

Abstract
The olfactory bulb (OB) is affected early in both Parkinson's (PD) and Alzheimer's disease (AD), evidenced by the presence of disease‐specific protein aggregates and an early loss of olfaction. Whereas previous studies showed amoeboid microglia in the classically affected brain regions of PD and AD patients, little was known about such changes in the OB. Using a morphometric approach, a significant increase in amoeboid microglia density within the anterior olfactory nucleus (AON) of AD and PD patients was observed. These amoeboid microglia cells were in close apposition to β‐amyloid, hyperphosphorylated tau or α‐synuclein deposits, but no uptake of pathological proteins by microglia could be visualized. Subsequent analysis showed (i) no correlation between microglia and α‐synuclein (PD), (ii) a positive correlation with β‐amyloid (AD), and (iii) a negative correlation with hyperphosphorylated tau (AD). Furthermore, despite the observed pathological alterations in neurite morphology, neuronal loss was not apparent in the AON of both patient groups. Thus, we hypothesize that, in contrast to the classically affected brain regions of AD and PD patients, within the AON rather than neuronal loss, the increased density in amoeboid microglial cells, possibly in combination with neurite pathology, may contribute to functional deficits.
Keywords: Alzheimer's disease, anterior olfactory nucleus, hyperphosphorylated tau, microglia, neuroinflammation, olfactory bulb, Parkinson's disease, α‐synuclein, β‐amyloid
Introduction
Neuroinflammatory processes have been implicated in the pathogenesis of various neurodegenerative disorders, including Parkinson's (PD) and Alzheimer's disease (AD) 23, 30. Under degenerative conditions, the resting microglial cells, that represent the brain's innate immune system, rapidly transform from morphologically ramified into amoeboid‐shaped cells. During this transition, they acquire specific functions like phagocytosis, while they can also secrete cytokines, chemokines, reactive oxygen species (ROS) and growth factors 38, 81. In vivo imaging as well as human post‐mortem studies have revealed the presence of reactive microglia and proinflammatory mediators at the classical pathological sites in the PD 22, 47, 48, 54 and in the AD brain 55, 78. In post‐mortem tissue, microglia with a reactive phenotype accumulate not only around α‐synuclein (α‐syn) aggregates in the substantia nigra (SN) of PD patients 47, but also around beta amyloid (Aβ) plaques and neurofibrillary tangles (NFT) in the hippocampus (HC) and entorhinal cortex (EC) of AD patients 16, 47, 58, 64. Moreover, polymorphisms in genes encoding various inflammatory cytokines have been associated with a greater risk to develop AD or PD 17, 26.
In vitro application of Aβ, or α‐syn, to cultured microglia influences their phagocytic properties, and possibly those of astroglial cells, while increasing their secretion of inflammatory mediators, such as tumor necrosis factor‐alpha (TNFα) and ROS 31, 57. Activation of microglia by pathological protein deposits has further been linked to increases in neurotoxicity and degeneration 4, 62, 85 as supported by in vivo data from related PD and AD mouse models 72, 73. Taken together, this suggests a putative role for activated microglia and inflammatory mediators in the mechanisms of protein pathology and/or neuronal loss in PD and AD.
Recently, attention has shifted away from the SN in PD, or the HC and EC in AD patients as the only pathological sites, to other affected brain regions. This shift was further encouraged by histopathological studies indicating that α‐syn pathology in PD, and the NFTs and Aβ plaques in AD, spread throughout the brain in a well‐defined anatomical sequence 6, 8. Further evidence for a “prion‐like” hypothesis for α‐syn spreading and a causal relationship with neuronal death was recently provided by a study showing cell‐to‐cell transmission of pathological α‐syn in wild‐type mice after a single intracerebral injection of synthetic α‐syn fibrils 45. In PD, α‐syn pathology in the olfactory bulb (OB) is an early event and present already during presymptomatic stages of the disease 7. The occurrence of NFTs in the OB of AD patients further seems to reflect early neuropathology as well, as it occurs in advance of, or in parallel to, the Aβ pathology in the EC 2, 37, 75.
Interestingly, in both PD and AD, neuropathology in the OB appears concentrated in the anterior olfactory nucleus (AON) mostly, a dispersed region within the granular cell layer of the OB and olfactory tract that is important for olfactory function 11, 43. Notably, these neuropathological changes are consistent with the characteristic olfactory impairments in de novo PD patients as well as hyposmia as an early symptom in AD 15, 19, 27, 60. Moreover, olfactory dysfunction seems to relate to disease progression in both the pre‐motor and motor phase of PD 3, 51.
Previously, we observed microgliosis and elevated expression of interleukin‐1 family members in the OB of 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridinel (MPTP)‐treated mice, supporting that in this animal model for PD, neuroinflammatory responses extend beyond the nigrostriatal areas 80. In the present study, we focus on the activational status of microglial cells in the human OB and their relationship with protein pathology or neuronal degeneration. Therefore, we assessed densities of amoeboid microglial cells, ramified microglial cells and neurons in the AON by using a morphometric approach in well‐characterized cohorts of AD and PD patients and control subjects, while neurite pathology was also investigated. Subsequently, correlations between pathological protein levels and the densities of amoeboid microglia were examined and we carefully studied colocalization of pathological protein aggregates and microglia or astrocytes, as an indicator for possible phagocytosis and local clearance of pathological protein deposits within the AON.
Materials and Methods
Human subjects
Human post‐mortem brain tissue was obtained from the Netherlands Brain Bank (NBB, Amsterdam, The Netherlands) or from the Department of Pathology, VU University Medical Center in Amsterdam, The Netherlands. In compliance with all local ethical and legal guidelines, all donors had given written informed consent for brain autopsy, for use of brain tissue and for allowing access to the neuropathological and clinical information for scientific research. The OB and ventral mesencephalon were included from seven clinically diagnosed and neuropathologically verified PD patients as well as the OB and HC from eight patients with clinically diagnosed and neuropathologically verified AD. The control group consisted of 11 subjects without neurological or psychiatric disease, of which all above mentioned brain regions were studied. The age of the PD patients ranged from 73 to 88 years, the age of the AD patients ranged from 62 to 86 years and the control subjects ranged from 66 to 93 years of age. The clinicopathological data, including cause of death and Braak staging for PD and AD of all donors are summarized in Table 1.
Table 1.
Patient information. Abbreviations: AD = Alzheimer's disease; C = control subject; D = clinical and neuropathological diagnosis; NFT = neurofibrillary tangles; PD = Parkinson's disease; PMD = post mortem delay
| Patient number | D | Gender | Age | PMD (h) | Brain weight (g) | Braak α‐synuclein stage | Braak NFT stage | Braak amyloid stage | Cause of death |
|---|---|---|---|---|---|---|---|---|---|
| 1 | C | M | 74 | 5:00 | 1125 | 0 | 3 | C | Brochus carcinoma |
| 2 | C | M | 80 | 7:15 | 1376 | 0 | 0 | 0 | Cachexia and dehydration |
| 3 | C | M | 91 | 8:00 | 1243 | 1 | 1 | B | Cardiac decompensation |
| 4 | C | F | 93 | 5:50 | 1145 | 0 | 2 | 0 | Mamma carcinoma |
| 5 | C | F | 93 | 4:25 | 1223 | 0 | 1 | A | Unknown |
| 6 | C | F | 85 | 5:00 | 1257 | 0 | 1 | B | Ruptured abdominal aneurysm |
| 7 | C | F | 85 | 4:40 | 1168 | 1 | 2 | A | Dehydration |
| 8 | C | M | 66 | 7:45 | 1590 | 0 | 0 | 0 | Ruptured abdominal aneurysm aorta |
| 9 | C | F | 84 | 4:45 | 1179 | 0 | 1 | 0 | Heart failure |
| 10 | C | F | 89 | 6:25 | 1210 | 0 | 2 | B | Old age |
| 11 | C | M | 84 | 5:35 | 1457 | 0 | 1 | A | Heart failure |
| 12 | PD | M | 88 | 5:50 | 1205 | 6 | 1 | C | Unknown |
| 13 | PD | F | 87 | 5:25 | 1195 | 6 | 2 | B | Pneumonia |
| 14 | PD | M | 84 | 6:05 | 1243 | 5 | 1 | C | Myocardial infarction |
| 15 | PD | M | 73 | 6:35 | 1572 | 3 | 1 | A | Aspiration pneumonia |
| 16 | PD | F | 84 | 7:25 | 1244 | 5 | 2 | B | Unknown |
| 17 | PD | M | 87 | 3:40 | 1205 | 5 | 1 | B | Cachexia and dehydration |
| 18 | PD | M | 80 | 7:05 | 1612 | 6 | 1 | B | Pneumonia |
| 19 | AD | M | 67 | 4:10 | 1387 | 0 | 5 | C | Cachexia |
| 20 | AD | M | 64 | 11:16 | 1066 | 0 | 6 | C | Cachexia and dehydration |
| 21 | AD | F | 86 | 5:55 | 950 | 0 | 4 | B | Cachexia |
| 22 | AD | M | 62 | 6:45 | 1011 | 0 | 6 | C | Aspiration pneumonia |
| 23 | AD | F | 83 | 4:55 | 1250 | 0 | 6 | C | Dehydration |
| 24 | AD | F | 84 | 4:50 | 1120 | 0 | 4 | C | Cardiac arrest |
| 25 | AD | F | 77 | 6:05 | 1059 | 0 | 5 | C | Pneumonia |
| 26 | AD | F | 73 | 5:55 | 1090 | 0 | 6 | C | Unknown |
Tissue processing
After autopsy, brain regions were dissected and immersion‐fixed in 4% formaldehyde for 4 weeks, after which they were embedded in paraffin. From the paraffin blocks that contained the ventral mesencephalon, which included the SN pars compacta and from the HC, 6 μm sections were cut on a microtome and dried in an oven overnight at 37°C before immunohistochemical analysis. From the entire OB, 20 μm horizontal sections were cut and dried, and every 10th section was used for immunohistochemical staining and morphometric analysis. Sections were mounted on positively charged glass slides (Menzel‐Glaser SuperFrost plus, Braunschweig, Germany).
