

Abstract
BRAF V600E mutations have been recently reported in glioneuronal tumors (GNTs). To evaluate the expression of the BRAF V600E mutated protein and its association with activation of the mammalian target of rapamycin (mTOR) pathway, immunophenotype and clinical characteristics in GNTs, we investigated a cohort of 174 GNTs. The presence of BRAF V600E mutations was detected by direct DNA sequencing and BRAF V600E immunohistochemical detection. Expression of BRAF‐mutated protein was detected in 38/93 (40.8%) gangliogliomas (GGs), 2/4 (50%) desmoplastic infantile gangliogliomas (DIGs) and 23/77 (29.8%) dysembryoplastic neuroepithelial tumors (DNTs) by immunohistochemistry. In both GGs and DNTs, the presence of BRAF V600E mutation was significantly associated with the expression of CD34, phosphorylated ribosomal S6 protein (pS6; marker of mTOR pathway activation) in dysplastic neurons and synaptophysin (P < 0.05). In GGs, the presence of lymphocytic cuffs was more frequent in BRAF‐mutated cases (31 vs. 15.8%; P = 0.001). The expression of both BRAF V600E and pS6 was associated with a worse postoperative seizure outcome in GNT (P < 0.001). Immunohistochemical detection of BRAF V600E‐mutated protein may be valuable in the diagnostic evaluation of these glioneuronal lesions and the observed association with mTOR activation may aid in the development of targeted treatment involving specific pathogenic pathways.
Keywords: BRAF, immunohistochemistry, inflammation, long‐term epilepsy associated tumors, mTOR, sequencing
Introduction
Glioneuronal tumors (GNTs), including gangliogliomas (GGs) and dysembryoplastic neuroepithelial tumors (DNTs), represent a well‐recognized cause of intractable epilepsy in children and young adults. Their cellular composition, characterized by mixed neuroepithelial cell types, including aberrantly shaped neuronal cells and glial elements, in coexistence with cortical dysplasia suggests a developmental pathogenesis for these lesions 12, 14, 31, 60. The possible origin of GGs from a dysplastic precursor lesion is also supported by the reported association with molecular alterations common to other developmental glioneuronal lesions, such as reelin and the mammalian target of rapamycin (mTOR) signaling pathways 11, 18, 39. mTOR is regulated by several proteins, including PI3 kinase, PDK1, PTEN, AKT and LKB1 [tumor suppressor liver kinase B1 54, 58]. Deregulation of the mTOR pathway, which is critical to cell growth and proliferation during the development of the cerebral cortex, has been linked to different malformations of cortical development (MCD) associated with epilepsy and neurobehavioral disabilities [for reviews, see 21]. Accordingly, GNTs have been included among the MCD, in the group of malformations [including focal cortical dysplasia (FCD) and brain lesions of tuberous sclerosis complex (TSC)], characterized by abnormal neuronal and glial proliferation 9. In our previous study, in which we examined a small cohort of GG and DNTs (including only simple DNTs), enhanced mTOR signaling pathway activation was only observed in GGs 18. However, recently, non‐specific forms of DNTs, representing a diagnostic challenge to neuropathologists, have also been reported and their pathogenesis and relationship with GGs is still a matter of discussion 17, 46, 59 [for review, see 60].
Recent studies have reported a mutation of the BRAF oncogene (a member of the RAF family of serine/threonine protein kinases involved in the RAS‐RAF‐MEK‐ERK‐MAP kinase signaling pathway) in up to 50% GGs 22, 26, 41, 56 and in few DNTs 20 and desmoplastic infantile gangliogliomas [DIGs 40], suggesting a link with other tumor entities, such as pleomorphic xanthoastrocytoma (PXA) and pilocytic astrocytoma (PA) [for reviews, see 36, 60].
In the present study, we collected and analyzed the histopathological and clinical data in a large cohort of 174 patients with GNTs.
Our aims were (i) to evaluate the presence and distribution, across GNT subtypes, of enhanced mTOR signaling pathway activation; (ii) to evaluate the incidence and distribution, across GNT subtypes, of BRAF V600E mutation detected by a BRAF V600E mutation‐specific antibody, as well as by direct DNA sequencing; and (iii) to compare the tumor immunohistochemical characteristics with molecular genetics profile and clinical features, including seizure outcome after surgery.
Material and Methods
Subjects
The GNT specimens included in this study (inclusion period: 1991–2011) were all obtained from the databases of the Departments of Neuropathology of the Academic Medical Center (University of Amsterdam) in Amsterdam, the University Medical Center in Utrecht and of the University College London Institute of Neurology Queen Square (London, UK). The extent of resection was determined by reviewing the operative report and postoperative brain scans.
We examined a total of 174 surgical specimens (n = 93 GGs, n = 4 DIGs, n = 77 DNTs; Table 1). Informed consent was obtained for the use of brain tissue and for access to medical records for research purposes. Tissue was obtained and used in a manner compliant with the Declaration of Helsinki. We reviewed all cases, and the diagnosis was confirmed according to the revised World Health Organization (WHO) classification of tumors of the nervous system 46.
Table 1.
Summary of clinical findings of patients. Abbreviations: CPS = complex partial seizures; GS = generalized seizures; SGS = secondary generalized seizures; SPS = simple partial seizures; HS = hippocampal sclerosis; FCD = focal cortical dysplasia; GNT = Glioneuronal tumor; PR = partial resection; GTR = gross‐total resection; MSR = mesial‐structures resection; F = frontal; T = temporal; BG = basal ganglia; O = occipital; P = parietal; SC = spinal cord; Cer = cerebellum; BS = brain stem; DIGs = desmoplastic infantile gangliogliomas; GGs = gangliogliomas; DNTs = dysembryoplastic neuroepithelial tumors
| Diagnosis | GGs (n = 93) | DIGs (n = 4) | DNTs (n = 77) |
|---|---|---|---|
| Male/Female | 48/45 | 2/2 | 41/36 |
| Mean age at surgery: years (range) | 24.6 (1–66) | 6.5 (1–12) | 24.7 (3–49) |
| Location | F : 5; F‐T : 2; T : 68; O : 2; P : 8; P‐T : 3; P‐O : 1; BG : 1; BS : 1; Cer : 1; SC : 1 | F : 1; T : 1; P : 1; BG : 1 | F : 4; P : 7; T : 65; O : 1 |
| Histological grading/subtype |
GG WHO I : 89 GG WHO III : 4 |
WHO I |
WHO I Complex DNT : 33; Simple DNT : 15; Diffuse DNT : 22; DNT/GG : 7 |
| Recurrence (yes/no) | 4/89 | 0/4 | 2/75 |
| Epilepsy (yes/no) | 87/6 | 3/1 | 77/0 |
| Seizure type | CPS (90%); SGS (20%); GS: 7%; SPS: 5% | CPS (100%); SGS (33%) | CPS (100%); SGS (5%); GS (37%); SP (2.5%) |
| Mean age at seizure onset: years (range) | 13.1 (0.25–52) | 3.7 (0.25–10) | 11.8 (0.3–29) |
| Duration of epilepsy: years (range) | 11.1 (0.66–41) | 2 (0.75–2) | 12.87 (0.91–40) |
| FCD IIIb: present/absent | 4/89 | 0/4 | 5/72 |
| Extent of resection | GTR (40%); GTR/MSR (51%); PR (9%) | PR (25%); GTR (75%) | GTR (20%); GTR/MSR (76%); PR (4%) |
| Hippocampus histology (yes/no): cases with hippocampal resection | 15/78 (2 HS‐ILAE type 1; 2 HS‐ILAE type 3; 11, no HS) | 0/4 | 11/66 (2 HS‐ILAE type 1; 1 HS‐ILAE type 3; 8, no HS) |
| Postoperative epilepsy: Engel's class | I (74%); II (14%); III (7%); IV (5%) | IV | I (81%); II (10%); III (5%); IV (4%) |
ILAE classification 16.
The clinical features of the included patients (such as age at surgery, duration of epilepsy and seizure type) are summarized in Table 1. One hundred sixty‐eight patients (96%) underwent resection of GNT for medically intractable epilepsy. The predominant type of seizure pattern was that of complex partial seizures, which were resistant to maximal doses of anti‐epileptic drugs (AEDs). The patients with epilepsy underwent presurgical evaluation 62 and the postoperative seizure outcome was classified according to Engel 29. Patients who were free of habitual preoperative seizures were classified as class I, and patients in class II were almost seizure free or had rare or nocturnal seizures only. The follow‐up period ranged from 1 to 15 years.
Tissue preparation
Formalin fixed, paraffin‐embedded (FFPE) tissue was sectioned at 6 μm and mounted on precoated glass slides (Star Frost, Waldemar Knittel GmbH, Braunschweig, Germany). Representative sections of all specimens were processed for hematoxylin & eosin (HE), luxol fast blue (LFB) and Nissl stains, as well as for a number of immunohistochemical markers (Table 2).
Table 2.
Immunohistochemistry: primary antibodies. Abbreviation: MHC = major histocompatibility complex; GFAP = glial fibrillary acidic protein
| Antigen | Primary antibody | Source | Dilution |
|---|---|---|---|
| GFAP | Polyclonal rabbit | DAKO, Glostrup, Denmark | 1:4000 |
| Neuronal nuclear protein (NeuN) | Mouse clone MAB377 | Chemicon, Temecula, CA, USA | 1:2000 |
| Synaptophysin | Mouse clone Sy38 | DAKO, Glostrup, Denmark | 1:200 |
| Ki67 | Mouse clone MIB‐1 | DAKO, Glostrup, Denmark | 1:200 |
| BRAF V600E | VE1 clone | a | 1:5 |
| IDH1 R132H | Clone H09 | Dianova, Hamburg, Germany | 1:75 |
| CD34 | Mouse clone QBEnd10 | Immunotech, Marseille, Cedex, France | 1:600 |
| Lin28A | Rabbit polyclonal | Cell Signaling Technology, Beverly, MA, USA | 1:200 |
| Phospho‐S6 ribosomal protein (pS6) | Monoclonal rabbit (Ser235/236) | Cell Signaling Technology, Beverly, MA, USA | 1:200 |
| Phospho‐S6 ribosomal protein (pS6) | Monoclonal rabbit (Ser240/244) | Cell Signaling Technology, Beverly, MA, USA | 1:2000 |
| Phospho‐tumor suppressor liver kinase B1 (pLKB1) | Monoclonal rabbit | Cell Signaling Technology, Beverly, MA, USA | 1:50 |
| Human leukocyte antigen (HLA‐DP, DQ, DR; MHC‐II) | Mouse clone CR3/43 | DAKO, Glostrup, Denmark | 1:100 |
| Human leukocyte antigen (HLA‐A, B, C; MHC‐I) | Mouse clone HC‐10 | b | 1:200 |
Kindly provided by D. Capper and A. von Deimling 19.
