

Abstract
Maturation of the auditory pathway is dependent on the central nervous system myelination and it can be affected by pathologies such as neonatal hypoxic ischemic (HI) encephalopathy. Our aim was to evaluate the functional integrity of the auditory pathway and to visualize, by histological and cellular methods, the damage to the brainstem using a neonatal rat model of HI brain injury. To carry out this morphofunctional evaluation, we studied the effects of the administration of the antioxidants nicotine, melatonin, resveratrol and docosahexaenoic acid after hypoxia‐ischemia on the inferior colliculus and the auditory pathway. We found that the integrity of the auditory pathway in the brainstem was altered as a consequence of the HI insult. Thus, the auditory brainstem response (ABR) showed increased I–V and III–V wave latencies. At a histological level, HI altered the morphology of the inferior colliculus neurons, astrocytes and oligodendricytes, and at a molecular level, the mitochondria membrane potential and integrity was altered during the first hours after the HI and reactive oxygen species (ROS) activity is increased 12 h after the injury in the brainstem. Following antioxidant treatment, ABR interpeak latency intervals were restored and the body and brain weight was recovered as well as the morphology of the inferior colliculus that was similar to the control group. Our results support the hypothesis that antioxidant treatments have a protective effect on the functional changes of the auditory pathway and on the morphological damage which occurs after HI insult.
Keywords: auditory brainstem response (ABR), auditory pathway, hypoxic ischemic injury, inferior colliculus, morphological brainstem damage, neuronal injury, rat, white matter injury
Introduction
Despite improvements in neonatology, perinatal hypoxic ischemic (HI) encephalopathy remains one of the main causes of disabilities in term‐born infants 54. This specific pathology underlies many neurological disorders such as learning difficulties, language and attention deficit, hyperactivity disorders and cerebral palsy 69. Moreover, it is also a notable risk factor for hearing impairments that affect neonates 12, 48.
The auditory brainstem response (ABR) has been shown to be very sensitive to low blood oxygen concentrations, which result in damage to the neurosensory cells of the Organ of Corti or loss of brainstem neurons, such as those of cochlear nuclei or the inferior colliculus (IC) 18, 30. This neuronal damage compromises nerve conduction in the brain, leading to acute auditory impairment 29. ABRs are the most sensitive and reliable methods used to evaluate such functional impairments 61, 66. In particular, the two most widely used parameters are wave V latency and its I–V interval because they intimately reflect neuronal conduction, brainstem conduction time, myelination and synaptic function, both in the brainstem and in the central auditory pathway 31.
Following a perinatal asphyxia event, ABR abnormalities have been reported, including an elevated response threshold, an increase in wave latencies, brainstem conduction time and interpeak intervals; reduced wave amplitudes and V/I amplitude ratio 22, 34, 75. These electrophysiological findings have been correlated with histopathological observations, which indicated that perinatal asphyxia often causes discrete lesions in brainstem auditory nuclei 65.
The effects of HI injury are caused by diverse mechanisms that ultimately produce cell damage 19. Thus, neural cells are particularly susceptible to energy failure (decreased ATP levels), cellular excitotoxicity and oxidative stress, which, in turn, can promote cell death 4, 68 and in this sense, death receptor‐activated pathways, altered mitochondrial function 50. HI events are also known to perturb neuron metabolism and depress synaptic function between cells 31. Although astrocytes can also be affected by HI, which can compromise their capacity for neurotransmitter uptake, they are more resistant to HI events than neurons 4, 13. In contrast, oligodendrocytes are particularly vulnerable to perinatal asphyxia which affects myelination, leading to white matter lesions and damaging gray matter oligodendrocyte progenitors 60. These cells are responsible for myelin production. Thus, reduced expression of myelin basic protein (MBP), the major myelin protein, is considered a hallmark of inflammation‐associated diffuse white matter damage in fetal rodents 70, 72 and in preterm infants 26.
One of the key events in HI pathogenesis is the early generation of reactive oxygen species (ROS) 42. Increased ROS production leads to lipid and protein oxidation, loss of endogenous antioxidants and damage to neurons 37, 46. Consequently, supplementation or treatment with antioxidants has been proposed to be neuroprotective and may be an appropriate target area for novel therapies 47, 49.
There are many possible candidates for antioxidant therapies. Here, we have chosen four agents that have shown good protective effects in brain damage after HI, but their effects on auditory impairments after HI injury are unknown. Nicotine (N) exerts its effects via specific nicotinic acetylcholine receptors (nAChRs), the stimulation of which has various effects, including antioxidant functions. In addition, nicotine can inhibit glutamate neurotoxicity and increase the expression of BCL‐2 and other antiapoptotic proteins. Melatonin (M) (N‐acetyl‐5‐methoxytryptamine) is an endogenous indolamine generated primarily by the pineal gland and released into the blood stream and cerebrospinal fluid. It exerts a wide range of physiological functions including the removal of free radicals 64 and the inhibition of the oxidation of biomolecules 56. Resveratrol (RV) (3,5,4′‐trihydroxystilbene) is a non‐flavonoid polyphenolic compound consisting of two aromatic rings attached by a methylene bridge. It is produced by 72 different plant species, including grapevines, pines, legumes, peanuts, soybeans and pomegranates, but the most common dietary source of resveratrol is red wine 44. The neuroprotective effects of RV are caused by its antioxidant activity associated with its stilbene structure with two phenol rings 45. DHA (docosahexaenoic acid) (22:6n‐3) is a long‐chain omega‐3 fatty acid, commonly found in fish such as salmon and tuna. DHA provides plasma membrane fluidity at synaptic regions, so it is crucial for maintaining membrane integrity and consequently, neuronal excitability and synaptic function 15, 76. A DHA‐enriched diet during pregnancy has been shown to provide neuroprotection against neonatal brain injury by inhibiting oxidative stress 62.
The aim of the present work was to evaluate morphofunctionally the effect of a panel of antioxidants on HI‐induced auditory deficits. To this end, we studied the effects of nicotine, melatonin, resveratrol and DHA on the neonatal auditory system via measurement of auditory‐evoked potentials and characterization of the morphological integrity of the IC.
Materials and Methods
Subjects
Seven‐day‐old Sprague‐Dawley rats were used for histological and functional studies that were carried out in compliance with the animal research regulations specified in the European Communities Directive [2010/63/EU] and were approved by The Basque Country University Animal Care and Use Committee. Meanwhile, flow cytometer experiments were studied at different points of time after the HI insult (3, 12 and 24 h).
Hypoxia ischemia
The HI event was induced in perinatal rat pups by the Rice–Vannucci method 58. Briefly, Sprague‐Dawley rat pups were anesthetized with isoflurane (3.5% for induction and 1.5% for maintenance) in oxygen. The left common carotid artery was permanently ligated with 6–9 surgical silk and cauterized to block blood flow through the carotid. Animals were returned to their mother for 2 h to recover from anesthesia and then placed in a bath at 36°C during the hypoxic event. They were locked in a glass bottle and were subjected to 8% oxygen in a nitrogen gas mixture for 135 minutes. In order to verify the occurrence of brain hypoxia, some of the animals were injected with Hypoxyprobe™‐1 just before the hypoxic event and were sacrificed immediately after the hypoxic stress. The rest of the animals were returned to their biological mothers until they were 14 days old. Following measurement of the ABR, they were euthanized.
Experimental groups
Pups were randomly assigned to six experimental groups (n = 8): control, HI and the four HI groups administered with the different drugs (Table 1). Drugs were intraperitoneally administered, and nicotine, resveratrol and DHA were administered before the hypoxic event while melatonin was administered 10 minutes after the hypoxic event (Table 1).
Table 1.