Immunohistochemistry
Sections were heated in a stove for 1 h at 56°C, before they were deparaffinized and rehydrated through a graded series of ethanol. For subsequent antigen retrieval, sections were rinsed in 0.01 M citrate buffer (pH 6.0) or in 10 mM Tris buffer (pH 9.0) containing 1 mM EDTA (Tris‐EDTA) and afterwards placed in preheated citrate buffer or Tris‐EDTA, respectively in a steamer for 30 minutes at 90–99°C. For α‐syn staining, antigen retrieval was performed using pretreatment with 100% formic acid for 10 minutes. After pretreatment, the sections were allowed to regain room temperature (RT), rinsed in Tris‐buffered saline (TBS), and incubated for 20 minutes in TBS containing 0.3% H2O2 and 0.1% sodiumazide. Non‐specific binding sites were blocked with 5% non‐fat dried milk in TBS containing 0.5% Triton (TBS‐T; blocking solution) for 30 minutes at RT. Subsequently, sections were incubated overnight at 4°C with mouse anti‐CD68, mouse anti‐Aβ, mouse anti‐α‐syn, or mouse anti‐hyperphosphorylated tau (HPtau) antibodies (see Table 2 for details on primary antibodies), diluted in blocking solution. Following this, sections were washed in TBS and incubated for 2 h at RT in biotinylated donkey anti‐mouse IgG's (1:400, Jackson Immunoresearch, Westgrove, PA, USA), followed by HRP‐labeled avidin‐biotin complex (ABC complex 1:400; Vector Laboratories, Burlingame, CA, USA) for 1 h at RT. Staining was visualized using 3,3‐diaminobenzidine (DAB, Sigma, St. Louis, MO, USA) as chromogen and counterstaining was performed with Nissl. After dehydration in graded ethanol solutions, the sections were cleared in xylene and coverslipped in Entellan (Merck, Darmstadt, Germany).
Table 2.
Primary antibodies used for single labeling
| Antigen | Species | Final dilution | Source |
|---|---|---|---|
| Human CD68 | Mouse | 1:400 | DAKO, clone KP1 |
| Human hyperphosphorylated Tau | Mouse | 1:1000 | Innogenetics, clone AT‐8 |
| Human β‐amyloid | Mouse | 1:500 | DAKO, MO872 |
| Human α‐synuclein | Mouse | 1:2000 | BD‐Bioscience, 610 786 |
| Cow GFAP | Rabbit | 1:2000 | DAKO, ZO334 |
Bodian silver staining
To examine possible neuropathological changes in the neurites and general anatomy of the neuronal network, a classic Bodian silver staining protocol was used 5. The silver solution (2 g albumin silver in 200 mL H2O) was applied overnight at 37°C to deparaffinized and rehydrated sections of the OB with copper sheets. Sections were washed in H2O and subsequently treated for 10 minutes with hydroquinone (2 g in 10 mL 37% formaldehyde and 200 mL H2O). Sections were again washed in H2O. To increase contrast, a 1% gold‐chloride solution was applied for 2–5 minutes. Thereafter, the sections were washed with H2O and incubated with 1.5% oxalic acid in H20 for 5 minutes. Surplus silver was removed with 5% sodium thiosulfate in H2O (5–10 minutes). Sections were dehydrated in graded ethanol solutions, cleared in xylene and coverslipped in Entellan 25.
Immunofluorescence
For double‐labeling of glial cells and pathological proteins, a combination of antibodies for microglia, that is, CD68, or astrocytes, that is, glial fibrillary acidic protein (GFAP), and for Aβ, HPtau, or α‐syn were used. Sections were sequentially incubated (CD68/HPtau, CD68/Aβ) or co‐incubated (CD68/α‐syn, GFAP/HPtau, GFAP/Aβ, GFAP/α‐syn) with the appropriate primary antibodies. Co‐incubations of sections with anti‐α‐syn and anti‐CD68 were pretreated with Tris‐EDTA (pH 9.0). Sections to be stained for all other antibody combinations were pretreated with citrate buffer (pH 6.0). All antibodies were diluted in the blocking solution as indicated above. After a 48 h incubation at 4°C, the sections were washed and subsequently incubated for 90 minutes at RT with appropriate Alexa Fluor 488 or Alexa Fluor labeled 594 IgG's or with streptavidin‐labeled Alexa Fluor 594 (1:400, Jackson Immunoresearch) when the primary antibodies were already biotinylated (see Table 3 for detailed information on primary antibodies and conjugates). After washing, the sections were coverslipped with Vectashield (Vector Laboratories, Burlingame, CA, U.S.A.). Sections were examined using a confocal laser scanning microscope (Leica TSC‐SP2‐AOBS; Leica Microsystems, Wetzlar, Germany).
Table 3.
Antibodies (ab's) and conjugates used for double labeling
| Antigen | Species | Final dilution | Source | Secondary ab's and conjugates |
|---|---|---|---|---|
| Human CD68 | Mouse | 1:300 | DAKO, clone KP1 | DoaM‐AF488 1:400 |
| Human α‐synuclein | Goat | 1:200 | Santa Cruz sc 7012 | DoaG‐AF594 1:400 |
| Cow GFAP | Rabbit | 1:2000 | DAKO, ZO334 + | DoaR‐AF594 1:400 |
| Human α‐synuclein | Goat | 1:200 | Santa Cruz sc 7012 | DoaG‐AF488 1:400 |
| Human CD68 | Mouse | 1:300 | DAKO, clone KP1 | DoaM‐AF488 1:400 |
| Biotinylated human hyperphosphorylated Tau | Mouse | 1:100 | Thermo Scientific | Streptavidin‐AF594 |
| Cow GFAP | Rabbit | 1:2000 | DAKO | DoaR‐AF488 1:400 |
| Human hyperphosphorylated tau | Mouse | 1:300 | Innogenetics | DoaM‐AF594 1:400 |
| Human CD68 | Mouse | 1:300 | DAKO, clone KP1 | DoaM‐AF488 1:400 |
| Biotinylated human β‐amyloid | Mouse | 1:400 | Covance, SIG39240 | Streptavidin‐AF594 |
| Cow GFAP | Rabbit | 1:2000 | DAKO, ZO334 | DoaR‐AF594 1:400 |
| Human β‐amyloid | Mouse | 1:1000 | Chemicon mab1561 | DoaM‐AF488 1:400 |
Delineation of the AON for quantification of microglial and neuronal cell numbers
The OB was identified by the presence of the glomerular cell layer; the AON was recognized as a clearly demarcated group of medium‐to‐large sized pyramidal neurons with a diameter of 15–20 μm in the granular cell layer of the OB in the Nissl‐stained horizontal sections 11, 43. AON parts that extended into the olfactory tracts and substantia perforata anterior were rare and excluded from analysis. Delineation of the total AON was performed in each section by outlining the separate groups of large neurons that were just slightly Nissl‐positive (Figure 1A, B).
