

Abstract
Primary aldosteronism (PA) is the most common cause of secondary hypertension. The hallmark of PA is adrenal production of aldosterone under suppressed renin conditions. PA subtypes include adrenal unilateral and bilateral hyperaldosteronism. Considerable progress has been made in defining the role for somatic gene mutations in aldosterone-producing adenomas (APA) as the primary cause of unilateral PA. This includes the use of next-generation sequencing (NGS) to define recurrent somatic mutations in APA that disrupt calcium signaling, increase aldosterone synthase (CYP11B2) expression, and aldosterone production. The use of CYP11B2 immunohistochemistry on adrenal glands from normal subjects, patients with unilateral and bilateral PA has allowed the identification of CYP11B2-positive cell foci, termed aldosterone-producing cell clusters (APCC). APCC lie beneath the adrenal capsule and like APA, many APCC harbor somatic gene mutations known to increase aldosterone production. These findings suggest that APCC may play a role in pathologic progression of PA. Herein, we provide an update on recent research directed at characterizing APCC and also discuss the unanswered questions related to the role of APCC in PA.
Keywords: primary aldosteronism, aldosterone-producing cell clusters, aldosterone-producing micronodules, Cytochrome P-450 CYP11B2
A. Overview of primary aldosteronism (PA)
Primary aldosteronism (PA), which is caused by inappropriate aldosterone production by the adrenal gland, is the most common form of secondary hypertension. PA accounts for 15–20% of patients with resistant hypertension as well as 7–10% of all hypertensive patients [1–3]. Chronic inappropriate elevations in aldosterone can increase pro-inflammatory cytokines [4] and cause oxidative stress [5], which in the long-term leads to tissue damage and fibrosis [6]. As a result PA patients have higher cardiovascular risk than those with essential hypertension suggesting an impact of inappropriate aldosterone that is beyond increased blood pressure [7–9]. Moreover, the increased risk of morbidity at least with regard to cardiac remodeling may be reversible through targeted management with surgery or medical treatment that includes the use of mineralocorticoid receptor antagonist [8,10]. With the above in mind, early identification and appropriate therapy in patients with PA are very important to prevent the cardiovascular complications.
The two principal forms of PA are unilateral disease due to aldosterone-producing adenoma (APA) or bilateral adrenal hyperaldosteronism (BHA) that most often results from hyperplasia or accumulation of aldosterone-producing cell clusters (APCC) [11]. In addition, there are less common subtypes of sporadic PA that include computed tomography (CT)-positive bilateral APA [12], CT-undetectable unilateral PA [13,14], and rarely adrenocortical cancer [15]. Recent research into the causes for each of these sporadic (non-familial) forms of PA have focused on somatic gene mutations.
The application of whole exome next-generation sequencing (NGS) to PA research has led to the discovery of recurrent somatic mutations in APA [16–19]. KCNJ5, potassium inwardly rectifying channel, subfamily J, member 5, is the most common gene with somatic mutations [20–24]. KCNJ5 variants cause abnormal sodium permeability [25] and activate voltage-dependent calcium channels, thereby leading to aldosterone overproduction [19]. Activating mutations in the calcium channel-gated channel subunit alpha 1D (CACNA1D) cause an increase in calcium influx and aldosterone production [16,17,20,26]. Inactivating mutations in the sodium/potassium transporting subunit alpha1 ATPase (ATP1A1) [17,18,20,27] have been proposed to cause pathologic production of aldosterone in APA via depolarization of adrenocortical cells, disruption of intracellular potassium (K+) balance, and decreased intracellular pH. Intriguingly, unlike somatic mutations in other aldosterone-driver genes, ATP1A1 mutations do not lead to increased intracellular calcium (Ca+) levels [28]. Rarer inactivating mutations have been observed in the plasma membrane Ca2+ transporting type 3 ATPase (ATP2B3), which decreases Ca2+ export [18,20,27]. β-catenin (CTNNB1) mutations were also identified in a small percentage of APA and a recent study suggests that these mutations may be associated with the process of tumorigenesis rather than direct activation of aldosterone production [29]. Recent studies have demonstrated a small percentage of APA harboring somatic mutations in the voltage-gated chloride channel type 2 (CLCN2) [30] and the calcium channel-gated channel subunit alpha 1H (CACNA1H) which were originally shown to cause germline PA [31–34]. Recent application of aldosterone synthase (CYP11B2)-guided capture of APA followed by higher depth gene-targeted NGS suggests that greater than 90% of APA have aldosterone-driver mutations [24,26]. Regardless of the sequencing technology used, defining the relative mutation prevalence has been complicated by a significant impact of age, sex and race on the gene mutated. However, KCNJ5, CACNA1D, and ATP1A1 are the dominant causes of APA despite the population being studied. The cellular origin of APA and the rationale for the distinct age, sex and race differences in mutation prevalence continue to be an area of active research. This review will focus on recent studies showing the presence of similar gene mutations in microscopic clusters of subscapular adrenal cells that express CYP11B2.
B. Discovery of aldosterone-producing cell clusters (APCC)
Aldosterone biosynthesis normally occurs in the zona glomerulosa (ZG) of the adrenal cortex (Figure 1) [11]. Aldosterone and cortisol synthesis share the early enzymatic reactions starting from cholesterol; however, CYP11B2 is only expressed in the ZG and is responsible for the terminal reactions in aldosterone synthesis. Conversely, CYP11B1 (11β-hydroxylase), the enzyme responsible for the final reaction in cortisol synthesis is expressed in the zona fasciculata (ZF) but not the ZG [35–39]. Under a typical unrestricted sodium diet, there is a limited area of CYP11B2-expressing cells in the human ZG [40]. With aging human adrenals develop a discontinuous ZG appearance [41] which contrasts with textbook depictions of the adrenal ZG or the continuous CYP11B2-positive ZG seen in some laboratory models, including the rat adrenal [11].
Figure 1.
