

Abstract
Prion diseases include sporadic, acquired and genetic forms linked to mutations of the prion protein (PrP) gene (PRNP). In subjects carrying the D178N PRNP mutation, distinct phenotypes can be observed, depending on the methionine/valine codon 129 polymorphism. We present here a 53‐year‐old woman with D178N mutation in the PRNP gene and homozygosity for valine at codon 129. The disease started at age 47 with memory deficits, progressive cognitive impairment and ataxia. The clinical picture slowly worsened to a state of akinetic mutism in about 2 years and the disease course was 6 years. The neuropathologic examination demonstrated severe diffuse cerebral atrophy with neuronal loss, spongiosis and marked myelin loss and tissue rarefaction in the hemispheric white matter, configuring panencephalopathic Creutzfeldt‐Jakob disease. PrP deposition was present in the cerebral cortex, basal ganglia and cerebellum with diffuse synaptic‐type pattern of immunoreactivity and clusters of countless, small PrP deposits, particularly evident in the lower cortical layers, in the striatum and in the molecular layer of the cerebellum. Western blot analysis showed the presence of type 1 PrPSc (Parchi classification). These findings underline the clear‐cut distinction between the neuropathological features of Creutzfeldt‐Jakob disease associated with D178N PRNP mutation and those of fatal familial insomnia.
Keywords: Creutzfeldt‐Jakob disease, familial, immunohistochemistry, neuropathology, prion protein gene, type 1 PrPSc
Introduction
Prion diseases are a heterogeneous group of fatal neurological disorders characterized by cerebral deposition of misfolded forms of the cellular prion protein (PrPC), referred to as PrPSc. They may be sporadic, genetic or acquired. The genetic forms include different clinicopathological entities caused by mutations of the prion protein gene (PRNP) 8.
In subjects carrying the D178N PRNP mutation, distinct phenotypes are determined by the methionine/valine (M/V) codon 129 polymorphism present on the mutated allele. D178N‐129 M haplotype causes fatal familial insomnia (FFI), while D178N‐129V haplotype determines Creutzfeldt‐Jakob disease (CJD) 6.
Several detailed reports are available for FFI that is characterized by insomnia early in the disease course and peculiar neuropathological and biochemical features, allowing its recognition as a separate nosological entity. At difference, descriptions of patients with D178N PRNP mutation on 129V allele are few and rather vague as regards to the neuropathological picture 1.
We report a patient with D178N PRNP mutation, homozygous for valine at codon 129, affected by a prion disease of unusually long duration with features of panencephalopathic CJD and a distinctive pattern of PrP deposition.
Material and Methods
Case report
A 47‐year‐old woman complained for impairment in left leg movements, followed by deficits of episodic memory and difficulty in instrumental activities. Five months later, she developed ataxia, dysarthria, myoclonic jerks and choreoathetoid movements. Ten months after the onset of symptoms, Electroencephalogram (EEG) showed non‐specific slowing of background activity. FLAIR and diffusion‐weighted magnetic resonance imaging (MRI) documented signal hyperintensity in the head of the caudate nucleus, the anterior part of the putamen and, to a lesser extent, the posterior thalamus, frontal cortex, cingulate gyrus and insula. Cerebrospinal fluid (CSF) examination demonstrated the presence of 14.3.3 and high levels of tau protein (3018 pg/mL; normal values 90–150 pg/mL). PRNP analysis revealed a GAC to AAC mutation at codon 178 resulting in aspartic acid to asparagine substitution, and homozygosity for valine at codon 129.
One year after the onset, the patient was still able to understand and execute simple commands, but at age 49 she was bedridden in a state of akinetic mutism.
Repeated polysomnographic studies revealed a severe nocturnal sleep fragmentation with continuous transitions between wakefulness and light sleep. Slow wave sleep, spindles and REM sleep were undetectable 2. Despite these marked alterations, long‐term actigraphic monitoring was consistent with the persistence of a rudimental circadian rhythm.
Tilt test and hypothalamic pituitary hormones 24 h assay were performed over time and were normal.
