

Abstract
Melanotic tumors of the nervous system show overlapping histological characteristics but differ substantially in their biological behavior. In order to achieve a better delineation of such tumors, we performed an in‐depth molecular characterization. Eighteen melanocytomas, 12 melanomas, and 14 melanotic and 14 conventional schwannomas (control group) were investigated for methylome patterns (450k array), gene mutations associated with melanotic tumors and copy number variants (CNVs). The methylome fingerprints assigned tumors to entity‐specific groups. Methylation groups also showed a substantial overlap with histology‐based diagnosis suggesting that they represent true biological entities. On the molecular level, melanotic schwannomas were characterized by a complex karyotype with recurrent monosomy of chromosome 22q and variable whole chromosomal gains and recurrent losses commonly involving chromosomes 1, 17p and 21. Melanocytomas carried GNAQ /11 mutations and presented with CNV involving chromosomes 3 and 6. Melanomas were frequently mutated in the TERT promoter, harbored additional oncogene mutations and showed recurrent chromosomal losses involving chromosomes 9, 10 and 6q, as well as gains of 22q. Together, melanotic nervous system tumors have several distinct mutational and chromosomal alterations and can reliably be distinguished by methylome profiling.
Keywords: 450k, copy number variants, GNA11, GNAQ, melanocytoma, melanoma, melanotic schwannoma, TERT promoter
Introduction
The central and peripheral nervous system (CNS/PNS) can be affected by a spectrum of melanotic lesions. A frequent diagnostic challenge is the differentiation of a metastatic melanoma, the most common melanotic tumor, from other primary melanotic tumors. The differential diagnosis for such lesions frequently includes melanotic schwannoma, melanocytoma and metastasis of melanoma 2, 19, 29.
Such tumors are a source of considerable diagnostic difficulty because they represent a histological continuum 36. Melanotic schwannomas, melanocytomas and melanomas share the histological characteristics of variable pigmentation. They show a variable growth pattern ranging from sheet‐like to lobular. The cellular heterogeneity of these tumors is often represented by epithelioid and/or spindle‐shaped cells. Even mitotic activity, a predicate of melanoma that is typically low or absent in melanocytomas and melanotic schwannomas, is not a certain distinctive feature in all instances 2, 24, 37. Thus, a histology‐based discrimination of these tumors might be highly complicated, if not impossible, in some instances.
Melanocytomas typically arise in the leptomeninges and preferentially occur in the posterior fossa and the upper spinal cord, probably because of the higher density of melanocytes at these sites 10. Melanotic schwannomas are often associated with spinal nerve roots, frequently in close proximity to the leptomeninges, but also occur in the PNS. Metastatic melanomas do not favor any anatomic site for manifestation whereas primary nervous system melanomas may have a slight predilection of the posterior fossa and upper spinal cord 18. Diagnostics are particularly hampered when leptomeningeal or nerval structures are involved.
Despite their overlapping histology and similar initial clinical presentation, the biological behavior of melanotic nervous system entities substantially differs. Melanocytomas and melanotic schwannomas predominantly have a more favorable clinical course compared with melanomas 26, 32, 33, 38. However, a recent study about melanotic schwannomas observed a malignant biological behavior in terms of local recurrence and metastatic spread in 35% and 44% of cases, respectively 33. Melanocytomas also bear to some extent the potential for malignant progression, even though to a lesser frequency compared with melanotic schwannomas 3, 15, 37. Thus, a sharp delineation of these melanotic tumors is of paramount clinical importance.
Recently, hotspot mutations in GNAQ and GNA11 have been revealed in melanocytomas 8, 17, 23. They were proposed as biomarkers separating melanocytomas from melanotic schwannomas 16. Moreover, the vast majority of uveal melanomas carry GNAQ or GNA11 hotspot mutations 11. Therefore, a close molecular relation was suggested between uveal melanomas and melanocytomas 30. In melanotic schwannomas, recurrent molecular alterations are not well established yet. They most frequently arise as sporadic cases, but are variably associated with the autosomal dominant disorder Carney complex 4.
