI] [PubMed] [Google Scholar]
- 15. Buss A, Pech K, Kakulas BA, Martin D, Schoenen J, Noth J, Brook GA (2007) Matrix metalloproteinases and their inhibitors in human traumatic spinal cord injury. BMC Neurol 7:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cajal Ramon YS (1928) Cajal's Degeneration and Regeneration of the Nervous System, translated by Raoul M. May. J DeFelipe, EG Jones (eds). Oxford University Press: New York. 1991. [Google Scholar]
- 17. Casha S, Yu WR, Fehlings MG (2005) FAS deficiency reduces apoptosis, spares axons and improves function after spinal cord injury. Exp Neurol 196:390–400. [DOI] [PubMed] [Google Scholar]
- 18. Choo AM, Liu J, Dvorak M, Tetzlaff W, Oxland TR (2008) Secondary pathology following contusion, dislocation, and distraction spinal cord injuries. Exp Neurol 212:490–506. [DOI] [PubMed] [Google Scholar]
- 19. Coleman MP, Perry VH (2002) Axon pathology in neurological disease: a neglected therapeutic target. Trends Neurosci 25: 532–537. [DOI] [PubMed] [Google Scholar]
- 20. Cornish R, Blumbergs PC, Manavis J, Scott G, Jones NR, Reilly PL (2000) Topography and severity of axonal injury in human spinal cord trauma using amyloid precursor protein as a marker of axonal injury. Spine (Phila Pa 1976) 25:1227–1233. [DOI] [PubMed] [Google Scholar]
- 21. Croul SE, Flanders AE (1997) Neuropathology of human spinal cord injury. Adv Neurol 72:317–323. [PubMed] [Google Scholar]
- 22. Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS (1997) Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med 3:73–76. [DOI] [PubMed] [Google Scholar]
- 23. Curtis R, Green D, Lindsay RM, Wilkin GP (1993) Up‐regulation of GAP‐43 and growth of axons in rat spinal cord after compression injury. J Neurocytol 22:51–64. [DOI] [PubMed] [Google Scholar]
- 24. Deumens R, Koopmans GC, Joosten EA (2005) Regeneration of descending axon tracts after spinal cord injury. Prog Neurobiol 77:57–89. [DOI] [PubMed] [Google Scholar]
- 25. Ensinger EM, Boekhoff TM, Carlson R, Beineke A, Rohn K, Tipold A, Stein VM (2010) Regional topographical differences of canine microglial immunophenotype and function in the healthy spinal cord. J Neuroimmunol 227:144–152. [DOI] [PubMed] [Google Scholar]
- 26. Fenrich KK, Skelton N, MacDermid VE, Meehan CF, Armstrong S, Neuber‐Hess MS, Rose PK (2007) Axonal regeneration and development of de novo axons from distal dendrites of adult feline commissural interneurons after a proximal axotomy. J Comp Neurol 502:1079–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fleming JC, Norenberg MD, Ramsay DA, Dekaban GA, Marcillo AE, Saenz AD et al (2006) The cellular inflammatory response in human spinal cords after injury. Brain 129:3249–3269. [DOI] [PubMed] [Google Scholar]
- 28. Fluehmann G, Doherr MG, Jaggy A (2006) Canine neurological diseases in a referral hospital population between 1989 and 2000 in Switzerland. J Small Anim Pract 47:582–587. [DOI] [PubMed] [Google Scholar]
- 29. Gensel JC, Donnelly DJ, Popovich PG (2011) Spinal cord injury therapies in humans: an overview of current clinical trials and their potential effects on intrinsic CNS macrophages. Expert Opin Ther Targets 15:505–518. [DOI] [PubMed] [Google Scholar]
- 30. Goldstein B, Little JW, Harris RM (1997) Axonal sprouting following incomplete spinal cord injury: an experimental model. J Spinal Cord Med 20:200–206. [DOI] [PubMed] [Google Scholar]
- 31. Gomes‐Leal W, Corkill DJ, Picanço‐Diniz CW (2005) Systematic analysis of axonal damage and inflammatory response in different white matter tracts of acutely injured rat spinal cord. Brain Res 1066:57–70. [DOI] [PubMed] [Google Scholar]
