

Abstract
Frontotemporal Lobar Degeneration (FTLD) is a clinically, pathologically and genetically heterogeneous group of disorders that affect principally the frontal and temporal lobes of the brain. There are three major associated clinical syndromes, behavioral variant frontotemporal dementia (bvFTD), semantic dementia (SD) and progressive non‐fluent aphasia (PNFA); three principal histologies, involving tau, TDP‐43 and FUS proteins; and mutations in three major genes, MAPT, GRN and C9orf72, along with several other less common gene mutations. All three clinical syndromes can exist separately or in combination with Amyotrophic Lateral Sclerosis (ALS). SD is exclusively a TDP‐43 proteinopathy, and PNFA may be so, with both showing tight clinical, histological and genetic inter‐relationships. bvFTD is more of a challenge with overlapping histological and genetic features, involvement of any of the three aggregating proteins, and changes in any of the three major genes. However, when ALS is present, all cases show a clear histological phenotype with TDP‐43 aggregated proteins, and familial forms are associated with expansions in C9orf72. TDP‐43 and FUS are nuclear carrier proteins involved in the regulation of RNA metabolism, whereas tau protein – the product of MAPT – is responsible for the assembly/disassembly of microtubules, which are vital for intracellular transport. Mutations in TDP‐43 and FUS genes are linked to clinical ALS rather than FTLD (with or without ALS), suggesting that clinical ALS may be a disorder of RNA metabolism. Conversely, the protein products of GRN and C9orf72, along with those of the other minor genes, appear to form part of the cellular protein degradation machinery. It is possible therefore that FTLD is a reflection of dysfunction within lysosomal/proteasomal systems resulting in failure to remove potentially neurotoxic (TDP‐43 and tau) aggregates, which ultimately overwhelm capacity to function. Spread of aggregates along distinct pathways may account for the different clinical phenotypes, and patterns of progression of disease.
Keywords: clinical phenotypes, frontotemporal lobar degeneration, genetics, neuropathology, pathogenesis
Introduction
Frontotemporal Lobar Degeneration (FTLD) refers to a clinically, pathologically and genetically heterogeneous group of disorders that affect principally the frontal and temporal lobes of the brain. After Alzheimer's disease, it is the second most common cause of dementia in younger people. Its prevalence has been estimated at 10.8 per 100 000 and lifetime risk at 1 in 742 33. FTLD is a “disorder of threes”: there are three major associated clinical syndromes, three principal histologies and three main genetic mutations.
Clinical Syndromes
The most common clinical syndrome, which accounts for more than half of cases 72, 77, is behavioral variant frontotemporal dementia (bvFTD), characterized by behavioral change and associated with atrophy of the frontal and anterior temporal lobes 116. Core behavioral features are social disinhibition, apathy, emotional blunting with loss of sympathy and empathy for others, repetitive, obsessive and stereotyped behaviors, and dietary changes such as gluttony and altered preference for sweet foods. These key characteristics form the basis for contemporary clinical diagnostic criteria 133. Patients vary in terms of the relative preponderance of these behaviors. Hence, some patients are profoundly apathetic whereas others are overactive and socially disinhibited 94. Other behavioral characteristics, seen in some patients, include hypersensitivity or hyposensitivity to pain, sounds and other sensory stimuli 10, 44, 45, 151. Moreover, although thought to be rare 106, it is now recognized that some bvFTD patients experience psychotic symptoms of delusions and hallucinations 92, 147, 158. Behavioral changes are accompanied by cognitive impairments in “frontal” executive functions such as abstraction, planning, attention, reasoning and judgment. Distinct behavioral profiles within bvFTD have been attributed to differential vulnerability within functional brain networks 132.
A second, less common, clinical syndrome is semantic dementia (SD), a disorder of conceptual knowledge 116. It is characterized by impaired understanding of the meaning of words, faces, objects and other sensory stimuli. SD is also known as semantic variant primary progressive aphasia 54 because of the prominence of language‐related problems. Neuroimaging shows bilateral, albeit often asymmetrical atrophy of the temporal lobes 65. Patients with predominantly left‐sided atrophy have particular difficulties with the understanding of words whereas right‐predominant patients show difficulties in face recognition 82, 153, 162, but in both there is a gradual loss of conceptual understanding that ultimately affects all sensory domains.
The third clinical syndrome, termed progressive non‐fluent Aphasia (PNFA), 116 or non‐fluent variant primary progressive aphasia 54, is a disorder of expressive language, associated with asymmetric atrophy of the left hemisphere. It is typically characterized by effortful speech and impaired use of grammar. The precise characteristics of the language disorder are not uniform across patients 58, 107, 138, 142. Some patients, for example, show effortful speech production with distortions of utterances (speech apraxia) whereas others do not.
The clinical syndromes of bvFTD, PNFA and SD may present in relatively pure form. Symptoms may, however, overlap 59. Thus, some patients who meet criteria for bvFTD also exhibit language features of PNFA or SD, whereas those presenting with a language disorder may develop “frontal” behavioral changes.
Histologies
The three histologies are characterized by abnormal neuronal, and sometimes glial, accumulations of aggregated proteins (Figures 1, 2, 3). In about 45% of cases, neuronal intracytoplasmic inclusions (NCI) are composed of the microtubule associated protein, tau 146. Such cases are termed FTLD‐tau. In about half of these tau‐positive cases, rounded bodies, known as Pick bodies, are seen (Figure 1A) and glial tau inclusions may also be present 146. In the remainder, the neuronal tau is present as neurofibrillary tangle‐like structures (NFT) or more amorphous deposits (pre‐tangles) (Figure 1B). Cases with Pick bodies and glial inclusions conform to the modern definition of Pick's disease.
Figure 1.
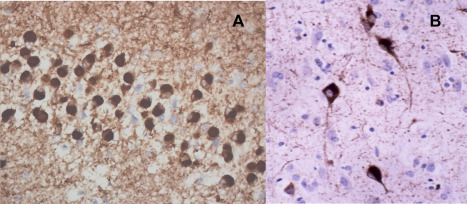
Tau inclusions (Pick bodies) (A) in missense MAPT mutations and sporadic bvFTD. Tangle‐like inclusions in neurons (B) and glia associated with intronic mutations in MAPT.
Figure 2.
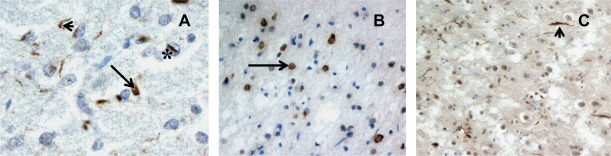
TDP‐43 inclusions. Subtype A (A) is characterized by neuronal cytoplasmic inclusions (arrow) and dystrophic neurites (arrowhead), and neuronal intranuclear inclusions (*) when GRN mutation is present. In Subtype B, NCI predominate, and in Subtype C, DN are mostly present.
Figure 3.
FUS pathology in aFTLD‐U (A–C) and Neuronal Intermediate Filament Inclusion Body Disease (D–F), as shown by immunostaining for FUS protein (A,D), transportin‐1 (B,E) and TAF15 (C,E).
In about 50% of FTLD cases, the RNA‐ and DNA‐binding protein, TDP‐43, is present within ubiquitinated NCI, neuritic processes (dystrophic neurites, DN) or neuronal intranuclear inclusions (NII) 35, 119, 146 (Figure 2). Such cases, formerly known as FTLD‐U are now termed FTLD‐TDP. The relative preponderance of pathological features varies, giving rise to a sub‐classification of FTLD‐TDP pathology 100. Subtype A is applied when NCI and DN are both commonly present (Figure 2A), type B when NCI predominate over DN (Figure 2B), type C when DN predominate over NCI (Figure 2C) and type D when NII (see Figure 2A) are most common type of histological change.
Most of the remaining 5% of cases show NCI, and sometimes NII, composed of the protein, Fused in Sarcoma (FUS) (also known as Translocation in Liposarcoma, TLS) (Figure 3). Such cases are described as FTLD‐FUS 99, 100, or FTLD‐FET ‐ in recognition that the NCI contain other members of the FET family of proteins, such as transportin‐1 (Figure 3B,E), TAF15 (Figure 3C,F) and Ewing's Sarcoma protein (EWS) 17, 36, 122. FTLD‐FUS encompasses 3 distinct histopathological variants; atypical FTLD‐U (Figure 3A–C), Neuronal Intermediate Filament Inclusion Body Disease (NIFID) (Figure 3D–F) and Basophilic Inclusion Body Disease (BIBD) 22, 23, 24, 112, 120, 121. Although for purposes of classification it is logical to lump these together into the broad‐brush category of FTLD‐FUS, given the common presence of FUS protein within inclusions, these are distinct diseases and their development presumably reflects mechanistic differences. The disorders clearly differ in neuropathology – the NCI in NIFID and aFTLD‐U do not contain RNA, NCI in NIFID contain internexin‐1 whereas those in aFTLD‐U and BIBD do not, vermiform NII are commonplace in NIFID and aFTLD‐U, but rare or absent in BIBD, loss of lower motor neurons is frequent in BIBD but less common in aFTLD‐U 22, 23, 24, 112, 120, 121 – all features pointing to distinctions in pathogenesis.
In a very small minority of cases no inclusions are seen (FTLD‐ni), though it is still unclear whether these are true members of FTLD family or simply represent clinical phenocopies.