Administered drugs with dose, dilution, administration time, mode and commercial house from each one. Each drug was administered intraperitoneally and was purchased from Sigma Aldrich Co., St. Louise, MO, USA. Abbreviations: DMSO = dimethyl sulfoxide; HSA = human serum albumin
| Drug | Dose (mg/kg) | Solvent | Administration time |
|---|---|---|---|
| Nicotine hydrogen tartrate | 1.2 14 | Saline | 2 h before hypoxia 14 |
| Melatonin | 15 9 | Saline and DMSO 5% | 10 min after hypoxia 3, 9, 11. |
| Resveratrol | 20 74 | DMSO | 10 min before hypoxia 25, 74 |
| Docosahexaenoic acid | 1 7 | HSA 25% diluted in normal saline | 10 min before hypoxia 51 |
The time intervals of treatment injections pre‐ and post‐HI and treatment doses were selected because of the existing good results for rat data on other brain areas after a perinatal asphyxia. DHA was previously described as neuroprotective antioxidant when administered 1 or 3h before the HI event 51, resveratrol when given 10–15 minutes before 25, 74, and nicotine 2 h before the damage 14. Meanwhile, melatonin is neuroprotective when administered just after the damage 3, 9, 11.
ABR measurements
Animals were anesthetized with ketamin‐xylacin (80 and 10 mg/kg, respectively) to measure the auditory‐evoked potentials. GSI Audera equipment with software version 1.0.3.4 was used to record the ABR. Measurements were performed in a sound‐proofed room to ensure minimization of background noise. ABR latencies and amplitudes were recorded using three gold‐plated disk electrodes placed at the middle forehead (positive), ipsilateral earlobe (negative) and middle body (ground), respectively.
Measurement acquisition was calibrated with the following parameters: sweep time of 10 ms with 150 and 3000 Hz filters for low and high frequencies, respectively. Stimulation was performed by means of 11.1/s clicks ABR and averages were taken of 2006 responses. We used continuous clicks at intensities of 100 dB. Repeated ABRs were recorded for both ears. The parameters analyzed were peak amplitudes (μV), peak latencies (ms) and intervals between peaks (ms).
Tissue processing for histological study
After ABR recordings, animals were weighed, sacrificed with sodium pentobarbital overdose and perfused with saline‐heparin followed by 4% formaldehyde prepared with fresh paraformaldehyde. The brain was removed and weighed before being fixed in 4% formaldehyde overnight. The brainstem was isolated, dehydrated and embedded in paraffin and then sectioned with a microtome (5 μm) at stereotaxic standard level of bregma 9.30 mm, at the level of the mesencephalon interaural 0.30 mm 52. Sections were collected on polylysine‐coated slides and processed for hematoxylin–eosin staining. The same procedure was carried out for Hypoxyprobe™‐1 staining, but P7 pups were used instead of using P14 animals in this case. For MBP expression analysis, at least three sections per brain were assayed for the central nuclei of the inferior colliculus (CIC) and three more sections per brain for the external cortex of the inferior colliculus (ECIC) 63.
Assessment of brain damage
To quantify cell damage in the IC with the hypoxia marker staining, astrogliosis or white matter injury assessments, we only used samples with a reduced infarct area or without it. To represent the infarct area that occurs in some animals, we used the hematoxylin–eosin staining.
Hypoxia marker
To evaluate the existence of hypoxia in the brain, we used the hypoxia marker Hypoxyprobe™‐1 (Hypoxyprobe Inc., Burlington MA, USA). Hypoxyprobe™‐1 (60 mg/kg) was intraperitoneally injected immediately after the HI event. Hypoxyprobe™‐1 distributes to all tissues including the brain but it only binds thiol‐containing proteins in those cells which have an oxygen concentration less than 14 μm. After blocking endogenous peroxidase with H2O2 (3%), dewaxed and rehydrated sections were incubated with Hypoxyprobe™‐1 MAb1 mouse primary antibody (1/50) for 1 h. Following abundant washing in saline, sections were indicated for 10 minutes with peroxidase‐conjugated anti‐mouse antiserum (1/500). The presence of bound primary antibody was manifest with diaminobenzidine and sections were finally weakly counterstained with hematoxylin.
Neuronal injury
The neuronal injury was evaluated in sections stained with hematoxylin–eosin (Sigma‐Aldrich Co., St. Louis, MO, USA).
Astrogliosis
Astrogliosis was evaluated using glial fibrillary acidic protein (GFAP) immunohistochemistry. Sections were rehydrated and blocked with endogenous peroxidase (1%), incubated with mouse anti‐GFAP primary antibody (1:500, Dako, Glostrup, Denmark) overnight and then with peroxidase‐labeled second antibody (HRP anti‐mouse 1:100, Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 1 h. Sections were stained with diaminobenzidine and counterstained with hematoxylin.
White matter injury assessment
For MBP immunohistochemistry, sections were incubated with mouse primary anti‐MBP antibody (1:200, Santa Cruz Biotechnology) overnight and then with peroxidase‐labeled second antibody (HRP anti‐mouse 1:100, Santa Cruz Biotechnology) for 1 h. The sections were stained with diaminobenzidine and counterstained with hematoxylin. White matter injury was analyzed by densitometry of MBP immunostaining using a computerized video‐camera‐based image analysis system (National Institutes of Health Image software, public domain, http://rsb.info.nih.gov/nih‐image/) as described by Liu et al 43. Unaltered TIFF images were digitized, segmented (using a consistent arbitrary threshold −50%) and binarized (black vs. white). Total black pixels per hemisphere were counted, and average values were calculated per brain, and expressed as pixels per hemisphere. Hemisphere areas were also outlined and measured for each section that was analyzed by densitometry. At least eight sections per brain were analyzed and only sections with obvious technical artifacts related to the staining procedure were excluded. Densitometry values were expressed as the ratio of left‐to‐right hemispheric measurements; for each brain sample (left hemisphere /right hemisphere), pixels of MBP were calculated.
Flow cytometry analysis
For the flow cytometry analysis, animals were sacrificed with pentobarbital sodium overdose and perfused with Ringer lactate solution. Ipsilateral brainstem sections were isolated and disaggregated with a blade in a lactate solution always kept in ice cold and then samples were placed in a cell strainer with 4 mL of collagenase [1.5 mg/mL diluted in Hank's (HBSS); Sigma‐Aldrich, St. Louis, MO, USA] in each dish and incubated at 37°C for 20 min. Cell suspension was washed with Hank's in a centrifugation at 1640 g for 5 minutes, and after removing the supernatant, the pellet was suspended in 5 mL of Hank's. Then, cell suspensions (600–1000 μL) were incubated with different fluorochromes. The samples were incubated for 30 minutes at 37°C and after washing twice with centrifugation for 5 minutes at 1640 g in Hank's solution, pellet suspended in 350 μL of Hank's were taken to the cytometry. Analyses were determined by an EPICS ELITE Flow Cytometry (Colter, Inc., Miami, FL, USA). To exclude debris and cellular aggregates, samples were gated based on light scattering properties, in side scattering (SSC), which correlates with cell complexity, and forward scattering (FSC), which correlates with cell size, and 10 000 events per sample within a gate (R1) were collected. Events within R1, which corresponded to individual cells, were plotted for their fluorescence. An unstained sample was used as a control to remove autofluorescence. Data analysis was performed using the Summit v4.3 software and statistical analyses with GraphPad prism 5 software.
To study the membrane integrity and potential, the level of cardiolipin was observed by using the fluorochrome nonyl acridine orange (NAO, Invitrogen, Leek, The Netherlands) and the level of Rhodamine 123 (Rh‐123, Invitrogen) a lipophilic cationic fluorochrome. In the first marker, cell suspensions were incubated with NAO (10−2 M) in phosphate buffered saline (PBS) at 4°C and in the dark conditions for 30 minutes and later cells were washed twice in buffer before loading to the flow cytometry. For Rhodamine 123 study, cell suspension was incubated with Rh123 (4 μL) in 100 μL of Hanks' solution for 30 minutes at 37°C, followed by washing and incubated for 30 minutes more at 37°C before taking to the flow cytometry for the analysis.
Intracellular ROS were detected using fluorochrome 2′,7′‐dichlorofluorescein diacetate (DCFH‐DA; Invitrogen). Cell suspension was incubated with DCFH‐DA fluorochrome (10 μM) in HBSS for 30 minutes at 37 C and taken directly to the flow cytometry for the analysis.