We conducted an in‐depth molecular characterization of a cohort comprising tissue of patients with melanotic schwannomas, melanocytomas or melanomas of the nervous system. Molecular analyses included targeted sequencing of genes associated with melanotic tumors, assessing chromosomal copy number aberrations and genome‐wide DNA methylation patterns. The goal was to reveal distinctive molecular characteristics that may serve as a basis for a molecular classification and may facilitate diagnostics of this heterogeneous group of tumors.
Materials and Methods
Tissue specimens and patient characteristics
Tumor tissues were obtained from the archives of the Institutes of Neuropathology at the University Medical Center Freiburg and Bonn (Germany), the Departments of Neuropathology at the University Medical Center Heidelberg, Tuebingen, Erlangen and Hannover (Germany), the Department of General Pathology at the University Medical Center Heidelberg (Germany), the Neurological Institute/Edinger Institute Frankfurt/Main (Germany), the Institute of Pathology at the University of Bern (Switzerland), and at the Hospitals Ludwigsburg and Stuttgart (Germany). Research use of tissues and anonymization of data were in accordance with local ethical approvals. The tumor collection was composed of 18 meningeal melanocytomas, 14 melanotic schwannomas including 1 Carney complex associated case, 12 melanomas (5 with a cutaneous primary tumor, 3 with a uveal primary tumor and 4 without known primary tumor) and 14 conventional schwannomas (the latter as control group). Detailed patient characteristics are listed in Supporting Information Table S1.
Histopathology
Each case was carefully reviewed by CK and DR. In general, all melanotic tumors of this series were tested positive for the expression of S100 and HMB‐45. Each diagnosis was verified by examination of hematoxylin and eosin (H&E) and Tibor Pap (TP) reticulin fiber impregnation‐stained sections according to the World Health Organization (WHO) classification of nervous system tumors before cases were selected for further analysis 19. Reticulin staining was divided into either a pericellular, perilobular or biphasic pattern, adapted from a previous study 16. Mitotic activity had to be low for both melanocytomas and melanotic schwannomas. In general, high cellularity, low nuclear pleomorphism, small eosinophilic nucleoli and lobular or sheet‐like growth patterns were considered factors favoring the diagnosis of melanocytoma whereas lower cellularity, increased nuclear pleomorphism, prominent macronuclei and a predominantly fascicular growth favored melanotic schwannoma. However, these criteria are somewhat subjective and not all cases could be classified using these criteria alone. Therefore, additional information about localization, involvement of peripheral nervous tissue and the clinical history were taken into account for the histological classification. The supplement “with increased proliferation” was assigned to melanocytomas if a proliferation index (Ki67) above 5% was detected 24, 25. However, none of the melanocytomas fulfilled the criteria for “Intermediate Grade” according to the WHO suggestions 19. All melanoma tissues of this series had, to some extent, contact or involvement to the leptomeningeal or CNS/PNS tissue. They were diagnosed if high cellularity, nuclear atypia and brisk mitotic activity were present. One tumor (67908) with a histological picture not distinguishable from melanocytoma was diagnosed as melanoma metastasis because of its parenchymal CNS localization and a clinical history of uveal melanoma. All melanomas were untreated prior to biopsy, apart from the uveal melanoma cases treated with adjuvant radiochemotherapy. Conventional schwannomas serving as control tumors were moderately cellular tumors without pigmentation. They presented with a prototypical fascicular growth and a dense pericellular reticulin network. Mitotic activity was low. Detailed clinical and histopathological tumor characteristics are listed in Supporting Information Tables S1 and S2.
Immunohistochemistry
Immunohistochemistry was conducted on 4‐μm‐thick formalin‐fixed, paraffin‐embedded (FFPE) tissue sections mounted on StarFrost Advanced Adhesive slides (Engelbrecht, Kassel, Germany) followed by drying at 80°C for 15 minutes. Antibody dilution was 1:100 for Ki67 (clone SP6, Cell Marque Corp., Rocklin, CA, USA). Staining was performed on a VentanaBenchMark XT immunostainer applying the ultraView Universal Alkaline Phosphatase Red Detection Kit (Ventana Medical Systems, Tucson, AZ, USA). For Ki67 analysis, tumor areas with the highest Ki67 labeling indices were evaluated for the fraction of positive cell nuclei by counting all cells excluding vascular cells and lymphocytes in one 200× microscopic field using a counting grid.