The patient died for respiratory failure after 6 years of disease. The autopsy was performed 24 h after death.
The patient's father died at age 58 after a 2‐year history of a neurological disorder very similar to that of the proband, and her paternal grandfather died after a rapidly progressive dementia in his 40s. Molecular genetic data are not available for them.
Neuropathologic study
The brain was taken at autopsy and the neuropathological study was carried out on Alcolin‐ (Diapath, Martinengo, BG, Italy) or formalin‐fixed sections of the cerebral hemispheres, cerebellum and brainstem, stained with hematoxylin‐eosin, cresyl violet for Nissl substance, Luxol fast blue for myelin, thioflavine S for amyloid and immunostained with antibodies against PrP (see below), glial fibrillary acidid protein (GFAP, polyclonal, DakoCytomation, Glostrup, Denmark, 1:800), myelin basic protein (monoclonal, DakoCytomation, 1:500) and neurofilaments (monoclonal, DakoCytomation, 1:800). The anti‐PrP monoclonal antibodies 3F4 (epitope at residues 109–112 of human PrP, DakoCytomation, 1:800) and 12F10 (1:200, Cayman Chemicals Ann Arbor, MI, USA) were used after specific pretreatment of the sections 3. The immunoreaction was visualized using the EnVision Plus/Horseradish Peroxidase system (DakoCytomation) and 3‐3'‐diaminobenzidine as chromogen.
Western blot analysis was carried out on frozen samples of several areas of the cerebral cortex, thalamus and cerebellum, using the above mentioned anti‐PrP antibodies, as previously described 3.
Results
The brain weight was 750 g before fixation. Striking cerebral atrophy with marked thinning of cortical gyri and enlargement of lateral ventricles was present (Figure 1A). The white matter of the cerebral hemispheres showed tissue rarefaction with diffuse myelin pallor (Figure 1B), loss of neurites (Figure 1C) with severe fibrous gliosis (Figure 1D), suggestive of panencephalopathic CJD.
Figure 1.
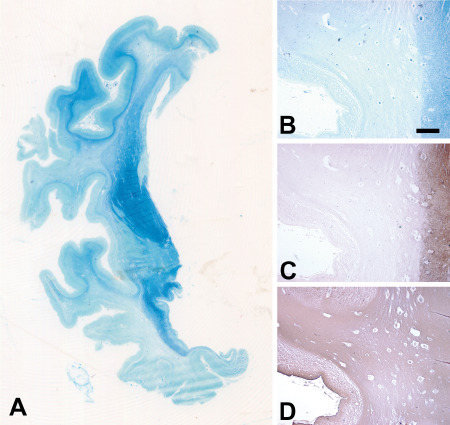
Macroscopic images of a coronal section of the left cerebral hemisphere at the level of the globus pallidum stained for myelin (Luxol fast blue) (A) and low magnification images of the middle temporal gyrus stained for myelin (B, Luxol fast blue), and immunostained for neurofilaments (C) and GFAP (D) show diffuse myelin pallor, axonal loss and fibrous gliosis in the emispheric white matter. U‐fibers, the internal capsule and the anterior commissure are relatively spared by myelin loss (A). Myelin loss (B) is associated with degeneration of neurites (C), while fibrous gliosis is diffuse (D). Scale bar in (B) = 1 mm (B, C and D are the same magnification).
The histological examination demonstrated severe neuronal loss, spongiosis and gliosis in the cerebral cortex, in the cerebellum and in the striatum (Figure 2A–C,F). In the medial thalamic nuclei, neuronal loss was more severe than in lateral ones (Figure 2J,K). Brainstem nuclei, such as substantia nigra, locus coeruleus and inferior olives were devoid of significant changes (Figure 2L,M).
Figure 2.