Genetic Mutations
FTLD is strongly familial, with up to 40% of patients having a history of a similar disorder within their family 137, indicating that genetic factors play a major role in its aetiology. A clearly autosomal dominant pattern of inheritance is documented in about 10% 137. Three major causal genes have been identified. Mutations in MAPT on chromosome 17 71, 160, not unexpectedly, drive FTLD‐tau pathology. FTLD‐TDP pathology, by contrast, is not, or perhaps only rarely, associated with mutations in TDP‐43 gene, TARDBP. Rather, it is represented by mutations in the progranulin gene (GRN) on chromosome 17 9, 34 or expansions in C9orf72 on chromosome 9 40, 134. Mutations in other genes have also, less commonly, been associated with FTLD. Of these, most notable are CHMP2B mutations on chromosome 3 20, 56, 148 occurring almost exclusively within a single pedigree within the Jutland region of Denmark 20, 56, 74. NCI are present in such cases, and although these are ubiquitinated the target protein remains to be identified 68. The classification, FTLD‐UPS, has been applied in recognition of the involvement of the ubiquitin proteasome system in the disease. The great majority of cases with FTLD‐FUS pathology appear to be sporadic. Interestingly, mutations in FUS do occur, but these are mostly associated with Amyotrophic Lateral Sclerosis (ALS) 90, 167, and isolated claims that these might be associated with bvFTD remain to be substantiated 57. Other rare genetic forms of FTLD involve mutations in valosin containing protein (VCP) 174, SQSTMI (also known as p62) 97, optineurin (OPTN) 104, ubiquilin 2 (UBQLN2) 41 and TANK binding‐kinase 1 (TBK1) 129. Although, collectively, such cases are numerically few, they do provide important clues to pathogenesis since all involve TDP‐43 proteinopathy and all have functions within the cell's protein degradation systems.
Demographics and Neurology
The distinct clinical syndromes, histologies and genetic mutations emphasize the heterogeneity within FTLD. There is variation too in demographic characteristics and neurological findings.
FTLD is typically thought of as an early‐onset dementia, and indeed the average age at onset of disease is typically around 60 years 46, 94. There is, however, wide variation. There are published reports of patients presenting in their twenties 30, 75, 152. Conversely, between 25 and 30% of patients develop symptoms after the age of 65 years 88, 94, 124. The mean duration of symptoms from onset until death is around 8 years 5 but again there is wide variation 28, 33, 64 from less than 2 to more than 20 years. Progression can be rapid or very slow.
Patients presenting with clinical syndromes of FTLD are typically physically well, with signs of parkinsonism (slowing, limb rigidity) emerging only relatively late in the disease course. Importantly, however, some patients (around 15%) develop motor neuron disease (MND) 115, providing clinical evidence of the link between the disorders 22, 116, 117. The form of MND is that of ALS), characterized by weakness, wasting and fasciculations of the muscles. Onset may involve the bulbar muscles, affecting speech production and swallowing, or the limbs, affecting mobility. The presence of ALS attenuates the disease course 55, 64, 79, 86 and accounts for patients with very short duration of disease. The presence of ALS is also associated with differences in gender distribution. FTLD disorders as a whole affect men and women equally 124, 141. In patients with accompanying ALS, however, there is a significant male bias 143. Moreover, the presence of ALS influences the likelihood of one clinical syndrome rather than another. It is most often seen in combination with bvFTD. An association with SD and PNFA can occur but it is rare 143.
ALS is not the only possible physical accompaniment of FTLD syndromes. There is a clinical overlap between bvFTD and the neurological disorders of progressive supranuclear palsy (PSP), characterized by ophthalmoplegia and falls, and corticobasal syndrome (CBS), characterized by limb rigidity and apraxia 14, 81, 89.
The clinical, pathological and genetic heterogeneity within FTLD has led authors to highlight the need for biomarkers to enable the different pathologies to be distinguished in vivo 73. Whilst this is undoubtedly an important goal, there are already some clues. Close examination reveals certain predictabilities. Pathological subtypes do have clinical and genetic associations. These are important because they may inform understanding of the mechanics of neurodegeneration.
Clinical—Pathological Correlates
The behavioral disorder of bvFTD can be associated with FTLD‐tau, FTLD‐TDP or FTLD‐FUS pathology (Table 1, Figure 5). Thus, the presence of a behavioral disorder per se does not distinguish between pathologies. However, when it occurs in combination with ALS, bvFTD is consistently associated with FTLD‐TDP rather than tau pathology 84, 140, 157. Thus, the presence of ALS is a strong predictor of TDP rather than other pathologies. By contrast, neurological signs of PSP (vertical gaze palsy) and CBS predict tau pathology 46, 84. A very early onset of disease, before the age of 40 years is a strong predictor of FUS pathology 120, 145, 156, 166.
Table 1.
Clinicopathological relationships.
| Clinical phenotype | Pathology (type and subtype) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Tau | TDP‐43 | FUS | |||||||
| A | B | C | D | aFTLD‐U | NIFID | BIBD | |||
| bvFTD | NFT | PB | NCI/DN | NCI | – | NII | NCI/NII | NCI | NCI |
| bvFTD/ALS | – | – | – | NCI | – | – | – | – | – |
| PNFA | – | PB | NCI/DN | – | – | – | – | – | – |
| SD | – | – | – | – | DN | – | – | – | – |
bvFTD = behavioral variant frontotemporal dementia; ALS = Amyotrophic Lateral Sclerosis; PNFA = Progressive Non‐Flent Aphasia; SD = Semantic dementia; aFTLD‐U = atypical FTLD‐U; NIFID = Neuronal Intermediate Filament Inclusion Body Disease; BIBD = Basophilic Inclusion Body Disease; NFT = neurofibrillary tangles; PB = Pick bodies; NCI = neuronal cytoplasmic inclusions; DN = dystrophic neurites; NII = neuronal intranuclear inclusions.
Figure 5.
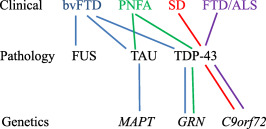
Clinicopathological‐genetic relationships.
[Correction added on 26 April 2017, after first online publication: Figure 5 has been replaced to correct an error in the alignment of the figure labels in the originally published version.]
SD (in its pure form) is consistently linked to TDP pathology 84, 140, 157. PNFA may be underpinned by either FTLD‐tau or FTLD‐TDP pathology 84, 140, 157 yet there are specific characteristics of the language disorder that favor one pathological type over the other. In particular, effortful speech production and speech apraxia is a predictor of tau rather than TDP‐43 pathology 80. Non‐fluent speech production arising secondary to severe anomia has been linked to TDP‐43 pathology 155.
BvFTD, PNFA and SD, as noted above, can all be associated with FTLD‐TDP pathology. Yet, interestingly, the pathological characteristics are not identical. Clinical phenotype predicts a specific sub‐type of TDP pathology (Table 1, Figure 5). PNFA is specifically associated with subtype A, in which there are numerous short DN and NCI, mostly in layer 2 of frontal and temporal cortex, associated with loss of neurones from that layer and a vacuolated appearance to the tissue (microvacuolation) 100. FTD, when combined clinically with ALS predicts subtype B histology, characterized by a predominance of NCI, again mostly in layer 2, but also involving those within deeper layers of the cortex. DN are relatively sparse 100. SD is associated with FTLD‐TDP type C, where the predominant pathology is that of long neuritic profiles traversing the entire depth of the cerebral cortical ribbon 100.
The histological variants of FTLD‐FUS are also associated with different clinical phenotypes. Patients with aFTLD‐U pathology present with a prominent behavioral disorder of bvFTD 93, 140, 145, 156, 166, whereas in NIFID and BIBD motor symptoms predominate 93. Patients with aFTLD typically show obsessional and stereotyped behaviors, which are more marked than in other forms of bvFTD 139, 156. They may also show psychotic symptoms 156. Caudate atrophy is commonly reported 83, 156. It is the aFTLD‐U form of FUS pathology that is particularly associated with very early onset of disease, in the 4th or 5th decade of life.
Familial vs. sporadic disease
FTLD may be familial or sporadic. The prevalence of familial disease differs according to clinical phenotype. bvFTD is most likely to be familial 137. SD, in its pure form, is rarely so 66, 137. There are, by implication, differences in type of pathology. TDP‐43 types A and B pathology are more likely to be found in familial disease than type C, reflecting the fact that SD is unique to type C pathology.
In terms of tau pathology, familial forms of FTLD can be associated with either neurofibrillary or Pick type of tau pathology, depending on the location of the genetic mutation (Figure 5). Sporadic forms are mostly associated with Pick body histology. Just occasionally, apparently sporadic, forms show globular tau‐immunoreactive oligodendroglial inclusions within white matter, leading to the pathological diagnosis of FTLD‐tau with globular glial inclusions.
Clinical—Genetic Correlates
Familial cases are most caused by mutation in one of 3 main genes, MAPT, GRN or C9orf72, although a proportion of cases still remains in whom there is a positive family history of dementia without known mutations. Clues to genotype can be deduced from clinical presentation.
bvFTD is commonly associated with mutations in each of the genes 159, so its presence, in itself, is non‐discriminatory (Figure 5). However, in patients with MAPT mutations prominent behavioral disinhibition is characteristic 76, 127, 159 whereas in GRN and C9orf72 cases apathy typically prevails 11, 159. Moreover, MAPT patients with intronic (splice site) mutations in exon 10 frequently exhibit semantic impairments in addition to their behavioral disorder 76, 127, a feature not seen in GRN, and only rarely in C9orf72, patients 159.
By contrast, mutations in GRN are associated with PNFA 26, 39, 95, 96, 128, 131, 168, 178 in up to a third of cases (Figure 5). The remaining two thirds presents with bvFTD. The distinct clinical presentations of bvFTD and PNFA may occur within the same family 11, 159 and there may be overlapping characteristics with disease progression. PNFA is absent in MAPT and rare in C9orf72 cases 159.
FTD‐ALS is associated with hexanucleotide expansions in the C9orf72 gene 15, 70, 158, 169, but not with MAPT or GRN mutations 96, 159 (Figure 5). Psychotic features (delusions and hallucinations) too are common in patients with C9orf72 expansions 43, 49, 87, 158. They occur to some degree in patients with GRN mutations 159, 170, but are rare or absent in sporadic FTD and in association with MAPT mutations 158, 159.
Consistent with the clinical differences MAPT, GRN and C9orf72 are associated with different patterns of atrophy can be detected on neuroimaging 176.
Genetic—pathological correlates
The tau gene (MAPT)
The tau gene (MAPT) has 15 coding regions (exons), and transcripts are alternatively spliced to produce 6 different isoforms ranging from 352 to 441 amino acids in length. Tau is normally located in axons and regulates microtubule assembly/disassembly, and axonal transport of proteins and organelles. All six isoforms play a role in maintenance of microtubular structure. If one or more fails, or if there is a stoichiometric imbalance in the different variants, microtubule formation will become more difficult or the stability of microtubules formed will become compromised. Any excess of tau (of any isoform composition) can be bundled into indigestible residues (neurofibrillary tangles or Pick bodies) that choke the cell, but may also induce neurotoxicity. All patients with MAPT mutations are characterized by the deposition of insoluble, aggregated tau proteins within neurons and glial cells in the cerebral cortex and other brain regions. Nevertheless, there are two histological patterns depending on the location of the mutational change in the gene.