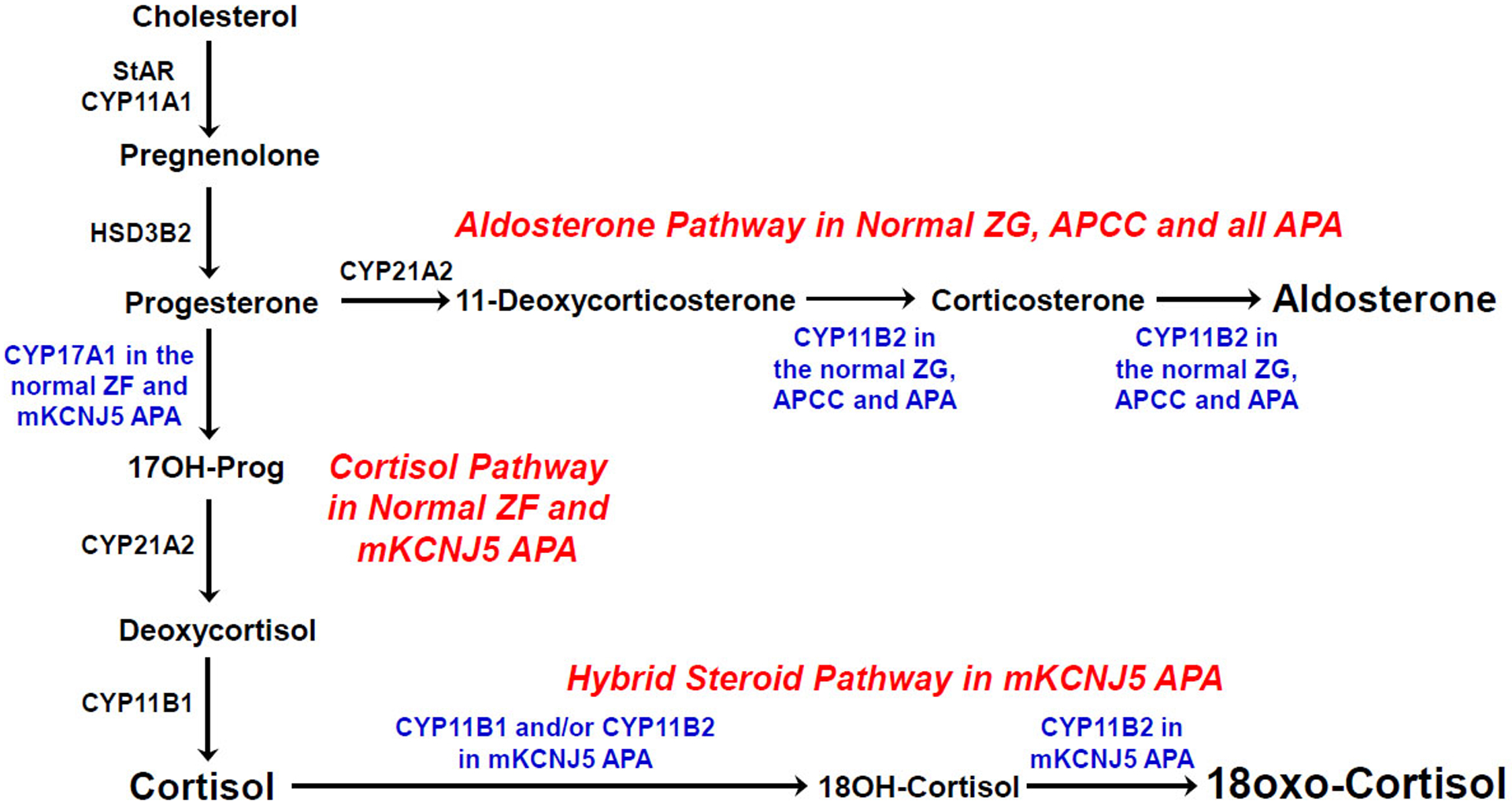
Steroidogenic pathways related to the production of aldosterone, cortisol and hybrid adrenal steroids. ZG, zona glomerulosa; ZF, zona fasciculata; StAR, steroidogenic acute regulatory protein; CYP11A1, cholesterol side-chain cleavage; CYP17A1, 17α-hydroxylase; HSD3B2, 3β-hydroxysteroid dehydrogenase type 2; CYP21A2, 21-hydroxylase; CYP11B1, 11β-hydroxylase; CYP11B2, aldosterone synthase; 17OH-Prog, 17-hydroxyprogesterone; 18OH-Cortisol, 18-hydroxycortisol; APCC, aldosterone-producing cell clusters; APA, aldosterone-producing adenoma; mKCNJ5, mutant KCNJ5
The availability of human CYP11B2 and CYP11B1 antibodies has allowed further studies on the expression patterns of CYP11B2 and CYP11B1 in normal and pathologic adrenals [37,42], although there had been attempts to compare histopathology of adrenal gland with the expression of steroidogenic enzymes by in situ hybridization in some previous studies [43,44]. Nishimoto et al. reported the successful immunohistochemical localization for the two CYP11B enzymes in human formalin-fixed paraffin-embedded (FFPE) samples using the first selective polyclonal antibodies for the human isozymes [37]. In addition to conventional adrenal zonation, the authors noted substantial numbers of normal adrenal tissues with CYP11B2-expressing cell clusters that extended from the ZG into the ZF, and they termed these cells APCC because of their positive CYP11B2 expression and their unique focal localization. Using in situ hybridization for CYP11B2, Boulkroun et al. described similar structures which they divided into three categories, including foci, megafoci, and APCC, which was based on the size of cell clusters and relative expression of a previously described ZG marker, Disable-2 (DAB2) [45]. There was recently a consensus meeting to address multiple pathology nomenclature issues related specifically to primary aldosteronism, including APCC. One term considered was aldosterone-producing micronodules, which takes into consideration APCC’s aldosterone production and the need for microscopy for their visualization.
Several studies reported that APCC are a common feature in normal and pathologic human adrenals. Using adrenals from renal transplant donors, 69% of 127 glands had at least one APCC [41], and 50% of 22 adrenals with APA expressing CYP11B2 showed an APCC in the tissues adjacent to the tumor [46]. Although the definition of APCC varies somewhat between laboratories, APCC seem to have distinctive features that distinguish them from APA (Figure 2) [37,47,48]. These include: 1) The size of APCC are smaller (approximately 0.2–1.5mm in length) than that of APA (typically more than 3mm in length), 2) The morphology of APCC cells appears to be normal, so they cannot be easily differentiated from adjacent cortical cells by hematoxylin-eosin (H&E). 3) immunohistochemistry (IHC) suggests APCC cells have high levels of CYP11B2 and low levels CYP11B1 or CYP17A1, both enzymes involved in cortisol production [38,41]. However, APCC can exhibit different enzyme expression patterns with some exhibiting homogeneous CYP11B2 expression while others show a polarity with decreasing expression as its cells migrate further into the cortex. This pattern of expression is associated with acquisition of CYP17A1 suggesting that some APCC cells undergo, at least in part, transition to a more ZF-like phenotype. This can be seen when examining the expression pattern of CYP11B2 and CYP17A1 within a representative APCC (Figure 2). The molecular mechanisms causing APCC morphological and steroidogenic variability are unknown but could well relate to their somatic mutation status, which will be discussed below. Understanding the similarities and differences distinguishing APCC and APA may provide a better understanding the underlying mechanism leading to PA.