Figure 1.
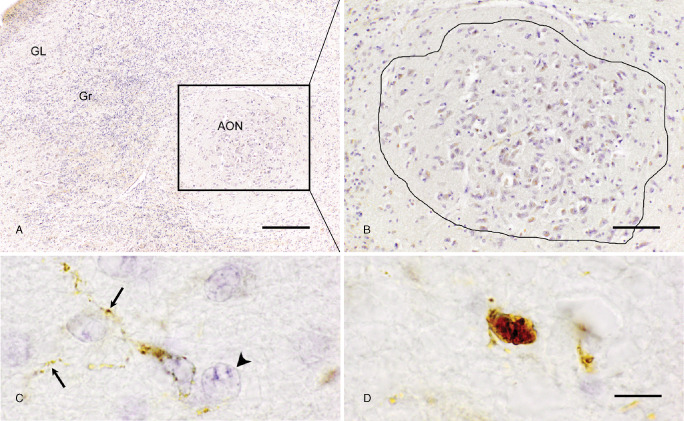
Delineation of the anterior olfactory nucleus (AON) and cell morphologies to be quantified. A. The AON (square) lies within the granular layer (Gr) of the OB; bar = 250 μm. B. Higher magnification of an AON, consisting of large, light Nissl positive, neurons. Line represents the delineation of an AON used for quantification of cells; bar = 100 μm. C. Ramified CD68‐positive microglial cell showing thin and radially projecting processes (arrows), AON neuron is identified by having a light Nissl stained cytoplasm and a clearly Nissle positive nucleolus (arrowhead). D. Amoeboid CD68 positive microglial cells showing a rounded morphology; bar (C,D) = 10 μm.
Identification criteria of ramified and amoeboid microglia, and of AON neurons
Microglial cells were identified by positive CD68‐immunoreactivity 32. While both microglial subtypes are characterized by a cytoplasmic staining, the ramified microglial cell type can be clearly distinguished as it has a small cell body and thin, radially projecting processes (Figure 1C). The amoeboid type of microglia is characterized by a densely CD68‐stained cell body typically surrounded by no or only very few short/stump processes (Figure 1D). CD68‐immunopositive cells wrapped around or touching blood vessels were not included to avoid overlap with blood‐derived monocytes. AON neurons were identified by their large size with little Nissl substance in their cytoplasm 41 and a big nucleus containing Nissl substance bound to nucleic acids (Figure 1C).
Quantitative analysis of microglial cells and neurons in the AON using a morphometric approach
Quantitative analysis of the density of microglial cells and neurons within the AON was performed using a computer‐assisted morphometry system, consisting of a Leica DMR Axioplan photomicroscope with a CCD color video camera (Optronics, 1200–1660 pixels, Goleta, CA, U.S.A.) and a LEP XY motorized stage with StereoInvestigator software version 9.0 (MicroBrightfield Inc., Colchester, VT, USA). The depth of the focal plane was measured with a Heidenhain MT12 microcator attached to the stage with a resolution of approximately 0.5 μm.
To prevent experimenter bias, all OB sections to be analyzed were coded. The AON was delineated at a final magnification of 100× in sections collected serially throughout the OB, yielding three to six sections in which the AON was visible per subject. The total number of ramified microglia, amoeboid microglia or neurons within the volume fraction of the AON examined was estimated using the optical fractionator 83 workflow of the StereoInvestigator software. As the anatomy of the AON can vary within and between subjects, quantification of the density of microglial cells and neurons in the entire AON of the OB thus needs to be standardized carefully. We therefore counted and subsequently calculated the density of microglial cells and neurons within the volume fraction of the AON examined in the OB (VolumeAON) using the following equation: cell density = (ΣQ‐* (1/tsf)*(1/asf))/VolumeAON, where ΣQ‐ is the number of cells counted in the 3D counting frames, asf is the area sampling fraction (counting frame area = 5625 μm2/grid size area = 8100 μm2, asf = 0.69). Tsf is the thickness sampling fraction, determined by ratio of the height of the optical dissector probe (ie, 10 μm) and the mean weighted thickness of the sections [13.9 μm, standard error of the mean (SEM) 0.4 μm] included in the analysis. Quantification of the microglial cells and neurons was performed using a 40× objective lens resulting in a final magnification of 400×.
Assessment of AD and PD neuropathology in the AON
The presence and severity of HPtau‐immunoreactive NFTs, Aβ‐immunoreactive senile plaques and α‐syn‐immunoreactive Lewy bodies/neurites (LBs/LNs) were analyzed by two trained investigators unaware of the Braak pathological score of each patient. A semi‐quantitative scoring of pathology in the AON was performed using a 200 × magnification. A final score ranging from 0 to 4 was assigned to the major histological signatures of AD and PD, as previously done by others 35, 59 with a score of 0 given to the AON devoid of HPtau positive NFTs, Aβ positive senile plaques or α‐syn positive LBs/LNs, a score of 1 was given to sparse pathology in the AON. A score of 2 corresponded to moderate deposition and a score of 3 to widespread depositions in the AON. Finally, a score of 4 indicated severe deposition of HPtau positive NFTs, extensive numbers of Aβ positive senile plaques or α‐syn positive LBs/LNs in the AON (see Table 4 for pathology score per case).
Table 4.
Pathology scores in AON of (1) PD patients and (2) AD patients. Abbreviations: AD = Alzheimer's disease; NFT = neurofibrillary tangles; n.a = not available; PD = Parkinson's disease
| (1) | ||||
|---|---|---|---|---|
| Patient number | Diagnosis | α‐synuclein score | NFT score | β‐amyloid score |
| 12 | PD | 1 | 1 | 3 |
| 13 | PD | 2 | 3 | n.a |
| 14 | PD | 3 | 2 | n.a |
| 15 | PD | 1 | 1 | 0 |
| 16 | PD | 3 | 1 | 0 |
| 17 | PD | 3 | 1 | 3 |
| 18 | PD | 2 | 1 | n.a |
| (2) | ||||
| Patient number | Diagnosis | β‐amyloid score | NFT score | α‐synuclein score |
| 19 | AD | 1 | 4 | 1 |
| 20 | AD | 3 | 4 | 3 |
| 21 | AD | 2 | 2 | 0 |
| 22 | AD | 4 | 3 | 0 |
| 23 | AD | 3 | 2 | 1 |
| 24 | AD | 2 | 2 | 0 |
| 25 | AD | 1 | 4 | 0 |
| 26 | AD | 0 | 4 | 0 |
Statistical analysis
The mean and SEM of microglial and neuron densities in the OB were calculated for each patient group. Normal distribution of the data was demonstrated by Kolmogorov–Smirnov significance tests. When normal distributions were not apparent, analysis was performed on log10 transformed data. For the cell density calculations, stepwise multiple regression analyses were performed to control for possible influences of age, gender, post‐mortem delay (PMD) and whole brain weight. Amoeboid microglia density showed an inverse correlation with overall brain weight (r = −0.65, P = 0.001). Therefore, this variable was taken as covariate in the subsequent one‐way independent multivariate analysis of covariance (MANCOVA), performed for amoeboid microglial density, ramified microglial density and neuronal density. The main analysis was followed up by Bonferroni corrected, pairwise comparisons. Correlation analysis was performed using Spearman rank correlation analysis. Statistical analyses were performed with the SPPS package version 20.0 (Statistical Product and Service Solutions, Chicago, IL, USA).
Results
Microglial phenotypes in classical pathological brain regions of AD and PD patients
Before studying microglial cell morphology in the OB, we examined microglial phenotypes in the SN of PD patients and in the HC of AD patients as activated microglial cells were reported to be present in these brain regions 47, 55. In the SN of control subjects, numerous neuromelanin‐pigmented, dopaminergic neurons were recognized by a dark discoloration around large neuronal nuclei while CD68‐positive microglia had long and fine processes, indicative of their ramified state (Figure 2A). In contrast, PD patients showed an extensive loss of neuromelanin‐containing, dopaminergic neurons in the SN (Figure 2B). Moreover, there was an increased appearance of CD68‐positive amoeboid microglia, the phenotype associated with microglial activation (Figure 2B). In the Ammon's horn of the HC of AD patients, similar results were obtained. Numerous and widespread microglial cells with ramified morphology were detected in control subjects, whereas in AD patients, additional clusters of amoeboid cells were seen, frequently localized around Aβ plaques (Figure 2C, D, respectively).
Figure 2.