Kindly provided by J. Neefjes (NKI, the Netherlands).
Immunohistochemistry
Single‐label immunohistochemistry (see Table 2) was performed, as previously described 4, using the Powervision kit (Immunologic, Duiven, the Netherlands) and 3,3‐diaminobenzidine as chromogen. Immunostaining for BRAF V600E (clone VE1, Spring Bioscience, Pleasanton, CA, USA) was performed on a Ventana BenchMark XT immunostainer (Ventana Medical Systems, Tucson, AZ, USA) as previously reported 41. Sections were counterstained with hematoxylin. As positive and negative controls, we used a tissue microarray of colon carcinoma specimens with known BRAF V600E status (66 wild‐type, 15 BRAF V600E).
For double‐labeling of VE1 with pS6 or phospho‐tumor suppressor liver kinase B1 (pLKB1), sections were, after incubation with the primary antibodies overnight at 4°C, incubated for 2 h at room temperature with Alexa Fluor® 568‐conjugated anti‐rabbit and Alexa Fluor® 488 anti‐mouse IgG (1:100, Molecular Probes, Leiden, the Netherlands). Sections were then analyzed by means of a laser scanning confocal microscope (Leica TCS Sp2, Wetzlar, Germany).
For double‐labeling of VE1 with NeuN (both mouse monoclonal antibodies), sections were incubated with the first primary antibody, anti‐NeuN, which was visualized with a polymer‐alkaline phosphatase (AP)‐labeled anti‐mouse antibody (BrightVision #DPVM55AP, Immunologic, Duiven, the Netherlands) and Vector Red (AP substrate kit III, #SK‐5100, Vector Labs, Burlingame, CA, USA) as chromogen. To remove the first primary antibody, sections were incubated at 121°C in citrate buffer (0.01 M, pH 6.0) for 10 minutes. Sections were then incubated for 1 h at room temperature with the second primary antibody (VE1). The second primary antibody was visualized with poly‐AP anti‐mouse antibody (BrightVision) and Vector Blue (AP substrate kit III, #SK‐5300, Vector Labs) as chromogen.
Evaluation of histology and immunohistochemistry
All labeled tissue sections were evaluated by two independent observers blinded to clinical data for the presence or absence of various histopathological parameters and specific immunoreactivity (IR) for the different markers. HE stained slides were used to evaluate the neuronal and glial components of the tumors, the presence of dysplastic neurons, calcifications, hemosiderin deposition and perivascular cuffs of lymphocytes. Ki67 staining was examined using an ocular grid and counting 1000 cells from representative fields of the tumor (the section border and hemorrhagic areas were omitted). The result was recorded as the Ki67 labeling index of the immunostained nuclei (number of labeled cells per total number of cells, excluding vascular cells and lymphocytes).
We also semi‐quantitatively evaluated the IR for the different markers, such as synaptophysin, glial fibrillary acidic protein (GFAP) and CD34, MHC‐I, MHC‐II, IDH1(R132H) and Lin28A. The intensity of the staining was evaluated as previously described 37, 52, using a semi‐quantitative scale ranging from 0 to 3 (0: negative; 1: weak; 2: moderate; 3: strong IR). All areas of the specimen were examined and the score represents the predominant cell staining intensity found in each case. The approximate proportion of cells showing IR for the different makers (1: single to 10%; 2: 11%–50%; 3: >50%) was also scored to give information about the relative number (“frequency” score) of positive cells within the tumor areas. In case of disagreement, independent re‐evaluation was performed by both observers to define the final score. As proposed before 5, 37, the product of these two values (intensity and frequency scores) was taken to give the overall score (immunoreactivity total score; IRS). For the pS6 staining, an immunoreactivity score of 2 or more was considered to be positive. To analyze the percentage of VE1 positive cells that express pS6 or pLKB1 (n = 8, 5 GGs, 3 DNTs), in each specimen, two representative, adjacent, non‐overlapping fields of the areas of interest were captured and digitized using a laser scanning confocal microscope. The total number of cells stained with pS6 (or pLKB1) or VE1, as well as the number of double‐labeled cells, were counted and percentages were calculated [expressed as mean ± standard error of mean (SEM)] of cells co‐expressing pS6 (or pLKB1) and VE1.
DNA extraction and BRAF V600E mutation analysis
Areas of representative tumor identified on HE stained sections by the neuropathologists (AE, WS, MT) were marked and tumor DNA was extracted from 10‐μm‐thick sections of FFPE tissue using BiOstic FFPE Tissue DNA Isolation kit (MO BIO, Carlsbad, CA, USA) according to the manufacturer's instructions. We analyzed a total of 168 cases (in 6 cases, the amount of representative tumor tissue available was not sufficient for the analysis). The sections in which the tumor area was below 60% were macrodissected. Polymerase chain reaction (PCR) amplifications and sequencing for codon 600 of BRAF were performed as previously described 56. Purified PCR products were sequenced using the Big Dye Terminator Cycle Sequencing Kit (PerkinElmer Biosystems, Foster City, CA, USA).
Statistical analysis
Statistical analyses were performed with SPSS for Windows (SPSS 20, SPSS Inc., Chicago, IL, USA). Continuous variables were described with mean and ranges; categorical variables with proportions and percentages. The two‐tailed Student's t‐test or the non‐parametric Kruskal–Wallis test followed by Mann–Whitney U‐test was used to assess differences between the groups. Correlation between BRAF V600E status with histological/immunohistochemical and clinical features (duration of epilepsy, seizure frequency, age at surgery, age at seizure onset, epilepsy outcome, etc.) were assessed using the Spearman's rank correlation test. A value of P < 0.05 was defined statistically significant. In addition, we performed an unsupervised analysis comparing tumor recurrence, postoperative seizure outcome and different tumor diagnosis in BRAF, CD34 and pS6 positive tumors with tumors negative for these markers. We analyzed all GNT together without considering DNT variants. The Pearson's chi‐square test or the Fisher's exact test was used to analyze the association of the immunostainings with the clinical features. The results are reported in Table 4.
Table 4.
BRAF V600E, CD34 and pS6 expression in glioneuronal tumors and clinical features: tumor recurrence and postoperative seizure outcome. Abbreviations: GGs = gangliogliomas; DNTs = dysembryoplastic neuroepithelial tumors
| Parameters (positive/negative) | Recurrence (No. of patients) | Postoperative seizure outcome | Diagnosis | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Without | With | P‐valuea | Engel I | Engel II‐IV | P‐valuea | GG | DNT | P‐valuea | |
| BRAF V600E | 63/105 | 0/6 | 0.088 | 43/90 | 17/16 | 0.040 | 38/55 | 23/54 | 0.137 |
| pS6‐DN | 96/72 | 2/4 | 0.406 | 61/72 | 29/4 | <0.001 | 83/10 | 12/65 | <0.001 |
| CD34 | 107/60 | 0/6 | 0.003 | 80/53 | 21/11 | 0.568 | 66/27 | 39/38 | 0.008 |
| pS6 & CD34 | 73/38 | 0/4 | 0.016 | 47/39 | 20/3 | 0.005 | 62/7 | 9/35 | <0.001 |
| BRAF V600E & pS6‐DN | 47/56 | 0/4 | 0.129 | 27/56 | 17/4 | <0.001 | 38/10 | 7/49 | <0.001 |
| BRAF V600E & CD34 | 55/53 | 0/6 | 0.028 | 39/49 | 14/9 | 0.157 | 32/22 | 22/37 | 0.020 |
| BRAF V600E & pS6‐DN & CD34 | 40/37 | 0/4 | 0.116 | 24/38 | 14/3 | 0.001 | 32/7 | 7/34 | <0.001 |
Fisher's exact test or Pearson's chi square test.
Results
Case material and histological features
Among the 174 patients included in the study (Table 1), 168 had a history of chronic pharmacoresistant epilepsy. Postoperatively, 133 patients (79%) were completely seizure free (Engel's class I). The mean age at diagnosis for DIG was 6.5 years and they were significantly younger (P < 0.05) than patients with GG (mean age: 24.7 years) and DNT (mean age: 24.7 years). There was no male or female predominance within each tumor group. All the DNTs and DIGs were supratentorial tumors. Of the 93 GGs, 90 tumors were supratentorial (96.77%), 1 from the posterior fossa and 2 from the spinal cord/brainstem. One hundred seventy‐three of the 174 patients were alive at the last follow‐up. Evidence of hippocampal sclerosis was detected in 15% of GGs and 14% of DNTs; in addition, in 4 GG and 5 DNTs, areas of dyslamination [FCD type IIIb 15] were observed (Table 1).