Figure 2.
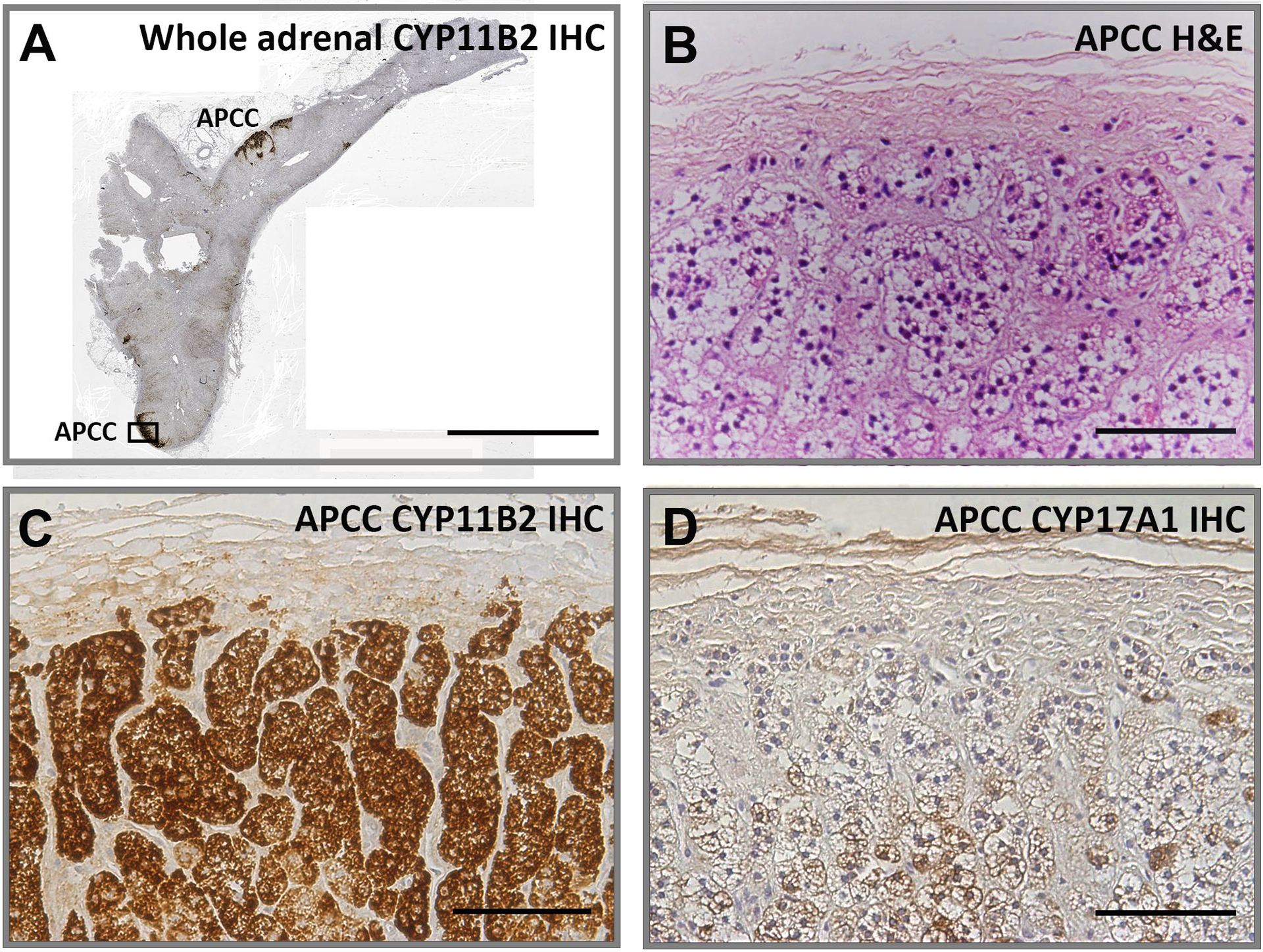
Visualization of representative adrenal with aldosterone-producing cell clusters (APCC) with immunohistochemistry. Panel A shows a whole adrenal with multiple APCC visualized following CYP11B2 IHC. The APCC indicated by the black box is the focus of Panels B, C and D. Panel B. Hematoxylin and eosin (H&E) staining, Panel C. CYP11B2 IHC. Panel D. CYP17A1 IHC. Panel A bar indicates 5 mm. Panel B, C and D bars indicate 100 μm.
C. Molecular characterization of APCC
CYP11B2 IHC-guided capture of APCC, ZG and ZF has allowed a transcriptome comparison and determination of additional gene differences beyond CYP11B2. Overall transcription profiles showed APCC to be highly similar to ZG as compared to ZF; however, even between APCC and ZG there were significant differences. These included APCC elevations in SLC35F1 (Solute carrier family 35, member F1), MC2R (ACTH receptor), and PPP4R4 (Serine/threonine-protein phosphatase 4 regulatory subunit 4). The role of these proteins in APCC function are not clear but the higher expression of the MC2R suggests further studies are needed to determine if APCC aldosterone production is responsive to circulating adrenocorticotrophic hormone (ACTH). These proteins may also be useful as additional IHC markers for APCC [49]. The transcriptome data did not show an elevation APCC DAB2 transcripts, unlike was seen for the protein levels in the previous finding [45]. The authors speculated that it was probably due to differences in the histologic definition of APCC between the two studies, and that the transcriptome study may include some of the DAB2-expressing megafoci and foci that were described by Boulkroun et al [45,49].
The ability to combine FFPE adrenal tissue, CYP11B2 IHC-guided APCC capture, and gene-targeted NGS have been instrumental in demonstrating the presence of aldosterone-driver APA somatic mutations in APCC [50]. In terms of somatic mutation prevalence, APCC exhibit distinct characteristics when compared to APA. Using an American cohort of normal adrenals, approximately 35% of 23 APCC were found to harbor known APA-associated driver mutations, including CACNA1D and ATP1A1, suggesting a potential precursor role of APCC in the development of certain forms of APA [49]. Intriguingly, however, APCC were not found to have KCNJ5 mutations, the most common gene mutated in APA. Similarly, Omata et al. demonstrated that APCC were common in Japanese non-hypertensive adrenals with 34% harboring known aldosterone-driver somatic mutations with the most frequent mutations in CACNA1D but no KCNJ5 mutations were detected [51].