Statistical analysis
Values are represented as means ± SD. Group differences were studied by one‐factor analysis of variance with Bonferroni–Dunn correction. The statistical analysis of data was performed using GraphPad prism 5 software version 5.01 (GraphPad Software, Inc., San Diego, CA, USA).
Results
Alterations in ABR
Wave amplitudes
The amplitudes of the peaks were different in most of the animals and the standard deviation was very high in the groups to compare them, so we did not consider them for analysis.
Wave latencies
In the pups with neonatal HI, the latency of waves I, II and III (0.36 ms ± 0.02; 0.99 ms ± 0.04; 1.84 ms ± 0.14, respectively) was similar to that of the control group. In contrast, wave IV and V latencies in the HI animals were significantly longer than in control animals, from 2.28 ms ± 0.06 to 2.32 ms ± 0.09 and from 2.75 ms ± 0.08 to 3.04 ms ± 0.11, respectively (Figure 1B). All antioxidant‐treated groups showed wave latencies that were similar to those of the control group with slight differences, which were not statistically significant (Figure 1A,B). As there were no differences between the left and right ears, we evaluated the data together.
Figure 1.
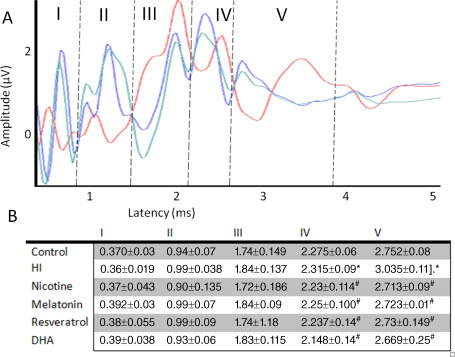
Typical auditory brainstem response (ABR) recording of 14‐day‐old rats (n = 8). A. Representative ABR responses of control (blue), hypoxia‐ischemia (HI) (red) and treated rats (green) (n = 8 in each group). A representative rat treated with melatonin was use for the illustration. B. The results of the ABR expressed as mean ± standard deviation in all experimental groups. *P < 0.05 vs. control and #P < 0.05 vs. HI, ANOVA.
Interpeak intervals
The HI event induced a statistically significant decline in ABR latency intervals (Figure 2) in the III–V and in the I–V intervals between the peaks, while there were no statistical differences between the treatments and control groups. In the I–III interval, there were no statistical differences between the peaks (mean latency; 1.4 ms in all groups). In contrast, wave V latency after HI tended to be slightly longer than in the control or treated groups and a statistically significant enhancement of the latency interval was observed in III–V intervals with a 35% decrease in the interpeak interval of the latency in almost all treated groups compared with the HI group. In the I–V interpeak interval, a 14% decrease was measured in the treated groups compared with the HI group.
Figure 2.
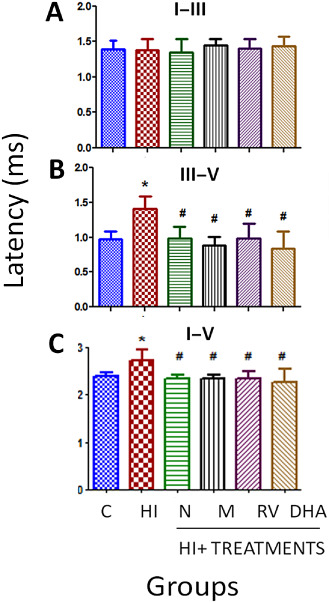
Interpeak latency of ABRs in 14‐day‐old control, HI and treated rats. Data are expressed as means ± SD (*P < 0.05 vs. controls and #P < 0.05 vs. HI, ANOVA). Abbreviations: C = control; HI = hypoxia‐ischemia; N = nicotine; M = melatonin; RV = resveratrol; DHA = docosahexaenoic acid.
Body weight
We compared the body weight of each animal in each group, and no differences were found among the different groups at postnatal day 7 (P7). However, after experiments (day 14), differences in body weight were observed (P < 0.05) (Figure 3). The HI group showed a 14% loss of body weight during these days, (29.13 g ± 2.2 vs. 25.19 g ± 1.34, control and HI groups, respectively), while all antioxidant‐treated groups showed body weights similar to the control group.
Figure 3.
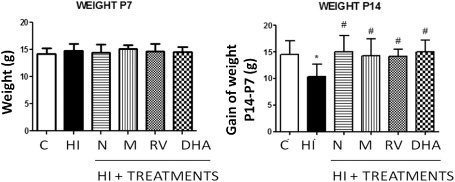
Body weight changes in 7‐ and 14‐day‐old neonatal rats (n = 8 for each group). A. Body weight (in g) of P7 animals before experimental intervention. B. Body weight of 14‐day‐old rats after hypoxia‐ischemia and recovery following antioxidant treatment. Body weight loss caused by hypoxia‐ischemia was blocked by all antioxidant treatments. Bar represents mean ± standard deviation. *P < 0.05 vs. control; #P < 0.05 vs. HI, ANOVA. Abbreviations: C = control; HI = hypoxia‐ischemia; N = nicotine; M = melatonin; RV = resveratrol; DHA = docosahexaenoic acid.
Assessment of brain damage
We also weighed brains after the sacrifice of the animals. There were statistical differences between the experimental groups (P < 0.05) (Figure 4) in that we found a 28% brain weight reduction in the HI group compared with the control group (14.49 g ± 2.54 vs. 10.40 g ± 2.25), while antioxidant treatment with nicotine, melatonin and resveratrol abolished this decrease. Treatment with DHA had a similar effect, but the difference was not significant.
Figure 4.
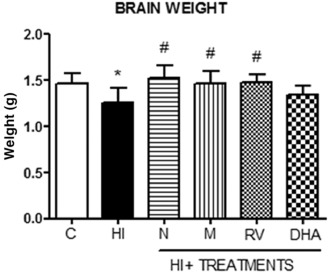
Brain weight of 14‐day‐old animals after hypoxia‐ischemia and recovery following administration of antioxidants (n = 8 for each group). Loss in brain weight caused by hypoxia‐ischemia was prevented by the different antioxidant treatments, with the exception of DHA. Bars represent mean ± standard deviation. *P < 0.05 vs. control; #P < 0.05 vs. HI, ANOVA. Abbreviations: C = control; HI = hypoxia‐ischemia; N = nicotine; M = melatonin; RV = resveratrol; DHA = docosahexaenoic acid.
We examined the presence of tissue hypoxia in samples stained with the immunohistochemical hypoxia marker Hypoxyprobe™‐1 (Figure 5). Examination of the histological sections at the midbrain levels 7 days after left carotid ligation and 2 h and 15 minutes of hypoxia revealed the absence of Hypoxyprobe™‐1 labeling in control midbrain sections (Figure 5A). However, intense hypoxia labeling was apparent in the HI group, especially in the ipsilateral side where morphological damage to the ischemic event is obvious (Figure 5B). In the treated groups, Hypoxyprobe™‐1‐labeled cells could also be observed but no morphological damage was apparent (Figure 5C). The image of the treated group (image of HI + nicotine rat) is a representative image for all of treatments. In more augmented images, reactive cells are observed in HI and treated groups while there is no evidence of this reactivity in the control group (Figure 5D–F).
Figure 5.
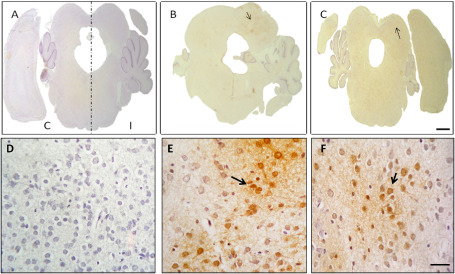
Representative stereo microscope image (A–C) and microphotographs (D–F) of Hypoxyprobe™‐1 labeling in midbrain sections (n = 8). A, D. No Hypoxyprobe™‐1 labeling was apparent in the control group; I = ipsilateral side; C = control side. B, E. Hypoxyprobe™‐1 labeling together with morphological damage was apparent on the ipsilateral side of the inferior colliculus in the HI group, with reactive cells (arrow). C, F. Hypoxyprobe™‐1‐labeled cells appear in the treatment group (nicotine), but there were no morphological differences between the ipsi‐ and contralateral sides. The contralateral side is to the left in all cases. Stereomicroscopic images, bar: 2.5 mm; microphotographs, bar: 500 μm.