The GGs were composed of dysplastic neuronal cells lacking uniform orientation, surrounded by neoplastic astrocytes (Figure 1A–F) and included 90 GG WHO grade I and 4 GG WHO grade III. Tumor recurrence with histological change occurred in two histologically benign GG (after partial resection), two GG WHO grade III and two DNT. The diagnosis at the second operation was anaplastic astrocytoma [n = 2; IDH1(R132H) negative; no P53 mutation], glioblastoma multiforme [n = 2; IDH1(R132H) negative; no P53 mutation] and DNT [n = 2 IDH1(R132H) negative; no. 1p19q co‐deletion]. Expression of the precursor cell marker, CD34 [13; Supporting Information Figure S1A–B], was observed in 71% of GGs (Figure 5A). The DIGs were characterized by a prominent desmoplastic stroma and displayed both astrocytic and ganglionic differentiation (Figure 2A–D). CD34 expression was observed in three of four DIGs (75%; Figure 5A). The DNTs were composed of a mixture of neuronal cells, astrocytes and a prominent population of oligodendroglia‐like cells (Figure 3A–B). Our series of DNTs consist of 77 specimens (WHO grade I) including 15 simple DNT, 33 complex DNT (characterized by the presence of specific glioneuronal elements) as well as the controversial non‐specific forms, including 22 diffuse DNT (with diffuse cortical infiltration pattern and relative lack of glioneuronal element or nodular growth pattern) and 7 mixed DNT/GG (Table 1), defined on the basis of the predominant growth pattern [59; applied criteria for the diffuse DNT variants are summarized in Supporting Information Table S1]. Previous molecular analysis reported lack of 1p19q co‐deletion in all cases. Expression of CD34 (Figures 4H and 5A) was observed in 51% of DNTs (Figure 5A). None of the 174 GNT cases displayed immunohistochemical detection of IDH1(R132H) or Lin28A 42 (not shown).
Figure 1.
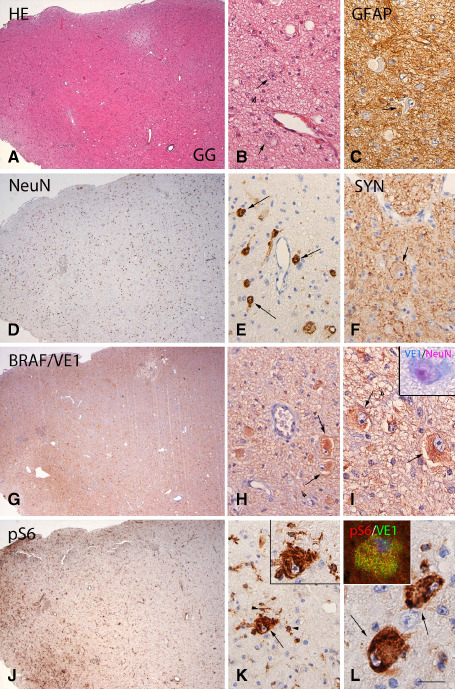
BRAF V600E mutated ganglioglioma (GG): histopathological features, VE1 and pS6 immunoreactivity. A, B. Hematoxylin & eosin (HE) staining of GG showing the mixture of neuronal cells, lacking uniform orientation (arrows in B) and glial cells. C. Glial fibrillary acidic protein (GFAP) immunoreactivity (IR) showing the astroglial component of the tumor. D, E. NeuN staining detects the neuronal component (nuclear staining, arrows in E) of GG. F. Synaptophysin expression along the cell borders of dysplastic neuron (arrow). G–I. BRAF V600E (VE1) immunostaining showing diffuse IR within the tumor area, with prominent expression in large dysplastic cells (arrows in H and I); inset in (I) shows VE1 IR (blue) in a dysplastic neuron (NeuN positive, red). J–L. Phosphorylated (p)‐S6 expression within the tumor area with IR in microglial cells (arrowheads in K) and dysplastic neurons (arrows in K and L); co‐localization of pS6 with VE1 is shown in the insert in (L). Scale bar in (L): A, D, G, J: 400 μm; B, C, E, F, H, K: 80 μm. I, L: 40 μm.
Figure 5.
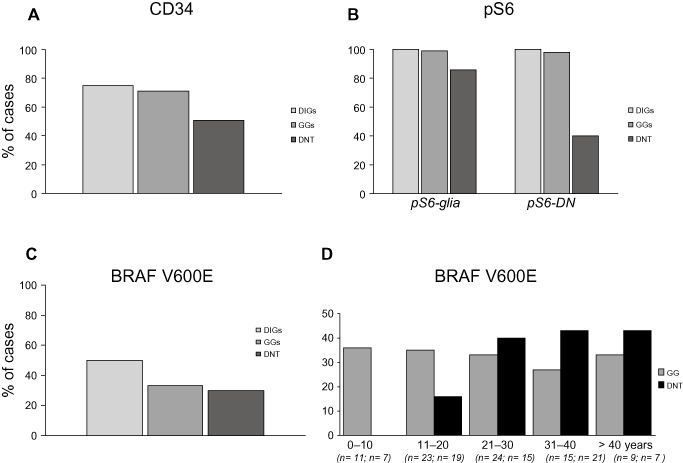
Frequency of CD34, pS6 expression (IR) and BRAF V600E mutation in desmoplastic infantile gangliogliomas (DIGs), gangliogliomas (GGs) and dysembryoplastic neuroepithelial tumors (DNTs). A. CD34 expression is detected in all three tumor types. B. DIGs and GGs show stronger expression of pS6 in both glial cells and dysplastic neurons (DN) compared to DNTs. C. BRAF V600E mutation is detected in all three tumor types. D. BRAF V600E mutation shows increase of frequency with the age in DNTs.
Figure 2.
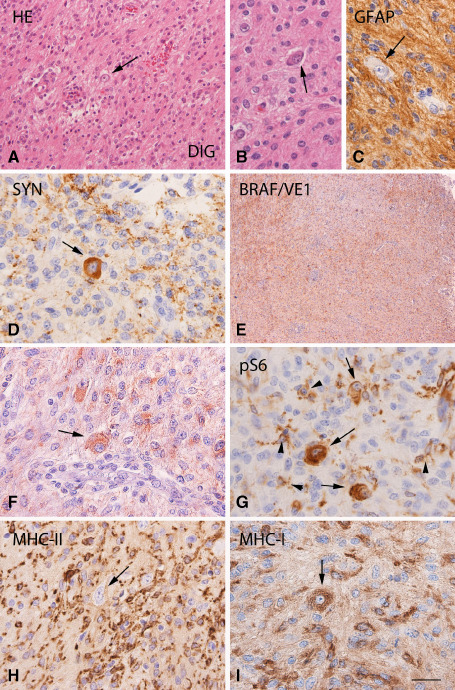
BRAF V600E mutated desmoplastic infantile ganglioglioma (DIG): histopathological features, VE1 and pS6, MHC‐II and MHC‐I immunoreactivity. A, B. Hematoxylin & eosin (HE) staining of DIG with a prominent population of neoplastic astrocytes and scattered dysplastic neurons (arrows). C. Glial fibrillary acidic protein (GFAP) immunoreactivity (IR) showing the astroglial component of the tumor. D. Synaptophysin IR in a dysplastic neuron (arrow). E, F. BRAF V600E (VE1) immunostaining showing diffuse IR within the tumor area, with expression also in large dysplastic cells (arrow in F). G. Phosphorylated (p)‐S6 expression within the tumor area with IR in microglial cells (arrowheads) and dysplastic neurons (arrows). H, I. Prominent MHC class II and I antigen (MHC‐II; MHC‐I) expression within the tumor area, with MHC‐I IR in large dysplastic cells (arrows in I). Scale bar in (I): A–H: 80 μm; B, C, D, F, G, I: 40 μm; E: 400 μm.
Figure 3.
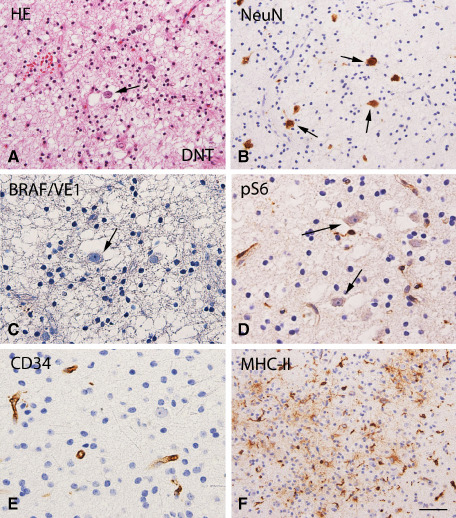
Dysembryoplastic neuroepithelial tumor (DNT; BRAF wild‐type status): histopathological features, VE1 and pS6, CD34 and MHC‐II immunoreactivity. A. Hematoxylin & eosin (HE) staining of DNT showing a typical heterogeneous cellular composition, with floating neurons (arrow) surrounded by a prominent population of oligodendroglia‐like cells. B. NeuN staining detects the neuronal component of DNT. C. No detectable BRAF V600E (VE1) immunoreactivity (IR) within the tumor area. D. No detectable phosphorylated (p)‐S6 IR in neuronal cells (arrows). E. CD34 shows IR only in blood vessels. F. MHC class II antigen (MHC‐II) expression within the tumor area (microglial cells). Scale bar in (F): A, B, F: 80 μm; C, D, E: 40 μm.
Figure 4.