Recent studies have investigated intra-APCC metabolic processes using novel tissue-based metabolomics methodologies, including in situ metabolomics by matrix-assisted laser desorption/ionization mass spectrometry imaging (MALDI-MSI), steroid metabolomics, energy pathway metabologenomics, and cellular reprogramming [52]. The application of MALDI-MSI to adrenal tissue sections allowed quantification of many small molecules across the tissue section at the same time [53]. Sun et al. showed that high resolution MALDI-MSI in conjunction to adrenal steroidogenic enzyme IHC could correlate metabolites and their associated metabolic pathways with the functional zonation of the normal adrenal gland [54]. Recently, Murakami et al. extended these studies to KCNJ5 and CACNA1D-mutated APA using MALDI-MSI and found a relationship between higher 18-oxocortisol levels, lower intensity of CYP11B1 staining and a higher possibility of postoperative biochemical cure for PA [55]. The same group has also used MALDI-MSI to study the metabolites of APCC in adrenals from PA patients [56]. Interestingly, they found two distinct populations of APCC, one of which was similar to that seen in APA. Application of these novel metabolomics approaches are expected to provide unexpected insights into the molecular characterization of APCC and their role in the pathophysiology of PA.
D. Steroid Biome of APCC
Steroid profiling using liquid chromatography-tandem mass spectrometry (LC-MS/MS) has evolved as a useful method for subtype classification of PA [57–61]. In particular, 18-oxocortisol has been previously reported as a biomarker for the differential diagnosis of PA [61]. Williams et al. recently reported that this hybrid steroid was significantly higher in peripheral and adrenal plasma from patients with KCNJ5-mutated than patients with other causes of PA, suggesting the usefulness of selective steroid profiling for PA [62]. Moreover, the peripheral steroid profiles and histologic features (including APA, hyperplasia and APCC) of unilateral PA patients who underwent adrenalectomy contributed to the predicted postoperative biochemical outcome [60]. APA harboring KCNJ5 mutations principally display a lipid rich clear cell (ZF-like) histology, whereas APA with CACNA1D, ATP1A1 and ATP2B3 mutations predominantly show a compact cell (ZG-like) predominate histology [63–67]. The histologic features of KCNJ5-mutated APA are also associated with the higher expression of the cortisol biosynthetic enzymes, CYP11B1 and CYP17A1 [63,65,66]. Both 18-oxocortisol and 18-hydroxycortisol are synthesized from cortisol by CYP11B2, while only the latter can be produced by CYP11B1 (Figure 1) [68,69]. The co-expression of enzymes from the ZF and ZG explain the ability of KCNJ5-mutated APA to produce cortisol that can then be converted by CYP11B2 to 18-oxocortisol or 18-hydroxycortisol.
Recently, several studies focused on defining APCC steroidogenic properties and particular whether or not they make aldosterone. Using in situ hybridization, Shigematsu et al. found the presence of APCC-like subcapsular micronodules, which showed abundant transcript expression for HSD3B2, CYP11B1, and CYP11B2, but not CYP17A1, thereby, indicating that the nodules have the steroidogenic enzymatic machinery to produce aldosterone [43]. Sugiura et al. were the first to demonstrate higher levels of aldosterone and 18-oxocortisol in APCC (and APA) in adrenals from PA patients using Fourier transform ion cyclotron resonance mass spectrometry and tandem mass spectrometry imaging [53]. Normal adrenals were recently studied and APCC were confirmed to have higher levels of aldosterone and 18-oxocortisol compared to the adjacent adrenal tissue [70]. These studies suggest that APCC synthesize aldosterone both in normal adrenals and under situations of suppressed renin as seen in adrenals from PA patients. Further research on the functional characteristic and significance of APCC aldosterone production is needed to better understand their role in PA.
E. APCC in preclinical and overt primary aldosteronism
As noted above, PA can primarily be categorized into APA-driven unilateral disease and BHA. Previous studies have suggested that APCC play a role in both PA subtypes but additional research is needed to establish the mechanisms whereby APCC could evolve to APA or what factors might increase APCC number to cause BHA. The hypothesis that APCC are the origin of APA can be supported to a certain extent by the observation that many APCC carry APA-associated mutations in CACNA1D and ATP1A1 [49]. However, the lack of APCC KCNJ5 mutations indicates that APCC, as currently localized by CYP11B2 IHC, are not the apparent precursor lesions for APA with KCNJ5 mutations. Nishimoto et al. suggested that APCC may be precursors of APA based on the presence of CYP11B2-positive micronodular lesions that they termed possible APCC-to-APA transitional lesions (pAATLs). Histologically and using CYP11B2 IHC pAATLs were composed of subcapsular APCC-like and inner APA-like portions [48,71]. Intriguingly, common APA somatic mutations, such as found in KCNJ5, were only observed in larger APCC/APA-like lesions but not in APCC or smaller APCC/APA-like lesions [71]. The authors suggest that the larger APCC/APA-like lesions may involve a with a secondary mutation in KCNJ5. In addition, Sugiura et al. also proposed a similar hypothesis of APCC transition to APA as follows: newly developed APCC produce aldosterone but retain regulation by the renin-angiotensin-system, with aging APCC accumulate aldosterone-driver mutations that lead to pAATL, and finally there is expansion of the mutation bearing cells to APA that then produce 18-oxocortisol [53]. The authors believe that together, these findings support a role of aldosterone-driver mutations in the shift of APCC to APA [71]. Concerns remain because both APCC and APA are common and it would be expected that independent APCC and APA could be in close proximity to one another forming the pAATL structure. Thus, more studies are needed to better understand the potential for APCC evolution to an 18-oxocortisol-producing adenoma. As noted above, 18-oxocortisol production is most associated with KCNJ5 mutations and to date these mutations are thought to be extremely rare in APCC. However, many APCC are mutation-negative and the potential for these to acquire KCNJ5 mutations remains to be determined.
The mechanisms leading to BHA remains poorly understood with the exception of the rare familial forms where germline mutations in CACNA1D, CACNA1H, KCNJ5 and CLCN2 have each been shown to cause bilateral adrenal aldosterone production [72–74]. The underlying causes of sporadic (non-familial) forms of CT-negative BHA often termed IHA still remain to be clarified. BHA has been particularly difficult to study at the molecular level because surgery is rarely used to treat BHA patients; thus, limiting adrenal tissue availability for histologic or molecular analyses. Because BHA is characterized as PA in the absence of an imaging detectable nodule, there are some similarities of BHA with unilateral CT-negative PA. As opposed to BHA, unilateral CT-negative PA is treated with adrenalectomy making these tissues available for study. A recent study showed that nearly half of these excised adrenals had increased CYP11B2-positive APCC when compared to normal adrenals [13]. In addition, the mutation spectrum for DNA isolated from these APCC and the CYP11B2-positive microAPA were similar to the spectrum seen for APCC, which is dominated by CACNA1D mutations donors [49]. This led the authors to suggest that the increased number or expanded size of APCC to microAPA were a possible source of aldosterone excess in these patients [13].