DNA extraction and Sanger sequencing
DNA was extracted from FFPE material. Areas with highest available tumor content were chosen. Extraction was carried out using the automated Maxwell system (Promega, Madison, WI, USA). For polymerase chain reaction (PCR), 20 ng of DNA and KOD Hot Start Master Mix (Merck, Darmstadt, Germany) were employed. Briefly, PCR was performed in a total reaction volume of 20 μL and was started with an initial polymerase activation step at 95°C for 2 minutes, followed by 35 cycles beginning with denaturation at 95°C for 20 s, annealing for 45 s at indicated temperatures, and extension at 70°C for 10 s, followed by a final extension at 70°C for 20 minutes with subsequent cooling to room temperature. The amplification product (2 μL) was submitted to bidirectional sequencing using the BigDye Terminator v3.1 Sequencing Kit (Applied Biosystems, Foster City, CA, USA). Mutations were identified by visual analysis of the sequence chromatograms using Sequence Pilot version 3.1 software (JSI‐Medisys, Kippenheim, Germany). Primer sequences, corresponding annealing temperatures, PCR product size and GenBank NCBI references are given in Supporting Information Table S5.
Methylome and copy number profiling
The Illumina Infinium HumanMethylation450 (450k) array was used to obtain the DNA methylation status of 482 421 CpG sites (Illumina, San Diego, CA, USA), according to the manufacturer's instructions at the Core Facility of the DKFZ. Probe filtering and genome‐wide copy number analysis were performed as previously described 13. Chromosomal copy number alterations were visualized by the IdeogramBrowser tool 22. For unsupervised hierarchical clustering, we selected the 6223 most variably methylated probes (SD >0.25) across the dataset. Euclidean distance and average linkage was used for ordering of the probes (y‐axis) and 1–Pearson correlation as distance measure and average linkage was used for clustering of samples (x‐axis).
Results
Unsupervised molecular profiling reveals distinct biological groups of melanotic tumors
We first analyzed the most variably methylated CpG sites (6223 individual CpG sites across the genome) in our cohort of melanotic tumors composed of 18 melanocytomas, 12 melanomas, and 14 melanotic schwannomas along with 14 conventional schwannomas. Unsupervised hierarchical clustering of these CpGs revealed two main methylation clusters each composed of two distinct methylation groups (Figure 1). Main cluster 1 was composed of conventional schwannomas (methylation group 1) and melanotic schwannomas (methylation group 2). Main cluster 2 was composed of melanocytomas (methylation group 3) and melanomas (methylation group 4). All but one sample was assigned to one of these four methylation groups. The single Carney complex associated melanotic schwannoma (ID 68318) clustered somewhat apart from the other melanotic schwannomas as a slight outlier within main cluster 1. Similar results were obtained when varying the number of CpGs used for the analysis (data not shown).
Figure 1.
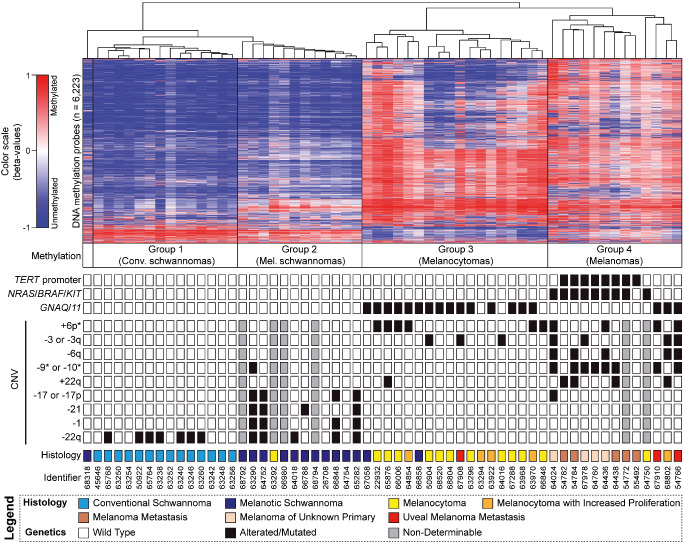
Molecular profiling of melanotic nervous system tumors. Heatmap depicting unsupervised hierarchical clustering of methylation levels of the top 6223 most variant probes (SD >0.25) in melanotic tumors of the nervous system. Each row represents a probe; each column represents a sample. The level of DNA methylation (beta value) is represented with a color scale as depicted. Conventional schwannomas served as control group. For each sample (n = 58), associated methylation group, results of oncogene sequencing, selected chromosomal copy number variants (CNVs) and histological diagnosis are indicated. * = partial losses included.