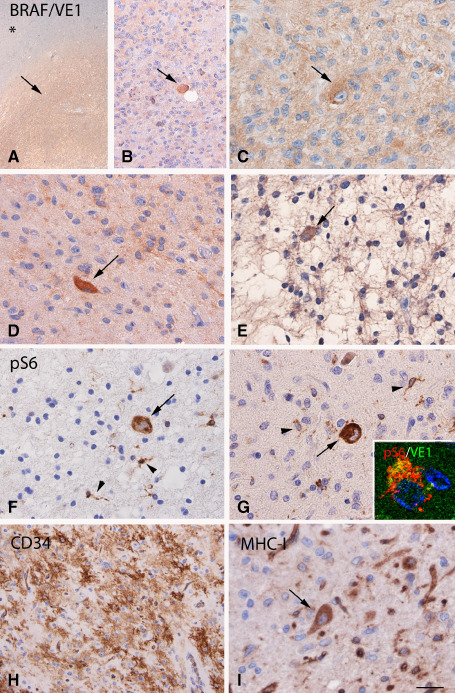
BRAF V600E mutated dysembryoplastic neuroepithelial tumor (DNT): VE1 and pS6, CD34 and MHC‐I. A–E. BRAF V600E (VE1) immunostaining, showing diffuse IR within the tumor area (arrow, but not in the adjacent non‐neoplastic tissue, asterisk in A), with expression also in cells with ganglionic appearance (arrows in B–D) and occasionally in floating neurons (arrow in E). F–G. Phosphorylated (p)‐S6 expression within the tumor area with IR in microglial cells (arrowheads) and dysplastic neurons (arrows); co‐localization of pS6 with VE1 is shown in the insert in (G). H. Strong CD34 IR within the tumor area. I. MHC class I antigen (MHC‐I) expression within the tumor area with expression also in dysplastic neurons (arrow). Scale bar in (I): A, 400 μm; B, H: 80 μm. C–G, I: 40 μm.
MHC‐I and MHC‐II IR in GNTs
As previously reported in GG specimens 51, MHC‐I (major histocompatibility complex class I) IR was detected in blood vessels, neurons and microglial cells, but not in astrocytes (Supporting Information Figures S1F–G and S4A). A similar IR pattern was observed in DIGs and DNTs (Figures 2I and 4I). The IRS of MHC‐I in both microglial and neuronal cells was found to be significantly higher in GG than in DNTs (Supporting Information Figure S4A). Variable amount of activated microglial cells positive for MHC‐II were observed within the tumor in a large majority of GNT specimens. The MHC‐II IRS was, however, significantly higher in GGs compared with DNTs (Supporting Information Figure S4A).
pS6 IR in GNTs
In agreement with previous studies 18, 51, expression of pS6 (a reliable marker of mTOR activation) was observed within GG samples in our cohort; pS6 IR was detected in microglial cells (as confirmed by co‐localization with microglial markers; not shown), as well as in the neuronal component of GGs (Figures 1J–L and 5). We used two antibodies directed against different phosphorylation sites of the S6 (Ser235/236 and Ser240/244; Table 2), taking into consideration that S6 can be phosphorylated at Ser235/236 not only by p70 ribosomal S6 protein kinase 1 but also by p90 ribosomal S6 kinase 3; however, similar IR patterns were observed using both these antibodies. As previously reported 18, cytoplasmic IR for p‐S6 was not detected in histologically normal cortex. In the present study, pS6 IR was also observed in DIGs (Figure 2G) and in a subpopulation of DNTs (Figures 4F–G and 5B; Supporting Information Figure S4B). The pS6 IRS in both microglial and neuronal cells was significantly higher in GGs compared to DNTs (Supporting Information Figure S4B and Table 4). The DNT cases with neuronal pS6 IR (n = 31) included 5 simple DNT, 17 complex DNT, 5 mixed DNT/GG and 4 diffuse DNT. Expression of pLKB1 was detected only in cases containing pS6 and BRAF V600E positive dysplastic neurons (Supporting Information Figure S2, GG).
Correlation of pS6 expression with clinical and histological/immunohistochemical features in GNTs
For both GGs and DNTs, we found no statistically significant association between pS6 IR and clinical features, such as age at surgery, gender, location of the tumor, tumor recurrence or duration of epilepsy (Supporting Information Table S2; Table 4). However, in GGs, a positive correlation was observed between the pS6 IRS (in both microglia and dysplastic neurons) and the frequency of dysplastic neurons, the presence of perivascular cuffs of lymphocytes (P < 0.001), as well as the expression of CD34 (P < 0.05). A significant association was observed (in both GG and DNT specimens) between pS6 IR (dysplastic neurons) and MHC‐II and MHC‐I IR (dysplastic neurons). The number of patients with DIGs was too small to perform meaningful statistical comparisons. Unsupervised analysis of clinical features (tumor recurrence and postoperative seizure outcome) in tumors with and without pS6 positive dysplastic neurons showed a worse postoperative seizure outcome in pS6 positive GNT (P < 0.001; Table 4).
BRAF V600E expression in GNTs
In agreement with recent studies 20, 22, 41, expression of BRAF‐mutated protein (using a BRAF V600E mutation‐specific antibody clone VE1) was observed in both GG, DNT and DIGs specimens; in our cohort, BRAF V600E IR was detected in 38/93 (40.8%) of GGs, in 23/77 (29.8%) of DNTs, as well as in 2 of 4 DIGs (Figure 5). We did not detect BRAF V600E expression in simple DNTs. BRAF V600E expression was observed in two of four GG WHO grade III. All six cases with tumor recurrence were BRAF wild type. The staining intensity ranged from weak to strong.
In all GGs and DIGs, the BRAF V600E‐mutated protein was predominantly localized within the neuronal component of the tumor (Figures 1 and 2) and we observed co‐localization of BRAF V600E with the neuronal marker NeuN (Figure 1I). In 2 of VE1 positive DIGs and in 25 of the 38 VE1 positive GGs, IR was also observed in astroglial cells, as well as in cells with ganglion appearance, expressing both GFAP and synaptophysin. In one DIG and in five GGs, we could detect only small clusters of VE1‐positive ganglionic cells, and in six GGs, only few VE1‐positive dysplastic neurons were observed (Supporting Information Figure S3A, GG).
In DNTs, diffuse BRAF V600E immunostaining was observed within the tumor area in the glial nodules, and IR was also detected in neuronal cells, including cells with gangliocytic appearance and only occasionally in floating neurons (Figure 4A–E). The DNTs that were positive for BRAF V600E expression included 8/33 complex DNT, 9/22 diffuse DNT and 6/7 mixed DNT/GG (within the GG component). The perilesional cortex did not display VE1 IR (DNT Figure 4A; Supporting Information Figure S3B, GG); non‐specific staining was only occasionally seen in macrophages.
In GNTs with BRAF V600E mutation, we observed co‐localization of VE1 with pS6 and pLKB1 (Figures 1L and 4G; Supporting Information Figure S2); 89 ± 8% and 82 ± 7% of the cells positive for VE1 co‐expressed pS6 and pLKB1, respectively.
Correlation between BRAF V600E protein expression and BRAF DNA sequencing
DNA sequencing was performed in 168 of 174 cases (93 GGs; 4 DIG; 71 DNTs). In 1 of 2 VE1‐positive DIG cases, in 27 of 38 VE1‐positive GG cases and in 21 of 23 VE1‐positive DNT cases, we detected the BRAF V600E mutation. None of the VE1‐negative GG or DNT cases showed the BRAF V600E mutation. The VE1‐positive cases (n = 13) failing to demonstrate BRAF V600E mutation were characterized by focal IR in small groups of ganglionic cells or in few dysplastic neurons. In these discordant cases, immunostaining and BRAF sequencing was repeated. In addition, we performed laser capture microdissection; however, the amount of DNA obtained from the few positive cells was not sufficient for additional analysis. Concordance between immunohistochemistry and sequencing was observed in 154/168 tumors (91.6%; 82/93 GGs, 88%; 3/4 DIG, 75%; 69/71 DNTs, 97.1%).
Correlation of BRAF V600E expression with clinical and histological/immunohistochemical features in GNTs
We performed a comparative analyses for GGs and DNTs (151 cases; 82 GG; 69 DNTs) in which immunohistochemistry and sequencing analysis for BRAF V600E mutation were concordant (Table 3). This analysis was not performed for DIGs because of the low number of cases.
Table 3.
Correlation of BRAF V600E expression with clinical data and histological/immunohistochemical features in GGs and DNTs. Abbreviation: nd = not done; GFAP = glial fibrillary acidic protein; GG = gangliogliomas; DNTs = dysembryoplastic neuroepithelial tumors; IRS = immunoreactivity score
| Parameter | BRAF V600E | BRAF wt | P‐valuea (correlation strength)b | |||
|---|---|---|---|---|---|---|
| GGs | DNTs | GGs | DNTs | GGs | DNTs | |
| Age at surgery (mean ± SEM) | 23.30 (± 2.57) | 30.71 (± 1.86) | 24.67 (± 1.57) | 22.90 (± 1.75) | 0.481 | 0.01 (0.296)** |
| Gender (male/female) | 15/12 | 10/11 | 25/30 | 25/23 | 0.396 | 0.737 |
| Location (temporal/other) | 22/5 | 20/1 | 40/15 | 38/10 | 0.329 | 0.120 |
| Duration of epilepsy (mean ± SEM) | 8.44 (± 1.51) | 17.06 (± 2.56) | 11.20 (± 1.42) | 12.50 (± 1.61) | 0.379 | 0.080 |
| Postoperative outcome (Engel's class I/other) | 19/6 | 19/2 | 39/11 | 33/15 | 0.848 | 0.055 (−0.232) |
| Histology (presence; % of cases) | ||||||
| Dysplastic/atypical neurons | 100 | 100 | 100 | 4.2 | nd | <0.001 (0.935)*** |
| Lymphocytic cuffs | 96.3 | 4.8 | 23.6 | 0 | <0.001 (0.684)*** | 0.132 |
| Calcifications | 37 | 38.1 | 23.6 | 20.8 | 0.209 | 0.137 |
| Hemosiderin deposition | 0 | 4.8 | 14.5 | 12.5 | 0.03 (−0.230)* | 0.335 |
| Ki67 (%) | 1.11 (± 0.08) | 1 | 1.15 (± 0.06) | 1.06 (± 0.03) | 0.635 | 0.248 |
| IRS | ||||||
| MHC‐I‐glia | 5.19 (± 0.45) | 2.00 (± 0.32) | 4.82 (± 0.32) | 1.20 (± 0.16) | 0.492 | 0.04 (0.250)* |
| MHC‐I‐DN | 4.26 (± 0.45) | 1.38 (± 0.36) | 3.75 (± 0.33) | 0.52 (± 0.16) | 0.571 | 0.020 (0.279)* |
| MHC‐II | 7.07 (± 0.43) | 3.90 (± 0.41) | 6.36 (± 0.31) | 2.85 (± 0.23) | 0.181 | 0.021 (0.278)* |
| CD34 | 5.11 (± 0.54) | 6.81 (± 0.56) | 2.47 (± 0.33) | 1.77 (± 0.40) | <0.001 (0.437)*** | <0.001 (0.650)*** |
| pS6‐glia | 4.30 (± 0.24) | 1.86 (± 0.23) | 3.40 (± 0.17) | 1.83 (± 0.13) | 0.001 (0.356)*** | 0.696 |
| pS6‐DN | 6.30 (± 0.42) | 0.90 (± 0.19) | 3.16 (± 0.28) | 0.42 (± 0.09) | <0.001 (0.574)*** | 0.023 (0.274)** |
| Others | ||||||
| Synaptophysin | 3.30 (± 0.51) | 1.14 (± 0.23) | 1.98 (± 0.28) | 0.48 (± 0.14) | 0.01 (0.285)** | 0.008 (0.318)** |
| NeuN | 7.00 (± 0.32) | nd | 3.60 (± 0.18) | nd | <0.001 (0.760)*** | nd |
| GFAP | 6.74 (± 0.45) | 4.86 (± 0.25) | 6.98 (± 0.32) | 4.7 (± 0.21) | 0.705 | 0.840 |
*P < 0.05; **P < 0.01; ***P < 0.001.