To directly study the histologic and molecular alterations in BHA adrenals, Omata et al. carried-out a multicenter study to obtain BHA adrenals from 15 PA patients [15]. CYP11B2 IHC was used to guide DNA capture and NGS analysis. All BHA adrenals exhibited at least one APCC or a microAPA, and the number of APCC in BHA adrenals was significantly higher than seen in age and sex matched adrenals from normotensive controls. Gene targeted NGS analysis of DNA from 99 APCC from the 15 BHA adrenals demonstrated that CACNA1D mutations (58%) were present at a higher prevalence than past seen in normal adrenals [15]. Interestingly, in vitro adrenal cell-based studies have shown that expression of mutated CACNA1D increases aldosterone production and this production could be significantly reduced by the calcium channel blocker, nifedipine [75]. The high prevalence of APCC with CACNA1D mutations suggests that research on the efficacy of calcium channel blockers as an inhibitor of BHA aldosterone overproduction should be further examined. More research is also needed to better determine if there is a causal relationship between APCC with somatic variants and the pathophysiology of BHA.
An alternative approach was taken by Meyer et al. to examine adrenals from patients with probable BHA [60]. This innovative approach was to examine the histology and CYP11B2-positive cells in adrenals from PA patients that had partial or absent biochemical cure following AVS-directed adrenalectomy of the dominant aldosterone-producing adrenal. In most referral centers these cases are rare, as adrenal vein sampling-driven adrenalectomy most often normalizes both aldosterone and renin levels. The study cohort included 43 adrenals patients who had limited biochemical cure. They found that almost half of the adrenals in this cohort exhibited hyperplastic regions of CYP11B2-expressing cells while slightly less than half had discrete solitary nodules. No aldosterone-driver mutation sequencing was done in this study. However, follow-up sequencing of the nodules and hyperplastic regions of these rare adrenal samples could improve our understanding of the role of somatic mutations in BHA.
There is growing interest in studying the progression of PA and recent reports have suggested that there is a continuum associated with development of renin-independent aldosterone production. This has led to the concept of a preclinical/subclinical forms of PA [76,77]. The presence of APCC in normal adrenals, their accumulation during the aging process and their aldosterone-driver mutations has raised the possibility that APCC might play a role in that continuum and contribute to preclinical forms of PA. The human adrenal cortex is composed of three dynamic zones with the regulation of the ZG and ZF well defined. Both can undergo significant expansion or atrophy based on the presence or absence of angiotensin II or ACTH. With aging there is a decrease in overall aldosterone production which is associated with a loss of the classical subscapular CYP11B2-postive ZG. Interestingly, as the CYP11B2-postive ZG becomes discontinuous, there is an increase in CYP11B2-positive APCC. The age-related increase of APCC has been confirmed in independent studies using adrenals from renal donors [41] as well as non-hypertensive autopsy cases of a Japanese cohort [47]. The age-dependent expansion of adrenal APCC correlated with a change of aldosterone physiology and autonomous aldosteronism as shown through dynamic testing of subjects with low and high sodium intake [41]. Together these findings suggest a potential role of APCC in the early stages of PA in aging adults [47] but additional studies are needed to elaborate that role.
F. Future directions
Herein we summarized the available information about APCC in normal and pathologic adrenal glands. There is accumulating evidence that APCC express CYP11B2, produce aldosterone, and that a significant proportion have somatic gene mutations seen in APA. Together, a model illustrating the potential role of APCC in adrenal pathology are presented in Figure 3. This model builds on the known features and presence of somatic aldosterone driver mutations that are found in APCC. While the concepts of APCC progression to APA and BHA are intriguing, more research is needed to clarify the mechanisms regulating aldosterone production in APCC. While many have CACNA1D mutations, it remains unclear whether or not angiotensin II or ACTH continue to partially regulate aldosterone production, which could play a role in any pathologic contribution to PA. Research directed at defining the mechanisms causing the age-associated accumulation of APCC and possible environmental genetic reasons to explain why some individuals accumulate large numbers of APCC and others very few. Even with highly sensitive gene targeted sequencing methods many APCC appear negative for the somatic mutations that cause APA. As technology develops, whole exome sequencing or whole genome sequencing may or reveal novel gene mutations that cause APCC CYP11B2 expression and aldosterone production. The variation of APCC somatic mutations suggest that like APA, APCC are likely to have distinct subtypes that could have varying roles in physiology or pathology. Combining NGS and MALDI-MSI to adrenal tissue sections that have APCC may help clarify subgroups and their relative ability to produce aldosterone. Finally, but most technically challenging, future clinical studies are needed to directly assess the role of APCC as the continuum leading to PA.
Figure 3.