Hematoxylin–eosin staining
Staining of brain sections with hematoxylin–eosin macroscopically revealed signs of early neuronal damage induced by the HI event, with the IC displaying significant evidence of infarction, whereas sections of non‐ischemic control animals or treated animals did not (Figure 6).
Figure 6.
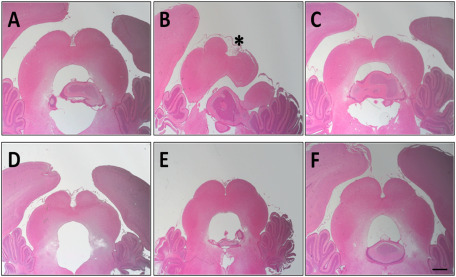
Representative stereo microscope photographs of the inferior colliculus of P14 perinatal rat brains (interaural distance 0.30 mm and bregma 9.30 mm) stained with hematoxylin–eosin (n = 8 for each group). A. Control group. B. Hypoxia‐ischemia (HI) group with the infarction area (indicated with an asterisk). C–F. HI + treatment: nicotine, melatonin, resveratrol and DHA, respectively. Bar: 1 mm.
In the IC (Figure 7), HI insult induced a significant alteration in neuron morphology, as indicated by an asterisk. Indeed, asphyctic animals showed swollen and deformed neurons in the CIC at the ipsilateral side, also with axonal prolongations and condensed cytoplasm. There were no signs of neuron damage in the slices from treated animals, in the CIC.
Figure 7.
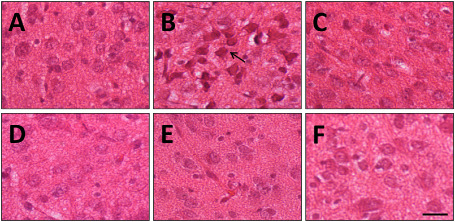
Representative microphotographs illustrating the alteration in neuron morphology in the central nuclei of the inferior colliculus in hematoxylin‐stained brain sections from P14 rat pups. A. Control. B. Hypoxia‐ischemia (HI). C–F. HI + treated groups, nicotine, melatonin, resveratrol and DHA, respectively. Condensate and bigger cells after HI (arrow) are apparent while after treatment administration the cells are similar to the control group. Bar: 100 μm.
Reactive astrogliosis
We next carried out GFAP immunohistochemistry to test if there was astrocyte reactivity in the IC after the HI event (Figure 8). Low levels of GFAP expression were detected in control animals (Figure 8A). In contrast, we found remarkable reactivity in the ipsilateral IC of HI animals (Figure 8B). Astrogliosis was diminished in all cases after treatment (Figure 8C–F).
Figure 8.
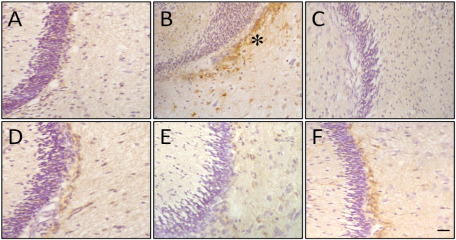
Increase in reactive gliosis [measured as glial fibrillary acidic protein (GFAP) expression] in injured animals and reduction of reactivity after treatment administration. Representative microphotographs illustrating GFAP immunostaining in brain sections from the central nucleus of the inferior colliculus (CIC) in control (A) hypoxia‐ischemia (HI) with reactive astrogliosis (asterisk) (B), HI + antioxidant treatment: nicotine, melatonin, resveratrol and DHA, respectively (C–F). Samples were obtained from P14 animals. Bar: 100 μm.
MBP immunoreactivity
MBP immunostaining was found to be reduced in subcortical white matter at the level of the ECIC and also in the CIC. We quantified the expression of MBP in the ipsilateral (L) and contralateral (R) hemispheres of the IC and established the MBP ratio as expression level in the left/right hemispheres. This L/R MBP ratio of HI pups was significantly decreased (P < 0.05) when compared with control animals in the ECIC (1.02 ± 0.22 vs. 0.53 ± 0.19 control and HI groups, respectively) and in the CIC (0.98 ± 0.06 vs. 0.68 ± 0.14, control and HI groups respectively). Treated animals showed a lesser degree of MBP loss in the ipsilateral hemisphere, with the L/ R MBP ratio being similar to that observed in the control group (Figure 9). Thus, antioxidant treatment resulted in the maintenance of MBP levels in the subcortical white matter of the ipsilateral hemisphere, in both anatomical regions.
Figure 9.
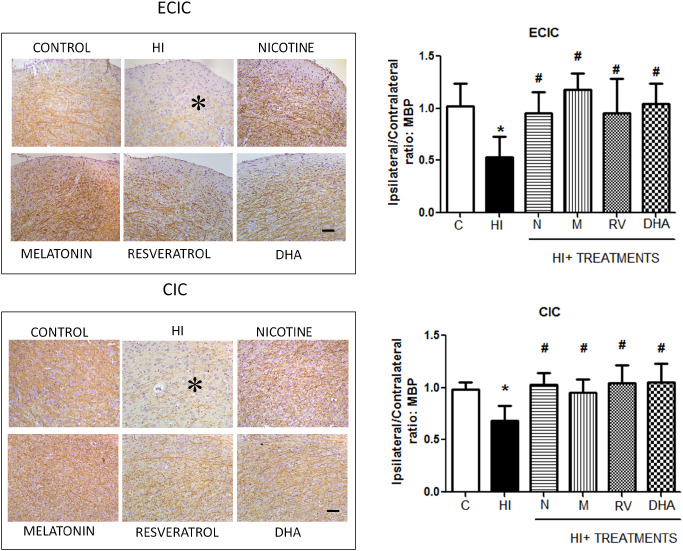
Loss of myelin basic protein (MBP) immunostaining at the level of the inferior colliculus in injured animals and recovery after antioxidant treatment. Microphotographs illustrating the disruption of MBP immunostaining in the external cortex of the inferior colliculus (ECIC) (B) (asterisk) compared with the control group and in the CIC (C). In (A) and (D), the histograms represent the ipsilateral/contralateral ratio of MBP expression in the ECIC and CIC, respectively. Bars represent the mean and SD (±standard deviation) with n = 8 for each group. *P < 0.05 vs. control; #P < 0.05 vs. HI, ANOVA. Bar: 100 μm.
Flow cytometer results
Just after the HI event, the membrane integrity is affected in the HI group and also in melatonin‐ and resveratrol‐treated groups, but treated groups recover the membrane integrity in 3 h, while the HI group has a statistical significant decrease in the percentage of positive cells to fluorochrome NAO compared with the control group. Thus, 12 h after the HI injury, all groups showed the same percentage of positive cells to this cardiolipin binding marker (Figure 10).
Figure 10.
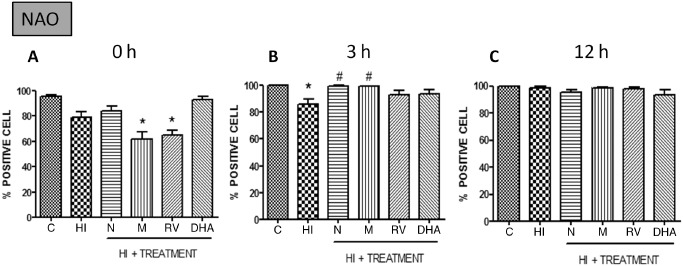
Percentage of positive cells with in vivo markers nonyl acridine orange (NAO) at different points after HI (n = 5). Data are expressed as means ± SEM (*P < 0.05 vs. controls and #P < 0.05 vs. HI, ANOVA). Abbreviations: C = control; HI = hypoxia‐ischemia; N = nicotine; M = melatonin; RV = resveratrol; DHA = docosahexaenoic acid.