Student's t‐test or Kruskal–Wallis test followed by Mann–Whitney U‐test.
Spearman's rank correlation coefficient.
For both GG and DNT, we found no statistically significant association between the BRAF status and several clinical and histopathological features, including gender, location of the tumor (temporal; extratemporal), duration of epilepsy, calcifications, hemosiderin deposition, presence of cortical dysplasia and proliferation index (Table 3). Patients with BRAF‐mutated DNTs were significantly older at surgery (30.7 ± 1.8 years) compared to DNTs with BRAF wild‐type status (22.9 ± 1.8 years).
In both GGs and DNTs, the presence of BRAF V600E mutation was significantly associated with the expression of CD34 (P < 0.001), pS6 in dysplastic neurons (GGs, P < 0.001; DNTs, P = 0.023) and synaptophysin (GGs, P = 0.01; DNTs, P = 0.008).
In GGs, the presence of lymphocytic cuffs was more frequent in BRAF‐mutated cases (31 vs. 15.8%; P < 0.001), whereas hemosiderin deposition was observed only in BRAF wild‐type GGs (0% vs. 8.6%; P = 0.03). In GGs, the BRAF V600E mutation was also significantly associated with the expression of pS6 in microglial cells (P = 0.001) and NeuN (P < 0.001).
BRAF V600E mutation in DNTs was significantly associated with the expression of MHC‐I in both microglia and neuronal cells (P = 0.04 and P = 0.02) as well as with MHC‐II (P = 0.021). Figure 6 shows the IRS for MHC‐I, MHC‐II, CD34, pS6, synaptophysin and GFAP in BRAF wild‐type or BRAF‐mutated GGs and DNTs.
Figure 6.
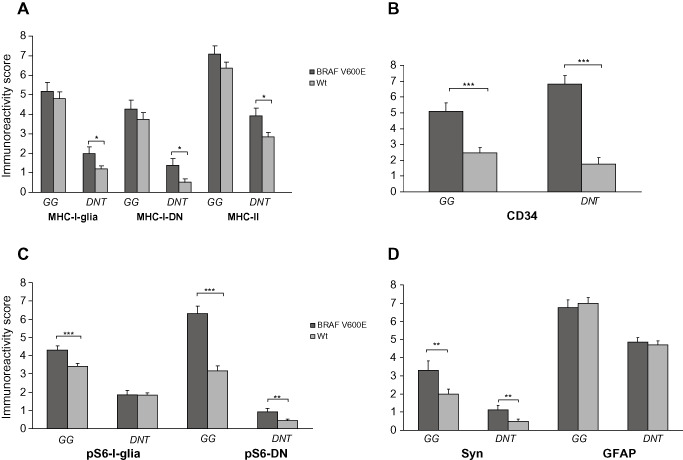
MHC‐I, MHC‐II, CD34, pS6, synaptophysin and glial fibrillary acidic protein (GFAP) immunoreactivity score (IRS) in BRAF wild‐type or BRAF‐mutated gangliogliomas (GGs) and dysembryoplastic neuroepithelial tumor (DNTs) . A. MHC‐I (glial/neuronal) and MHC‐II IR scores (total score; mean ± SEM) in BRAF wild‐type (Wt) or BRAF‐mutated (BRAF V600E) GGs and DNTs. B. CD34 IR scores (total score; mean ± SEM) in BRAF wild‐type (Wt) or BRAF‐mutated (BRAF V600E) GGs and DNTs. C. pS6 (glial/neuronal) IR scores (total score; mean ± SEM) in BRAF wild‐type (Wt) or BRAF‐mutated (BRAF V600E) GGs and DNTs. D. Synaptophysin (Syn) and GFAP IR scores (total score; mean ± SEM) in BRAF wild‐type (Wt) or BRAF‐mutated (BRAF V600E) GGs and DNTs. Syn = synaptophysin; DN = dysplastic neuron; *P < 0.05; **P < 0.01; ***P < 0.001.
Unsupervised analysis of clinical features (tumor recurrence and postoperative seizure outcome) in tumors with and without positive BRAF V600E staining showed a worse postoperative seizure outcome in BRAF V600E‐positive GNTs (P < 0.05) and this association was stronger in GNT expressing also pS6 within the neuronal component of the tumor (P < 0.001; Table 4). There were no statistical differences in the age at surgery and the duration of epilepsy between BRAF V600E positive (age at surgery: 26.3 ± 1.7; duration of epilepsy: 12.9 ± 1.2) and negative GNTs (age at surgery: 23.1 ± 1.1; duration of epilepsy: 11.2 ± 0.9). The expression of BRAF V600E, pS6 and CD34 was significantly higher in GGs compared to DNTs. The expression of CD34 did not influence the seizure outcome. In our cohort, all six cases with recurrence were BRAF wild‐type/CD34 negative tumors, whereas no significant differences were detected for pS6 or the combination of BRAF V600E with pS6 and/or CD34 (Table 4).
Discussion
In the present study, we evaluate a large cohort of GNTs and provide evidence of mTOR signaling pathway activation and of BRAF V600E mutation in a large percentage of these tumors, including GGs, DNTs and DIGs. The significance of these findings, including their pathogenic and diagnostic implications, as well as correlations to histopathological and clinical features, such as patients' epileptogenesis and clinical course, are discussed below.
mTOR signaling pathway across GNT subtypes
mTOR signaling pathway functions as a key regulator of cell growth, proliferation, differentiation and survival during brain development and increasing evidence supports the role of this signaling pathway in a wide variety of neurological disorders, including brain tumors and MCD 1, 21, 23, 67. Immunohistochemical studies provided evidence for mTOR hyperactivation in enlarged, dysmorphic neurons and balloon/giant cells in FCD type II, hemimegalencephaly and cortical tubers in TSC 6, 37, 49. Thus, the activation of this signaling pathway, evidenced by the detection of S6 phosphorylation, could serve as a biomarker for a group of histopathologically related epileptogenic glioneuronal lesions [for review, see 21]. GNTs, particularly GGs (characterized by the presence of dysmorphic ganglion cells), share pathologic features with FCD and TSC. In our previous study, we demonstrated activation of several components of this signaling pathway (including the downstream effector and a hallmark of mTORC1 activation, pS6), in a small cohort (n = 9) of GGs 18.
In the present study, we provide additional evidence of mTOR overactivation in a large cohort of GGs (n = 93). We confirmed the consistent pS6 expression in the neuronal component of the tumors (dysmorphic neurons) and provide evidence of pS6 expression in the neuronal component of DIGs. In contrast to our previous study, including a small cohort of simple forms of DNTs 18, here we provide evidence of mTOR activation in DNTs (40% of cases) in both pediatric and adult patients. Although the histopathological classification of GNT remains a matter of debate and the definition of “non‐specific” DNT variants (such as diffuse DNT) is still controversial, our observations indicate that the phosphorylation of S6, as a marker of mTOR activation, in neurons represents a common feature of GNTs, supporting the potential etiological relationship between GGs and DNTs.
Interestingly, in GGs, we observed a positive correlation between the pS6 expression and the presence of perivascular cuffs of lymphocytes, as well as with the MHC‐II and MHC‐I expression within the tumor. Whether the activation of mTOR signaling (involving both neuron and microglial cells) could contribute to the sustained inflammatory reaction in glioneuronal lesions is still unclear and requires further functional studies. In addition, expression of pS6 in both microglial and neuronal cells was more prominent in CD34‐positive GGs. In agreement with previous studies, CD34 positivity was observed in >70% GGs, as well as in DNTs 17, 20, 25, 59. The association between pS6 and CD34 may reflect the presence of a population of immature cells (glioneuronal precursor cells) in tumors, characterized by constitutive activation of the mTOR signaling cascade.
The mechanisms underlying activation and regulation of the mTOR signaling in GNTs are still a matter of discussion. Although mutational analysis of TSC1 and TSC2 was not performed in the GNT specimens examined in this study, previous studies have not identified mutations in GGs 50 and only a somatic mutation in intron 32 of the TSC2 gene was reported in one GG patient in glial cells, but not in dysplastic neurons 10. The secretion of growth factors, such as vascular endothelial growth factor (VEGF), in the microenvironment of these tumors has also been suggested to contribute to the activation of the mTOR pathway 35, 55, 61.