Most of the MAPT mutations are missense, deletion or silent mutations within coding regions of exons 1, 9, 11, 12 and 13, and affect all 6 isoforms of tau generating mutated tau molecules that (variably) lose their ability to interact with microtubules and promote axonal transport 60, 61, 67, 69, 113, 135, 136. Some also increase the propensity of the mutated tau to self‐aggregate into fibrils that form characteristic pathological structures, usually Pick‐like bodies (Figure 1A) within the brain 52, 60, 61, 114, 118, 135. Conversely, those with mutations in and around the stem loop structure produce an excess of 4R tau; hence, the insoluble tau aggregates are composed predominantly of 4R tau isoforms 29, 42, 69, 127, appearing as neurofibrillary tangle‐like structures within large and smaller pyramidal cells of cortical layers III and V, and also prominent within glial cells in the deep white matter, globus pallidus and internal capsule 42, 127 (Figure 1B).
The progranulin gene (GRN)
GRN contains 13 exons and encodes a full‐length protein (PGRN, predicted molecular weight 68 kDa) secreted as a glycosylated 85 kDa precursor which can be cleaved by elastase‐like activity into a series of 6 kDa cysteine‐rich fragments called granulins (GRNs) 63, 180. Both PGRN and the GRNs are biologically active with roles in tissue remodeling processes 4, 170. In peripheral tissues, PGRN is involved in development, wound repair and inflammation, activating signaling cascades that control cell cycle progression and cell motility 62. In the brain, PGRN is expressed in both neurons and microglia, particularly so in the latter following brain injury. GRN mutations in FTLD include missense mutations generating premature stop codons, insertion or deletion mutations resulting in frameshifts, or changes within initiation codons precluding transcription 9, 34. Nonetheless, all mutations, irrespective of type, ultimately generate the same functional effect – a null allele – with at least 50% loss of translated protein, causing haploinsufficiency. Most transcripts are immediately destroyed through nonsense‐mediated decay, and it is likely that none, or perhaps only a small proportion, is ever translated into mutant protein, thereby explaining the lack of mutated PGRN protein within NCI and DN 9. Intriguingly, whilst heterozygous mutations in GRN result in FTLD, homozygous GRN mutations are associated with the lysosomal storage disease, Neuronal Ceroid Lipofuscinosis (NCL) 150, implying a role for PGRN in endosomal/lysosomal/autophagosomal pathways.
Consistent with observations that GRN mutations do not result in the translation of mutated PGRN protein, the pathological changes in FTLD are witnessed as brain accumulations of TDP‐43, principally in the cerebral cortex and hippocampus, in the form of NCI and DN 9, 34 (Figure 2). NII of a “cat's eye” or “lentiform” appearance (Figure 2) have also been described 9, 34, 98, 128, 154.
C9orf72 expansion
The third major genetic cause of familial FTLD with or without ALS in combination, or familial ALS itself, is a hexanucleotide repeat expansion in C9orf72 gene 40, 134. Little is known about the normal function of C9orf72 protein, though homology suggests it may be part of the DENN (Differentially Expressed in Normal and Neoplastic cells) family of proteins which are GTD/GDP exchange factors that activate Rab‐GTPases. Consequently, the exact pathological mechanism(s) underlying the expansion in C9orf72 remains uncertain. A loss of function effect (haploinsufficiency) consequent on a reduced output of C9orf72 protein has been suggested 40, 173. Alternatively, the formation of both sense and antisense nuclear RNA foci has been demonstrated, both in human disease 31, 32, 40, 108 and in fly models 108. These may sequester RNA transcripts 40, 108, or other endogenous RNA binding proteins 31, 32, thereby interfering with the transcriptome. Lastly, a non‐ATG mediated (RAN) sense and antisense translation of the expansion itself leads to formation and cellular (usually cytoplasmic) accumulation of the dipeptide repeat proteins (DPR), poly‐GA, poly‐GR, poly‐GP, poly‐PA and poly‐PR, of presumed variable length 6, 37, 103, 110, 111, 181, any, or all, of which could confer neurotoxicity 91, 105, 109, 144, 175, 179. None of these three possible mechanisms is likely to be mutually exclusive and all could play some part in driving the TDP‐43 proteinopathy that characterizes the disorder.
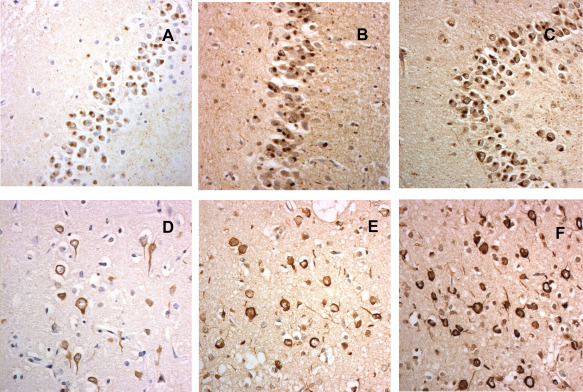
Expansions in C9orf72 are invariably associated with two histopathological changes. Firstly, there is TDP‐43 proteinopathy, type A in patients with bvFTD alone or type B when bvFTD is combined with MND (Figure 5). Secondly, RAN translation of the expansion results in the deposition of aggregates of p62‐positive, TDP‐43‐negative dipeptide repeat proteins (DPR) within the cytoplasm, and sometimes nucleus, of affected cells (Figure 4). DPR derived from sense translation (poly‐GA, poly‐GP and poly‐GR) seem to be more abundant than those derived from antisense translation (poly‐PA and poly‐PR) 38, 101. The presence of the pathognomonic p62‐containing DPR in C9orf72 expansion bearers suggests an underlying dysfunction of the ubiquitin‐proteasome system (UPS), which accords well with observations that mutations in genes coding for other members of this system (ie SQSTM1, UBQLN2, OPTN, VCP, TBK1 and CHMP2B) are all associated with bvFTD, and all except CHMP2B 68 with TDP‐43 proteinopathy. Indeed, TMEM106B, variations in which have been associated with bvFTD and TDP‐43 pathology 168, is also claimed to have functions within lysosome/endosome system. Hence, FTLD associated with TDP‐43 proteinopathy may reflect dysfunctions within protein degradation systems of the brain and failure to clear neurotoxic TDP‐43 aggregates. Whilst mutations in MAPT are clearly not associated with TDP‐43 proteinopathy, and no clear role in regulation of protein degradation pathways has been assigned to tau protein, this form of FTLD could, as with Alzheimer's disease and other tauopathies, again derive from failures of the lysosomal/proteasomal system to clear neurotoxic (phosphorylated, oligomeric) species of tau, which eventually overwhelm the neuron leading to its demise.
Figure 4.
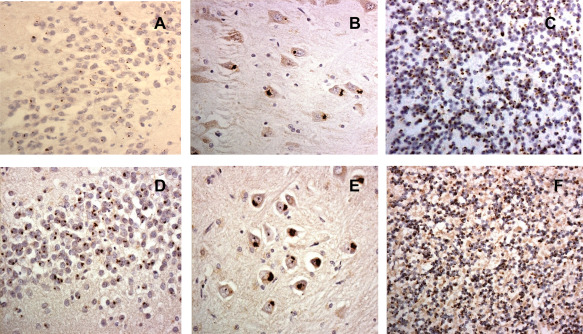
Dipeptide repeat proteins in hippocampus (A,B,D,E) and cerebellum (C,F) as observed in p62 immunostaining (A–C) and with antibody against poly (GA) (D–F).
How, or indeed whether, DPR are related to TDP‐43 proteinopathy remains unclear. Despite strong experimental evidence pointing the finger at DPR, especially those containing arginine residues, poly‐GR and poly‐PR 109, the clinical phenotype of bvFTD, bvFTD with ALS or ALS itself seems more closely related to the presence and brain distribution of TDP‐43 pathological changes than of DPR 37, 38, 53, 101. DPR are present in similar brain distributions in patients with bvFTD, bvFTD with ALS or ALS alone, and are widely present in affected and unaffected (by TDP‐43) regions of cerebral cortex, and numerous in granule cells of the cerebellum, CA subregions of the hippocampus and subcortical structures such as thalamus (Figure 4) 38, 53. Such a widespread distribution in “clinically silent” regions argues against a pathogenic role. Moreover, the extent of TDP‐43 pathological changes in bearers of C9orf72 expansion is identical in degree and distribution to that in patients with GRN mutation or no known mutation at all 38. Thus DPR may simply be pathological bystanders with no functional consequence. Nevertheless, as with other aggregating proteins such as Aβ or α‐synuclein, it remains possible that more soluble, and potentially neurotoxic, forms of DPR exist, and that the aggregated proteins seen in neurons represents a “neuroprotective” action on the part of the neuron in an attempt to package these out of harm's way. In this context the absence of DPR in spinal neurone with TDP‐43 proteinopathy in ALS 38, 53 may be relevant. However, the presence of such soluble (oligomeric) forms of DPR in human brain tissue has not been substantiated by western blotting. This may not be surprising given the extremely hydrophobic nature of the DPR which would “naturally” lead them to rapidly aggregate rapidly.
Some support for a pathogenic role of DPR comes from the observation that brains of preclinical C9orf72 expansion carriers and people with psychiatric or systemic disease in whom disease has been prematurely terminated due to accident or physical illness, show widespread DPR and RNA foci in the absence or near absence of TDP‐43 pathology 8, 50, 130, 171. The presence of DPR within the brain may predate that of TDP‐43 by many years 171. Rare cases such as these are useful in confirming that DPR accumulation precedes TDP‐43 pathology, but do not provide irrefutable evidence for a pathogenic role of DPR, especially in the most clinically common C9orf72 phenotypes. It remains possible that DPR are evidence of a functional/physiological deficit brought about by the effects of the gene expansion per se.