As in the case of the membrane integrity study, the membrane potential is affected in the HI groups with a statistically significant decline in the percentage of positive cells in the Rhodamine 123 in vivo marker. This reduction is remarkable just after the damage and also 3 h after the damage but it seems that the membrane potential returns to normal after 12 h, as in the previous case. Antioxidant treatments act different in the first hours after the injury but they also recover the normal percentage of positive cells and fluorescence 12 h after the HI event (Figure 11).
Figure 11.
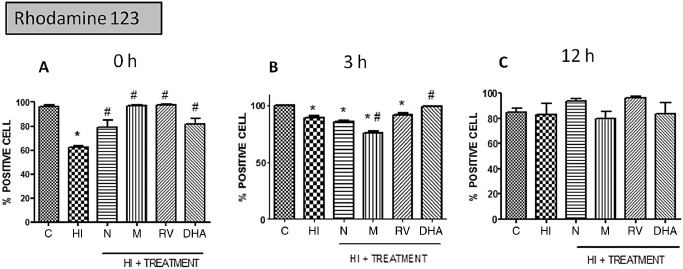
Percentage of positive cells with in vivo markers Rhodamine 123 at different points after HI (n = 5) in P7 rats. Data are expressed as means ± SEM (*P < 0.05 vs. controls and #P < 0.05 vs. HI, ANOVA). Abbreviations: C = control; HI = hypoxia‐ischemia; N = nicotine; M = melatonin; RV = resveratrol; DHA = docosahexaenoic acid.
According to the ROS, there is also a statistical significant decrease in the HI and DHA‐treated groups compared with the control group just after the damage. Three hours after the damage, all of the groups have more or less the same positive cells of DCFH marker, so the HI group and DHA group have increased the positive cells during this hour. However, the main difference compared with the previous results is that 12 h after the injury in all groups except from the melatonin‐treated group, the ROS production has increased by almost 20% compared with the control group (Figure 12).
Figure 12.
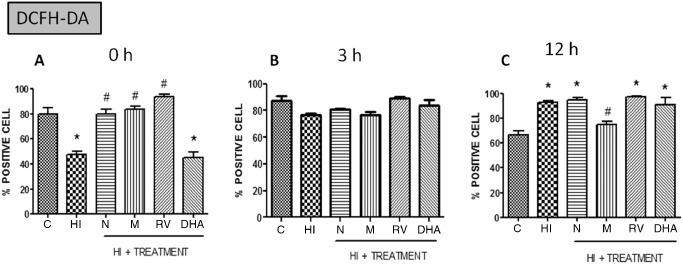
Percentage of positive cells with in vivo markers DCFH‐DA at different points after HI (n = 5) in P7 rats. Data are expressed as means ± SEM (*P < 0.05 vs. controls and #P < 0.05 vs. HI, ANOVA). Abbreviations: C = control; HI = hypoxia‐ischemia; N = nicotine; M = melatonin; RV = resveratrol; DHA = docosahexaenoic acid.
Discussion
The present work supports the hypothesis that hypoxia ischemia induces significant functional, histological and cellular damage to the brainstem and auditory system. We provide additional evidence that antioxidant treatment can be a powerful tool in order to minimize HI‐associated damage.
Hypoxia ischemia is one of the most important causes of damage to the inner ear and can develop many hearing disorders such as sudden sensorineural hearing loss, presbycusis and noise‐induced hearing loss that are suspected to be related to alterations in blow flow 59. Diverse studies have characterized the damage to neurons in the hippocampus, striatum and cortex after a perinatal asphyxia event 1, 4, but relatively little attention has been given to the auditory system. According to these studies, hair cell loss is correlated with the deficiency in both oxygen and glucose in the cochlea 20.
The ABR is an important diagnostic method to evaluate brainstem functionality and is widely used to detect HI auditory impairments 38, 65, 77. In this study, we have used the ABR to observe the existence of changes in auditory function in response to HI. There were differences in the amplitude of the ABR between the animals (the intensity of the response was different among animals in the same group). However, the latency of the peaks showed the same pattern in all animals, indicating that ABR wave latency is a stable and objective parameter. Alterations in this pattern could therefore be indicative of brain damage.
In the ABR, the latency of the wave V and the I–V interval latency are the two most widely used variables that reflect neural conduction. These are associated with myelination and synaptic function in the brainstem or central auditory pathway 28, 32. We observed a significant increase both in the I–V and in the III–V intervals, although the increase in the III–V interval was larger than that of the I–III interval. It seems that wave V following perinatal HI may be particularly vulnerable to physiological changes 66. Thus, our finding corroborates the findings of others in that the more central part (III–V interval) of the auditory pathway is more affected by HI insult than the more peripheral part (I–III interval) of the pathway 33. The progressive increase in ABR latencies and interpeak intervals may be attributed to a cumulative decrease in the efficacy of synaptic transmission at high stimulus rates, resulting in prolonged synaptic delays along the brainstem auditory pathway 35.
The brainstem receives blood from different sources but is mainly supplied by the vertebral artery, and the common carotid arteries play a minor role in this region. For this reason, it has been suggested that the brainstem is largely unaffected in the Rice–Vannucci model, which is a perinatal asphyxia model broadly used to investigate cerebral damage because the maturity of the CNS of P7 rats is similar to term human babies 21, 71. Nevertheless, Tomimatsu has demonstrated reduced blood flow to the ipsilateral IC during the HI event with this method, thereby validating its extensive use as a model of perinatal asphyxia by many research teams including our own 4, 19, 24.
To evaluate the effects of HI, here we have also measured the parameters of animal body and brain weight. Neonatal hypoxia‐ischemia not only causes brain damage and neurological deficits but also decreases somatic growth. Accordingly, we found somatic growth retardation in HI rats at P14 weeks compared with the control group, and that antioxidant treatments significantly improved body weight. Moreover, we also show that antioxidant‐treated pups maintained brain weight values, suggesting a beneficial effect of these compounds. The decrease in brain weight was likely caused by the loss of cerebral tissue, an observation that corroborates that of other authors who described the formation of brain cavities associated with long HI episodes. Indeed, some authors observed an important variability in the extension of the damaged area derived from HI insult, which leads to ipsilateral cerebral infarction 57 and manifests as a dramatic loss of cerebral tissue 8, 67. In response to the administration of antioxidant treatments, neurons in the IC appear to be less damaged and this may underlie the decreased infarct volume of antioxidant‐treated animals after a HI event.
In addition to considering functional perturbations, we consider that an optimal method to evaluate neuroprotection is the combination of functional approaches with histological evaluation. With this view in mind, we prospectively studied the effects of neonatal hypoxia ischemia on the three neural cell types: neurons, astrocytes and oligodendrocytes.
Depending on its severity and duration, brain injury may cause either infarction or selective neuronal death. Neurons are known to be the brain cells that are most sensitive to the lack of oxygen; they also exhibit selective vulnerability 24, 36. Moreover, in this study, we found that neuronal injury was more susceptible in the IC than in the rest of the brainstem, such as cochlear nucleus or vestibular nucleus. Indeed, it is widely known that astrocytes are generally more resistant to ischemia and other stressors than neurons because they are able to survive and function for extended periods under hypoxic conditions 20. However, astrocyte reactivity was observed in the form of GFAP upregulation during the HI event in the IC, while this reactivity was seen to be ameliorated in the rest of antioxidant‐treated groups.
White matter injury is a clinical hallmark of hypoxic ischemic encephalopathy (HIE) 27, 71. We also observed a reduction in MBP expression in the HI group and recuperation in the treated groups, results that corroborate the findings of other authors 2. MBP is the major myelin protein in the brain, constituting 30% of all myelin proteins. It is known to play a major role in myelin compaction during central nervous system development by intercalating between phospholipidic sheets and interacting with lipids and proteolipids. White matter injury is a clinical hallmark of preterm HIE 27, 71. Oligodendrocytes are the major cellular component of white matter and are the cells responsible for myelin formation in the central nervous system 6. Thus, damage to these cells can lead to a dysfunction that alters the formation of the myelin sheath after hypoxia‐ischemia 2. Maturation of oligodendroglia is known to be altered in HIE because of three mechanisms: microglial activation, excitotoxicity and free radical attack. Excitotoxicity likely leads to oligodendrocyte injury by promoting Ca2+ influx, and as a result, the generation of reactive oxygen and nitrogen species (ROS/RNS) 71.