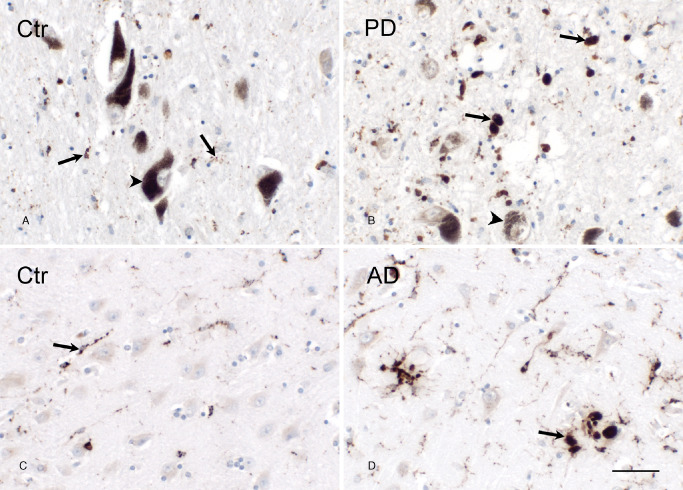
Appearance of amoeboid microglia in the SN of PD patients and in the HC of AD patients. A. Representative image of the SN of a control subject. Neuromelanin pigmented neurons were recognized by a dark coloring around large neuronal nuclei (arrowhead). CD68 positive ramified microglia (arrow) were observed randomly within the SN. B. Representative image of the SN of a PD patient. Degraded neuromelanin pigmented neurons were present in the SN (arrowhead). CD68 positive amoeboid microglia (arrow) appeared at the degenerative site. C. Representative image of CD68 immunoreactivity in the Ammon's horn of a control subject. CD68 positive ramified microglia (arrow) were detected. D. Representative image of the Ammon's horn of an AD patient. Numerous clustered CD68 positive amoeboid microglia (arrow) appeared; bar (A–D) = 50 μm.
HPtau‐, Aβ and α‐syn pathology in the OB
The presence of disease‐specific proteopathy was subsequently determined in the OB of PD and AD patients. Aberrant α‐syn (Figure 3A), Aβ (Figure 3C) and HPtau (Figure 3E) immunoreactivity were less present or absent in the AON of control subjects. In contrast, α‐syn staining was prominent in the AON of PD patients (Figure 3B), whereas in AD patients, Aβ and HPtau protein was abundantly expressed in the AON (Figure 3D, F, respectively). Furthermore, only a few AD and PD patients presented some α‐syn or Aβ pathology in the AON, respectively, whereas all PD patients showed HPtau pathology, although to a lesser extent than in AD patients.
Figure 3.
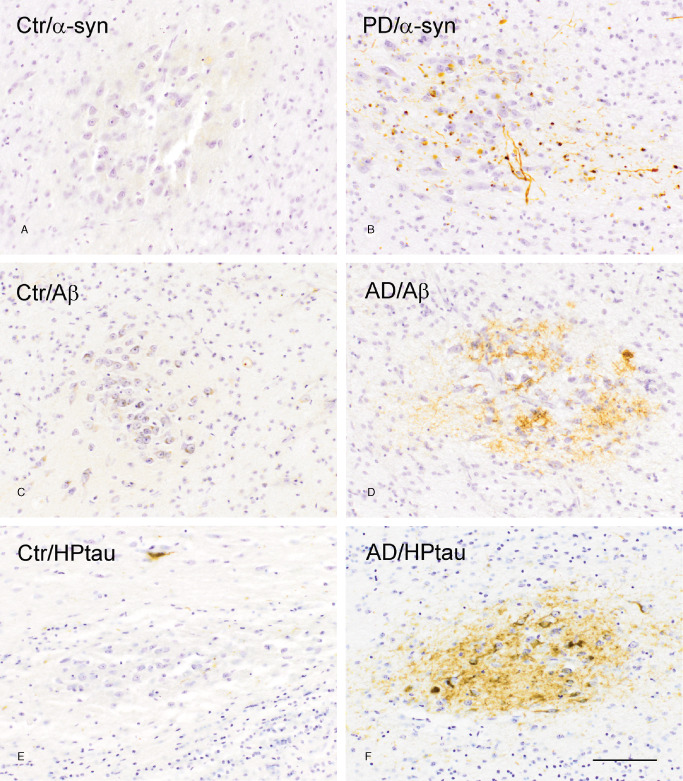
Protein pathology in the AON of control subjects, PD and AD patients. (A) α‐syn, (C) Aβ and (E) HPtau immunoreactivity in the AON of control subjects is minimal compared to (B) α‐syn immunoreactivity in the AON of PD patients, and (D) Aβ and (F) HPtau immunoreactivity in the AON of AD patients; bar (A–F) = 100 μm.
Microglial phenotypes in the AON
In the OB of control subjects and of PD and AD patients, CD68 positive microglia with a ramified morphology were present in various layers of the OB, and also in the AON (Figure 4A–C). In control subjects, the presence of amoeboid microglia was less prominent (Figure 4A) when compared to the increased appearance of microglia with an amoeboid morphology within the AON of PD (Figure 4B) and AD patients (Figure 4C) which was a consistent finding.
Figure 4.
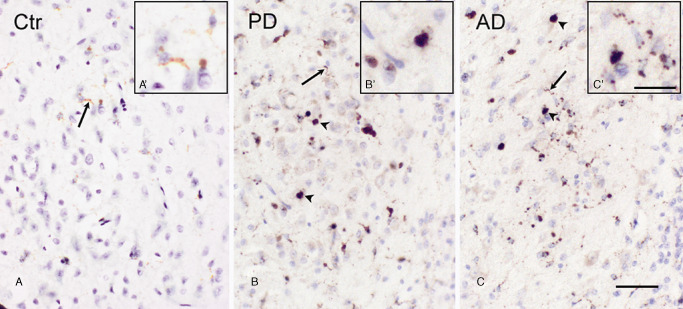
CD68 immunoreactivity in the AON of control subjects, PD patients and AD patients. A. a control subject showing CD‐68 positive ramified microglial cells (arrow), (A′) magnification of ramified microglial cells. B. A PD patient showing CD‐68 positive ramified (arrow) and amoeboid (arrowhead) microglial phenotypes, (B′) magnification of amoeboid microglial cell. C. An AD patient, showing CD‐68 positive ramified (arrow) and amoeboid (arrowhead) microglial phenotypes, (C′) magnification of amoeboid and ramified microglial cells; bar (A–C) = 50 μm, bar (A′–C′) = 25 μm.
Quantitative analysis of microglial cells and neurons in the AON
We used an unbiased random systematic sampling method to examine microglial and neuron densities in the AON of control subjects, PD and AD patients. Age, gender and PMD had no influence on any of the densities examined. The multivariate results of the MANCOVA indicated a significant effect of brain weight covariate (F(3,20) = 4.76, P =0.012). Nonetheless, a main effect for group was reported as well (F(6,38) = 4.04, P = 0.003). Subsequent univariate analysis revealed that a group effect was only significant for the number of amoeboid microglial cells per mm3 (F(2,22) = 10.86, P = 0.001; mean control subjects 6.65 × 103 ± 1.61, mean PD patients 25.54 × 103 ± 7.05, mean AD patients 39.26 × 103 ± 6.75). No significant differences were apparent for the number of ramified microglia per mm3 (F(2,22) = 1.89, P = 0.174; mean control subjects 28.79 × 103 ± 2.67, mean PD patients 24.04 × 103 ± 4.91, mean AD patients 19.42 × 103 ± 2.79). For the number of neurons per mm3, statistical significance was also not reached (F(2,22) = 2.01, P = 0.157; mean control subjects 72.06 × 103 ± 4.82, mean PD patients 66.79 × 103 ± 5.19, mean AD patients 61.28 × 103 ± 4.16). Compared to control subjects, increases in the density of amoeboid microglia were significant for both PD (P = 0.002) and AD patients (P = 0.003). These differences were quite prominent, with approximately four‐ to sixfold increases in amoeboid microglia densities for PD and AD patients, respectively (Figure 5).
Figure 5.
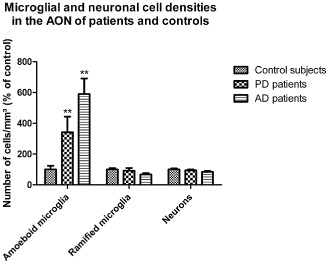
Quantification of cell densities within in the AON of control subjects, PD and AD patients. Quantification by a morphometric approach resulted in significant enhance densities of amoeboid microglia in the AON of PD and AD patients compared to control subjects (P = 0.002, P = 0.003, respectively). The densities of ramified microglia and neurons within the AON were not different between control subjects and PD or AD patients. Data represent mean ± SEM, **P < 0.01 (vs. control subjects).