A possible explanation for these discrepancies between experimental and human studies may lie with the simplistic model systems that have been used to date in which transient over‐expression of DPR in cell cultures artificially elevates the rate of DPR production, thereby quickly overwhelming the cells. In human disease, DPR accumulation proceeds at a much slower rate, possibly over decades as is likely with Aβ accumulation in Alzheimer's disease, so that the neurons are able to manage this effectively, and counteract any detrimental effect. In this regard, some recent studies using mammalian models may be relevant. Transgenic mice carrying a bacterial artificial chromosome (BAC) containing the human C9orf72 gene with a disease‐relevant expansion were found to develop DPR pathology and RNA foci though no behavioral abnormalities developed and no neurodegeneration or TDP‐43 pathology was present 125, 126. In contrast, when an adeno‐associated virus was used to express the expanded repeat mice did develop behavioral abnormalities and neurodegeneration and showed TDP‐43 pathology in addition to DPR pathology and RNA foci 27. Taken together, these observations from mammalian models support the conclusion that RNA foci and DPR develop early but are insufficient to drive neurodegeneration in the absence of TDP‐43 pathology.
One way of reconciling these apparently anomalous findings would be to regard the effects of the expansion as a “gatekeeper” to disease whose physiological/metabolic effects make key regions of brain, such as the frontal and temporal cortex, vulnerable to the same factors that induce TDP‐43 proteinopathy in sporadic forms of FTLD‐TDP. This concept is in keeping with observations that the expansion in C9orf72 has no direct bearing on the specific histological or biochemical subtype, those patients with C9orf72 expansion bearing type A histology showing a TDP‐43 C‐terminal banding pattern indistinguishable from that in patients with GRN mutation, or without known mutation, with type A histology, and those C9orf72 patients with type B histology a pattern akin to that in patients with sporadic bvFTD with ALS 164. However, such a mechanism would require expansions in C9orf72 essentially to exert a loss of function effect, and that sporadic forms of bvFTD and bvFTD with ALS displaying TDP‐43 type A and type B histologies, respectively, would also suffer loss of C9orf72 function. While there is some evidence to suggest this is so in C9orf72 expansion carriers at least 40, 173, the absence of well‐characterized antibodies to the protein has hindered our lack of understanding of the normal role of C9orf72, and how changes in expression may affect protein degradation and influence disease risk. Although RAN translation in C9orf72 expansions may drive DPR formation unambiguous evidence that these latter molecules are anything other than pathological bystanders, and are capable of inducing pathogenic effects and contributing to the generation of clinical change, in the context of human disease, is still lacking.
Functional consequences
TDP‐43 is a 43 kDa nuclear protein, 414 amino acids long, first identified as a binding partner to the TAR DNA element of the human immunodeficiency virus. The TDP‐43 gene (TARDBP), located on chromosome 1p36.2, contains 6 exons and is ubiquitously expressed. It has 2 highly conserved RNA recognition motifs (RRM1 and RRM2) and a C‐terminal, glycine rich tail, which mediates protein‐protein interactions, including interactions with other heterogeneous ribonuclear protein (hnRNP) family members. Under physiological conditions, TDP‐43 is mostly present within the nucleus, though low levels occur within the cytoplasm 7, 21, 163, 165, 177. It is believed that TDP‐43 shuttles continuously between nucleus and cytoplasm, a process regulated by nuclear localization, and nuclear export, signal motifs. When NCI are present in the cell, TDP‐43 is no longer detectable immunohistochemically in the nucleus, possibly having been sequestered into NCI (Figure 2). Perturbation of trafficking of TDP‐43 between nucleus and cytoplasm may be a functional consequence of its incorporation into these cytoplasmic aggregates 177, though it is still unclear whether disease is due to a toxic gain of function relating to these pathological aggregates or stems from loss of normal nuclear functions, or some combination of the two.
In some cases, ubiquitinated, Pick‐body‐like inclusions are present in neurons of frontal and temporal cortex, and basal ganglia. These inclusions are negative for tau, TDP‐43 and α‐synuclein but (variably) immunopositive for all class IV intermediate filaments (IF), light, medium and heavy neurofilament subunits 13, 22, 23, 24, 78 and α‐internexin 25, 121. Consequently, the disorder is known as neuronal intermediate filament inclusion disease, NIFID, and classified as FTLD‐IF 109.
TDP‐43 and FUS are both transcription factors involved in the transport of mRNA to dendritic spines for local translation in relation to synaptic activity. This process requires the involvement of several types of RNA‐containing granules – ribonuclear protein particles (RNP), processing bodies (PB) and stress granules (SG) 16. However, whereas TDP‐43 appears to be associated with PB, FUS relates to SG 16, 172. PB are associated with mRNA decay processes and decapping enzyme1 protein – a marker for PB – is absent from NCI in BIBD 47, explaining the lack of TDP‐43 in such inclusions. In contrast, NCI in BIBD contain mRNA‐binding proteins poly(A) binding protein 1 and T cell intracellular antigen 1, indicating their association with SG rather than RNP or PB 47. Hence, TDP‐43 and FUS play different, through complementary, roles in the processing of RNA. The fact that mutations in TARDBP and FUS are associated with ALS and not bvFTD, alone or in combination with MND 12, 51, 57, 85, 90, 161, suggests that pure ALS, when characterized by TDP‐43 or FUS proteinopathy, is driven by functional changes in RNA metabolism – a notion put forward many years ago 102
Mechanistic considerations
Apart from being diagnostically relevant, and of clear value in the classification of disease, the histopathological changes in TDP‐43 that characterize many cases of FTLD may also inform pathogenesis. Although the use of “generic” antibodies to TDP‐43 have proved helpful in characterizing the morphological changes of FTLD‐TDP to subtype TDP‐43 proteinopathy, they may mask subtle biochemical and proteolytic differences that could provide clues to neural pathways involved in the disease, and highlight mechanistic differences between disorders. Hence, while immunoblotting of sarkosyl‐insoluble fractions from FTLD‐TDP and ALS cases demonstrates hyperphosphorylated full‐length TDP‐43 at 45 kDa, smearing substances, and fragments at 18–25 and 35 kDa in common across all histological subtypes, the use of phosphospecific antibodies, enables at least three C‐terminal banding patterns to be distinguished, which correspond to pathological phenotypes 164. Moreover, the insoluble TDP‐43 aggregates appear to have prion‐like properties, whereby TDP‐43 can induce aggregates characteristic of its own particular C‐terminal subtype via a proposed templated self‐seeding mechanism 48, 123, 149. Indeed, TDP‐43 pathology in ALS, at least, appears to progress in a hierarchical fashion that permits the recognition of 4 neuropathological stages 18, 19, consistent with the hypothesis that TDP‐43 pathology is propagated along axonal pathways, Collectively, these observations raise the possibility that each histopathological subtype may be associated with a specific pattern of TDP‐43 mismetabolism and proteolysis, and that each clinical syndrome develops from the templating and spread of such aggregates along its own discrete series of pathways.
The different morphological appearances and topographic patterns associated with each histological subtype have neuroanatomical implications. Subtype A, in which there is widespread NCI and DN within layer 2 of the cortex (Figure 2A) suggests degeneration within connecting corticocortical pathways, this being the layer where nerve terminal from intracortical projection fibres terminate. Type B histology, where NCI within cells of origin in layers 2, 3 and 5 predominate over DN (Figure 2B), implies involvement of cortical efferent pathways to subcortical regions. In type C histology (Figure 2C), the predominance of DN as long neuritic profiles could reflect damage to cortical afferents originating in the bilaterally damaged temporal lobes.
Such interpretations have relevance in terms of clinical phenotype. Sub‐type A is particularly associated with PNFA, which affects the left hemisphere extensively and is accompanied by loss of white matter connectivity in dorsal pathways 2, 3. There is prominent involvement of arcuate fasciculus, the traditional language pathways associated with phonological processing. PNFA patients may know a word but have difficulty accessing the pattern of sounds with which it is associated, in keeping with lack of cortico‐cortical connections. In sub‐type B, the involvement of cortical efferent pathways to subcortical regions is consistent with the specific association of this subtype with FTD‐ALS. It could be argued that FTD‐ALS represents a “cortical” extension of MND‐type pathology. Type C histology is specifically linked to SD. From a clinical perspective, SD might be construed as a disorder of “connectivity.” Patients with SD hear words, perceive objects and experience other sensory inputs normally. The problem is that those normal percepts have lost their connotative associations: they no longer “connect up” to convey meaning. The anterior temporal lobes are thought to be crucial for the integration of information from different modalities. Neuroimaging studies in SD using tractography 1, 2 also provide evidence of impaired connectivity. They have shown extensive loss of white matter connectivity from ventrorostral temporal lobes, affecting uncinate and inferior longitudinal fasciculus and including pathways to the supramarginal gyrus and the posterior superior temporal gyrus, classical language areas. The imaging findings have been interpreted 1 in terms of degeneration of axons whose cell bodies arise in the anterior temporal lobe and project to posterior language areas. The findings suggest a mechanism whereby a primary ventrorostral temporal lesion may result in a loss of input to language areas that are not themselves damaged.
Diagnostic and therapeutic issues
As summarized above, certain forms of FTLD, particularly the language variants, “obey” strict rules as regards their clinical, histological and genetic characteristics. SD and some forms of PNFA reflect TDP‐43 proteinopathy. Where there is autosomal inheritance of disease in PNFA, this relates to mutations in GRN. Similarly, when MND is present combined with bvFTD (and rarely SD or PNFA), TDP‐43 underlies this, and inherited forms are always associated with expansions in C9orf72. Should therapeutic options based on a correction of TDP‐43 abnormalities become available, choice of treatment could be based on these clinical and/or genetic features. The major challenge to diagnosis and therapy lies with bvFTD, since here there are potentially three histologies, and 3 genes (at least), that could drive the disease process. Distinguishing which might be responsible to direct treatment is not easy, unless there is a genetic cause. Whilst a validated clinical genetic test for C9orf72 is now widespread and relatively inexpensive, screening for MAPT or GRN mutations is more challenging, both in time and cost, given that a mutation could putatively exist in most of 15 exons in MAPT or 13 exons in GRN. FTLD‐FUS is essentially non‐genetic, ruling out one line of diagnostic enquiry. Better ways of predicting disease, either employing patterns of atrophy based on MRI image, or PET ligands that specifically bind to the relevant aggregated protein, would help reduce diagnostic uncertainty for bvFTD at least, though in this respect more precise neuropsychological profiling may help refine diagnosis and point to relevant therapy. Clues like the association between MAPT mutations and semantic impairment, very early onset with FUS pathology, psychosis and ALS with C9orf72 expansion with TDP‐43 proteinopathy, can provide helpful pointers.