The role of mitochondria in apoptosis seems to be very important. During prolonged hypoxia, the mitochondria in the majority of neonatal neurons were slowly depolarized, and during a brief period of hypoxia followed by reoxygenation, the majority of the neonatal mitochondria demonstrated a partial depolarization followed by recovery, which correlates with the previous studies 41. In the study of mitochondrial integrity, we observed a decrease in the percentage of positive cells to NAO in the HI group in the first hours in the brainstem, which supports the idea that the oxidation of cardiolipin is implicated in mitochondrial dysfunction and can be the consequence of release of cytochrome C from the mitochondria to the cytoplasm, a process that is involved in the apoptotic cell death cascade 16, 17, 19, 55. Moreover, the same results were found in the percentage of positive cells to Rhodamine 123, suggesting that both membrane potential and integrity are altered during the HI event in the first moments.
ROS play a critical role in HI injury. ROS that were generated during the HI event caused cell damage not only via direct action on the cell but also via activation of inflammatory pathway 79. We observed an increase in ROS production 12 h after the HI event; this could be because mitochondria are both a source and a target of ROS 78, so after the mitochondrial first injury, there is an increase of ROS production and that can lead to cell death 40.
Taking into account that antioxidant treatments have been previously described as a type of neuroprotective therapy, we consider if it could also be effective at this level. Antioxidant effect was observed in all treatments but the protective mechanism is unknown. In the functional and histological studies, all treatments show neuroprotective effect, while these results are not so clear in the molecular studies. The mitochondrial membrane seem to act in the same way in all treated groups after 12 h, but ROS production after 12 h to the injury is only decreased in the melatonin‐treated group, suggesting that the antioxidant effect of melatonin is faster compared with the rest of the studied groups.
Melatonin is one of the most well‐known neurohormones that are used as a neuroprotective agent for several brain injuries 1, 10. It has been shown to exhibit neuroprotective effects against transient or permanent ischemic brain injury 39, 53, making it a potentially good tool for the treatment of neonatal hypoxia‐ischemia 9, 73. RVT and DHA treatments have also been demonstrated to be effective against HI brain injury in the neonatal rat model, reducing infarct volume and neuronal loss, minimizing lipid and DNA peroxidation, blocking some apoptotic pathways, decreasing inflammation, inhibiting free radical production and increasing the production of some antioxidant enzymes such as GPx and SOD 5. Apart from the well‐known antioxidant treatments for the HI injury, activation of neuronal nAChRs by nicotine has also been suggested to protect neurons against hypoxic insult 23. Our findings confirm this potentially protective effect of nicotine by showing reduced brain damage following HI, when nicotine is administered.
Conclusions
Taken as a whole, the present prospective study presents for the first time a correlation between the functional, morphological and molecular aspects underlying the antioxidant‐induced amelioration of HI‐induced brainstem damage. Thus, antioxidant treatments were found to provide effective neuroprotection to the immature auditory system before a perinatal HI event. To our knowledge, this is the first study that demonstrates the neuroprotective effects of nicotine, melatonin, resveratrol and DHA on the functional changes of the auditory pathway and on the morphological damage, which occurs after neonatal HI insult.
Acknowledgments
This work was supported by grants from the Basque Country Government (IT773/13) and Fundación Jesus de Gangoiti Barrera.
References
- 1. Alonso‐Alconada D, Alvarez A, Lacalle J, Hilario E (2012) Histological study of the protective effect of melatonin on neural cells after neonatal hypoxia‐ischemia. Histol Histopathol 27:771–783. [DOI] [PubMed] [Google Scholar]
- 2. Alonso‐Alconada D, Hilario E, Alvarez FJ, Alvarez A (2012) Apoptotic cell death correlates with ROS overproduction and early cytokine expression after hypoxia‐ischemia in fetal lambs. Reprod Sci 19:754–763. [DOI] [PubMed] [Google Scholar]
- 3. Alonso‐Alconada D, Alvarez A, Arteaga O, Martínez‐Ibargüen A, Hilario E (2013) Neuroprotective effect of melatonin: a novel therapy against perinatal hypoxia‐ischemia. Int J Mol Sci 14:9379–9395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alvarez‐Diaz A, Hilario E, de Cerio FG, Valls‐i‐Soler A, Alvarez‐Diaz FJ (2007) Hypoxic‐ischemic injury in the immature brain—key vascular and cellular players. Neonatology 92:227–235. [DOI] [PubMed] [Google Scholar]
- 5. Arteaga O, Revuelta M, Montalvo H, Cañavate ML, Alonso‐Alconada D, Martinez‐Ibargüen A et al (2014) Neuroprotective effect of antioxidants in neonatal rat brain after hypoxia‐ischemia. Microscopy 6:335–343. [Google Scholar]
- 6. Barres BA (2008) The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60:430–440. [DOI] [PubMed] [Google Scholar]
- 7. Berman DR, Mozurkewich E, Liu Y, Barks J (2009) Docosahexaenoic acid pretreatment confers neuroprotection in a rat model of perinatal cerebral hypoxia‐ischemia. Am J Obstet Gynecol 200:305.e1–305.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Biran V, Joly LM, Heron A, Vernet A, Vega C, Mariani J et al (2006) Glial activation in white matter following ischemia in the neonatal P7 rat brain. Exp Neurol 199:103–112. [DOI] [PubMed] [Google Scholar]
- 9. Carloni S, Perrone S, Buonocore G, Longini M, Proietti F, Balduini W (2008) Melatonin protects from the long‐term consequences of a neonatal hypoxic‐ischemic brain injury in rats. J Pineal Res 44:157–164. [DOI] [PubMed] [Google Scholar]
- 10. Carloni S, Albertini MC, Galluzzi L, Buonocore G, Proietti F, Balduini W (2014) Melatonin reduces endoplasmic reticulum stress and preserves sirtuin 1 expression in neuronal cells of newborn rats after hypoxia‐ischemia. J Pineal Res 57:192–199. [DOI] [PubMed] [Google Scholar]
- 11. Cetinkaya M, Alkan T, Ozyener F, Kafa IM, Kurt MA, Koksal N (2011) Possible neuroprotective effects of magnesium sulfate and melatonin as both pre‐ and post‐treatment in a neonatal hypoxic‐ischemic rat model. Neonatology 99:302–310. [DOI] [PubMed] [Google Scholar]
- 12. Chayasirisobhon S, Yu L, Griggs L, Westmoreland SJ, Leu N (1996) Recording of brainstem evoked potentials and their association with gentamicin in neonates. Pediatr Neurol 14:277–280. [DOI] [PubMed] [Google Scholar]
- 13. Chen Y, Swanson RA (2003) Astrocytes and brain injury. J Cereb Blood Flow Metab 23:137–149. [DOI] [PubMed] [Google Scholar]
- 14. Chen Y, Nie H, Tian L, Tong L, Yang L, Lao N et al (2013) Nicotine‐induced neuroprotection against ischemic injury involves activation of endocannabinoid system in rats. Neurochem Res 38:364–370. [DOI] [PubMed] [Google Scholar]