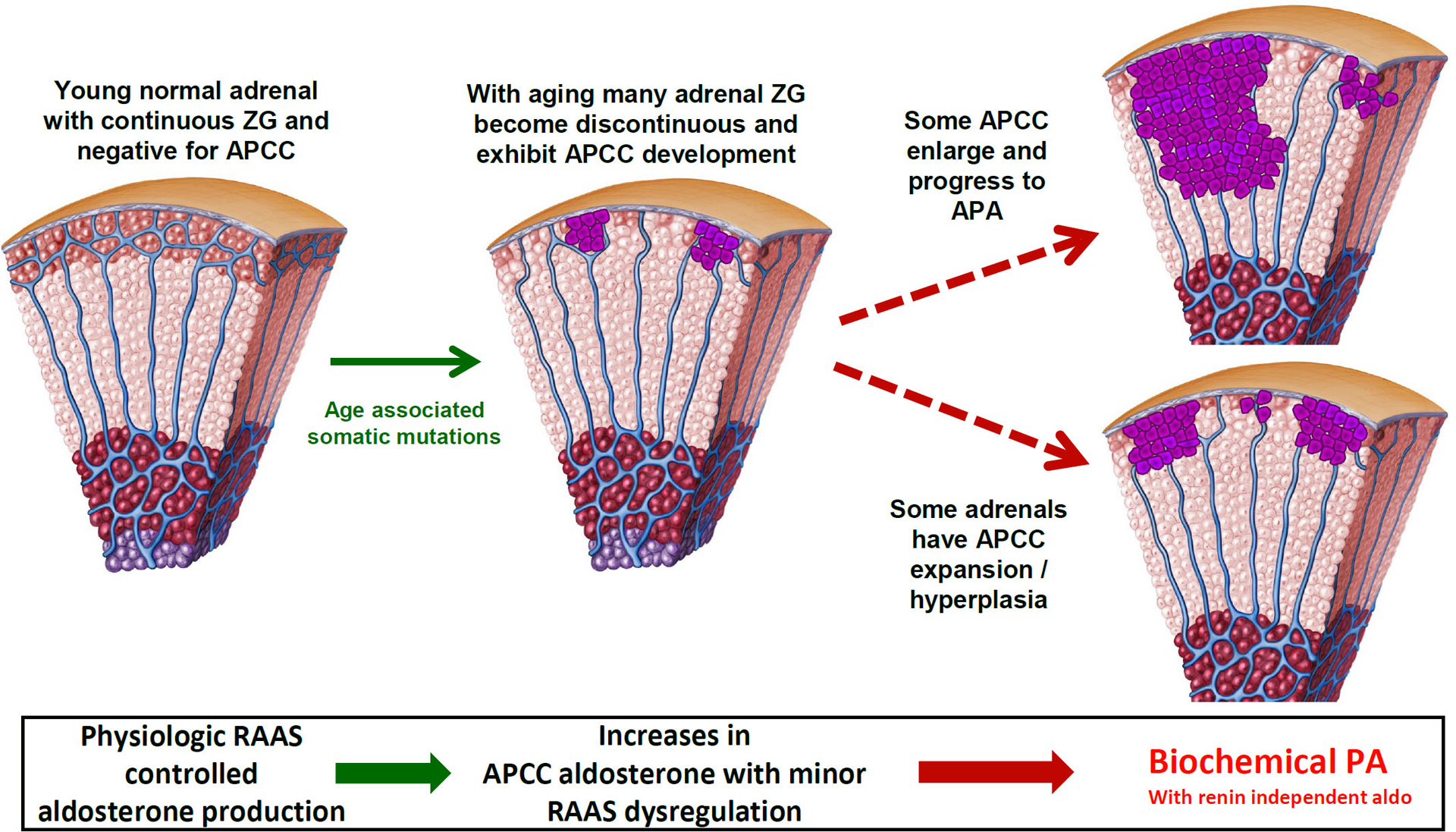
Potential role of APCC in adrenal progression to primary aldosteronism (PA). Continuous CYP11B2 expression in the zona glomerulosa (ZG) is often seen in young adrenal glands, whereas many older adrenals exhibit a loss of normal ZG CYP11B2 expression and the appearance of aldosterone-producing cell clusters (APCC). Because APCC have mutations in CACNA1D and ATP1A1, they may act as the precursors to APA. However, the lack of mutations in KCNJ5 suggest that APCC may not be the cellular origins of these tumors. RAAS, renin-angiotensin-aldosterone system
Sources of Funding
This work was supported by a grant from the National Institutes of Diabetes and Digestive and Kidney Disease (DK106618) to W.E. Rainey.
Footnotes
Disclosure Statement: The authors have nothing to disclose.
References
- 1.Monticone S, D’Ascenzo F, Moretti C et al. Cardiovascular events and target organ damage in primary aldosteronism compared with essential hypertension: a systematic review and meta-analysis. Lancet Diabetes Endocrinol 2018; 6: 41–50 [DOI] [PubMed] [Google Scholar]
- 2.Kayser SC, Dekkers T, Groenewoud HJ et al. Study heterogeneity and estimation of prevalence of primary aldosteronism: a systematic review and meta-regression analysis. J Clin Endocrinol Metab 2016; 101: 2826–2835 [DOI] [PubMed] [Google Scholar]
- 3.Williams B, MacDonald TM, Morant SV et al. Endocrine and haemodynamic changes in resistant hypertension, and blood pressure responses to spironolactone or amiloride: The PATHWAY-2 mechanisms substudies. Lancet Diabetes Endocrinol 2018; 6: 464–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim JS, Park S, Park SI et al. Cardiac dysfunction in association with increased inflammatory markers in primary aldosteronism. Endocrinol Metab (Seoul) 2016; 31: 567–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zia AA, Kamalov G, Newman KP et al. From aldosteronism to oxidative stress: the role of excessive intracellular calcium accumulation. Hypertens Res 2010; 33: 1091–1101 [DOI] [PubMed] [Google Scholar]
- 6.Herrada AA, Campino C, Amador CA et al. Aldosterone as a modulator of immunity: implications in the organ damage. J Hypertens 2011; 29: 1684–1692 [DOI] [PubMed] [Google Scholar]
- 7.Yang Y, Reincke M, Williams TA. Prevalence, diagnosis and outcomes of treatment for primary aldosteronism. Best Pract Res Clin Endocrinol Metab 2019, DOI: 10.1016/j.beem.2019.101365: 101365. [DOI] [PubMed] [Google Scholar]
- 8.Hundemer GL, Curhan GC, Yozamp N et al. Cardiometabolic outcomes and mortality in medically treated primary aldosteronism: a retrospective cohort study. Lancet Diabetes Endocrinol 2018; 6: 51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rossi GP. Primary aldosteronism: JACC state-of-the-art review. J Am Coll Cardiol 2019; 74: 2799–2811 [DOI] [PubMed] [Google Scholar]
- 10.Rossi GP, Cesari M, Cuspidi C et al. Long-term control of arterial hypertension and regression of left ventricular hypertrophy with treatment of primary aldosteronism. Hypertension 2013; 62: 62–69 [DOI] [PubMed] [Google Scholar]
- 11.Gomez-Sanchez CE, Kuppusamy M, Reincke M et al. Disordered CYP11B2 expression in primary aldosteronism. Horm Metab Res 2017; 49: 957–962 [DOI] [PubMed] [Google Scholar]
- 12.Wu VC, Chueh SC, Chang HW et al. Bilateral aldosterone-producing adenomas: differentiation from bilateral adrenal hyperplasia. QJM 2008; 101: 13–22 [DOI] [PubMed] [Google Scholar]
- 13.Yamazaki Y, Nakamura Y, Omata K et al. Histopathological classification of cross-sectional image-negative hyperaldosteronism. J Clin Endocrinol Metab 2017; 102: 1182–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Omura M, Sasano H, Fujiwara T et al. Unique cases of unilateral hyperaldosteronemia due to multiple adrenocortical micronodules, which can only be detected by selective adrenal venous sampling. Metabolism 2002; 51: 350–355 [DOI] [PubMed] [Google Scholar]
- 15.Omata K, Satoh F, Morimoto R et al. Cellular and genetic causes of idiopathic hyperaldosteronism. Hypertension 2018; 72: 874–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scholl UI, Goh G, Stolting G et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet 2013; 45: 1050–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Azizan EA, Poulsen H, Tuluc P et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet 2013; 45: 1055–1060 [DOI] [PubMed] [Google Scholar]
- 18.Beuschlein F, Boulkroun S, Osswald A et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet 2013; 45: 440–444, 444e441–442 [DOI] [PubMed] [Google Scholar]
- 19.Choi M, Scholl UI, Yue P et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 2011; 331: 768–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernandes-Rosa FL, Williams TA, Riester A et al. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension 2014; 64: 354–361 [DOI] [PubMed] [Google Scholar]
- 21.Williams TA, Monticone S, Mulatero P. KCNJ5 mutations are the most frequent genetic alteration in primary aldosteronism. Hypertension 2015; 65: 507–509 [DOI] [PubMed] [Google Scholar]
- 22.Taguchi R, Yamada M, Nakajima Y et al. Expression and mutations of KCNJ5 mRNA in Japanese patients with aldosterone-producing adenomas. J Clin Endocrinol Metab 2012; 97: 1311–1319 [DOI] [PubMed] [Google Scholar]
- 23.Hong AR, Kim JH, Song YS et al. Genetics of aldosterone-producing adenoma in Korean patients. PLoS One 2016; 11: e0147590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nanba K, Omata K, Else T et al. Targeted molecular characterization of aldosterone-producing adenomas in white Americans. J Clin Endocrinol Metab 2018; 103: 3869–3876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scholl UI. Unanswered questions in the genetic basis of primary aldosteronism. Horm Metab Res 2017; 49: 963–968 [DOI] [PubMed] [Google Scholar]