In parallel, we analyzed the mutational status of genes, which are known to be involved in the development of melanotic tumors. The genes sequenced for hotspot mutations included TERT promoter, NRAS, KRAS, BRAF and KIT, which are known oncogenes in melanomas, as well as GNAQ and GNA11 being known oncogenes in melanocytomas and uveal melanomas.
TERT promoter mutations and activating mutations in NRAS, BRAF or KIT were present exclusively in methylation group 4 (melanomas). All cases in methylation group 2 (melanotic schwannomas) lacked hotspot mutations except for a single case that carried a KRAS G60D mutation. GNAQ/11 mutations occurred in a mutually exclusive manner and were found exclusively in methylation groups 3 (15/18 melanocytomas) and 4 (3/13 melanomas). Details of sequencing results are listed in Supporting Information Table S3.
Next, we examined the tumors for chromosomal copy number variants (CNVs) (Figure 2, Supporting Information Table S4). Several recurrent variations showed a group specific distribution. Gain of chromosome 6p was present only in a subset of tumors of methylation group 3 (6/18) and group 4 (4/11). Loss of one copy of chromosome 3 or 3q was only seen in methylation group 3 (3/18) and group 4 (4/11). Loss of chromosome 6q was exclusively present and loss of chromosome 9 or 10 (including partial losses) was almost exclusively present in tumors of methylation group 4 (5/11). Gain of chromosome 22 or 22q was present in 4/11 tumors of methylation group 4 and in 1 tumor of methylation group 3. Tumors of methylation group 2 (melanotic schwannomas) often showed a complex karyotype with recurrent whole chromosomal gains involving chromosomes 4–9 (5/8). Loss of chromosome 17 or 17p assigned almost specific to methylation group 2 whereas monosomy of chromosome 1 (4/8) or chromosome 21 (4/8) was seen only in tumors of group 2. Loss of chromosome 22q was exclusively present in tumors of methylation group 1 (7/14) and methylation group 2 (5/8). An overview of molecular characteristics is given in Figure 1.
Figure 2.
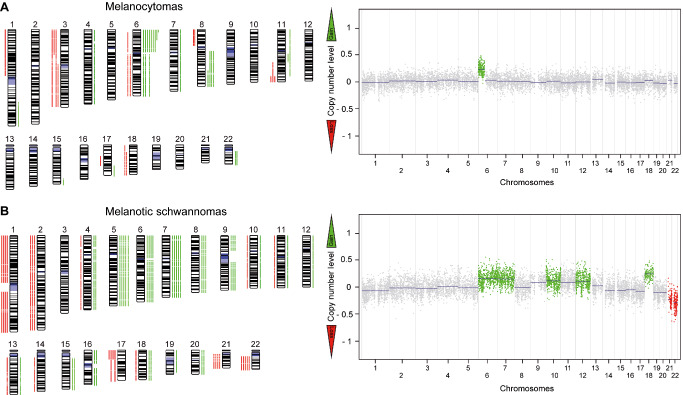
Numerical chromosomal alterations in melanotic nervous system tumors. Chromosomal ideograms summarizing the chromosomal copy number aberrations of 18 melanocytomas (A) and 9 melanotic schwannomas (B) identified by 450k processed data. Green bars indicate areas with copy number gain and red bars indicate areas with copy number loss. A representative copy number plot is given for each tumor type.