Correlation between amoeboid microglia density and protein pathology scores in the AON
The local protein pathology in the AON of the patient groups was scored semi‐quantitatively (Table 4). These scores were correlated to the amoeboid microglia densities within the respective patient group. Aβ immunoreactivity scores in the AON of AD patients correlated positively with the density of amoeboid microglia (rho = 0.764, n = 8, P = 0.027*). Interestingly, HPtau immunoreactivity within the AON of AD patients showed a significant negative correlation with the density of amoeboid microglia (rho = −0.756, n = 8, P = 0.03*). We also correlated α‐syn pathology scores with amoeboid microglia within AD patients and found no correlation [rho = −0.261, n = 8, P = 0.053 not significant (NS)]. Within PD patients, α‐syn immunoreactivity scores in the AON showed no significant correlation with the density of amoeboid microglia (rho = 0.170, n = 7, P = 0.715 NS). HPtau and Aβ immunoreactivity scores within PD patients were taken into account but showed no correlation with amoeboid microglia (rho = 0.178, n = 7, P = 0.702 NS and rho = 0.894, n = 4, P = 0.106 NS, respectively)
No colocalization of CD68 or GFAP with pathological proteins
The presence of amoeboid microglia near pathological protein aggregates might reflect a role in phagocytosis of aberrant proteins. Using confocal microscopy, CD68 immunopositive microglia with an amoeboid morphology were found to surround α‐syn aggregates in the AON (Figure 6A) and SN (Figure 6B) of PD patients as well as Aβ and HPtau in the AON (Figure 6C, E, respectively) and HC (Figure 6D, F, respectively) of AD patients. Based upon these double‐labeling studies, neither colocalization between Aβ or HPtau and CD68 positive amoeboid microglia was observed in the AON of AD patients, nor did α‐syn colocalize with CD68 in PD patients. Similarly, colabeling with GFAP showed that the pathological proteins in the AON (Figure 7A) and SN (Figure 7B) of PD patients, and in the AON (Figure 7C, E) and HC (Figure 7D, F) of AD patients, were not colocalized with astrocytes.
Figure 6.
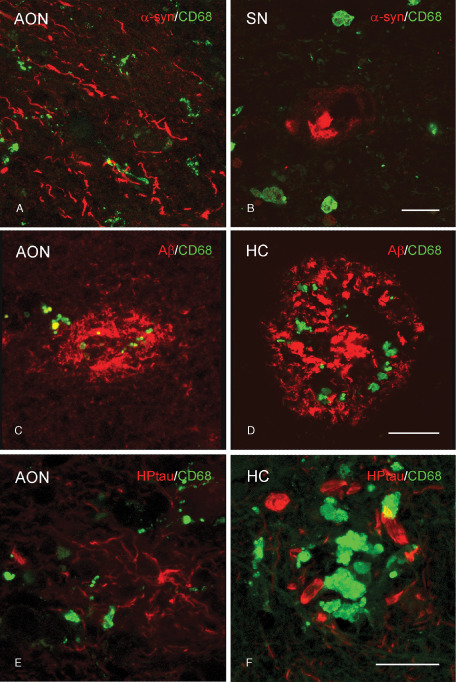
Absence of colocalization of CD68 positive microglia and protein aggregates in PD and AD patients. Representative images of confocal laser scanning microscopy revealed (A,B) no colocalization of CD68 (green) with α‐syn (red) in the (A) AON or (B) SN of PD patients. C,D. In AD patients, CD68 (green) colocalized with Aβ (red) in the (C) AON but not in the (D) HC. E,F. In AD patients no colocalization of CD68 (green) with HPtau (red) in the (E) AON or (F) HC was found; bar (A–F) = 20 μm.
Figure 7.
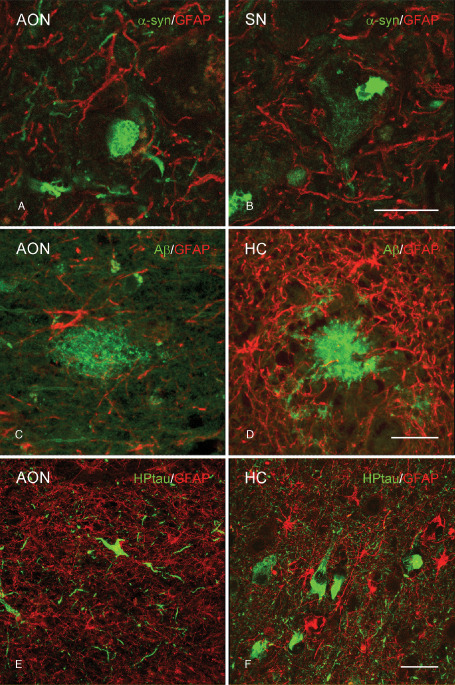
Absence of colocalization of GFAP positive astrocytes and protein aggregates in PD and AD patients. Representative images of confocal laser scanning microscopy revealed (A,B) no colocalization of GFAP (red) with α‐syn (green) in the (A) AON or (B) SN of PD patients. C,D. In AD patients, GFAP (red) did not colocalize with Aβ (green) in the (C) AON or (D) HC. E,F. In AD patients no colocalization of GFAP (red) and HPtau (green) in the (E) AON or (F) HC was observed; bar (A–D) = 20 μm, bar (E–F) = 40 μm.
Neurites in the AON
Within the AON, Bodian silver staining revealed normal appearing neurites in control subjects (Figure 8A) whereas in the AON of the AD and PD patients, a typical pathological pattern was found with morphologically altered neurites (Figure 8B, C).
Figure 8.
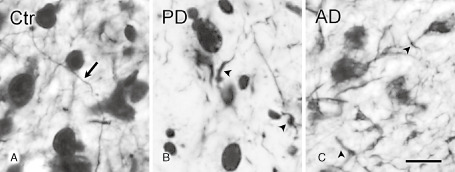
Bodian staining in the AON of control subjects, PD and AD patients. A. In the AON of control subjects, Bodian staining showed normal appearing neurites (arrow). B. In PD patients, and (C) in AD patients, Bodian staining showed morphologically altered neurites, and disconnected fiber parts (arrowhead); bar (A–C) = 20 μm.
Discussion
In the present study, we show the presence of amoeboid and ramified microglia phenotypes in the OB of PD and AD patients, a non‐traditional pathological site, which is affected early in both disorders. While a significant increase was found in the densities of amoeboid microglia in the AON in both disorders, indicative of an activated state of the cells, the absence of colocalization with pathological protein aggregates indicates that microglia in the AON do not exert extensive phagocytic activity toward these disease‐specific protein deposits. Although neurites appeared morphologically different in PD and AD patients, this was not paralleled by overt neuronal loss in the AON.
The accumulation of disease‐specific proteins in the AON of our verified PD and AD patient cohorts is consistent with previous studies demonstrating the presence of Aβ plaques and NFTs in the OB of AD patients, and of α‐syn inclusions in the OB of PD patients 8, 9, 50, 63, 75. The localization of these pathological proteins mainly within the AON suggests a selective vulnerability of this region that is functionally involved in the processing of olfactory information 61.
Neuroinflammatory processes, including microglial activation, have consistently been shown to play a role in neurodegeneration. While microgliosis had been observed in the OB of MPTP‐treated mice 37, 75, 80, it was still unknown whether microglial cells within the OB were altered in the human disorder as well, and which phenotypes were involved. Our state‐of‐the‐art quantification revealed a substantial and specific increase in amoeboid microglial density, suggesting that these cells are activated in the AON of PD and AD patients. As similar observations have been made in another non‐traditional pathological site in PD, the hippocampus 34, microglial activation may not be solely occurring in the nigrostriatal region, but may rather arise at several pathological sites in PD. Moreover, the density of amoeboid microglial cells in the AON of PD patients did not correlate with the extent of α‐syn pathology, suggesting that the increased microglial activation in this region is not proportional to the extent of α‐syn deposition, but rather that a local presence of α‐syn pathology is already sufficient to evoke microgliosis 71. Within other brain regions, this may be different 82 as in the SN of PD patients a positive correlation has been found before between microglial cell activation and α‐syn deposition 12. Hence, microglia may respond differentially to α‐syn pathology in the AON compared to the SN of PD patients because of differences in microglial cell subtypes 18, 24. The concomitant differences in local environment may have various consequences for neuronal functioning 18.