Whether seizure activity contributes to the mTOR activation has also to be taken into consideration. In this respect, mTOR dysregulation has also been demonstrated in acquired forms of epilepsy (in both humans and animal models), supporting the role of mTOR as a key regulator of cellular changes involved in epileptogenesis [for reviews, see 34, 48, 63]. Interestingly, in our cohort, the neuronal expression of pS6 was associated with a worse postoperative seizure outcome. However, expression of pS6 was observed also in GGs without history of seizure and pS6 IR was not detected in all DNTs associated with seizure activity, or in normal cortex adjacent to the epileptogenic lesions. Moreover, for both GGs and DNTs, we found no statistically significant association between pS6 IR and the duration of epilepsy. It would be interesting to identify the mechanisms that regulate the mTOR signaling to explain the observed differences in activation of this pathway in the different GNTs. Several other pathways such as the ERK1/2‐, the LKB1‐AMPK‐ and the Wnt‐signaling pathway are known to regulate the TSC1/TSC2 complex 8, 38, 43, 47 and GGs have been reported in Peutz‐Jeghers patients with a mutation of the LKB1 gene 24, 53. Previous studies support the biological relationship between LKB1 and mTOR regulation 54, 58 and more recent studies indicate that BRAF V600E mutant cells have a dysfunctional LKB1‐AMP‐activated protein kinase (AMPK)‐mTOR signaling 30, 68. Interestingly, in BRAF‐mutated papillary thyroid carcinoma, a positive association between BRAF V600E mutation and mTOR pathway activation has been reported, possibly mediated by phosphorylation of LKB1Ser428 32. Accordingly, we observed increased expression of pLKB1 in dysplastic neurons of BRAF V600E tumors and the presence of BRAF V600E mutation was significantly associated with the expression of pS6 in dysplastic neurons. Thus, these observations suggest BRAF‐induced phosphorylation of LKB1 as possible mechanism contributing to mTOR activation in BRAF V600E‐mutated GNTs, possibly through uncoupling of the LKB1‐AMPK‐mTOR signaling.
BRAF V600E mutation across GNT subtypes
In the present study, we confirmed the occurrence of BRAF V600E mutation in a large cohort of non‐IDH1(R132H)‐mutated GNTs, including both GGs and DNTs.
In GGs, the mutated BRAF protein was detected in 38/93 (40%) of GGs. In a recent study (including 71 GGs), this mutation was identified by immunohistochemistry in up to 58% of GGs 41. In agreement with this study 41, in our cohort, the mutated protein was detected in both the glial and the neuronal components of the tumor, strongly supporting the involvement of neuronal component in the pathogenesis of BRAF mutated GGs. In several cases, we only detected BRAF IR in small nests or sparse dysplastic neurons with ganglioid morphology and this distribution may explain the difference in frequency of the BRAF mutation reported in different cohorts of GGs 26, 41, 56, as well as the VE1‐positive GG cases failing to demonstrate BRAF V600E mutation by DNA sequencing. Thus, in these cases, the VE1 antibody may represent a more specific and sensitive tool compared to sequencing, emphasizing the importance of performing DNA sequencing from macrodissected tissue, or even, when possible, to apply single‐cell laser‐capture microdissection 41. In addition, we also reported the presence of BRAF V600E mutation in DIG, rare entities within the spectrum of GNTs 46, 60.
In previous studies, including only few DNTs, BRAF V600E mutation was not detected 26, 56. In a recent study, including 20 DNTs and focusing on pediatric tumors, this mutation was identified by immunohistochemistry in up to 30% of DNTs 20. Similarly, in our larger cohort of 77 DNTs (both adult and pediatric cases), we could confirm the presence of mutated BRAF protein 29.8% of the cases. In DNTs, diffuse immunostaining was observed within the tumor area in the glial nodules and we observed an excellent concordance with DNA sequencing (98.2%). As in GGs, the mutated protein was also detected in neuronal cells, including cells with ganglionic appearance and only occasionally in floating neurons. Interestingly, we did not detect BRAF V600E mutation in simple DNTs, but only in complex and non‐specific forms. The presence of BRAF mutation in about 30% of DNTs, based on two independent studies [20; present study], strongly supports a relationship between DNTs and GGs, pointing to the pathogenic role of BRAF in different entities within the large spectrum of GNTs together with other lower grade tumors, such pilocytic astrocytomas (approximately 10% of cases) and PXA [60% of cases 28, 56]. The search for the underlying mechanisms of low‐grade glial/GNT development in BRAF wild‐type cases represents a major challenge for future studies.
Several histopathological features were evaluated in our cohort and we found a statistically significant association between the BRAF status and synaptophysin in both GGs and DNTs as well as for BRAF V600E mutation with NeuN in GGs. This finding was not so surprising, considering the neuronal expression of BRAF V600E‐mutated protein. Accordingly, Koelsche et al 41 also reported a more frequent expression of synaptophysin in BRAF‐mutated GGs. Our study confirms the predominant expression of BRAF V600E‐mutated protein in the neuronal component of GNTs, including also DNTs and DIGs.
GGs with BRAF V600E mutation showed lymphocytic infiltrates more frequently. This association has also been reported by Koelsche et al in GNTs 41, as well as in other BRAF‐mutated tumors 27, 44. In addition, DNTs with BRAF V600E mutation showed higher expression of MHC‐II and MHC‐I. MHC‐I expression in neurons and microglial cells appears to be a feature of the immune response occurring in epileptogenic glioneuronal lesions, including GNTs [51; present study]. These observations, suggest a possible role of the BRAF‐mutated protein in the regulation of the immune response. Accordingly, a potential immunogenicity of BRAF‐mutated protein has been shown in melanoma patients and suggested as a potential target for immunotherapy 2, 33. The potential immunogenicity of BRAF‐mutated protein in GNTs requires further investigation and could possibly be related to the above discussed positive association between the BRAF V600E mutation and mTOR pathway activation, which is known to influence both the innate and the adaptive immune response 45, 57, 66. This is particularly interesting in view of the observation that proinflammatory molecules may alter neuronal excitability and, in experimental models, have been shown to decrease the seizure threshold 7, 64, 65.
GGs and DNTs with BRAF V600E mutation showed a more frequent expression of CD34. This is in contrast to the two other recent studies in which CD34 was not differentially expressed in BRAF wild‐type and ‐mutated GG and DNT tumors 20, 41. However, the study of Chappe et al 20 focused on pediatric tumors, whereas our study consists of a larger cohort, including several adult DNT and GG cases. Thus, both CD34 and pS6 represent two markers that characterize GNTs with BRAF V600E mutation and these markers are particularly highly expressed in GGs. In addition, our results suggest a prognostic value for BRAF V600E and pS6 as potential indicators of worse postoperative seizure outcome.
Similar to Koelsche et al 41, we observed an association between BRAF wild‐type and hemosiderin deposition in GGs (but not in DNTs). However, in our cohort, hemosiderin deposition was not associated with older patient age. In our series of GGs, we did not detect a correlation between the BRAF status and the age at surgery or other clinical variables, such as duration of epilepsy or seizure outcome after surgery. In our retrospective study, we could not determine whether GGs or DNTs with BRAF V600E mutation would display a tendency to malignant transformation. In our cohort, the number of patients with tumor recurrence (all BRAF V600E negative tumors) was too small to evaluate the possible prognostic value of the BRAF status on recurrence‐free survival, as recently reported in GG 22.
In conclusion, our findings support the pathogenic role of BRAF V600E mutation in different entities within the large spectrum of GNTs, with a cellular distribution that points to the involvement of the neuronal component in the pathogenesis of BRAF‐mutated tumors. The immunophenotype of these tumors is characterized by CD34 expression, indicating the presence of glioneuronal precursor cells, and phosphorylation of S6, indicating activation of the mTOR signaling cascade. Thus, immunohistochemical detection of BRAF V600E‐mutated protein may be valuable in the diagnostic evaluation of these glioneuronal lesions and the observed association with mTOR activation may aid in the development of targeted treatment involving specific pathogenic pathways.
Supporting information
Figure S1. BRAF V600E mutated ganglioglioma (GG): CD34, MHC‐II and MHC‐I immunoreactivity. A, B. Prominent CD34 immunoreactivity (IR) within the tumor area. C–E. Prominent MHC class II antigen (MHC‐II) expression within the tumor area, with IR in microglial cells (arrows in D and E) and occasionally in large dysplastic neurons (arrowhead in E). F, G. Prominent MHC class I antigen (MHC‐I) expression within the tumor area, with IR in large dysplastic neurons (arrows in G). Scale bar in (G): A, C, F: 400 μm; B, G: 80 μm; D, E: 40 μm.
{kind=link}
Figure S2. BRAF V600E mutated ganglioglioma (GG): pLKB1. A–C. Phosphorylated (p)‐LKB1 immunoreactivity (IR) within the tumor area in large dysplastic neurons (arrows in A; B, C); co‐localization of pLKB1 with VE1 is shown in the insert in (A). Scale bar in (C): A. 40 μm; B, C. 20 μm.
{kind=link}
Figure S3. VE1 immunoreactivity in ganglioglioma (GG) and in control cortex. A. GG with only few VE1‐positive dysplastic neurons (arrows and insert). B. Perilesional cortex without VE1 immunoreactivity (IR). Scale bar in (B): A. 80 μm; B. 160 μm.
{kind=link}
Figure S4. MHC‐I, MHC‐II and pS6 immunoreactivity score (IRS) in GGs and DNTs. A. MHC‐I (glial/neuronal) and MHC‐II IR scores (total score; mean ± SEM) in GGs and DNTs. B. pS6 (glial/neuronal) IR scores (total score; mean ± SEM) in GGs and DNTs (***P < 0.001). Desmoplastic infantile gangliogliomas (DIGs); gangliogliomas (GGs); dysembryoplastic neuroepithelial tumors (DNTs); dysplastic neurons (DN).
Table S1. Criteria to distinguish diffuse DNTs from other DNT types.