Comparison of molecular vs. histological classification
The molecular findings revealed four distinct biological groups. Comparing these groups with histological diagnosis, a large overlap was evident: conventional schwannomas (group 1), melanotic schwannomas (group 2), melanocytomas (group 3) and melanomas (group 4). However, there were some discrepant cases.
One histologically diagnosed melanocytoma (ID 63292) clustered together with melanotic schwannomas (methylation group 2). This tumor was wild type for GNAQ/11 but carried a KRAS mutation. KRAS mutations occur in about 2% of melanomas but have also been found in rare cases of schwannoma 1, 27. CNVs were undeterminable in this case due to poor DNA quality.
Methylation group 3 (melanocytomas) included two histologically diagnosed melanotic schwannomas (ID 66858, ID 67058). Both tumors carried hotspot mutation in GNAQ or GNA11. Furthermore, they lacked CNVs associated with melanotic schwannomas, that is, chromosomal losses of 17p, 21 or 22q, further supporting the classification as melanocytoma. Methylation group 3 also included one tumor diagnosed as uveal melanoma metastasis (ID 67908). Of note, this tumor was indeed histologically indistinguishable from melanocytoma but was diagnosed as melanoma metastasis because of its parenchymal localization and a clinical history of uveal melanoma in that patient. CNV analysis of this tumor revealed monosomy of chromosome 3, a strong biomarker for metastatic disease in uveal melamomas 11. However, clinical follow‐up data and the primary tumor of the patient were not available. Two more melanocytomas (ID 64016 and ID 68802) presented with monosomy of chromosome 3. On the one hand, chromosome 3 monosomy in these cases might further indicate the close molecular relation to uveal melanoma or might even hint to misdiagnosed uveal melanoma metastasis. On the other hand, two other cases with clinical history of uveal melanoma (ID 67910, ID 54866) clustered together with melanomas in methylation group 4. Reasonably, further investigation will be required to reliably decipher this molecular puzzle.
Methylation group 4 (melanomas) included two histologically diagnosed melanocytomas (ID 64750, ID 68802). One of these tumors (ID 64750) carried a BRAF V600E mutation supporting the classification as melanoma. CNV and clinical follow‐up data were not available for this case. The other tumor (ID 68802) carried a GNA11 mutation, which is found in both melanocytomas and uveal melanomas 35. CNV analysis of this tumor revealed a loss of 6q, which was exclusively found in the melanoma cluster and is known to be present in a subset of uveal melanomas, which further supports the classification as melanoma 20. Clinical follow‐up data were not available for this case.
Discussion
The substantial histological overlap makes molecular classification attractive for the diagnostics of melanotic tumors of the nervous system. One diagnostically useful molecular alteration discovered in recent years is the recurrence of GNAQ/11 mutations in uveal melanomas, blue naevi, leptomeningeal or uveal melanocytomas, and in such melanomas that developed from these benign precursor lesions 8, 17, 21, 23, 34. GNAQ/11 mutations are absent in melanotic schwannomas, making them highly specific in this differential diagnosis 16. Of note, GNAQ/11 mutations rather activate the Hippo than the MAPK signaling pathway and thus provide novel therapeutic opportunities in such mutated tumors 7, 39. Cutaneous melanomas frequently carry mutations in BRAF or NRAS whereas acral and mucosal melanomas often show activating mutations or amplification of KIT. These genetic alterations are virtually never seen in melanocytomas and melanotic schwannomas 8, 17, 23, 30. However, it must be considered that all so far examined childhood CNS melanomas or patients with the neurocutaneous melanocytosis syndrome carried NRAS mutations at the mutational hotspot codon 61 17, 26. Furthermore, hotspot mutations in the promoter region of TERT strongly argue for cutaneous melanoma because they are absent in uveal melanomas, melanocytomas and melanotic schwannomas 6, 9, 12, 14. Mutational analyses in the present study were in line with these previous reports with a very few exceptions, most likely due to histologically misdiagnosed cases. The same is true for copy number profiles in which also several entity‐specific alterations can be observed. Thus, mutational and copy number analyses are clearly helpful in the differential diagnosis of melanotic tumors but lack sufficient sensitivity to clarify all cases.