- 32. Guzmán‐Lenis MS, Navarro X, Casas C (2009) Drug screening of neuroprotective agents on an organotypic‐based model of spinal cord excitotoxic damage. Restor Neurol Neurosci 27:335–349. [DOI] [PubMed] [Google Scholar]
- 33. Hagg T, Oudega M (2006) Degenerative and spontaneous regenerative processes after spinal cord injury. J Neurotrauma 23:264–280. [DOI] [PubMed] [Google Scholar]
- 34. Higo N, Nishimura Y, Murata Y, Oishi T, Yoshino‐Saito K, Takahashi M et al (2009) Increased expression of the growth‐associated protein 43 gene in the sensorimotor cortex of the macaque monkey after lesioning the lateral corticospinal tract. J Comp Neurol 516:493–506. [DOI] [PubMed] [Google Scholar]
- 35. Hsu JY, McKeon R, Goussev S, Werb Z, Lee JU, Trivedi A, Noble‐Haeusslein LJ (2006) Matrix metalloproteinase‐2 facilitates wound healing events that promote functional recovery after spinal cord injury. J Neurosci 26:9841–9850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Imbschweiler I, Seehusen F, Peck CT, Omar M, Baumgärtner W, Wewetzer K (2011) Increased p75 neurotrophin receptor expression in the canine distemper virus model of multiple sclerosis identifies aldynoglial Schwann cells that emerge in response to axonal damage. Glia 60:358–371. [DOI] [PubMed] [Google Scholar]
- 37. Jeffery ND, Lakatos A, Franklin RJ (2005) Autologous olfactory glial cell transplantation is reliable and safe in naturally occurring canine spinal cord injury. J Neurotrauma 22:1282–1293. [DOI] [PubMed] [Google Scholar]
- 38. Jeffery ND, Smith PM, Lakatos A, Ibanez C, Ito D, Franklin RJ (2006) Clinical canine spinal cord injury provides an opportunity to examine the issues in translating laboratory techniques into practical therapy. Spinal Cord 44:584–593. [DOI] [PubMed] [Google Scholar]
- 39. Jeffery ND, Hamilton L, Granger N (2011) Designing clinical trials in canine spinal cord injury as a model to translate successful laboratory interventions into clinical practice. Vet Rec 168: 102–107. [DOI] [PubMed] [Google Scholar]
- 40. Jeong DK, Taghavi CE, Song KJ, Lee KB, Kang HW (2011) Organotypic human spinal cord slice culture as an alternative to direct transplantation of human bone marrow precursor cells for treating spinal cord injury. World Neurosurg 75:533–539. [DOI] [PubMed] [Google Scholar]
- 41. Kakulas BA (1999) The applied neuropathology of human spinal cord injury. Spinal Cord 37:79–88. [DOI] [PubMed] [Google Scholar]
- 42. Kamada T, Koda M, Dezawa M, Anahara R, Toyama Y, Yoshinaga K et al (2011) Transplantation of human bone marrow stromal cell‐derived Schwann cells reduces cystic cavity and promotes functional recovery after contusion injury of adult rat spinal cord. Neuropathology 31:48–58. [DOI] [PubMed] [Google Scholar]
- 43. Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T (2005) In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat Med 11:572–577. [DOI] [PubMed] [Google Scholar]
- 44. Kocsis JD, Lankford KL, Sasaki M, Radtke C (2009) Unique in vivo properties of olfactory ensheathing cells that may contribute to neural repair and protection following spinal cord injury. Neurosci Lett 456:137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K et al (1990) Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci U S A 87:1561–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Krassioukov AV, Ackery A, Schwartz G, Adamchik Y, Liu Y, Fehlings MG (2002) An in vitro model of neurotrauma in organotypic spinal cord cultures from adult mice. Brain Res Brain Res Protoc 10:60–68. [DOI] [PubMed] [Google Scholar]