Conclusion
While it is clear that FTLD is a beast with many heads, their faces bear distinguishing features that point to distinctive origins and properties. SD (especially) and PNFA have tight clinical, histological and genetic inter‐relationships, which ease diagnostic uncertainty and facilitate potential therapeutic strategy. BvFTD remains a challenge, where the overlapping clinical and histological properties are as yet only partially resolved by genetic analysis or neuropsychological assessment. Future advances in blood biomarkers and brain imaging may help to dissect underlying processes and direct therapy. It remains to be seen whether we can devise a single magic bullet that will slay the heart of the beast, and in so doing unselectively take with it all of its varying heads, irrespective of their complexion, or whether a more targeted approach will be required to identify and take out the individual malign elements that cripple the beast as a whole.
Conflict of Interest
The authors have no Conflicts of Interest to declare.
References
- 1. Acosta‐Cabronero J, Patterson K, Fryer TD, Hodges JR, Pengas G, Williams GB et al (2011) Atrophy, hypometabolism and white matter abnormalities in semantic dementia tell a coherent story. Brain 134:2025–2035. [DOI] [PubMed] [Google Scholar]
- 2. Agosta F, Henry RG, Migliaccio R, Neuhaus J, Miller BL, Dronkers NF et al (2010) Language networks in semantic dementia. Brain 133:286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Agosta F, Galantucci S, Canu E, Cappa SF, Magnani G, Franceschi M et al (2013) Disruption of structural connectivity along the dorsal and ventral language pathways in patients with nonfluent and semantic variant primary progressive aphasia: A DT MRI study and a literature review. Brain Lang 127:157–166. [DOI] [PubMed] [Google Scholar]
- 4. Ahmed Z, Mackenzie IR, Hutton ML, Dickson DW (2007) Progranulin in frontotemporal lobar degeneration and neuroinflammation. J Neuroinflammation 4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Armstrong RA (2016) Survival in the pre‐senile dementia frontotemporal lobar degeneration with TDP‐43 proteinopathy: Effects of genetic, demographic and neuropathological variables. Folia Neuropathol 54:137–148. [DOI] [PubMed] [Google Scholar]
- 6. Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus‐Hernandez M et al (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77:639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ayala YM, Misteli T, Baralle FE (2008) TDP‐43 regulates retinoblastoma protein phosphorylation through the repression of cyclin dependent kinase 6 expression. Proc Natl Acad Sci USA 105:3785–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baborie A, Griffiths TD, Jaros E, Perry R, McKeith IG, Burn DJ et al (2015) Accumulation of dipeptide repeat proteins predates that of TDP‐43 in Frontotemporal Lobar Degeneration associated with hexanucleotide repeat expansions in C9ORF72 gene. Neuropath Appl Neurobiol 41:601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baker M, Mackenzie IRA, Pickering‐Brown SM, Gass J, Rademakers R, Lindholm C et al (2006) Mutations in Progranulin cause tau‐negative frontotemporal dementia linked to chromosome 17. Nature 442:916–919. [DOI] [PubMed] [Google Scholar]
- 10. Bathgate D, Snowden JS, Varma A, Blackshaw A, Neary D (2001) Behaviour in frontotemporal dementia: A comparison with Alzheimer's disease and vascular dementia. Acta Neurol Scand 103:367–378. [DOI] [PubMed] [Google Scholar]
- 11. Beck J, Rohrer JD, Campbell T, Isaacs A, Morrison KE, Goodall EF et al (2008) A distinct clinical, neuropsychological and radiological phenotype is associated with progranulin gene mutations in a large UK series. Brain 131:706–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Benajiba L, Le Ber I, Camuzat A, Lacoste M, Thomas‐Anterion C, Couratier P et al (2009) TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol 65:470–473. [DOI] [PubMed] [Google Scholar]
- 13. Bigio EH, Lipton AM, White CL, III , Dickson DW, Hirano A (2003) Frontotemporal and motor neurone degeneration with neurofilament inclusion bodies: Additional evidence for overlap between FTD and ALS. Neuropathol Appl Neurobiol 29:239–253. [DOI] [PubMed] [Google Scholar]
- 14. Boeve BF (2007) Links between frontotemporal lobar degeneration, corticobasal degeneration, progressive supranuclear palsy and amyotrophic lateral sclerosis. Alzheimer Dis Assoc Disord 21:S31–S38. 3 [DOI] [PubMed] [Google Scholar]
- 15. Boeve BF, Boylan KB, Graff‐Radford NR, DeJesus‐Hernandez M, Knopman DS, Pedraza O et al (2012) Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 135:765–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bramham CR, Wells DG (2007) Dendritic mRNA: transport, translation and function. Nature Rev Neurosci 8:776–789. [DOI] [PubMed] [Google Scholar]
- 17. Brellstaff J, Lashley T, Holton JL, Lees AJ, Rossor MN, Bandopadhyay R et al (2011) Transportin 1: A marker for FTLD‐FUS. Acta Neuropathol 122:591–600. [DOI] [PubMed] [Google Scholar]
- 18. Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M et al (2013) Stages of pTDP‐43 pathology in amyotrophic lateral sclerosis. Ann Neurol 74:20–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brettschneider J, Del Tredici K, Irwin DJ, Grossman M, Robinson JL, Toledo JB et al (2014) Sequential distribution of pTDP‐43 pathology in behavioural variant frontotemporal dementia (bvFTD). Acta Neuropathol 127:423–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brown J, Ashworth A, Gydesen S, Sorensen A, Rossor M, Hardy J et al (1995) Familial non‐specific dementia maps to chromosome 3. Hum Mol Genet 4:1625–1628. [DOI] [PubMed] [Google Scholar]
- 21. Buratti E, Baralle FE (2008) Multiple roles of TDP‐43 in gene expression, splicing regulation and human disease. Front Bioscience 13:867–878. [DOI] [PubMed] [Google Scholar]
- 22. Burrell JR, Halliday GM, Kril JJ, Ittner LM, Gotz J, Kiernan MC et al (2016) The frontotemporal dementia‐motor neuron disease continuum. Lancet 388:919–931. [DOI] [PubMed] [Google Scholar]
- 23. Cairns NJ, Perry RH, Jaros E, Burn D, McKeith IG, Lowe JS et al (2003) Patients with a novel neurofilamentopathy: Dementia with neurofilament inclusions. Neurosci Lett 341:177–180. [DOI] [PubMed] [Google Scholar]
- 24. Cairns NJ, Grossman M, Arnold SE, Burn DJ, Jaros E, Perry RH et al (2004) Clinical and neuropathologic variation in neuronal intermediate filament inclusion disease. Neurology 63:1376–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cairns NJ, Zhuckareva V, Uryu K, Zhang B, Bigio E, Mackenzie IR et al (2004) Alpha‐internexin is present in the pathological inclusions of neuronal intermediate filament inclusion disease. Am J Pathol 164:2153–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen‐Plotkin AS, Martinez‐Lage M, Sleiman PMA, Hu W, Greene R, Wood EM et al (2011) Genetic and clinical features of progranulin‐associated frontotemporal lobar degeneration. Arch Neurol 68:488–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes‐Casey M et al (2015) Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP‐43 pathology, neuronal loss, and behavioral deficits. Science 348:1151–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chiu WZ, Kaat LD, Seelaar H, Rosso SM, Boon AJ, Kamphorst W et al (2010) Survival in progressive supranuclear palsy and frontotemporal dementia. J Neurol Neurosurg Psychiatry 81:441–445. [DOI] [PubMed] [Google Scholar]
- 29. Clark LN, Poorkaj P, Wzsolek Z, Geschwind DH, Nasreddine ZS, Miller B et al (1998) Pathogenic implications of mutations in the tau gene in pallido‐ponto‐nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc Natl Acad Sci USA 95:13103–13107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Coleman LW, Digre KB, Stephenson GM, Townsend JJ (2002) Autopsy proven, sporadic Pick disease with onset at age 25 years. Arch Neurol 59:856–859. [DOI] [PubMed] [Google Scholar]
- 31. Cooper‐Knock J, Walsh MJ, Higginbottom A, Highley RJ, Dickman MJ, Edbauer D et al (2014) Sequestration of multiple RNA recognition motif‐containing proteins by C9orf72 repeat expansions. Brain 137:2040–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cooper‐Knock J, Higginbottom A, Stopford MJ, Highley RJ, Ince PG, Wharton SB et al (2015) Antisense RNA foci in the motor neurones of C9ORF72‐ALS patients are associated with TDP‐43 proteinopathy. Acta Neuropathol 130:63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Coyle‐Gilchrist IT, Dick KM, Patterson K, Vazquez‐Rodriquez P, Wehmann E, Wilcox A et al (2016) Prevalence, characteristics and survival of frontotemporal lobar degeneration syndromes. Neurology 86:1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D et al (2006) Null mutations in progranulin cause ubiquitin‐positive frontotemporal dementia linked to chromosome 17q21. Nature 442:920–924. [DOI] [PubMed] [Google Scholar]
- 35. Davidson Y, Kelley T, Mackenzie IR, Pickering‐Brown S, Du Plessis D, Neary D et al (2007) Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA‐binding protein, TDP‐43. Acta Neuropathol 113:521–533. [DOI] [PubMed] [Google Scholar]