Genome‐wide methylation analysis using DNA from FFPE material appears to be the most reliable molecular tool to differentiate this heterogeneous tumor group. Cluster analyses of DNA methylation profiles using 450k arrays sharply classified all tumors of this study. This classification by DNA methylation profile showed a substantial overlap with histology. Most importantly, the classification according to methylation profiles was supported by the results of mutational and copy number analyses, even in cases with discrepant histology (illustrative case presented in Supporting Information Figure S1). Because of the lack of clinical follow‐up data for most of the cases, it is not possible to establish the molecular classification presented here as unambiguously correct. However, the concordance of methylation and genomic profiles suggests that the found molecular groups represent true biological entities as already reported for other nervous system tumors 13, 31.
In this context, the question arises whether bona fide primary CNS melanomas differ from melanomas of other or unknown sites. In principal, primary CNS melanoma is a diagnosis of exclusion. The current series included four melanomas of unknown primary. We cannot exclude that one or more of these tumors were primary CNS melanomas. However, the mutational profiles of these tumors were closely related to the genotype of cutaneous melanomas 5. It will be necessary to analyze well‐documented primary CNS melanomas on the molecular level particularly with respect to the more favorable clinical course of some primary CNS melanoma compared with melanoma metastasis 2. It will also be of interest to extend the proposed molecular classification by including pediatric neurocutaneous melanocytosis syndrome associated lesions to clarify their relation to the entities analyzed here.
It is assumed that melanotic nervous system tumors arise from neural crest descendants like melanocytes or Schwann cells. Methylation profiles are known to be cell type and tissue specific 28. Apparently, different methylation profiles of melanocytoma and melanotic schwannoma may therefore indicate a different cell of origin for these tumors. Melanotic schwannomas formed a cluster separate from conventional schwannomas, but their profiles were more similar to those of conventional schwannomas than to those of melanocytomas or melanomas. Profiles of melanocytomas and melanomas were also unique but showed more similarities with each other than with those of schwannomas. This may argue for a common origin of conventional and melanotic schwannomas on the one side and melanocytoma and melanoma on the other side.
In conclusion, we have presented an integrated molecular analysis that sheds new light on the molecular background of melanotic tumors of the nervous system, revealed novel insights regarding their evolutionary lineage and provided evidence for DNA methylation profiling as a useful diagnostic tool.
Disclosure/Conflict of Interest
The authors declare that they have no conflict of interest.
Supporting information
Figure S1. Clinicopathological and molecular findings in a melanotic tumor (15).
Table S1. Patients' characteristics.
Table S2. Histopathological tissue characteristics.
Table S3. Oncogene sequencing results.
Table S4. Chromosomal imbalances in melanotic nervous system tumors.
Table S5. Primer sequences and PCR conditions.
Acknowledgments
MP's work was supported by the German Research Fundation (DFG) (CRC992).
References
- 1. Bertola DR, Pereira AC, Brasil AC, Suzuki L, Leite C, Falzoni R et al (2012) Multiple, diffuse schwannomas in a RASopathy phenotype patient with germline KRAS mutation: a causal relationship? Clin Genet 81:595–597. [DOI] [PubMed] [Google Scholar]
- 2. Brat DJ, Giannini C, Scheithauer BW, Burger PC (1999) Primary melanocytic neoplasms of the central nervous systems. Am J Surg Pathol 23:745–754. [DOI] [PubMed] [Google Scholar]
- 3. Bydon A, Gutierrez JA, Mahmood A (2003) Meningeal melanocytoma: an aggressive course for a benign tumor. J Neurooncol 64:259–263. [DOI] [PubMed] [Google Scholar]