- 47. Kreutzer M, Seehusen F, Kreutzer R, Pringproa K, Kummerfeld M, Claus P et al (2012) Axonopathy is associated with complex axonal transport defects in a model of multiple sclerosis. Brain Pathol 22:454–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kummerfeld M, Meens J, Haas L, Baumgärtner W, Beineke A (2009) Generation and characterization of a polyclonal antibody for the detection of Theiler's murine encephalomyelitis virus by light and electron microscopy. J Virol Methods 160:185–188. [DOI] [PubMed] [Google Scholar]
- 49. Kwon BK, Hillyer J, Tetzlaff W (2010) Translational research in spinal cord injury: a survey opinion from the SCI community. J Neurotrauma 27:21–33. [DOI] [PubMed] [Google Scholar]
- 50. Lampert PW (1967) A comparative electron microscopic study of reactive, degenerating, regenerating, and dystrophic axons. J Neuropathol Exp Neurol 26:345–368. [DOI] [PubMed] [Google Scholar]
- 51. Leppik IE, Patterson EN, Coles LD, Craft EM, Cloyd JC (2011) Canine status epilepticus: a translational platform for human therapeutic trials. Epilepsia 8:31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Levine JM, Ruaux CG, Bergman RL, Coates JR, Steiner JM, Williams DA (2009) Matrix metalloproteinase‐9 activity in the cerebrospinal fluid and serum of dogs with acute spinal cord trauma from intervertebral disk disease. Am J Vet Res 67:283–287. [DOI] [PubMed] [Google Scholar]
- 53. Levine JM, Levine GJ, Porter BF, Topp K, Noble‐Haeusslein LJ (2011) Naturally occurring disk herniation in dogs: an opportunity for pre‐clinical spinal cord injury research. J Neurotrauma 28:675–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li GL, Farooque M, Holtz A, Olsson Y (1995) Changes of beta‐amyloid precursor protein after compression trauma to the spinal cord: an experimental study in the rat using immunohistochemistry. J Neurotrauma 12:269–277. [DOI] [PubMed] [Google Scholar]
- 55. Li GL, Farooque M, Holtz A, Olsson Y (1996) Increased expression of growth‐associated protein 43 immunoreactivity in axons following compression trauma to rat spinal cord. Acta Neuropathol 92:19–26. [DOI] [PubMed] [Google Scholar]
- 56. Mahad DJ, Ziabreva I, Campbell G, Lax N, White K, Hanson PS et al (2009) Mitochondrial changes within axons in multiple sclerosis. Brain 132:1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Markus S, Failing K, Baumgärtner W (2002) Increased expression of pro‐inflammatory cytokines and lack of up‐regulation of anti‐inflammatory cytokines in early distemper CNS lesions. J Neuroimmunol 125:30–41. [DOI] [PubMed] [Google Scholar]
- 58. Nagano S, Kim SH, Tokunaga S, Arai K, Fujiki M, Misumi K (2011) Matrix metalloprotease‐9 activity in the cerebrospinal fluid and spinal injury severity in dogs with intervertebral disc herniation. Res Vet Sci 91:482–485. [DOI] [PubMed] [Google Scholar]
- 59. Nikić I, Merkler D, Sorbara C, Brinkoetter M, Kreutzfeldt M, Bareyre FM et al (2011) A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med 17:495–499. [DOI] [PubMed] [Google Scholar]
- 60. Noble LJ, Donovan F, Igarashi T, Goussev S, Werb Z (2002) Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J Neurosci 22:7526–7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Olby N (2010) The pathogenesis and treatment of acute spinal cord injuries in dogs. Vet Clin North Am Small Anim Pract 40:791–807. [DOI] [PubMed] [Google Scholar]
- 62. Pan JZ, Ni L, Sodhi A, Aguanno A, Young W, Hart RP (2002) Cytokine activity contributes to induction of inflammatory cytokine mRNAs in spinal cord following contusion. J Neurosci Res 68:315–322. [DOI] [PubMed] [Google Scholar]