In AD patients, a significant positive correlation was observed between the density of amoeboid microglial cells and the level of Aβ deposits in the AON. These observations are in line with previous correlations reported between microglial activation and the severity of Aβ deposition in the HC 77. Unexpectedly, HPtau levels and amoeboid microglia density in the AON of AD patients showed a significant negative correlation. The level of HPtau was, at least in most of the individual AD patients, inversely related to the level of Aβ scores within the AON of the same patient. This “within patient effect” could explain the negative correlation between HPtau and amoeboid microglia.
The lack of a positive correlation between α‐syn or HPtau deposition and amoeboid microglial densities within the AON of PD or AD patients, respectively, suggests that irrespective of the level of these pathological proteins, microglial cells are affected, and become activated. This does not seem to hold for Aβ deposition that does correlate with amoeboid microglial density, and may be explained by the fact that Aβ is deposited outside the cell, and could thus act as a “dose‐dependent” stimulus for microglial activation, whereas α‐syn and HPtau are mostly intracellularly deposited 13, 65, 66, but can be externalized to have paracrine effects 28, 42, for example, activate local microglial cells.
Since a common hypothesis focuses on a predominantly detrimental role of activated microglia, we quantified the number of Nissl‐positive neurons within the AON but no differences were observed in the densities of large AON neurons between PD patients, AD patients and control subjects. In contrast to the extensive neurodegeneration in the SN of PD and HC of AD patients, our present data indicate that activated microglial cells in the AON are not related to any local neuronal loss. Although we cannot exclude a limited vulnerability of AON neurons 10, it is worth noting that the OB is part of the neurogenic pathway 14 and new‐born neurons residing in the OB might, at least in theory, compensate for PD and AD related neuronal degeneration 39, 46, 76. Moreover, the number of dopaminergic neurons is increased in the glomerular layer of the OB of PD patients, indeed suggesting that some compensatory mechanisms may occur in the AON 33, 50. As the Bodian silver staining did reveal a typical neuropathological pattern with altered neurite morphology, indicative of dystrophic neurites 5, 53 within the AON of these patients, it is tempting to speculate that these structural changes may affect AON functionality and could contribute to the olfactory deficits in PD and AD patients. This notion agrees with more general concepts proposing that network dysfunction rather than neuronal death per se, likely underlies several of the clinical manifestations in neurodegenerative diseases, including the cognitive decline in AD 49, 56. On the other hand, HPtau, Aβ and α‐syn deposits in the olfactory bulb can affect normal neurotransmitter release and hence cause disturbances in local information processing 50.
We also investigated whether amoeboid microglial cells are engaged in phagocytosis in the AON and SN of PD patients, and in the AON and HC of AD patients. Microglia are commonly seen as scavengers of the central nervous system (CNS), given their enhanced phagocytic properties upon activation in vitro 79. Indeed, several in vitro studies have presented evidence of phagocytosis of PD or AD related pathological proteins by microglial cells 29, 70. However, few studies have focused on pathological protein phagocytosis by microglia in post‐mortem PD or AD tissue. In our study, we could not observe clear immunohistochemical colocalization of microglia with pathological proteins in several affected brain regions. Although the interpretation of such data should be done with care, it suggests that microglia either do not readily phagocytose pathological proteins or ingested pathological proteins are quickly degraded within microglia 1. In addition, our data did not support the alternative option, that is, that phagocytosis is performed by astrocytes, that can also have scavenger properties under specific conditions 44, 84.
In agreement with little phagocytic activity of glial cells in PD and AD brain, it has been shown that pathological protein aggregates in an AD mouse model were also surrounded by microglia and astroglia, but were not internalized by these cells 67. In fact, in vitro studies suggest that isolated, and healthy, human microglia cells take up Aβ less readily in contrast to peripheral macrophages 20. In addition, macrophages derived from AD patients indeed have poorer Aβ phagocytic properties when compared to macrophages isolated from control subjects 21. Lately, attention has focused on the possibility that the altered morphology of microglial cells during aging or under neurodegenerative conditions is a consequence of microglial senescence rather than a reflection of their activational state 68. Using similar immunohistochemical techniques, the so‐called dystrophic microglial cells, rather than “activated” microglial cells, were found to have impaired phagocytic function 69. The concept of dystrophic microglia that develops due to ageing contributes to the idea that microgliosis per se does not cause neuronal death, but acts as a bystander in disease progression, as it may have lost its neuroprotective nature 52, 69.
An alternative explanation for the putative impairment in, or absence of, phagocytic capacity, is that effective phagocytosis by microglial and astroglial cells will depend on the aggregation status of the protein to be eliminated. This would imply that phagocytosis of large aggregated proteins, occurring in progressive or end stage of the disease and probably present in our patient material, is reduced, while truncated or monomeric forms can be cleared 36, 57, 74. This inability of microglia and astrocytes to take appropriate care of pathological protein aggregates 40 would then lead to progressive pathological protein accumulation within the CNS and could induce a feed‐forward loop by which further activation of microglia is induced. This may eventually promote more neuronal dysfunction and/or demise.
In summary, we have shown an increase in the density of amoeboid microglia within the AON of the OB, a region beyond the traditional sites of PD and AD neuropathology, occurring irrespective of the type of pathological protein deposited. Correlation analysis between amoeboid microglia density and level of pathological protein deposition within the AON suggests a protein‐dependent effect on the amount of activated microglial cells. This is not reflected at the level of phagocytosis since no clear colocalization of pathological proteins with amoeboid microglia or, alternatively, astrocytes was observed. Finally, although a causal relation needs to be proven, we hypothesize that it is not cell loss, but rather an increased density of amoeboid microglial cells, possibly in combination with neurite pathology, that may contribute to the functional deficits in the AON of PD and AD patients, such as hyposmia.
Disclosure Statement
The authors declare that they have no conflict of interest.
Acknowledgments
We thank Dr I. Huitinga of the Netherlands Brain Bank for the provision of human post‐mortem brain material and Michiel Kooreman for technical support. We gratefully acknowledge the patients and their families for the brain donations. The Stichting Internationaal Parkinson Fonds (IPF; KJD, PJL, A‐MvD), and Internationale Stichting Alzheimer Onderzoek (ISAO; KJD, PJL) are kindly acknowledged for financial support.