- 15. Davis‐Bruno K, Tassinari MS (2011) Essential fatty acid supplementation of DHA and ARA and effects on neurodevelopment across animal species: a review of the literature. Birth Defects Res B Dev Reprod Toxicol 92:240–250. [DOI] [PubMed] [Google Scholar]
- 16. Fernandez‐Gomez FJ, Galindo MF, Gomez‐Lazaro M, González‐García C, Ceña V, Aguirre N, Jordán J (2005) Involvement of mitochondrial potential and calcium buffering capacity in minocycline cytoprotective actions. Neuroscience 133:959–967. [DOI] [PubMed] [Google Scholar]
- 17. Ferriero DM (2001) Oxidant mechanisms in neonatal hypoxia‐ischemia. Dev Neurosci 23:198–202. [DOI] [PubMed] [Google Scholar]
- 18. Freeman DT (1991) Computer recognition of brain stem auditory evoked potential wave V by a neural network. Proc Annu Symp Comput Appl Med Care 305–309. [PMC free article] [PubMed] [Google Scholar]
- 19. Goni‐de‐Cerio F, Alvarez A, Caballero A, Mielgo VE, Alvarez FJ, Rey‐Santano MC et al (2007) Early cell death in the brain of fetal preterm lambs after hypoxic‐ischemic injury. Brain Res 1151:161–171. [DOI] [PubMed] [Google Scholar]
- 20. Gross J, Oltze H, Mazurek B (2014) Differential expression of transcription factors an inflammation‐, ROS‐, and cell death‐related genes in organotypic cultures in the modiolus, the organ of corti and stria vascularis of newborn rats. Cell Mol Neurobiol 4:523–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hagberg H, Bona E, Gilland E, Puka‐Sundvall M (1997) Hypoxia‐ischaemia model in the 7‐day‐old rat: possibilities and shortcomings. Acta Paediatr Suppl 422:85–88. [DOI] [PubMed] [Google Scholar]
- 22. Hecox KE, Cone B (1981) Prognostic importance of brainstem auditory evoked responses after asphyxia. Neurology 31:1429–1434. [DOI] [PubMed] [Google Scholar]
- 23. Hejmadi MV, Dajas‐Bailador F, Barns SM, Jones B, Wonnacott S (2003) Neuroprotection by nicotine against hypoxia‐induced apoptosis in cortical cultures involves activation of multiple nicotinic acetylcholine receptor subtypes. Mol Cell Neurosci 24:779–786. [DOI] [PubMed] [Google Scholar]
- 24. Hilario E, Rey‐Santano MC, Goni‐de‐Cerio F, Alvarez FJ, Gastiasoro E, Mielgo VE et al (2005) Cerebral blood flow and morphological changes after hypoxic‐ischaemic injury in preterm lambs. Acta Paediatr 94:903–911. [DOI] [PubMed] [Google Scholar]
- 25. Huang SS, Tsai MC, Chih CL, Hung LM, Tsai SK (2001) Resveratrol reduction of infarct size in Long‐Evans rats subjected to focal cerebral ischemia. Life Sci 69:1057–1065. [DOI] [PubMed] [Google Scholar]
- 26. Inder TE, Anderson NJ, Spencer C, Wells S, Volpe JJ (2003) White matter injury in the premature infant: a comparison between serial cranial sonographic and MR findings at term. AJNR Am J Neuroradiol 24:805–809. [PMC free article] [PubMed] [Google Scholar]
- 27. Jellema RK, Wolfs TG, Lima Passos V, Zwanenburg A, Ophelders DR, Kuypers E et al (2013) Mesenchymal stem cells induce T‐cell tolerance and protect the preterm brain after global hypoxia‐ischemia. PLoS ONE 8:e73031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jiang ZD (1998) Maturation of peripheral and brainstem auditory function in the first year following perinatal asphyxia: a longitudinal study. J Speech Lang Hear Res 41:83–93. [DOI] [PubMed] [Google Scholar]
- 29. Jiang ZD, Brosi DM, Shao XM, Wilkinson AR (2000) Maximum length sequence brainstem auditory evoked responses in term neonates who have perinatal hypoxia‐ischemia. Pediatr Res 48:639–645. [DOI] [PubMed] [Google Scholar]
- 30. Jiang ZD, Yin R, Shao XM, Wilkinson AR (2004) Brain‐stem auditory impairment during the neonatal period in term infants after asphyxia: dynamic changes in brain‐stem auditory evoked response to clicks of different rates. Clin Neurophysiol 115:1605–1615. [DOI] [PubMed] [Google Scholar]
- 31. Jiang ZD, Liu XY, Shi BP, Lin L, Bu CF, Wilkinson AR (2008) Brainstem auditory outcomes and correlation with neurodevelopment after perinatal asphyxia. Pediatr Neurol 39:189–195. [DOI] [PubMed] [Google Scholar]
- 32. Jiang ZD, Brosi DM, Chen C, Wilkinson AR (2009) Brainstem response amplitudes in neonatal chronic lung disease and differences from perinatal asphyxia. Clin Neurophysiol 120:967–973. [DOI] [PubMed] [Google Scholar]
- 33. Jiang ZD, Brosi DM, Chen C, Wilkinson AR (2009) Impairment of perinatal hypoxia‐ischemia to the preterm brainstem. J Neurol Sci 287:172–177. [DOI] [PubMed] [Google Scholar]
- 34. Jiang ZD, Wu YY, Wilkinson AR (2009) Age‐related changes in BAER at different click rates from neonates to adults. Acta Paediatr 98:1284–1287. [DOI] [PubMed] [Google Scholar]
- 35. Jiang ZD, Brosi DM, Wilkinson AR (2010) Differences in impaired brainstem conduction between neonatal chronic lung disease and perinatal asphyxia. Clin Neurophysiol 121:725–733. [DOI] [PubMed] [Google Scholar]
- 36. Johnston RV, Grant DA, Wilkinson MH, Walker AM (1998) Repetitive hypoxia rapidly depresses arousal from active sleep in newborn lambs. J Physiol 510:651–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kelly FJ (1993) Free radical disorders of preterm infants. Br Med Bull 49:668–678. [DOI] [PubMed] [Google Scholar]
- 38. Kileny P, Connelly C, Robertson C (1980) Auditory brainstem responses in perinatal asphyxia. Int J Pediatr Otorhinolaryngol 2:147–159. [DOI] [PubMed] [Google Scholar]
- 39. Kilic U, Kilic E, Reiter RJ, Bassetti CL, Hermann DM (2005) Signal transduction pathways involved in melatonin‐induced neuroprotection after focal cerebral ischemia in mice. J Pineal Res 38:67–71. [DOI] [PubMed] [Google Scholar]
- 40. Lara‐Celador I, Castro‐Ortega L, Alvarez A, Goñi‐de‐Cerio F, Lacalle J, Hilario E (2012) Endocannabinoids reduce cerebral damage after hypoxic‐ischemic injury in perinatal rats. Brain Res 1474:91–99. [DOI] [PubMed] [Google Scholar]
- 41. Larsen GA, Skjellegrind HK, Vinje ML, Berg‐Johnsen J (2008) Mitochondria are more resistant to hypoxic depolarization in the newborn than in the adult brain. Neurochem Res 33:1894–1900. [DOI] [PubMed] [Google Scholar]
- 42. Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S et al (2009) Hypoxia‐inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15:501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu Y, Silverstein FS, Skoff R, Barks JD (2002) Hypoxic‐ischemic oligodendroglial injury in neonatal rat brain. Pediatr Res 51:25–33. [DOI] [PubMed] [Google Scholar]
- 44. Liu BL, Zhang X, Zhang W, Zhen HN (2007) New enlightenment of French paradox: resveratrol's potential for cancer chemoprevention and anti‐cancer therapy. Cancer Biol Ther 6:1833–1836. [DOI] [PubMed] [Google Scholar]
- 45. Lopez‐Miranda V, Soto‐Montenegro ML, Vera G, Herradon E, Desco M, Abalo R (2012) Resveratrol: a neuroprotective polyphenol in the Mediterranean diet. Rev Neurol 54:349–356. [PubMed] [Google Scholar]
- 46. Margaill I, Plotkine M, Lerouet D (2005) Antioxidant strategies in the treatment of stroke. Free Radic Biol Med 39:429–443. [DOI] [PubMed] [Google Scholar]