- 63. Petzold A (2005) Neurofilament phosphoforms: surrogate markers for axonal injury, degeneration and loss. J Neurol Sci 233:183–198. [DOI] [PubMed] [Google Scholar]
- 64. Povlishock JT, Christman CW (1995) The pathobiology of traumatically induced axonal injury in animals and humans: a review of current thoughts. J Neurotrauma 12:555–564. [DOI] [PubMed] [Google Scholar]
- 65. Povlishock JT, Jenkins LW (1995) Are the pathobiological changes evoked by traumatic brain injury immediate and irreversible? Brain Pathol 5:415–426. [DOI] [PubMed] [Google Scholar]
- 66. Povlishock JT, Becket DP, Cheng CLY, Vaughan GW (1983) Axonal change in minor head injury. J Neuropathol Exp Neurol 42:225–242. [DOI] [PubMed] [Google Scholar]
- 67. Profyris C, Cheema SS, Zang D, Azari MF, Boyle K, Petratos S (2004) Degenerative and regenerative mechanisms governing spinal cord injury. Neurobiol Dis 15:415–436. [DOI] [PubMed] [Google Scholar]
- 68. Puckett WR, Hiester ED, Norenberg MD, Marcillo AE, Bunge RP (1997) The astroglial response to Wallerian degeneration after spinal cord injury in humans. Exp Neurol 148:424–432. [DOI] [PubMed] [Google Scholar]
- 69. Puff C, Krudewig C, Imbschweiler I, Baumgärtner W, Alldinger S (2009) Influence of persistent canine distemper virus infection on expression of RECK, matrix‐metalloproteinases and their inhibitors in a canine macrophage/monocytic tumour cell line (DH82). Vet J 182:100–107. [DOI] [PubMed] [Google Scholar]
- 70. Purdy PD, White CL 3rd, Baer DL, Frawley WH, Reichard RR, Pride GL Jr et al (2004) Percutaneous translumbar spinal cord compression injury in dogs from an angioplasty balloon: MR and histopathologic changes with balloon sizes and compression times. AJNR Am J Neuroradiol 25:1435–1442. [PMC free article] [PubMed] [Google Scholar]
- 71. Rosenberg LJ, Wrathall JR (1997) Quantitative analysis of acute axonal pathology in experimental spinal cord contusion. J Neurotrauma 14:823–838. [DOI] [PubMed] [Google Scholar]
- 72. Schmitt AB, Buss A, Breuer S, Brook GA, Pech K, Martin D et al (2000) Major histocompatibility complex class II expression by activated microglia caudal to lesions of descending tracts in the human spinal cord is not associated with a T cell response. Acta Neuropathol 100:528–536. [DOI] [PubMed] [Google Scholar]
- 73. Schmitt AB, Breuer S, Polat L, Pech K, Kakulas B, Love S et al (2003) Retrograde reactions of Clarke's nucleus neurons after human spinal cord injury. Ann Neurol 54:534–539. [DOI] [PubMed] [Google Scholar]
- 74. Schwab ME, Bartholdi D (1996) Degeneration and regeneration of axons in the lesioned spinal cord. Physiol Rev 76:319–370. [DOI] [PubMed] [Google Scholar]
- 75. Seehusen F, Baumgärtner W (2010) Axonal pathology and loss precede demyelination and accompany chronic lesions in a spontaneously occurring animal model of multiple sclerosis. Brain Pathol 20:551–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shi F, Zhu H, Yang S, Liu Y, Feng Y, Shi J et al (2009) Glial response and myelin clearance in areas of Wallerian degeneration after spinal cord hemisection in the monkey Macaca fascicularis. J Neurotrauma 26:2083–2096. [DOI] [PubMed] [Google Scholar]
- 77. Shichinohe H, Kuroda S, Tsuji S, Yamaguchi S, Yano S, Lee JB et al (2008) Bone marrow stromal cells promote neurite extension in organotypic spinal cord slice: significance for cell transplantation therapy. Neurorehabil Neural Repair 22:447–457. [DOI] [PubMed] [Google Scholar]