References
- 1. Akiyama H, Schwab C, Kondo H, Mori H, Kametani F, Ikeda K, McGeer PL (1996) Granules in glial cells of patients with Alzheimer's disease are immunopositive for C‐terminal sequences of β‐amyloid protein. Neurosci Lett 206:169–172. [DOI] [PubMed] [Google Scholar]
- 2. Attems J, Lintner F, Jellinger KA (2005) Olfactory involvement in aging and Alzheimer's disease: an autopsy study. J Alzheimers Dis 7:149–158. [DOI] [PubMed] [Google Scholar]
- 3. Berendse HW, Roos DS, Raijmakers P, Doty RL (2011) Motor and non‐motor correlates of olfactory dysfunction in Parkinson's disease. J Neurol Sci 310:21–24. [DOI] [PubMed] [Google Scholar]
- 4. Block ML, Hong JS (2005) Microglia and inflammation‐mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 76:77–98. [DOI] [PubMed] [Google Scholar]
- 5. Bodian D (1936) A new method for staining nerve fibers and nerve endings in mounted paraffin sections. Anat Rec 65:89–97. [Google Scholar]
- 6. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol (Berl) 82:239–259. [DOI] [PubMed] [Google Scholar]
- 7. Braak H, Del Tredici K, Bratzke H, Hamm‐Clement J, Sandmann‐Keil D, Rüb U (2002) Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson's disease (preclinical and clinical stages). J Neurol 249:1–5. [DOI] [PubMed] [Google Scholar]
- 8. Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197–211. [DOI] [PubMed] [Google Scholar]
- 9. Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K (2004) Stages in the development of Parkinson's disease‐related pathology. Cell Tissue Res 318:121–134. [DOI] [PubMed] [Google Scholar]
- 10. Braak H, Rüb U, Schultz C, Del Tredici K (2006) Vulnerability of cortical neurons to Alzheimer's and Parkinson's diseases. J Alzheimers Dis 9:35–44. [DOI] [PubMed] [Google Scholar]
- 11. Brunjes PC, Illig KR, Meyer EA (2005) A field guide to the anterior olfactory nucleus (cortex). Brain Res Rev 50:305–335. [DOI] [PubMed] [Google Scholar]
- 12. Croisier E, Moran LB, Dexter DT, Pearce RKB, Graeber MB (2005) Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha‐synuclein deposition. J Neuroinflammation 2:14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Crowther RA (1993) Tau protein and paired helical filaments of Alzheimer's disease. Curr Opin Struct Biol 3:202–206. [Google Scholar]
- 14. Curtis MA, Kam M, Nannmark U, Anderson MF, Axell MZ, Wikkelso C et al (2007) Human neuroblasts migrate to the olfactory bulb via a lateral ventricular extension. Science 315:1243–1249. [DOI] [PubMed] [Google Scholar]
- 15. Devanand DP, Michaels‐Marston KS, Liu X, Pelton GH, Padilla M, Marder K et al (2000) Olfactory deficits in patients with mild cognitive impairment predict Alzheimer's disease at follow‐up. Am J Psychiatry 157:1399–1405. [DOI] [PubMed] [Google Scholar]
- 16. DiPatre PL, Gelman BB (1997) Microglial cell activation in aging and Alzheimer disease: partial linkage with neurofibrillary tangle burden in the hippocampus. J Neuropathol Exp Neurol 56:143–149. [DOI] [PubMed] [Google Scholar]
- 17. Dodel RC, Lohmüller F, Du Y, Eastwood B, Gocke P, Oertel WH, Gasser T (2001) A polymorphism in the intronic region of the IL‐1alpha gene and the risk for Parkinson's disease. Neurology 56:982–983. [DOI] [PubMed] [Google Scholar]
- 18. Doorn KJ, Lucassen PJ, Boddeke HW, Prins M, Berendse HW, Drukarch B, Van Dam AM (2012) Emerging roles of microglial activation and non‐motor symptoms in Parkinson's disease. Prog Neurobiol 98:222–238. [DOI] [PubMed] [Google Scholar]
- 19. Doty RL, Hawkes CH, Berendse HW (2011) Olfactory dysfunction in Parkinson's disease and related disorders. In: Non‐Dopamine Lesions in Parkinson's Disease, Halliday GM, Barker RA, Rowe DB (eds), pp. 65–91. Oxford University Press Inc., New York. [Google Scholar]
- 20. Familian A, Eikelenboom P, Veerhuis R (2007) Minocycline does not affect amyloid‐β phagocytosis by human microglial cells. Neurosci Lett 416:87–91. [DOI] [PubMed] [Google Scholar]
- 21. Fiala M, Liu PT, Espinosa‐Jeffrey A, Rosenthal MJ, Bernard G, Ringman JM et al (2007) Innate immunity and transcription of MGAT‐III and Toll‐like receptors in Alzheimer's disease patients are improved by bisdemethoxycurcumin. Proc Natl Acad Sci USA 104:12849–12854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A et al (2006) In vivo imaging of microglial activation with [11C](R)‐PK11195 PET in idiopathic Parkinson's disease. Neurobiol Dis 21:404–412. [DOI] [PubMed] [Google Scholar]
- 23. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140:918–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Graeber MB, Streit WJ (2010) Microglia: biology and pathology. Acta Neuropathol (Berl) 119:89–105. [DOI] [PubMed] [Google Scholar]
- 25. Gregory GE, Greenway AR, Lord KA (1980) Alcoholic Bouin fixation of insect nervous systems for Bodian silver staining. I,II,III. Biotechnic & Histochemistry 55:143–165. [DOI] [PubMed] [Google Scholar]
- 26. Grimaldi LME, Casadei VM, Ferri C, Veglia F, Licastro F, Annoni G et al (2000) Association of early onset Alzheimer's disease with an interleukin(−alpha gene polymorphism. Ann Neurol 47:361–365. [PubMed] [Google Scholar]
- 27. Haehner A, Hummel T, Reichmann H (2011) Olfactory loss in Parkinson's disease. Park Dis Article ID 450939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hansen C, Angot E, Bergström AL, Steiner JA, Pieri L, Paul G et al (2011) Alpha‐synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest 121:715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hashioka S, Miklossy J, Schwab C, Klegeris A, McGeer PL (2008) Adhesion of exogenous human microglia and THP‐1 cells to amyloid plaques of postmortem Alzheimer's disease brain. J Alzheimers Dis 14:345–352. [DOI] [PubMed] [Google Scholar]
- 30. Hirsch EC, Vyas S, Hunot S (2012) Neuroinflammation in Parkinson's disease. Parkinsonism Relat Disord 18:210–212. [DOI] [PubMed] [Google Scholar]
- 31. Hjorth E, Frenkel D, Weiner H, Schultzberg M (2010) Effects of immunomodulatory substances on phagocytosis of Aβ142 by human microglia. International J Alzheimers Dis Article ID 798424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Holness CL, Simmons DL (1993) Molecular cloning of CD68, a human macrophage marker related to lysosomal glycoproteins. Blood 81:1607–1613. [PubMed] [Google Scholar]
- 33. Huisman E, Uylings H, Hoogland PV (2004) A 100% increase of dopaminergic cells in the olfactory bulb may explain hyposmia in Parkinson's disease. Mov Disord 19:687–692. [DOI] [PubMed] [Google Scholar]
- 34. Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y (2003) Distribution of major histocompatibility complex class II‐positive microglia and cytokine profile of Parkinson's disease brains. Acta Neuropathol (Berl) 106:518–526. [DOI] [PubMed] [Google Scholar]
- 35. Irwin DJ, White MT, Toledo JB, Xie SX, Robinson JL, Deerlin VV et al (2012) Neuropathologic substrates of Parkinson's disease dementia. Ann Neurol 72:587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Koenigsknecht J, Landreth G (2004) Microglial phagocytosis of fibrillar β‐amyloid through a 1 integrin‐dependent mechanism. J Neurosci 24:9838–9846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kovacs T, Cairns NJ, Lantos PL (1999) beta‐amyloid deposition and neurofibrillary tangle formation in the olfactory bulb in ageing and Alzheimer's disease. Neuropathol Appl Neurobiol 25:481–491. [DOI] [PubMed] [Google Scholar]
- 38. Kreutzberg GW (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19:312–318. [DOI] [PubMed] [Google Scholar]
- 39. Kuhn HG, Palmer TD, Fuchs E (2001) Adult neurogenesis: a compensatory mechanism for neuronal damage. Eur Arch Psychiatry Clin Neurosci 251:152–158. [DOI] [PubMed] [Google Scholar]
- 40. Lai AY, McLaurin JA (2012) Clearance of amyloid‐β peptides by microglia and macrophages: the issue of what, when and where. Future Neurol 7:165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lavelle A (1956) Nucleolar and Nissl substance development in nerve cells. J Comp Neurol 104:175–205. [DOI] [PubMed] [Google Scholar]
- 42. Lee HJ, Patel S, Lee SJ (2005) Intravesicular localization and exocytosis of α‐synuclein and its aggregates. J Neurosci 25:6016–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lei H, Mooney R, Katz LC (2006) Synaptic integration of olfactory information in mouse anterior olfactory nucleus. J Neurosci 26:12023–12032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lööv C, Hillered L, Ebendal T, Erlandsson A (2012) Engulfing astrocytes protect neurons from contact‐induced apoptosis following injury. PLoS One 7:e33090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VMY (2012) Pathological α‐synuclein transmission initiates Parkinson‐like neurodegeneration in nontransgenic mice. Science 338:949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marlatt MW, Lucassen PJ (2010) Neurogenesis and Alzheimer's disease: biology and pathophysiology in mice and men. Curr Alzheimer Res 7:113–125. [DOI] [PubMed] [Google Scholar]