- 36. Davidson YS, Robinson AC, Hu Q, Mishra M, Baborie A, Jaros E et al (2013) Nuclear Carrier and RNA Binding Proteins in Frontotemporal Lobar Degeneration associated with Fused in Sarcoma (FUS) pathological changes. Neuropathol Appl Neurobiol 39:157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Davidson Y, Barker H, Robinson AC, Troakes C, Smith B, Al‐Sarraj S et al (2014) Brain distribution of dipeptide repeat proteins in Frontotemporal Lobar Degeneration and Motor Neurone Disease associated with expansions in C9ORF72 . Acta Neuropathol Commun 2:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Davidson Y, Robinson AC, Liu X, Wu D, Troakes C, Rollinson S et al (2016) Neurodegeneration in frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9orf72 is linked to TDP‐43 pathology and not associated with aggregated forms of dipeptide repeat proteins. Neuropathol Appl Neurobiol 42:242–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Davion S, Johnson N, Weintraub S, Mesulam M‐M, Engberg A, Michra M et al (2007) Clinicopathologic correlation in PGRN mutations. Neurology 69:1113–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron 72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N et al (2011) Mutations in UBQLN2 cause dominant X‐linked juvenile and adult‐onset ALS and ALS/dementia. Nature 477:211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. De Silva R, Lashley T, Strand K, Shiarli AM, Shi J, Tian J et al (2006) An immunohistochemical study of cases of sporadic and inherited Frontotemporal lobar degeneration using 3R‐ and 4R‐specific tau monoclonal antibodies. Acta Neuropathol 111:329–340. [DOI] [PubMed] [Google Scholar]
- 43. Devenney E, Hornberger M, Irish M, Mioshi E, Burrell J, Tan R et al (2014) Frontotemporal dementia associated with the C9ORF72 mutation. A unique clinical profile. JAMA Neurol 71:331–339. [DOI] [PubMed] [Google Scholar]
- 44. Fletcher PD, Downey LE, Golden HL, Clark CN, Slattery CF, Paterson RW et al (2015) Auditory hedonic phenotypes in dementia: A behavioural and neuroanatomical analysis. Cortex 67:95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fletcher PD, Downey LE, Golden HL, Clark CN, Slattery CF, Paterson RW et al (2015) Pain and temperature processing in dementia: A clinical and neuroanatomical analysis. Brain 138:3360–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Forman MS, Farmer J, Johnson JK, Clark CM, Arnold SE, Coslett HB et al (2006) Frontotemporal dementia: Clinicopathological correlations. Ann Neurol 59:952–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fujita K, Ito H, Nakano S et al (2008) Immunohistochemical identification of messenger RNA‐related proteins in basophilic inclusions of adult‐onset atypical motor neuron disease. Acta Neuropathol 118:439–445. [DOI] [PubMed] [Google Scholar]
- 48. Furukawa Y, Kaneko K, Watanabe S, Yamanaka K, Nukina N (2011) A seeding reaction recapitulates intracellular formation of sarkosyl‐insoluble TAR DNA binding protein‐43 inclusions. J Biol Chem 286:18664–18672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Galimberti D, Fenoglio C, Serpente M, Villa C, Bonsi R, Arighi A et al (2013) Autosomal dominant frontotemporal lobar degeneration due to the C9ORF72 hexanucleotide repeat expansion: Late‐onset psychotic clinical presentation. Biol Psychiatry 74:384–391. [DOI] [PubMed] [Google Scholar]
- 50. Gami P, Murray C, Schottlaender L, Bettencourt C, De Pablo Fernandez E, Mudanohwo E et al (2015) A 30‐unit hexanucleotide repeat expansion in C9orf72 induces pathological lesions with dipeptide‐repeat proteins and RNA foci, but not TDP‐43 inclusions and clinical disease. Acta Neuropathol 130:599–601. [DOI] [PubMed] [Google Scholar]
- 51. Gitcho MA, Baloh AH, Chakraverty S, Mayo K, Norton JB, Levitch D et al (2008) TDP‐43 A315T mutation in familial motor neurone disease. Ann Neurol 63:535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Goedert M, Jakes R, Crowther RA (1999) Effects of frontotemporal dementia FTDP‐17 mutations on heparin‐induced assembly of tau filaments. FEBS Lett 450:306–311. [DOI] [PubMed] [Google Scholar]
- 53. Gomez‐Deza J, Lee Y‐B, Troakes C, Nolan M, Al‐Sarraj S, Gallo J‐M et al (2015) Dipeptide repeat protein inclusions are rare in the spinal cord and almost absent from motor neurons in C9ORF72 mutatant amyotrophic lateral sclerosis and are unlikely to cause their neurodegeneration. Acta Neuropathol Commun 3:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gorno‐Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF et al (2011) Classification of primary progressive aphasia and its variants. Neurology 76:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Govaarts R, Beeldman E, Kamelmacher MJ, von Tol MJ, van den Berg LH, van der Kooi AJ et al (2016) The frontotemporal syndrome of ALS is associated with poor survival. J Neurol 263:2476–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gydesen S, Hagen S, Klinken L, Abelskov J, Sørensen SA (1987) Neuropsychiatric studies in a family with presenile dementia different from Alzheimer and Pick disease. Acta Psychiatr Scand 76:276–284. [DOI] [PubMed] [Google Scholar]
- 57. Hardy J, Rogaeva E (2014) Motor Neurone Disease and frontotemporal dementia: Sometimes related, sometimes not. Exp Neurol 262:75–83. [DOI] [PubMed] [Google Scholar]
- 58. Harris JM, Gall C, Thompson JC, Richardson AM, Neary D, du Plessis D et al (2013) Classification and pathology of primary progressive aphasia. Neurology 81:1832–1839. [DOI] [PubMed] [Google Scholar]
- 59. Harris JM, Jones M, Gall C, Richardson AM, Neary D, du Plessis D et al (2016) Co‐occurrence of language and behavioural change in frontotemporal lobar degeneration. Dement Geriatr Cogn Dis Extra 6:205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hasegawa M, Smith MJ, Goedert M (1998) Tau proteins with FTDP‐17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett 437:207–210. [DOI] [PubMed] [Google Scholar]
- 61. Hayashi S, Toyoshima Y, Hasegawa M, Umeda Y, Wakabayashi K, Tokiguchi S et al (2002) Late‐onset frontotemporal dementia with a novel exon 1 (Arg5His) tau gene mutation. Ann Neurol 51:525–530. [DOI] [PubMed] [Google Scholar]
- 62. He Z, Bateman A (2003) Progranulin (granulin‐epithelin precursor, PC‐cell‐derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med 1:600–612. [DOI] [PubMed] [Google Scholar]
- 63. He Z, Ong CH, Halper J, Bateman A (2003) Progranulin is a mediator of the wound response. Nature Med 9:225–229. [DOI] [PubMed] [Google Scholar]
- 64. Hodges JR, Davies R, Xuereb J, Kril J, Halliday G (2003) Survival in frontotemporal dementia. Neurology 61:349–354. [DOI] [PubMed] [Google Scholar]
- 65. Hodges JR, Patterson K (2007) Semantic dementia: A unique clinicopathological syndrome. Lancet Neurol 6:1004–1014. [DOI] [PubMed] [Google Scholar]
- 66. Hodges JR, Mitchell J, Dawson K, Spillantini MG, Xuereb JH, McMonagle P et al (2010) Semantic dementia: demography, familial factors and survival in a consecutive series of 100 cases. Brain 133:300–306. [DOI] [PubMed] [Google Scholar]
- 67. Hogg M, Grujic ZM, Baker M, Demirci S, Guillozet AL, Sweet AP et al (2003) The L266V tau mutation is associated with frontotemporal dementia and Pick‐like 3R and 4R tauopathy. Acta Neuropathol 106:323–336. [DOI] [PubMed] [Google Scholar]
- 68. Holm IE, Englund E, Mackenzie IRA, Johannsen P, Isaacs AM (2007) A reassessment of the neuropathology of frontotemporal dementia linked to chromosome 3 (FTD‐3). J Neuropathol Exp Neurol 66:884–891. [DOI] [PubMed] [Google Scholar]
- 69. Hong M, Zhukareva V, Vogelsberg‐Ragaglia V, Wszolek Z, Reed L, Miller BI (1998) Mutation‐specific functional impairments in distinct tau isoforms of hereditary FTDP‐17. Science 282:1914–1917. [DOI] [PubMed] [Google Scholar]
- 70. Hsiung G‐YR, DeJesus‐Hernandez M, Feldman HH, Sengdy P, Bouchard‐Kerr P, Dwosh E et al (2012) Clinical and pathological features of familial Frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain 135:709–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H et al (1998) Association of missense and 5'‐splice mutations in tau with the inherited FTDP‐17. Nature 393:702–705. [DOI] [PubMed] [Google Scholar]
- 72. Ioannidis P, Konstantinopoulou E, Maiovis P, Karacostas D (2012) The frontotemporal dementias in a tertiary referral center: Classification and demographic characteristics in a series of 232 cases. J Neurol Sci 318:171–173. [DOI] [PubMed] [Google Scholar]
- 73. Irwin DJ, Cairns NJ, Grossman M, McMillan CT, Lee EB, Van Deerlin VM et al (2015) Frontotemporal lobar degeneration: Defining phenotypic diversity through personalized medicine. Acta Neuropathol 129:469–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Isaacs AM, Johannsen P, Holm I, Nielsen JE, FReJA Consortium (2011) Frontotemporal dementia caused by CHMP2B mutations. Curr Alzheimer Res 8:246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jacob J, Revesz T, Thom M, Rossor MN (1999) A case of sporadic Pick disease with onset at 27 years. Arch Neurol 56:1289–1291. [DOI] [PubMed] [Google Scholar]
- 76. Janssen JC, Warrington EK, Morris HR, Lantos P, Brown J, Revesz T et al (2002) Clinical features of Frontotemporal dementia due to the intronic tau 10(+16) mutation. Neurology 58:1161–1168. [DOI] [PubMed] [Google Scholar]
- 77. Johnson J, Diehl‐Schmid J, Mendez M, Neuhaus J, Shapira J, Forman MS et al (2005) Frontotemporal lobar degeneration: demographic characteristics of 353 patients. Arch Neurol 62:925–930. [DOI] [PubMed] [Google Scholar]
- 78. Josephs KA, Holton JL, Rossor MN, Braendgaard H, Ozawa T, Fox NC (2003) Neurofilament inclusion body disease: a new proteinopathy?. Brain 126:2291–2303. [DOI] [PubMed] [Google Scholar]
- 79. Josephs KA, Knopman DS, Whitwell JL, Boeve BF, Parisi JE, Petersen RC et al (2005) Survival in two variants of tau‐negative frontoemporal lobar degeneration: FTLS‐U vs FTLD‐MND. Neurology 65:645–647. [DOI] [PubMed] [Google Scholar]
- 80. Josephs KA, Duffy JR, Strand EA, Whitwell JL, Layton KF, Parisi JE et al (2006) Clinicopathological correlates of progressive aphasia and apraxia of speech. Brain 129:1385–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Josephs KA, Petersen RC, Knopman DS, Boeve BF, Whitwell JL, Duffy JR et al (2006) Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology 66:41–48. [DOI] [PubMed] [Google Scholar]
- 82. Josephs KA, Whitwell JL, Vemuri P, Senjem ML, Boeve BF, Knopman DS et al (2008) The anatomic correlate of prosopagnosia in semantic dementia. Neurology 71:1628–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Josephs KA, Whitwell JL, Parisi JE, Petersen RC, Boeve BF, Jack CR, Jr et al (2010) Caudate atrophy on MRI is a characteristic feature of FTLD‐FUS. Eur J Neurol 17:969e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Josephs KA, Hodges JR, Snowden JS, Mackenzie IR, Neumann M, Mann DM et al (2011) Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol 122:137–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C et al (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nature Genet 40:572–574. [DOI] [PubMed] [Google Scholar]