Table S2. Correlation of pS6 IRS with clinical data and histological/immunohistochemical features in GGs and DNTs.
Acknowledgments
This work was supported by the National Epilepsy Fund—“Power of the Small”, the Hersenstichting Nederland (NEF 012‐12) and KIKA (Stichting Kinderen Kankervrij; AE, AP). We are grateful to Prof. von Deimling (Department of Neuropathology, Ruprecht‐Karls‐Universität, Heidelberg) for providing the VE1 hybridoma supernatant (clone VE1). We are grateful to G.K.J. Hooijer and J.A. Reinten for skillful technical assistance and Prof. C.J.M. van Noesel for providing the tissue microarray of colon carcinoma specimens.
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Disclosure: D.C. has applied for a patent on the diagnostic use of BRAF V600E mutant‐specific antibody VE1. All terms are being managed by the German Cancer Research Center in accordance with its conflict of interest policies. The other authors do not have conflict of interest to disclose.
References
- 1. Alayev A, Holz MK (2013) mTOR signaling for biological control and cancer. J Cell Physiol 228:1658–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Andersen MH, Fensterle J, Ugurel S, Reker S, Houben R, Guldberg P et al (2004) Immunogenicity of constitutively active V599EBRaf. Cancer Res 64:5456–5460. [DOI] [PubMed] [Google Scholar]
- 3. Anjum R, Blenis J (2008) The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol 9:747–758. [DOI] [PubMed] [Google Scholar]
- 4. Aronica E, Gorter JA, Jansen GH, van Veelen CW, van Rijen PC, Ramkema M, Troost D (2003) Expression and cell distribution of group I and group II metabotropic glutamate receptor subtypes in taylor‐type focal cortical dysplasia. Epilepsia 44:785–795. [DOI] [PubMed] [Google Scholar]
- 5. Aronica E, Gorter JA, Redeker S, Ramkema M, Spliet WG, van Rijen PC et al (2005) Distribution, characterization and clinical significance of microglia in glioneuronal tumours from patients with chronic intractable epilepsy. Neuropathol Appl Neurobiol 31:280–291. [DOI] [PubMed] [Google Scholar]
- 6. Aronica E, Boer K, Baybis M, Yu J, Crino PB (2007) Co‐expression of cyclin D1 and phosphorylated ribosomal S6 proteins in hemimegalencephaly. Acta Neuropathol 114:287–293. [DOI] [PubMed] [Google Scholar]
- 7. Aronica E, Ravizza T, Zurolo E, Vezzani A (2012) Astrocyte immune responses in epilepsy. Glia 60:1258–1268. [DOI] [PubMed] [Google Scholar]
- 8. Astrinidis A, Henske EP (2005) Tuberous sclerosis complex: linking growth and energy signaling pathways with human disease. Oncogene 24:7475–7481. [DOI] [PubMed] [Google Scholar]
- 9. Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB (2012) A developmental and genetic classification for malformations of cortical development: update 2012. Brain 135(Pt 5):1348–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Becker AJ, Lobach M, Klein H, Normann S, Nothen MM, von Deimling A et al (2001) Mutational analysis of TSC1 and TSC2 genes in gangliogliomas. Neuropathol Appl Neurobiol 27:105–114. [DOI] [PubMed] [Google Scholar]
- 11. Becker AJ, Blumcke I, Urbach H, Hans V, Majores M (2006) Molecular neuropathology of epilepsy‐associated glioneuronal malformations. J Neuropathol Exp Neurol 65:99–108. [DOI] [PubMed] [Google Scholar]
- 12. Blumcke I, Wiestler OD (2002) Gangliogliomas: an intriguing tumor entity associated with focal epilepsies. J Neuropathol Exp Neurol 61:575–584. [DOI] [PubMed] [Google Scholar]
- 13. Blumcke I, Giencke K, Wardelmann E, Beyenburg S, Kral T, Sarioglu N et al (1999) The CD34 epitope is expressed in neoplastic and malformative lesions associated with chronic, focal epilepsies. Acta Neuropathol 97:481–490. [DOI] [PubMed] [Google Scholar]
- 14. Blümcke I, Vinters HV, Armstrong D, Aronica E, Thom M, Spreafico R (2009) Malformations of cortical development and epilepsies: neuropathological findings with emphasis on focal cortical dysplasia. Epileptic Disord 11:181–193. [DOI] [PubMed] [Google Scholar]
- 15. Blumcke I, Thom M, Aronica E, Armstrong DD, Vinters HV, Palmini A et al (2011) The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia 52:158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blumcke I, Thom M, Aronica E, Armstrong DD, Bartolomei F, Bernasconi A et al (2013) International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: a task force report from the ILAE Commission on Diagnostic Methods. Epilepsia 54:1315–1329. [DOI] [PubMed] [Google Scholar]
- 17. Bodi I, Selway R, Bannister P, Doey L, Mullatti N, Elwes R, Honavar M (2012) Diffuse form of dysembryoplastic neuroepithelial tumour: the histological and immunohistochemical features of a distinct entity showing transition to dysembryoplastic neuroepithelial tumour and ganglioglioma. Neuropathol Appl Neurobiol 38:411–425. [DOI] [PubMed] [Google Scholar]
- 18. Boer K, Troost D, Timmerman W, Spliet WGM, van Rijen PC, Aronica E (2010) Pi3K‐mTOR signaling and AMOG expression in epilepsy‐associated glioneuronal tumors. Brain Pathol 20:234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Capper D, Preusser M, Habel A, Sahm F, Ackermann U, Schindler G et al (2011) Assessment of BRAF V600E mutation status by immunohistochemistry with a mutation‐specific monoclonal antibody. Acta Neuropathol 122:11–19. [DOI] [PubMed] [Google Scholar]
- 20. Chappe C, Padovani L, Scavarda D, Forest F, Nanni‐Metellus I, Loundou A et al (2013) Dysembryoplastic neuroepithelial tumors share with pleomorphic xanthoastrocytomas and gangliogliomas BRAF mutation and expression. Brain Pathol 23:574–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Crino PB (2011) mTOR: a pathogenic signaling pathway in developmental brain malformations. Trends Mol Med 17:734–742. [DOI] [PubMed] [Google Scholar]
- 22. Dahiya S, Haydon DH, Alvarado D, Gurnett CA, Gutmann DH, Leonard JR (2013) BRAF(V600E) mutation is a negative prognosticator in pediatric ganglioglioma. Acta Neuropathol 125:901–910. [DOI] [PubMed] [Google Scholar]
- 23. Dazert E, Hall MN (2011) mTOR signaling in disease. Curr Opin Cell Biol 23:744–755. [DOI] [PubMed] [Google Scholar]
- 24. De Tommasi A, Luzzi S, D'Urso PI, De Tommasi C, Resta N, Ciappetta P (2008) Molecular genetic analysis in a case of ganglioglioma: identification of a new mutation. Neurosurgery 63:976–980; discussion 80. [DOI] [PubMed] [Google Scholar]
- 25. Deb P, Sharma MC, Tripathi M, Sarat Chandra P, Gupta A, Sarkar C (2006) Expression of CD34 as a novel marker for glioneuronal lesions associated with chronic intractable epilepsy. Neuropathol Appl Neurobiol 32:461–468. [DOI] [PubMed] [Google Scholar]
- 26. Dougherty MJ, Santi M, Brose MS, Ma C, Resnick AC, Sievert AJ et al (2010) Activating mutations in BRAF characterize a spectrum of pediatric low‐grade gliomas. Neuro Oncol 12:621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Edlundh‐Rose E, Egyhazi S, Omholt K, Mansson‐Brahme E, Platz A, Hansson J, Lundeberg J (2006) NRAS and BRAF mutations in melanoma tumours in relation to clinical characteristics: a study based on mutation screening by pyrosequencing. Melanoma Res 16:471–478. [DOI] [PubMed] [Google Scholar]
- 28. Eisenhardt AE, Olbrich H, Roring M, Janzarik W, Anh TN, Cin H et al (2011) Functional characterization of a BRAF insertion mutant associated with pilocytic astrocytoma. Int J Cancer 129:2297–2303. [DOI] [PubMed] [Google Scholar]
- 29. Engel JJ (1993) Outcome with respect to epileptic seizures. In: Surgical Treatment of the Epilepsies, Engel JJ (ed.), pp. 609–621. Raven Press: New York. [Google Scholar]
- 30. Esteve‐Puig R, Canals F, Colome N, Merlino G, Recio JA (2009) Uncoupling of the LKB1‐AMPKalpha energy sensor pathway by growth factors and oncogenic BRAF. Plos One 4:e4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fauser S, Becker A, Schulze‐Bonhage A, Hildebrandt M, Tuxhorn I, Pannek HW et al (2004) CD34‐immunoreactive balloon cells in cortical malformations. Acta Neuropathol 108:272–278. [DOI] [PubMed] [Google Scholar]
- 32. Faustino A, Couto JP, Populo H, Rocha AS, Pardal F, Cameselle‐Teijeiro JM et al (2012) mTOR pathway overactivation in BRAF mutated papillary thyroid carcinoma. J Clin Endocrinol Metab 97:E1139–E1149. [DOI] [PubMed] [Google Scholar]
- 33. Fensterle J, Becker JC, Potapenko T, Heimbach V, Vetter CS, Brocker EB, Rapp UR (2004) B‐Raf specific antibody responses in melanoma patients. BMC Cancer 4:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Galanopoulou AS, Gorter JA, Cepeda C (2012) Finding a better drug for epilepsy: the mTOR pathway as an antiepileptogenic target. Epilepsia 53:1119–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Han S, Santos TM, Puga A, Roy J, Thiele EA, McCollin M et al (2004) Phosphorylation of tuberin as a novel mechanism for somatic inactivation of the tuberous sclerosis complex proteins in brain lesions. Cancer Res 64:812–816. [DOI] [PubMed] [Google Scholar]