- 26.Nanba K, Omata K, Gomez-Sanchez CE et al. Genetic characteristics of aldosterone-producing adenomas in blacks. Hypertension 2019; 73: 885–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dutta RK, Welander J, Brauckhoff M et al. Complementary somatic mutations of KCNJ5, ATP1A1, and ATP2B3 in sporadic aldosterone producing adrenal adenomas. Endocr Relat Cancer 2014; 21: L1–4 [DOI] [PubMed] [Google Scholar]
- 28.Stindl J, Tauber P, Sterner C et al. Pathogenesis of adrenal aldosterone-producing adenomas carrying mutations of the Na(+)/K(+)-ATPase. Endocrinology 2015; 156: 4582–4591 [DOI] [PubMed] [Google Scholar]
- 29.Wu VC, Wang SM, Chueh SJ et al. The prevalence of CTNNB1 mutations in primary aldosteronism and consequences for clinical outcomes. Sci Rep 2017; 7: 39121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dutta RK, Arnesen T, Heie A et al. A somatic mutation in CLCN2 identified in a sporadic aldosterone-producing adenoma. Eur J Endocrinol 2019; 181: K37–K41 [DOI] [PubMed] [Google Scholar]
- 31.Scholl UI, Stolting G, Schewe J et al. CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat Genet 2018; 50: 349–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernandes-Rosa FL, Daniil G, Orozco IJ et al. A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nat Genet 2018; 50: 355–361 [DOI] [PubMed] [Google Scholar]
- 33.Daniil G, Fernandes-Rosa FL, Chemin J et al. CACNA1H mutations are associated with different forms of primary aldosteronism. EBioMedicine 2016; 13: 225–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scholl UI, Stolting G, Nelson-Williams C et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife 2015; 4: e06315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hattangady NG, Olala LO, Bollag WB et al. Acute and chronic regulation of aldosterone production. Mol Cell Endocrinol 2012; 350: 151–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev 2011; 32: 81–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishimoto K, Nakagawa K, Li D et al. Adrenocortical zonation in humans under normal and pathological conditions. J Clin Endocrinol Metab 2010; 95: 2296–2305 [DOI] [PubMed] [Google Scholar]
- 38.Gomez-Sanchez CE, Qi X, Velarde-Miranda C et al. Development of monoclonal antibodies against human CYP11B1 and CYP11B2. Mol Cell Endocrinol 2014; 383: 111–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakamura Y, Maekawa T, Felizola SJ et al. Adrenal CYP11B1/2 expression in primary aldosteronism: immunohistochemical analysis using novel monoclonal antibodies. Mol Cell Endocrinol 2014; 392: 73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ogishima T, Suzuki H, Hata J et al. Zone-specific expression of aldosterone synthase cytochrome P-450 and cytochrome P-45011 beta in rat adrenal cortex: histochemical basis for the functional zonation. Endocrinology 1992; 130: 2971–2977 [DOI] [PubMed] [Google Scholar]
- 41.Nanba K, Vaidya A, Williams GH et al. Age-related autonomous aldosteronism. Circulation 2017; 136: 347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rainey WE. Adrenal zonation: clues from 11beta-hydroxylase and aldosterone synthase. Mol Cell Endocrinol 1999; 151: 151–160 [DOI] [PubMed] [Google Scholar]
- 43.Shigematsu K, Kawai K, Irie J et al. Analysis of unilateral adrenal hyperplasia with primary aldosteronism from the aspect of messenger ribonucleic acid expression for steroidogenic enzymes: a comparative study with adrenal cortices adhering to aldosterone-producing adenoma. Endocrinology 2006; 147: 999–1006 [DOI] [PubMed] [Google Scholar]
- 44.Enberg U, Volpe C, Hoog A et al. Postoperative differentiation between unilateral adrenal adenoma and bilateral adrenal hyperplasia in primary aldosteronism by mRNA expression of the gene CYP11B2. Eur J Endocrinol 2004; 151: 73–85 [DOI] [PubMed] [Google Scholar]
- 45.Boulkroun S, Samson-Couterie B, Dzib JF et al. Adrenal cortex remodeling and functional zona glomerulosa hyperplasia in primary aldosteronism. Hypertension 2010; 56: 885–892 [DOI] [PubMed] [Google Scholar]
- 46.Nanba K, Tsuiki M, Sawai K et al. Histopathological diagnosis of primary aldosteronism using CYP11B2 immunohistochemistry. J Clin Endocrinol Metab 2013; 98: 1567–1574 [DOI] [PubMed] [Google Scholar]
- 47.Nishimoto K, Seki T, Hayashi Y et al. Human adrenocortical remodeling leading to aldosterone-producing cell cluster generation. Int J Endocrinol 2016; 2016: 7834356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nishimoto K, Seki T, Kurihara I et al. Case report: nodule development from subcapsular aldosterone-producing cell clusters causes hyperaldosteronism. J Clin Endocrinol Metab 2016; 101: 6–9 [DOI] [PubMed] [Google Scholar]
- 49.Nishimoto K, Tomlins SA, Kuick R et al. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. Proc Natl Acad Sci U S A 2015; 112: E4591–4599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Omata K, Tomlins SA, Rainey WE. Aldosterone-producing cell clusters in normal and pathological states. Horm Metab Res 2017; 49: 951–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Omata K, Anand SK, Hovelson DH et al. Aldosterone-producing cell clusters frequently harbor somatic mutations and accumulate with age in normal adrenals. J Endocr Soc 2017; 1: 787–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Papathomas TG, Sun N, Chortis V et al. Novel methods in adrenal research: a metabolomics approach. Histochem Cell Biol 2019; 151: 201–216 [DOI] [PubMed] [Google Scholar]