- 86. Kansal K, Mareddy M, Sloane KL, Minc AA, Rabins PV, McGready JB et al (2016) Survival in frontotemporal dementia phenotypes: A meta‐analysis. Dement Geriatr Cogn Disord 41:109–122. [DOI] [PubMed] [Google Scholar]
- 87. Kertesz A, Ang LC, Jesso S, MacKinley J, Baker M, Brown P et al (2013) Psychosis and hallucinations in frontotemporal dementia with the C9ORF72 mutation: A detailed clinical cohort. Cogn Behav Neurol 26:146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Knopman DS, Roberts RO (2011) Estimating the number of persons with frontotemporal lobar degeneration in the US population. J Mol Neurosci 45:330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kobylecki C, Jones M, Thompson JC, Richardson AM, Neary D, Mann DM et al (2015) Cognitive‐behavioural features of progressive supranuclear palsy syndrome overlap with frontotemporal dementia. J Neurol 262:916–222. [DOI] [PubMed] [Google Scholar]
- 90. Kwiatkowski TJ, Bosco DA, LeClerc AL, Tamrazian E, Vanderburg CR, Russ C et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208. [DOI] [PubMed] [Google Scholar]
- 91. Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T et al (2014) Poly‐dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 345:1139–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Landqvist WM, Gustafson L, Passant U, Englund E (2015) Psychotic symptoms in frontotemporal dementia: A diagnostic dilemma?. Int Psychogeriatr 27:531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lashley T, Rohrer JD, Brandopadhyay R, Fry C, Ahmed Z, Isaacs AM et al (2011) A comparative clinical, pathological, biochemical and genetic study of fused in sarcoma proteinopathies. Brain 134:2548–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Le Ber I, Guedj E, Gabelle A, Verpillat P, Volteau M, Thomas‐Anterion C et al (2006) Demographic, neurological and behavioural characteristics and brain perfusion SPECT in frontal variant of frontotemporal dementia. Brain 129:3051–3065. [DOI] [PubMed] [Google Scholar]
- 95. Le Ber I, van der Zee J, Hannequin D, Gijselinck I, Campion D, Puel M et al (2007) Progranulin null mutations in both sporadic and familial Frontotemporal dementia. Hum Mutation 28:846–855. [DOI] [PubMed] [Google Scholar]
- 96. Le Ber I, Camuzat A, Hannequin D, Pasquier F, Guedj E, Rovelet‐Lecrux A et al (2008) Phenotype variability in progranulin mutation carriers: A clinical, neuropsychological, imaging and genetic study. Brain 131:732–746. 2008; [DOI] [PubMed] [Google Scholar]
- 97. Le Ber I, Camuzat A, Guerreiro R, Bouya‐Ahmed K, Bras J, Nicolas G et al (2013) SQSTM1 mutations in French patients with Frontotemporal Dementia or Frontotemporal Dementia with Amyotrophic Lateral Sclerosis. JAMA Neurol 70:1403–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mackenzie IRA, Baker M, Pickering‐Brown S, Hsiung GY, Lindholm C, Dwosh E et al (2006) The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain 129:3081–3090. [DOI] [PubMed] [Google Scholar]
- 99. Mackenzie IRA, Neumann M, Bigio E, Cairns NJ, Alafuzoff I, Krill J et al (2009) Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: Consensus recommendations. Acta Neuropathol 117:15–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Mackenzie IRA, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E et al (2011) A harmonized classification system for FTLD‐TDP pathology. Acta Neuropathol 122:111–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Mackenzie IR, Arzberger T, Kremmer E, Troost D, Lorenzl S, Mori K et al (2013) Dipeptide repeat protein pathology in C9ORF72 mutation cases: Clinico‐pathological correlations. Acta Neuropathol 126:859–879. [DOI] [PubMed] [Google Scholar]
- 102. Mann DMA, Yates PO (1974) Motor Neurone Disease: The nature of the pathogenetic mechanism. J Neurol Neurosurg Psychiat 37:1036–1046. [Google Scholar]
- 103. Mann DMA, Rollinson S, Robinson A, Callister J, Snowden JS, Gendron T et al (2013) Dipeptide repeat proteins are present in the p62 positive inclusions in patients with Frontotemporal Lobar Degeneration and Motor Neurone Disease associated with expansions in C9ORF72. Acta Neuropathol Comm 1:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y et al (2010) Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465:223–226. [DOI] [PubMed] [Google Scholar]
- 105. May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM et al (2014) C9orf72 FTLD/ALS‐associated Gly‐Alal dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol 128:485–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Mendez MF, Shapira JS, Woods RJ, Licht EA, Saul RE (2008) Psychotic symptoms in frontotemporal dementia: Prevalence and review. Dement Geriatr Cogn Disord 25:206–211. [DOI] [PubMed] [Google Scholar]
- 107. Mesulam M‐M, Wieneke C, Thompson C, Rogalski E, Weintraub S (2012) Quantitative classification of primary progressive aphasia at early and mild impairment stages. Brain 135:1537–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mizielinska S, Lashley T, Norona FE, Clayton EL, Ridler CE, Fratta P et al (2013) C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol 126:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Mizielinska S, Gronke S, Niccoli T, Ridler CE, Clayton EL, Devoy A et al (2014) C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine‐rich proteins. Science 345:1192–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Mori K, Arzberger T, Grasser FA, Gijselinck I, May S, Rentzsch K et al (2013) Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol 126:881–894. [DOI] [PubMed] [Google Scholar]
- 111. Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E et al (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide‐repeat proteins in FTLD/ALS. Science 339:1335–1338. [DOI] [PubMed] [Google Scholar]
- 112. Munoz D, Neumann M, Kusaka H, Yokota O, Ishihara K, Terada S et al (2009) FUS pathology in Basophilic Inclusion Body disease (BIBD). Acta Neuropathol 118:617–627. [DOI] [PubMed] [Google Scholar]
- 113. Murrell JR, Spillantini MG, Zolo P, Guazzelli M, Smith MJ, Hasegawa M et al (1999) Tau gene mutation G389R causes a tauopathy with abundant pick body‐like inclusions and axonal deposits. J Neuropathol Exp Neurol 58:1207–1226. [DOI] [PubMed] [Google Scholar]
- 114. Nacharaju P, Lewis J, Easson C, Yen S, Hackett J, Hutton M et al (1999) Accelerated filament formation from tau protein with specific FTDP‐17 missense mutations. FEBS Lett 447:195–199. [DOI] [PubMed] [Google Scholar]
- 115. Neary D, Snowden JS, Mann DMA, Northen B, Goulding PJ, MacDermott N (1990) Frontal lobe dementia and motor neurone disease. J Neurol Neurosurg Psychiatry 53:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S et al (1998) Frontotemporal lobar degeneration. A consensus on clinical diagnostic criteria. Neurology 51:1546–1554. [DOI] [PubMed] [Google Scholar]
- 117. Neary D, Snowden JS, Mann DMA (2007) Frontotemporal lobar degeneration: Clinical and pathological relationships. Acta Neuropathol 114:31–38. [DOI] [PubMed] [Google Scholar]
- 118. Neumann M, Schulz‐Schaeffer W, Crowther RA, Smith MJ, Spillantini MG, Goedert M et al (2001) Pick's disease associated with the novel tau gene mutation K369I. Ann Neurol 50:503–513. [DOI] [PubMed] [Google Scholar]
- 119. Neumann M, Sampathu DM, Kwong LK, Chou TT, Micsenyi M, Truax A et al (2006) TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. [DOI] [PubMed] [Google Scholar]
- 120. Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IRA (2009) A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132:2922–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Neumann M, Roeber S, Kretzschmar H, Mackenzie IRA (2009) Abundant FUS pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol 118:605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Neumann M, Bentmann E, Dormann D, Jawaid A, DeJesus‐Hernandez M, Ansorge O et al (2011) FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain 134:2595–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Nonaka T, Masuda‐Suzukake M, Arai T, Hasegawa Y, Akatsu H, Obi T et al (2013) Prion‐like properties of pathological TDP‐43 aggregates from diseased brains. Cell Rep 4:124–134. [DOI] [PubMed] [Google Scholar]
- 124. Onyike CU, Diehl‐Schmid J (2013) The epidemiology of frontotemporal dementia. Int Rev Psych 25:130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. O'Rourke JG, Bogdanik L, Muhammad AK, Gendron TF, Kim KJ, Austin A et al (2015) C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron 88:892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Peters OM, Cabrera GT, Tran H, Gendron TF, McKeon JE, Metterville J et al (2015) Human C9ORF72 Hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron 88:902–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Pickering Brown SM, Richardson AM, Snowden JS, McDonagh AM, Burns A, Braude W et al (2002) Inherited frontotemporal dementia in 9 British families associated with intronic mutations in the tau gene. Brain 125:732–751. [DOI] [PubMed] [Google Scholar]
- 128. Pickering‐Brown SM, Rollinson S, Du Plessis D, Morrison KE, Varma A, Richardson AMT et al (2008) Frequency and clinical characteristics of progranulin mutation carriers in the Manchester Frontotemporal Lobar Degeneration cohort: Comparison to patients with MAPT and no known mutations. Brain 131:721–731. [DOI] [PubMed] [Google Scholar]