- 47. McMahon S, Charbonneau M, Grandmont S, Richard DE, Dubois CM (2006) Transforming growth factor beta1 induces hypoxia‐inducible factor‐1 stabilization through selective inhibition of PHD2 expression. J Biol Chem 281:24171–24181. [DOI] [PubMed] [Google Scholar]
- 48. Misra PK, Katiyar CP, Kapoor RK, Shukla R, Malik GK, Thakur S (1997) Brainstem auditory evoked response in neonates with birth asphyxia. Indian Pediatr 34:199–205. [PubMed] [Google Scholar]
- 49. Moosmann B, Behl C (2002) Antioxidants as treatment for neurodegenerative disorders. Expert Opin Investig Drugs 11:1407–1435. [DOI] [PubMed] [Google Scholar]
- 50. Northington FJ, Ferriero DM, Graham EM, Traystman RJ, Martin LJ (2001) Early neurodegeneration after hypoxia‐ischemia in neonatal rat is necrosis while delayed neuronal death is apoptosis. Neurobiol Dis 8:207–219. [DOI] [PubMed] [Google Scholar]
- 51. Pan HC, Kao TK, Ou YC, Yang DY, Yen YJ, Wang CC et al (2009) Protective effect of docosahexaenoic acid against brain injury in ischemic rats. J Nutr Biochem 20:715–725. [DOI] [PubMed] [Google Scholar]
- 52. Paxinos G (1986) The Rat Brain in Stereotaxic Coordinates, 2nd edn. Academic Press: San Diego, California, USA. [Google Scholar]
- 53. Pei Z, Ho HT, Cheung RT (2002) Pre‐treatment with melatonin reduces volume of cerebral infarction in a permanent middle cerebral artery occlusion stroke model in the rat. Neurosci Lett 318:141–144. [DOI] [PubMed] [Google Scholar]
- 54. Pin TW, Eldridge B, Galea MP (2009) A review of developmental outcomes of term infants with post‐asphyxia neonatal encephalopathy. Eur J Paediatr Neurol 13:224–234. [DOI] [PubMed] [Google Scholar]
- 55. Ramirez MR, Muraro F, Zylbersztejn DS, Abel CR, Arteni NS, Lavinsky D et al (2003) Neonatal hypoxia‐ischemia reduces ganglioside, phospholipid and cholesterol contents in the rat hippocampus. Neurosci Res 46:339–347. [DOI] [PubMed] [Google Scholar]
- 56. Reiter RJ (1998) Melatonin, active oxygen species and neurological damage. Drug News Perspect 11:291–296. [DOI] [PubMed] [Google Scholar]
- 57. Renolleau S, Aggoun‐Zouaoui D, Ben‐Ari Y, Charriaut‐Marlangue C (1998) A model of transient unilateral focal ischemia with reperfusion in the P7 neonatal rat: morphological changes indicative of apoptosis. Stroke 29:1454–1460. discussion 1461. [DOI] [PubMed] [Google Scholar]
- 58. Rice JE 3rd, Vannucci RC, Brierley JB (1981) The influence of immaturity on hypoxic‐ischemic brain damage in the rat. Ann Neurol 9:131–141. [DOI] [PubMed] [Google Scholar]
- 59. Ross HM, Pawlina W (2011) Histology: A Text and Atlas, 6th edn. Lippincott Williams & Wilkins: Baltimore. [Google Scholar]
- 60. Rothstein RP, Levison SW (2005) Gray matter oligodendrocyte progenitors and neurons die caspase‐3 mediated deaths subsequent to mild perinatal hypoxic/ischemic insults. Dev Neurosci 27:149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Smit AL, Seehase M, Stokroos RJ, Jellema RK, Felipe L, Chenault MN et al (2013) Functional impairment of the auditory pathway after perinatal asphyxia and the short‐term effect of perinatal propofol anesthesia in lambs. Pediatr Res 74:34–38. [DOI] [PubMed] [Google Scholar]
- 62. Suganuma H, Okumura A, Kitamura Y, Shoji H, Shimizu T (2013) Effect of hypoxic‐ischemic insults on the composition of fatty acids in the brain of neonatal rats. Ann Nutr Metab 62:123–128. [DOI] [PubMed] [Google Scholar]
- 63. Sun W, Manohar S, Jayaram A, Kumaraguru A, Fu Q, Li J, Allman B (2011) Early age conducive hearing loss causes audiogenic seizure and hyperacusis behaviour. Hear Res 282:178–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tan DX, Manchester LC, Terron MP, Flores LJ, Tamura H, Reiter RJ (2007) Melatonin as a naturally occurring co‐substrate of quinone reductase‐2, the putative MT3 melatonin membrane receptor: hypothesis and significance. J Pineal Res 43:317–320. [DOI] [PubMed] [Google Scholar]
- 65. Tomimatsu T, Fukuda H, Endoh M, Mu J, Watanabe N, Kohzuki M et al (2002) Effects of neonatal hypoxic‐ischemic brain injury on skilled motor tasks and brainstem function in adult rats. Brain Res 926:108–117. [DOI] [PubMed] [Google Scholar]
- 66. Tomimatsu T, Fukuda H, Endoh M, Mu J, Kanagawa T, Hosono T et al (2003) Long‐term neuroprotective effects of hypothermia on neonatal hypoxic‐ischemic brain injury in rats, assessed by auditory brainstem response. Pediatr Res 53:57–61. [DOI] [PubMed] [Google Scholar]
- 67. Towfighi J, Yager JY, Housman C, Vannucci RC (1991) Neuropathology of remote hypoxic‐ischemic damage in the immature rat. Acta Neuropathol 81:578–587. [DOI] [PubMed] [Google Scholar]
- 68. Vexler ZS, Ferriero DM (2001) Molecular and biochemical mechanisms of perinatal brain injury. Semin Neonatol 6:99–108. [DOI] [PubMed] [Google Scholar]
- 69. Volpe JJ (1998) Brain injury in the premature infant: overview of clinical aspects, neuropathology, and pathogenesis. Semin Pediatr Neurol 5:135–151. [DOI] [PubMed] [Google Scholar]
- 70. Volpe JJ (2008) Neonatal encephalitis and white matter injury: more than just inflammation? Ann Neurol 64:232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Volpe JJ, Kinney HC, Jensen FE, Rosenberg PA (2011) The developing oligodendrocyte: key cellular target in brain injury in the premature infant. Int J Dev Neurosci 29:423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang X, Hagberg H, Zhu C, Jacobsson B, Mallard C (2007) Effects of intrauterine inflammation on the developing mouse brain. Brain Res 1144:180–185. [DOI] [PubMed] [Google Scholar]
- 73. Welin AK, Svedin P, Lapatto R, Sultan B, Hagberg H, Gressens P et al (2007) Melatonin reduces inflammation and cell death in white matter in the mid‐gestation fetal sheep following umbilical cord occlusion. Pediatr Res 61:153–158. [DOI] [PubMed] [Google Scholar]
- 74. West T, Atzeva M, Holtzman DM (2007) Pomegranate polyphenols and resveratrol protect the neonatal brain against hypoxic‐ischemic injury. Dev Neurosci 29:363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wilkinson AR, Jiang ZD (2006) Brainstem auditory evoked response in neonatal neurology. Semin Fetal Neonatal Med 11:444–451. [DOI] [PubMed] [Google Scholar]
- 76. Wurtman RJ (2008) Synapse formation and cognitive brain development: effect of docosahexaenoic acid and other dietary constituents. Metabolism 57 (Suppl. 2):S6–S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yasuhara A, Kinoshita Y, Hori A, Iwase S, Kobayashi Y (1986) Auditory brainstem response in neonates with asphyxia and intracranial haemorrhage. Eur J Pediatr 145:347–350. [DOI] [PubMed] [Google Scholar]
- 78. Zhao J, Bolton EM, Bradley JA, Lever AM (2009) Lentiviral‐mediated overexpression of Bcl‐xL protects primary endothelial cells from ischemia/reperfusion injury‐induced apoptosis. J Heart Lung Transplant 28:936–943. [DOI] [PubMed] [Google Scholar]
- 79. Zhao WY, Han S, Zhang L, Zhu YH, Wang LM, Zeng L (2013) Mitochondria‐targeted antioxidant peptide SS31 prevents hypoxia/reoxygenation‐induced apoptosis by down‐regulating p66Shc in renal tubular epithelial cells. Cell Physiol Biochem 32:591–600. [DOI] [PubMed] [Google Scholar]