- 4. Carney JA (1990) Psammomatous melanotic schwannoma. A distinctive, heritable tumor with special associations, including cardiac myxoma and the Cushing syndrome. Am J Surg Pathol 14:206–222. [PubMed] [Google Scholar]
- 5. Egberts F, Bergner I, Kruger S, Haag J, Behrens HM, Hauschild A, Rocken C (2014) Metastatic melanoma of unknown primary resembles the genotype of cutaneous melanomas. Ann Oncol 25:246–250. [DOI] [PubMed] [Google Scholar]
- 6. Egberts F, Kruger S, Behrens HM, Bergner I, Papaspyrou G, Werner JA et al (2014) Melanomas of unknown primary frequently harbor TERT‐promoter mutations. Melanoma Res 24:131–136. [DOI] [PubMed] [Google Scholar]
- 7. Feng X, Degese MS, Iglesias‐Bartolome R, Vaque JP, Molinolo AA, Rodrigues M et al (2014) Hippo‐independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio‐regulated rho GTPase signaling circuitry. Cancer Cell 25:831–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gessi M, Hammes J, Lauriola L, Dorner E, Kirfel J, Kristiansen G et al (2013) GNA11 and N‐RAS mutations: alternatives for MAPK pathway activating GNAQ mutations in primary melanocytic tumours of the central nervous system. Neuropathol Appl Neurobiol 39:417–425. [DOI] [PubMed] [Google Scholar]
- 9. Gessi M, van de Nes J, Griewank K, Barresi V, Buckland ME, Kirfel J et al (2014) Absence of TERT promoter mutations in primary melanocytic tumors of the central nervous system. Neuropathol Appl Neurobiol 40:794–797. [DOI] [PubMed] [Google Scholar]
- 10. Goldgeier MH, Klein LE, Klein‐Angerer S, Moellmann G, Nordlund JJ (1984) The distribution of melanocytes in the leptomeninges of the human brain. J Invest Dermatol 82:235–238. [DOI] [PubMed] [Google Scholar]
- 11. Harbour JW (2012) The genetics of uveal melanoma: an emerging framework for targeted therapy. Pigment Cell Melanoma Res 25:171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A et al (2013) TERT promoter mutations in familial and sporadic melanoma. Science 339:959–961. [DOI] [PubMed] [Google Scholar]
- 13. Hovestadt V, Remke M, Kool M, Pietsch T, Northcott PA, Fischer R et al (2013) Robust molecular subgrouping and copy‐number profiling of medulloblastoma from small amounts of archival tumour material using high‐density DNA methylation arrays. Acta Neuropathol 125:913–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA (2013) Highly recurrent TERT promoter mutations in human melanoma. Science 339:957–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim OH, Kim SJ, Choo HJ, Lee SJ, Lee IS, Kim JY, Kim H (2013) Spinal meningeal melanocytoma with benign histology showing leptomeningeal spread: case report. Korean J Radiol 14:470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kusters‐Vandevelde HV, van Engen‐van Grunsven IA, Kusters B, van Dijk MR, Groenen PJ, Wesseling P, Blokx WA (2010) Improved discrimination of melanotic schwannoma from melanocytic lesions by combined morphological and GNAQ mutational analysis. Acta Neuropathol 120:755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kusters‐Vandevelde HV, Klaasen A, Kusters B, Groenen PJ, van Engen‐van Grunsven IA, van Dijk MR et al (2010) Activating mutations of the GNAQ gene: a frequent event in primary melanocytic neoplasms of the central nervous system. Acta Neuropathol 119:317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liubinas SV, Maartens N, Drummond KJ (2010) Primary melanocytic neoplasms of the central nervous system. J Clin Neurosci 17:1227–1232. [DOI] [PubMed] [Google Scholar]
- 19. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A et al (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mensink HW, Kilic E, Vaarwater J, Douben H, Paridaens D, de Klein A (2008) Molecular cytogenetic analysis of archival uveal melanoma with known clinical outcome. Cancer Genet Cytogenet 181:108–111. [DOI] [PubMed] [Google Scholar]
- 21. Mudhar HS, Doherty R, Salawu A, Sisley K, Rennie IG (2013) Immunohistochemical and molecular pathology of ocular uveal melanocytoma: evidence for somatic GNAQ mutations. Br J Ophthalmol 97:924–928. [DOI] [PubMed] [Google Scholar]