- 47. McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988) Reactive microglia are positive for HLA−DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology 38:1285–1286. [DOI] [PubMed] [Google Scholar]
- 48. Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, Nagatsu T (1994) Interleukin‐1 beta, interleukin‐6, epidermal growth factor and transforming growth factor‐alpha are elevated in the brain from parkinsonian patients. Neurosci Lett 180:147–150. [DOI] [PubMed] [Google Scholar]
- 49. Mucke L, Selkoe DJ (2012) Neurotoxicity of amyloid β protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med 2: a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mundiñano IC, Caballero MC, Ordóñez C, Hernandez M, DiCaudo C, Marcilla I et al (2011) Increased dopaminergic cells and protein aggregates in the olfactory bulb of patients with neurodegenerative disorders. Acta Neuropathol (Berl) 122:61–74. [DOI] [PubMed] [Google Scholar]
- 51. Murphy C, Gilmore MM, Seery CS, Salmon DP, Lasker BR (1990) Olfactory thresholds are associated with degree of dementia in Alzheimer's disease. Neurobiol Aging 11:465–469. [DOI] [PubMed] [Google Scholar]
- 52. Njie MG, Boelen E, Stassen FR, Steinbusch HWM, Borchelt DR, Streit WJ (2010) Ex vivo cultures of microglia from young and aged rodent brain reveal age‐related changes in microglial function. Neurobiol Aging 33:195.1–195.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Oide T, Kinoshita T, Arima K (2006) Regression stage senile plaques in the natural course of Alzheimer's disease. Neuropathol Appl Neurobiol 32:539–556. [DOI] [PubMed] [Google Scholar]
- 54. Ouchi Y, Yagi S, Yokokura M, Sakamoto M (2009) Neuroinflammation in the living brain of Parkinson's disease. Parkinsonism Relat Disord 15:200–204. [DOI] [PubMed] [Google Scholar]
- 55. Overmyer M, Helisalmi S, Soininen H, Laakso M, Riekkinen P Sr, Alafuzoff I (1999) Reactive microglia in aging and dementia: an immunohistochemical study of postmortem human brain tissue. Acta Neuropathol (Berl) 97:383–392. [DOI] [PubMed] [Google Scholar]
- 56. Palop JJ, Chin J, Mucke L (2006) A network dysfunction perspective on neurodegenerative diseases. Nature 443:768–773. [DOI] [PubMed] [Google Scholar]
- 57. Park JY, Paik SR, Jou I, Park SM (2008) Microglial phagocytosis is enhanced by monomeric α‐synuclein, not aggregated α‐synuclein: implications for Parkinson's disease. Glia 56:1215–1223. [DOI] [PubMed] [Google Scholar]
- 58. Perlmutter LS, Scott SA, Barron E, Chui HC (1992) MHC class II positive microglia in human brain: association with Alzheimer lesions. J Neurosci Res 33:549–558. [DOI] [PubMed] [Google Scholar]
- 59. Pletnikova O, West N, Lee MK, Rudow GL, Skolasky RL, Dawson TM et al (2005) Aβ deposition is associated with enhanced cortical α‐synuclein lesions in Lewy body diseases. Neurobiol Aging 26:1183–1192. [DOI] [PubMed] [Google Scholar]
- 60. Ponsen MM, Stoffers D, Booij J, van Eck Smit BLF, Wolters EC, Berendse HW (2004) Idiopathic hyposmia as a preclinical sign of Parkinson's disease. Ann Neurol 56:173–181. [DOI] [PubMed] [Google Scholar]
- 61. Price JL (1990) Olfactory system. In: The Human Nervous System. Paxinos G, Mai JK (eds.), pp. 979–998. Academic Press: San Diego. [Google Scholar]
- 62. Reynolds AD, Glanzer JG, Kadiu I, Ricardo Dukelow M, Chaudhuri A, Ciborowski P et al (2008) Nitrated alpha synuclein activated microglial profiling for Parkinson's disease. J Neurochem 104:1504–1525. [DOI] [PubMed] [Google Scholar]
- 63. Saiz‐Sanchez D, Ubeda‐Banon I, de la Rosa‐Prieto C, Argandona‐Palacios L, Garcia‐Munozguren S, Insausti R, Martinez‐Marcos A (2010) Somatostatin, tau, and β‐amyloid within the anterior olfactory nucleus in Alzheimer disease. Exp Neurol 223:347–350. [DOI] [PubMed] [Google Scholar]
- 64. Sasaki A, Kawarabayashi T, Murakami T, Matsubara E, Ikeda M, Hagiwara H et al (2008) Microglial activation in brain lesions with tau deposits: comparison of human tauopathies and tau transgenic mice TgTauP301L. Brain Res 1214:159–168. [DOI] [PubMed] [Google Scholar]
- 65. Selkoe DJ (1991) The molecular pathology of Alzheimer's disease. Neuron 6:487–498. [DOI] [PubMed] [Google Scholar]
- 66. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) Alpha‐synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 95:6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Stalder M, Deller T, Staufenbiel M, Jucker M (2001) 3D‐Reconstruction of microglia and amyloid in APP23 transgenic mice: no evidence of intracellular amyloid. Neurobiol Aging 22:427–434. [DOI] [PubMed] [Google Scholar]
- 68. Streit WJ, Sammons NW, Kuhns AJ, Sparks DL (2004) Dystrophic microglia in the aging human brain. Glia 45:208–212. [DOI] [PubMed] [Google Scholar]
- 69. Streit WJ, Braak H, Xue QS, Bechmann I (2009) Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol (Berl) 118:475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Strohmeyer R, Kovelowski C, Mastroeni D, Leonard B, Grover A, Rogers J (2005) Microglial responses to amyloid β peptide opsonization and indomethacin treatment. J Neuroinflammation 2:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Su X, Maguire‐Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ (2008) Synuclein activates microglia in a model of Parkinson's disease. Neurobiol Aging 29:1690–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Su X, Federoff HJ, Maguire‐Zeiss KA (2009) Mutant alpha‐synuclein overexpression mediates early proinflammatory activity. Neurotox Res 16:238–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tan J, Town T, Paris D, Mori T, Suo Z, Crawford F et al (1999) Microglial activation resulting from CD40‐CD40L interaction after β‐amyloid stimulation. Science 286:2352–2355. [DOI] [PubMed] [Google Scholar]
- 74. Thal DR, Schultz C, Dehghani F, Yamaguchi H, Braak H, Braak E (2000) Amyloid β‐protein (A+β)‐containing astrocytes are located preferentially near N‐terminal‐truncated A+β deposits in the human entorhinal cortex. Acta Neuropathol (Berl) 100:608–617. [DOI] [PubMed] [Google Scholar]
- 75. Tsuboi Y, Wszolek ZK, Graff‐Radford NR, Cookson N, Dickson DW (2003) Tau pathology in the olfactory bulb correlates with Braak stage, Lewy body pathology and apolipoprotein ε4. Neuropathol Appl Neurobiol 29:503–510. [DOI] [PubMed] [Google Scholar]
- 76. van den Berge SA, van Strien ME, Korecka JA, Dijkstra AA, Sluijs JA, Kooijman L et al (2011) The proliferative capacity of the subventricular zone is maintained in the parkinsonian brain. Brain 134:3249–3263. [DOI] [PubMed] [Google Scholar]
- 77. Van Everbroeck B, Dobbeleir I, De Waele M, De Leenheir E, Lübke U, Martin JJ, Cras P (2004) Extracellular protein deposition correlates with glial activation and oxidative stress in Creutzfeldt‐Jakob and Alzheimer's disease. Acta Neuropathol (Berl) 108:194–200. [DOI] [PubMed] [Google Scholar]
- 78. Versijpt JJ, Dumont F, Van Laere KJ, Decoo D, Santens P, Audenaert K et al (2003) Assessment of neuroinflammation and microglial activation in Alzheimer's disease with radiolabelled PK11195 and single photon emission computed tomography. Eur Neurol 50:39–47. [DOI] [PubMed] [Google Scholar]
- 79. Vilhardt F (2005) Microglia: phagocyte and glia cell. Int J Biochem Cell Biol 37:17–21. [DOI] [PubMed] [Google Scholar]
- 80. Vroon A, Drukarch B, Bol JGJM, Cras P, Brevé JJP, Allan SM et al (2007) Neuroinflammation in Parkinson's patients and MPTP‐treated mice is not restricted to the nigrostriatal system: microgliosis and differential expression of interleukin‐1 receptors in the olfactory bulb. Exp Gerontol 42:762–771. [DOI] [PubMed] [Google Scholar]
- 81. Walter L, Neumann H (2009) Role of microglia in neuronal degeneration and regeneration. Semin Immunopathol 31:513–525. [DOI] [PubMed] [Google Scholar]
- 82. Watson MB, Richter F, Lee SK, Gabby L, Wu J, Masliah E et al (2012) Regionally‐specific microglial activation in young mice over‐expressing human wildtype alpha‐synuclein. Exp Neurol 237:318–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. West MJ, Slomianka L, Gundersen HJG (1991) Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec 231:482–497. [DOI] [PubMed] [Google Scholar]
- 84. Wyss‐Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F et al (2003) Adult mouse astrocytes degrade amyloid‐β in vitro and in situ. Nat Med 9:453–457. [DOI] [PubMed] [Google Scholar]
- 85. Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML et al (2005) Aggregated alpha‐synuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB J 19:533–542. [DOI] [PubMed] [Google Scholar]