- 78. Smith PM, Jeffery ND (2006) Histological and ultrastructural analysis of white matter damage after naturally‐occurring spinal cord injury. Brain Pathol 16:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Spitzbarth I, Bock P, Haist V, Stein VM, Tipold A, Wewetzer K et al (2011) Prominent microglial activation in the early proinflammatory immune response in naturally occurring canine spinal cord injury. J Neuropathol Exp Neurol 70:703–714. [DOI] [PubMed] [Google Scholar]
- 80. Stavridis SI, Dehghani F, Korf HW, Hailer NP (2005) Characterisation of transverse slice culture preparations of postnatal rat spinal cord: preservation of defined neuronal populations. Histochem Cell Biol 123:377–392. [DOI] [PubMed] [Google Scholar]
- 81. Stein VM, Puff C, Genini S, Contioso VB, Baumgärtner W, Tipold A (2011) Variations on brain microglial gene expression of MMPs, RECK, and TIMPs in inflammatory and non‐inflammatory diseases in dogs. Vet Immunol Immunopathol 144:17–26. [DOI] [PubMed] [Google Scholar]
- 82. Stone JR, Singleton RH, Povlishock JT (2001) Intra‐axonal neurofilament compaction does not evoke local axonal swelling in all traumatically injured axons. Exp Neurol 172:320–331. [DOI] [PubMed] [Google Scholar]
- 83. Tobias CA, Shumsky JS, Shibata M, Tuszynski MH, Fischer I, Tessler A, Murray M (2003) Delayed grafting of BDNF and NT‐3 producing fibroblasts into the injured spinal cord stimulates sprouting, partially rescues axotomized red nucleus neurons from loss and atrophy, and provides limited regeneration. Exp Neurol 184:97–113. [DOI] [PubMed] [Google Scholar]
- 84. Tsunoda I, Fujinami RS (2002) Inside‐out versus outside‐in models for virus induced demyelination: axonal damage triggering demyelination. Springer Semin Immunopathol 24:105–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Tsunoda I, Kuang LQ, Libbey JE, Fujinami RS (2003) Axonal injury heralds virus‐induced demyelination. Am J Pathol 162:1259–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Uchida K, Baba H, Maezawa Y, Kubota C (2002) Progressive changes in neurofilament proteins and growth‐associated protein‐43 immunoreactivities at the site of cervical spinal cord compression in spinal hyperostotic mice. Spine (Phila Pa 1976) 27:480–486. [DOI] [PubMed] [Google Scholar]
- 87. Ulrich R, Seeliger F, Kreutzer M, Germann PG, Baumgärtner W (2008) Limited remyelination in Theiler's murine encephalomyelitis due to insufficient oligodendroglial differentiation of nerve/glial antigen 2 (NG2)‐positive putative oligodendroglial progenitor cells. Neuropathol Appl Neurobiol 34:603–620. [DOI] [PubMed] [Google Scholar]
- 88. van den Berg ME, Castellote JM, Mahillo‐Fernandez I, de Pedro‐Cuesta J (2010) Incidence of spinal cord injury worldwide: a systematic review. Neuroepidemiology 34:184–192. [DOI] [PubMed] [Google Scholar]
- 89. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real‐time quantitative RT‐PCR data by geometric averaging of multiple internal control genes. Genome Biol 18:RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wewetzer K, Radtke C, Kocsis J, Baumgärtner W (2011) Species‐specific control of cellular proliferation and the impact of large animal models for the use of olfactory ensheathing cells and Schwann cells in spinal cord repair. Exp Neurol 229: 80–87. [DOI] [PubMed] [Google Scholar]
- 91. Yong VW (2005) Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci 6:931–944. [DOI] [PubMed] [Google Scholar]
- 92. Zhang H, Chang M, Hansen CN, Basso DM, Noble‐Haeusslein LJ (2011) Role of matrix metalloproteinases and therapeutic benefits of their inhibition in spinal cord injury. Neurother 8:206–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zörner B, Schwab ME (2010) Anti‐Nogo on the go: from animal models to a clinical trial. Ann N Y Acad Sci 1198:E22–E34. [DOI] [PubMed] [Google Scholar]