- 36. Horbinski C (2013) To BRAF or not to BRAF: is that even a question anymore? J Neuropathol Exp Neurol 72:2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Iyer A, Zurolo E, Spliet WG, van Rijen PC, Baayen JC, Gorter JA, Aronica E (2010) Evaluation of the innate and adaptive immunity in type I and type II focal cortical dysplasias. Epilepsia 51:1763–1773. [DOI] [PubMed] [Google Scholar]
- 38. Jozwiak J, Wlodarski P (2006) Hamartin and tuberin modulate gene transcription via beta‐catenin. J Neurooncol 79:229–234. [DOI] [PubMed] [Google Scholar]
- 39. Kam R, Chen J, Blumcke I, Normann S, Fassunke J, Elger CE et al (2004) The reelin pathway components disabled‐1 and p35 in gangliogliomas—a mutation and expression analysis. Neuropathol Appl Neurobiol 30:225–232. [DOI] [PubMed] [Google Scholar]
- 40. Koelsche C, Sahm F, Paulus W, Mittelbronn M, Giangaspero F, Antonelli M et al (2013) BRAF V600E expression and distribution in desmoplastic infantile astrocytoma/ganglioglioma. Neuropathol Appl Neurobiol. doi: 10.1111/nan.12072. [DOI] [PubMed] [Google Scholar]
- 41. Koelsche C, Wohrer A, Jeibmann A, Schittenhelm J, Schindler G, Preusser M et al (2013) Mutant BRAF V600E protein in ganglioglioma is predominantly expressed by neuronal tumor cells. Acta Neuropathol 125:891–900. [DOI] [PubMed] [Google Scholar]
- 42. Korshunov A, Ryzhova M, Jones DT, Northcott PA, van Sluis P, Volckmann R et al (2012) LIN28A immunoreactivity is a potent diagnostic marker of embryonal tumor with multilayered rosettes (ETMR). Acta Neuropathol 124:875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kwiatkowski DJ, Manning BD (2005) Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet 14(Spec No. 2):R251–R258. [DOI] [PubMed] [Google Scholar]
- 44. Li WQ, Kawakami K, Ruszkiewicz A, Bennett G, Moore J, Iacopetta B (2006) BRAF mutations are associated with distinctive clinical, pathological and molecular features of colorectal cancer independently of microsatellite instability status. Mol Cancer 5:2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lim HK, Choi YA, Park W, Lee T, Ryu SH, Kim SY et al (2003) Phosphatidic acid regulates systemic inflammatory responses by modulating the Akt‐mammalian target of rapamycin‐p70 S6 kinase 1 pathway. J Biol Chem 278:45117–45127. [DOI] [PubMed] [Google Scholar]
- 46. Louis DN, Ohgaki H, Wiestler OD, Cavanee WK (2007) WHO Classification of Tumours of the Central Nervous System. IARC: Lyon. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mak BC, Kenerson HL, Aicher LD, Barnes EA, Yeung RS (2005) Aberrant beta‐catenin signaling in tuberous sclerosis. Am J Pathol 167:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McDaniel SS, Wong M (2011) Therapeutic role of mammalian target of rapamycin (mTOR) inhibition in preventing epileptogenesis. Neurosci Lett 497:231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Orlova KA, Tsai V, Baybis M, Heuer GG, Sisodiya S, Thom M et al (2010) Early progenitor cell marker expression distinguishes type II from type I focal cortical dysplasias. J Neuropathol Exp Neurol 69:850–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Parry L, Maynard JH, Patel A, Hodges AK, von Deimling A, Sampson JR, Cheadle JP (2000) Molecular analysis of the TSC1 and TSC2 tumour suppressor genes in sporadic glial and glioneuronal tumours. Hum Genet 107:350–356. [DOI] [PubMed] [Google Scholar]
- 51. Prabowo AS, Iyer AM, Anink JJ, Spliet WG, van Rijen PC, Aronica E (2013) Differential expression of major histocompatibility complex class I in developmental glioneuronal lesions. J Neuroinflammation 10:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ravizza T, Boer K, Redeker S, Spliet WG, van Rijen PC, Troost D et al (2006) The IL‐1beta system in epilepsy‐associated malformations of cortical development. Neurobiol Dis 24:128–143. [DOI] [PubMed] [Google Scholar]
- 53. Resta N, Lauriola L, Puca A, Susca FC, Albanese A, Sabatino G et al (2006) Ganglioglioma arising in a Peutz‐Jeghers patient: a case report with molecular implications. Acta Neuropathol 112:106–111. [DOI] [PubMed] [Google Scholar]
- 54. Rosner M, Hanneder M, Siegel N, Valli A, Fuchs C, Hengstschlager M (2008) The mTOR pathway and its role in human genetic diseases. Mutat Res 659:284–292. [DOI] [PubMed] [Google Scholar]
- 55. Schick V, Majores M, Engels G, Hartmann W, Elger CE, Schramm J et al (2007) Differential Pi3K‐pathway activation in cortical tubers and focal cortical dysplasias with balloon cells. Brain Pathol 17:165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold‐Mende C et al (2011) Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra‐cerebellar pilocytic astrocytoma. Acta Neuropathol 121:397–405. [DOI] [PubMed] [Google Scholar]
- 57. Schmitz F, Heit A, Dreher S, Eisenacher K, Mages J, Haas T et al (2008) Mammalian target of rapamycin (mTOR) orchestrates the defense program of innate immune cells. Eur J Immunol 38:2981–2992. [DOI] [PubMed] [Google Scholar]
- 58. Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, Cantley LC (2004) The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 6:91–99. [DOI] [PubMed] [Google Scholar]
- 59. Thom M, Toma A, An S, Martinian L, Hadjivassiliou G, Ratilal B et al (2011) One hundred and one dysembryoplastic neuroepithelial tumors: an adult epilepsy series with immunohistochemical, molecular genetic, and clinical correlations and a review of the literature. J Neuropathol Exp Neurol 70:859–878. [DOI] [PubMed] [Google Scholar]
- 60. Thom M, Blumcke I, Aronica E (2012) Long‐term epilepsy‐associated tumors. Brain Pathol 22:350–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Trinh XB, Tjalma WA, Vermeulen PB, Van den Eynden G, Van der Auwera I, Van Laere SJ et al (2009) The VEGF pathway and the AKT/mTOR/p70S6K1 signalling pathway in human epithelial ovarian cancer. Br J Cancer 100:971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. van Veelen CW, Debets RM, van Huffelen AC, de Boas W, Binnie CD, Storm van Leeuwen W et al (1990) Combined use of subdural and intracerebral electrodes in preoperative evaluation of epilepsy. Neurosurgery 26:93–101. [DOI] [PubMed] [Google Scholar]
- 63. Vezzani A (2012) Before epilepsy unfolds: finding the epileptogenesis switch. Nat Med 18:1626–1627. [DOI] [PubMed] [Google Scholar]
- 64. Vezzani A, French J, Bartfai T, Baram TZ (2011) The role of inflammation in epilepsy. Nat Rev Neurol 7:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vezzani A, Auvin S, Ravizza T, Aronica E (2012) Glia‐neuronal interactions in ictogenesis and epileptogenesis: role of inflammatory mediators. In: Jasper's Basic Mechanisms of the Epilepsies, 4th edn. Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado‐Escueta AV (eds), pp. 1–21. National Center for Biotechnology Information (USA): Bethesda, MD. [PubMed] [Google Scholar]
- 66. Weichhart T, Saemann MD (2009) The multiple facets of mTOR in immunity. Trends Immunol 30:218–226. [DOI] [PubMed] [Google Scholar]
- 67. Zemke D, Azhar S, Majid A (2007) The mTOR pathway as a potential target for the development of therapies against neurological disease. Drug News Perspect 20:495–499. [DOI] [PubMed] [Google Scholar]
- 68. Zheng B, Jeong JH, Asara JM, Yuan YY, Granter SR, Chin L, Cantley LC (2009) Oncogenic B‐RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol Cell 33:237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. BRAF V600E mutated ganglioglioma (GG): CD34, MHC‐II and MHC‐I immunoreactivity. A, B. Prominent CD34 immunoreactivity (IR) within the tumor area. C–E. Prominent MHC class II antigen (MHC‐II) expression within the tumor area, with IR in microglial cells (arrows in D and E) and occasionally in large dysplastic neurons (arrowhead in E). F, G. Prominent MHC class I antigen (MHC‐I) expression within the tumor area, with IR in large dysplastic neurons (arrows in G). Scale bar in (G): A, C, F: 400 μm; B, G: 80 μm; D, E: 40 μm.
Figure S2. BRAF V600E mutated ganglioglioma (GG): pLKB1. A–C. Phosphorylated (p)‐LKB1 immunoreactivity (IR) within the tumor area in large dysplastic neurons (arrows in A; B, C); co‐localization of pLKB1 with VE1 is shown in the insert in (A). Scale bar in (C): A. 40 μm; B, C. 20 μm.
Figure S3. VE1 immunoreactivity in ganglioglioma (GG) and in control cortex. A. GG with only few VE1‐positive dysplastic neurons (arrows and insert). B. Perilesional cortex without VE1 immunoreactivity (IR). Scale bar in (B): A. 80 μm; B. 160 μm.
Figure S4. MHC‐I, MHC‐II and pS6 immunoreactivity score (IRS) in GGs and DNTs. A. MHC‐I (glial/neuronal) and MHC‐II IR scores (total score; mean ± SEM) in GGs and DNTs. B. pS6 (glial/neuronal) IR scores (total score; mean ± SEM) in GGs and DNTs (***P < 0.001). Desmoplastic infantile gangliogliomas (DIGs); gangliogliomas (GGs); dysembryoplastic neuroepithelial tumors (DNTs); dysplastic neurons (DN).
Table S1. Criteria to distinguish diffuse DNTs from other DNT types.
Table S2. Correlation of pS6 IRS with clinical data and histological/immunohistochemical features in GGs and DNTs.