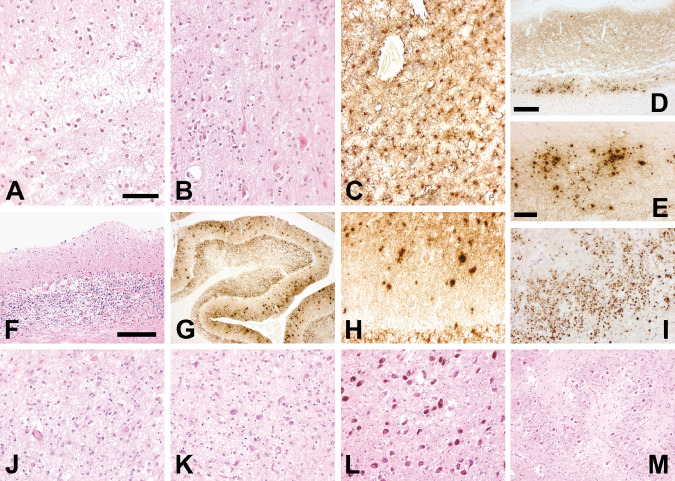
The microscopic neuropathologic analysis shows the presence of severe neuronal loss and spongiform changes in the cerebral cortex that are more marked in the frontal [A: hematoxylin and eosin (H&E)] than in the occipital cortex (B, H&E) and are associated with astrogliosis (C: GFAP immunostaining, frontal cortex). The pattern of PrPSc deposition is defined by diffuse, finely granular synaptic‐like immunoreactivity and by numerous focal deposits that are abundant in the lower cortical layers (D: 3F4 immunostaining, frontal cortex) and in the entorhinal cortex where they form large clusters (E: 3F4 immunostaining, frontal cortex; I: 3F4 immunostaining, entorhinal cortex). In the cerebellum, loss of Purkinje and granular neurons, astrogliosis (F: H&E) and PrP build up are present. Focal PrP deposits are numerous in the molecular layer (G, H: 3F4 immunostaining). The PrP deposits are not fluorescent after thioflavine S (not shown). Neuronal loss is more severe in the medial (J: H&E) than in the lateral thalamic nuclei (K: H&E). Brainstem nuclei such as locus coeruleus (L: H&E) and inferior olive (M: H&E) are spared by the pathologic process. Scale bars: in (A) 100 μm (A, B, C are the same magnification); in (D) 300 μm (D, G are the same magnification); in (E) 50 μm (E, H, I, J, K are the same magnification); in (F) 200 μm (F, L, M are the same magnification).
PrP deposition was present in the cerebral cortex, striatum and cerebellum with diffuse synaptic‐type pattern of immunoreactivity. In the same areas, countless, focal PrP deposits were evident (Figure 2D,E,G–I). Their size ranged from about 15 μm in the molecular layer of the cerebellum to 2–3 μm in the entorhinal cortex were they formed large clusters. They were not fluorescent after thioflavine S.
Western blot analysis showed the presence of type 1 PrPSc (Parchi classification) 7 with slight predominance of the diglycosylated form in all brain areas examined (Figure 3).
Figure 3.
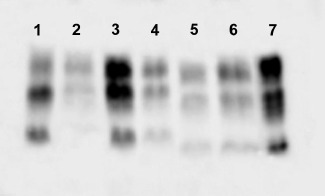
Western blot analysis of cerebellum (lane 2), frontal cortex (lane 3) and thalamus (lane 4) of the patient with PRNP D178N/129VV mutation performed with anti‐PrP antibody (3F4) after PK digestion showed the presence of type 1 PrPSc (for comparison, a sporadic CJD 129VV with type 1 PrP in lane 1). At difference, frontal cortex (lane 5) and thalamus (lane 6) of a patient with FFI show the presence of type 2 PrPSc. (for comparison, a sporadic CJD sporadic CJD 129VV with type 2 PrP in lane 7).
Discussion
Examination of the clinical and pathological features of this patient reveals many differences compared to FFI and a distinct pattern of PrP deposition.
From the clinical standpoint, our patient did not show early insomnia nor alterations of the hypothalamic‐pituitary‐adrenal axis that are the hallmarks of FFI.