- 22. Muller A, Holzmann K, Kestler HA (2007) Visualization of genomic aberrations using Affymetrix SNP arrays. Bioinformatics 23:496–497. [DOI] [PubMed] [Google Scholar]
- 23. Murali R, Wiesner T, Rosenblum MK, Bastian BC (2012) GNAQ and GNA11 mutations in melanocytomas of the central nervous system. Acta Neuropathol 123:457–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Navas M, Pascual JM, Fraga J, Pedrosa M, Shakur S, Carrasco R et al (2009) Intracranial intermediate‐grade meningeal melanocytoma with increased cellular proliferative index: an illustrative case associated with a nevus of Ota. J Neurooncol 95:105–115. [DOI] [PubMed] [Google Scholar]
- 25. Quatresooz P, Pierard‐Franchimont C, Pierard GE (2008) Highlighting the immunohistochemical profile of melanocytomas: review. Oncol Rep 19:1367–1372. [PubMed] [Google Scholar]
- 26. Raizer JJ, Hwu WJ, Panageas KS, Wilton A, Baldwin DE, Bailey E et al (2008) Brain and leptomeningeal metastases from cutaneous melanoma: survival outcomes based on clinical features. Neuro‐Oncol 10:199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Serrano C, Simonetti S, Hernandez‐Losa J, Valverde C, Carrato C, Bague S et al (2013) BRAF V600E and KRAS G12S mutations in peripheral nerve sheath tumours. Histopathology 62:499–504. [DOI] [PubMed] [Google Scholar]
- 28. Shiota K, Kogo Y, Ohgane J, Imamura T, Urano A, Nishino K et al (2002) Epigenetic marks by DNA methylation specific to stem, germ and somatic cells in mice. Genes Cells 7:961–969. [DOI] [PubMed] [Google Scholar]
- 29. Smith AB, Rushing EJ, Smirniotopoulos JG (2009) Pigmented lesions of the central nervous system: radiologic‐pathologic correlation. Radiographics 29:1503–1524. [DOI] [PubMed] [Google Scholar]
- 30. Solus JF, Kraft S (2013) Ras, Raf, and MAP kinase in melanoma. Adv Anat Pathol 20:217–226. [DOI] [PubMed] [Google Scholar]
- 31. Sturm D, Witt H, Hovestadt V, Khuong‐Quang DA, Jones DT, Konermann C et al (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425–437. [DOI] [PubMed] [Google Scholar]
- 32. Torres‐Mora J, Dry S, Li X, Binder S, Amin M, Folpe AL (2014) Malignant melanotic schwannian tumor: a clinicopathologic, immunohistochemical, and gene expression profiling study of 40 cases, with a proposal for the reclassification of “melanotic schwannoma”. Am J Surg Pathol 38:94–105. [DOI] [PubMed] [Google Scholar]
- 33. Vallat‐Decouvelaere AV, Wassef M, Lot G, Catala M, Moussalam M, Caruel N, Mikol J (1999) Spinal melanotic schwannoma: a tumour with poor prognosis. Histopathology 35:558–566. [DOI] [PubMed] [Google Scholar]
- 34. Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O'Brien JM et al (2009) Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 457:599–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T et al (2010) Mutations in GNA11 in uveal melanoma. N Engl J Med 363:2191–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wadasadawala T, Trivedi S, Gupta T, Epari S, Jalali R (2010) The diagnostic dilemma of primary central nervous system melanoma. J Clin Neurosci 17:1014–1017. [DOI] [PubMed] [Google Scholar]
- 37. Wang F, Qiao G, Lou X, Song X, Chen W (2011) Malignant transformation of intracranial meningeal melanocytoma. Case report and review of the literature. Neuropathology 31:414–420. [DOI] [PubMed] [Google Scholar]
- 38. Wang H, Zhang S, Wu C, Zhang Z, Qin T (2013) Melanocytomas of the central nervous system: a clinicopathological and molecular study. Eur J Clin Invest 43:809–815. [DOI] [PubMed] [Google Scholar]
- 39. Yu FX, Luo J, Mo JS, Liu G, Kim YC, Meng Z et al (2014) Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell 25:822–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Clinicopathological and molecular findings in a melanotic tumor (15).
Table S1. Patients' characteristics.
Table S2. Histopathological tissue characteristics.
Table S3. Oncogene sequencing results.
Table S4. Chromosomal imbalances in melanotic nervous system tumors.
Table S5. Primer sequences and PCR conditions.