- 53.Sugiura Y, Takeo E, Shimma S et al. Aldosterone and 18-oxocortisol coaccumulation in aldosterone-producing lesions. Hypertension 2018; 72: 1345–1354 [DOI] [PubMed] [Google Scholar]
- 54.Sun N, Wu Y, Nanba K et al. High-resolution tissue mass spectrometry imaging reveals a refined functional anatomy of the human adult adrenal gland. Endocrinology 2018; 159: 1511–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Murakami M, Rhayem Y, Kunzke T et al. In situ metabolomics of aldosterone-producing adenomas. JCI Insight 2019; 4: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun N, Meyer LS, Feuchtinger A et al. Mass spectrometry imaging establishes 2 distinct metabolic phenotypes of aldosterone-producing cell clusters in primary aldosteronism. Hypertension 2020, DOI: 10.1161/HYPERTENSIONAHA.119.14041: HYPERTENSIONAHA11914041 [DOI] [PubMed] [Google Scholar]
- 57.Lenders JWM, Williams TA, Reincke M et al. Diagnosis of endocrine disease: 18-oxocortisol and 18-hydroxycortisol: is there clinical utility of these steroids? Eur J Endocrinol 2018; 178: R1–R9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Holler F, Heinrich DA, Adolf C et al. Steroid profiling and immunohistochemistry for subtyping and outcome prediction in primary aldosteronism-a review. Curr Hypertens Rep 2019; 21: 77. [DOI] [PubMed] [Google Scholar]
- 59.Eisenhofer G, Dekkers T, Peitzsch M et al. Mass spectrometry-based adrenal and peripheral venous steroid profiling for subtyping primary aldosteronism. Clin Chem 2016; 62: 514–524 [DOI] [PubMed] [Google Scholar]
- 60.Meyer LS, Wang X, Susnik E et al. Immunohistopathology and steroid profiles associated with biochemical outcomes after adrenalectomy for unilateral primary aldosteronism. Hypertension 2018; 72: 650–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nakamura Y, Satoh F, Morimoto R et al. 18-Oxocortisol measurement in adrenal vein sampling as a biomarker for subclassifying primary aldosteronism. J Clin Endocrinol Metab 2011; 96: E1272–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Williams TA, Peitzsch M, Dietz AS et al. Genotype-specific steroid profiles associated with aldosterone-producing adenomas. Hypertension 2016; 67: 139–145 [DOI] [PubMed] [Google Scholar]
- 63.Azizan EA, Lam BY, Newhouse SJ et al. Microarray, qPCR, and KCNJ5 sequencing of aldosterone-producing adenomas reveal differences in genotype and phenotype between zona glomerulosa- and zona fasciculata-like tumors. J Clin Endocrinol Metab 2012; 97: E819–829 [DOI] [PubMed] [Google Scholar]
- 64.Dekkers T, ter Meer M, Lenders JW et al. Adrenal nodularity and somatic mutations in primary aldosteronism: one node is the culprit? J Clin Endocrinol Metab 2014; 99: E1341–1351 [DOI] [PubMed] [Google Scholar]
- 65.Monticone S, Castellano I, Versace K et al. Immunohistochemical, genetic and clinical characterization of sporadic aldosterone-producing adenomas. Mol Cell Endocrinol 2015; 411: 146–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ono Y, Yamazaki Y, Omata K et al. Histological characterization of aldosterone-producing adrenocortical adenomas with different somatic mutations. J Clin Endocrinol Metab 2019, DOI: 10.1210/clinem/dgz235: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamazaki Y, Omata K, Tezuka Y et al. Tumor cell subtypes based on the intracellular hormonal activity in KCNJ5-mutated aldosterone-producing adenoma. Hypertension 2018; 72: 632–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Freel EM, Shakerdi LA, Friel EC et al. Studies on the origin of circulating 18-hydroxycortisol and 18-oxocortisol in normal human subjects. J Clin Endocrinol Metab 2004; 89: 4628–4633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mulatero P, Curnow KM, Aupetit-Faisant B et al. Recombinant CYP11B genes encode enzymes that can catalyze conversion of 11-deoxycortisol to cortisol, 18-hydroxycortisol, and 18-oxocortisol. J Clin Endocrinol Metab 1998; 83: 3996–4001 [DOI] [PubMed] [Google Scholar]
- 70.Takeo E, Sugiura Y, Uemura T et al. Tandem mass spectrometry imaging reveals distinct accumulation patterns of steroid structural isomers in human adrenal glands. Anal Chem 2019; 91: 8918–8925 [DOI] [PubMed] [Google Scholar]
- 71.Nishimoto K, Koga M, Seki T et al. Immunohistochemistry of aldosterone synthase leads the way to the pathogenesis of primary aldosteronism. Mol Cell Endocrinol 2017; 441: 124–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fernandes-Rosa FL, Boulkroun S, Zennaro MC. Somatic and inherited mutations in primary aldosteronism. J Mol Endocrinol 2017; 59: R47–R63 [DOI] [PubMed] [Google Scholar]
- 73.Monticone S, Buffolo F, Tetti M et al. Genetics in endocrinology: the expanding genetic horizon of primary aldosteronism. Eur J Endocrinol 2018; 178: R101–R111 [DOI] [PubMed] [Google Scholar]
- 74.Seidel E, Schewe J, Scholl UI. Genetic causes of primary aldosteronism. Exp Mol Med 2019; 51: 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xie CB, Shaikh LH, Garg S et al. Regulation of aldosterone secretion by Cav1.3. Sci Rep 2016; 6: 24697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brown JM, Vaidya A. The spectrum of subclinical primary aldosteronism and incident hypertension. Ann Intern Med 2018; 168: 755–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brown JM, Robinson-Cohen C, Luque-Fernandez MA et al. The spectrum of subclinical primary aldosteronism and incident hypertension: a cohort study. Ann Intern Med 2017; 167: 630–641 [DOI] [PMC free article] [PubMed] [Google Scholar]