Biochemically, we detected abundant type 1 PrPSc (Parchi classification) 7, at difference with scarce, type 2 PrPSc usually found in FFI. However, a common feature between the present case and FFI is the predominance of the diglycosylated isoform of PrP.
Neuropathologically, neuronal loss and gliosis were not limited to the few brain structures (thalamus, inferior olives) affected in FFI. The inferior olive did not show significant neuronal loss in our case, despite the long duration of the disease and the striking cerebral atrophy.
Finally, while in FFI PrP, immunolabeling is very slight or even absent, our patient showed abundant and diffuse PrP immunostaining of the synaptic type, associated with large clusters of tiny, focal PrP deposits. This pattern is very atypical and is not a common feature of panencephalopathic CJD 5. A similar neuropathological scenario has been described in a patient with V180I PRNP mutation and panencephalopathic CJD 4. However, the biochemical features of the patient reported here diverged from those of V180I CJD who showed an immunoblot profile with type 2 PrP and absence of the diglycosylated form. This is most interesting considering the proximity of the two mutations.
On the other hand, the presence of clusters of small focal PrP deposits and the involvement of the cerebellum with abundant PrP deposition differentiate the patient reported here from the sporadic CJD patients homozygous for valine at codon 129 of PRNP with type 1 PrP (VV1) who are characterized by severe pathology in the cerebral cortex with only faint synaptic PrP staining and sparing of the cerebellum where PrP deposition is absent.
In conclusion, our findings confirm and document the difference between the clinical, neuropathological and biochemical features of the prion disease associated with D178N‐129V PRNP mutation and those of FFI, pointing also to a clear‐cut distinction between D178N/129VV CJD and sporadic VV1 CJD patients.
Acknowledgments
The work was supported by the Italian Ministry of Health, by the Italian Ministry of Education, Universities and Research (PRIN 2008HC7XR8_002) and by the Prion Encephalopathies Italian Association AIEnP.
References
- 1. Capellari S, Strammiello R, Saverioni D, Kretzschmar H, Parchi P (2011) Genetic Creutzfeldt‐Jakob disease and fatal familial insomnia: insights into phenotypic variability and disease pathogenesis. Acta Neuropathol 121:21–37. [DOI] [PubMed] [Google Scholar]
- 2. Dossena S, Imeri L, Mangieri M, Garofoli A, Ferrari L, Senatore A et al (2008) Mutant prion protein expression causes motor and memory deficits and abnormal sleep patterns in a transgenic mouse model. Neuron 60:598–609. [DOI] [PubMed] [Google Scholar]
- 3. Giaccone G, Canciani B, Puoti G, Rossi G, Goffredo D, Iussich S et al (2000) Creutzfeldt‐Jakob disease: Carnoy's fixative improves the immunohistochemistry of the proteinase K‐resistant prion protein. Brain Pathol 10:31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Iwasaki Y, Mori K, Ito M, Nagaoka M, Ieda T, Kitamoto T et al (2011) An autopsied case of V180I Creutzfeldt‐Jakob disease presenting with panencephalopathic‐type pathology and a characteristic prion protein type. Neuropathology 31:540–548. [DOI] [PubMed] [Google Scholar]
- 5. Jansen C, Head MW, Rozemuller AJM, Ironside JW (2009) Panencephalopathic Creutzfeldt‐Jakob disease in the Netherlands and the UK: clinical and pathological characteristics of nine patients. Neuropathol Appl Neurobiol 35:272–282. [DOI] [PubMed] [Google Scholar]
- 6. Lugaresi E, Medori R, Montagna P, Baruzzi A, Cortelli P, Lugaresi A et al (1986) Fatal familial insomnia and dysautonomia with selective degeneration of thalamic nuclei. N Engl J Med 315:997–1003. [DOI] [PubMed] [Google Scholar]
- 7. Parchi P, Giese A, Capellari S, Brown P, Schulz‐Schaeffer W, Windl O et al (1999) Classification of sporadic Creutzfeldt‐Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46:224–233. [PubMed] [Google Scholar]
- 8. Prusiner SB (1998) Prions. Proc Natl Acad Sci USA 95:13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]