- 129. Pottier C, Bieniek KF, Finch N, van de Vorst M, Baker M, Perkersen R et al (2015) Whole‐genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol 130:77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Proudfoot M, Gutowski NJ, Edbauer D, Hilton DA, Stephens M, Rankin J et al (2014) Early dipeptide repeat pathology in a frontotemporal dementia kindred with C9ORF72 mutation and intellectual disability. Acta Neuropathol 127:451–458. 2014 [DOI] [PubMed] [Google Scholar]
- 131. Rademakers R, Baker M, Gass J, Adamson J, Huey ED, Momeni P et al (2007) Phenotypic variability associated with progranulin haploinsufficiency in patients with the common 1477C→T (Arg493X) mutation: An international initiative. Lancet Neurol 6:857–868. [DOI] [PubMed] [Google Scholar]
- 132. Ranasinghe KG, Rankin KP, Pressman PS, Perry DC, Lobach IV, Seeley WW et al (2016) Distinct subtypes of behavioral variant frontotemporal dementia based on patterns of network degeneration. JAMA Neurol 73:1078–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J et al (2011) Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134:2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Renton AE, Majounie E, Waite A, Simon‐Sanchez J, Rollinson S, Gibbs JR et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron 72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Rizzini C, Goedert M, Hodges J, Smith MJ, Jakes R, Hills R et al (2000) Tau gene mutation K257T causes a tauopathy similar to Pick's disease. J Neuropathol Exp Neurol 59:990–1001. [DOI] [PubMed] [Google Scholar]
- 136. Rizzu P, Van Swieten JC, Joosse M, Hasegawa M, Stevens M, Tibben A et al (1999) High prevalence of mutations in the microtubule‐associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet 64:414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Rohrer JD, Guerreiro R, Vandrovcova J, Uphill J, Reiman D, Beck J et al (2009) The heritability and genetics of frontotemporal lobar degeneration. Neurology 73:1451–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Rohrer JD, Rosser M, Warren JD (2010) Syndromes of nonfluent primary progressive aphasia. A clinical and neurolinguistics analysis. Neurology 75:603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Rohrer JD, Lashley T, Holton J, Revesz T, Urwin H, Isaacs A et al (2010) The clinical and neuroanatomical phenotype of FUS associated frontotemporal lobar degeneration. J Neurol Neurosurg Psychiatr 82:1405–1407. [DOI] [PubMed] [Google Scholar]
- 140. Rohrer JD, Lashley T, Schott JM, Warren JE, Mead S, Isaacs AM et al (2011) Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain 134:2565–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Rosso SM, Donker Kaat L, Baks T, Joose M, de Koning I, Pijnenburg Y et al (2003) Frontotemporal dementia in the Netherlands: Patient characteristics and prevalence estimates from a population‐based study. Brain 126:2016–2022. [DOI] [PubMed] [Google Scholar]
- 142. Sajjadi SA, Patterson K, Arnold RJ, Watson PC, Nestor PJ (2012) Primary progressive aphasia: A tale of two syndromes and the rest. Neurology 78:1670–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Saxon JA, Harris JM, Thompson JC, Jones M, Richardson AMT, Langheinrich T et al (2017) Semantic dementia, progressive non‐fluent aphasia and their association with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Schludi MH, May S, Grässer FA, Rentzsch K, Kremmer E, Küpper C et al (2015) Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta Neuropathol 130:537–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Seelaar H, Klijnsma KY, de Koning I, van der Lugt A, Chiu WZ, Azmani A et al (2011) Frequency of ubiquitin and FUS‐positive, TDP‐43‐negative frontotemporal lobar degeneration. J Neurol 257:747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Shi J, Shaw CL, Richardson AMT, Bailey K, Julien C, Stopford C et al (2005) Histopathological changes underlying frontotemporal lobar degeneration with clinicopathological correlation. Acta Neuropathol 110:501–512. [DOI] [PubMed] [Google Scholar]
- 147. Shinagawa S, Nakajima S, Plitman E, Graff‐Guerrero A, Mimura M, Nakayama K et al (2014) Psychosis in frontotemporal dementia. J Alzheimers Dis 42:485–499. [DOI] [PubMed] [Google Scholar]
- 148. Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H et al (2005) Mutations in the endosomal ESCRTIII‐complex subunit CHMP2B in frontotemporal dementia. Nature Genet 37:806–808. [DOI] [PubMed] [Google Scholar]
- 149. Smethurst P, Newcombe J, Troakes C, Simone R, Chen Y‐R, Patani R et al (2016) In vitro prion‐like behaviour of TDP‐43 in ALS. Neurobiol Dis 6:236–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M et al (2012) Strikingly different clinicopathological phenotypes determined by progranulin‐mutation dosage. Am J Hum Genet 90:1102–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Snowden JS, Bathgate B, Varma A, Blackshaw A, Gibbons ZC, Neary D (2001) Distinct behavioural profiles in frontotemporal dementia and semantic dementia. J Neurol Neurosurg Psychiatry 70:323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Snowden JS, Neary D, Mann DMA (2004) Autopsy proven, sporadic frontotemporal dementia, due to microvacuolar histology, with onset at 21 years of age. J Neurol Neurosurg Psychiatry 75:1337–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Snowden JS, Thompson JC, Neary D (2004) Knowledge of famous faces and names in semantic dementia. Brain 127:860–872. [DOI] [PubMed] [Google Scholar]
- 154. Snowden JS, Pickering‐Brown SM, Mackenzie IR, Richardson AM, Varma A, Neary D et al (2006) Progranulin gene mutations associated with frontotemporal dementia and progressive non‐fluent aphasia. Brain 129:3091–3102. [DOI] [PubMed] [Google Scholar]
- 155. Snowden JS, Pickering‐Brown SM, Du Plessis D, Mackenzie IRA, Varma A, Mann DMA et al (2007) Progressive anomia revisited: focal degeneration associated with progranulin gene mutation. Neurocase 13:366–377. [DOI] [PubMed] [Google Scholar]
- 156. Snowden JS, Hu Q, Rollinson S, Halliwell N, Robinson A, Davidson YS et al (2011) The most common type of FTLD‐FUS (aFTLD‐U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS gene. Acta Neuropathol 122:99–110. [DOI] [PubMed] [Google Scholar]
- 157. Snowden JS, Thompson JC, Stopford CL, Richardson AMT, Gerhard A, Neary D et al (2011) Clinical diagnosis of early‐onset dementias: Diagnostic accuracy and clinico‐pathological relationships. Brain 134:2478–2492. [DOI] [PubMed] [Google Scholar]
- 158. Snowden JS, Rollinson S, Thompson JC, Harris JM, Stopford CL, Richardson AM et al (2012) Distinct clinical and pathological characteristics in patients with frontotemporal dementia and C9ORF72 mutations. Brain 135:693–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Snowden JS, Adams J, Harris J, Thompson JC, Rollinson S, Richardson A et al (2015) Distinct clinical and pathological phenotypes in frontotemporal dementia associated with MAPT, PGRN and C9orf72 mutations. Amyotroph Lat Sclerosis Frontotemporal Degeneration 16:497–505. [DOI] [PubMed] [Google Scholar]
- 160. Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B (1998) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA 95:7737–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B et al (2008) TDP‐43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Thompson SA, Patterson K, Hodges JR (2003) Left/right asymmetry of atrophy in semantic dementia: Behavioral‐cognitive implications. Neurology 61:1196–1203. [DOI] [PubMed] [Google Scholar]
- 163. Thorpe JR, Tang H, Atherton J, Cairns NJ (2008) Fine structural analysis of the neuronal inclusions in frontotemporal lobar degeneration with TDP‐43 proteinopathy. J Neural Transm 115:1661–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Tsuji H, Arai T, Kametani F, Nonaka T, Yamashita M, Suzukake M et al (2012) Molecular analysis and biochemical classification of TDP‐43 proteinopathy. Brain 135:3380–3391. [DOI] [PubMed] [Google Scholar]
- 165. Turner BJ, Baumer D, Parkinson NJ, Scaber J, Ansorge O, Talbot K (2008) TDP‐43 expression in mouse models of amyotrophic lateral sclerosis and spinal muscular atrophy. BMC Neuroscience 9:104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Urwin H, Josephs KA, Rohrer JD, Mackenzie IR, Neumann M, Authier A et al (2010) FUS pathology defines the majority of tau‐and TDP‐43‐negative frontotemporal lobar degeneration. Acta Neuropathol 120:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211. 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Van Deerlin VM, Sleiman PM, Martinez‐Lage M, Chen‐Plotkin A, Wang LS, Graff‐Radford NR et al (2010) Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP‐43 inclusions. Nature Genet 42:234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Van Langenhove T, van der Zee J, Gijselinck I, Engelborghs S, Vandenberghe R, Vandenbulcke M et al (2013) Distinct clinical characteristics of C9orf72 expansion carriers compared with GRN, MAPT, and nonmutation carriers in a Flanders‐Belgian FTLD cohort. JAMA Neurol 70:365–373. [DOI] [PubMed] [Google Scholar]
- 170. Van Swieten JC, Heutink P (2008) Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia. Lancet Neurol 7:965–974. [DOI] [PubMed] [Google Scholar]
- 171. Vatsavayai SC, Yoon SJ, Gardner RC, Gendron TF, Vargas JN, Trujillo A et al (2016) Timing and significance of pathological features in C9orf72 expansion‐associated frontotemporal dementia. Brain 139:3202–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Wang IF, Wu LS, Chang HY, Shen CK (2008) TDP‐43, the signature protein of FTLD‐U, is a neuronal activity‐responsive factor. J Neurochem 105:797–806. [DOI] [PubMed] [Google Scholar]
- 173. Waite AJ, Bäumer D, East S, Neal J, Morris HR, Ansorge O et al (2014) Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging 35:1779.e5–1779.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D et al (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin‐containing protein. Nature Genet 36:377–381. [DOI] [PubMed] [Google Scholar]
- 175. Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y et al (2014) Antisense proline‐arginine RAN dipeptides linked to C9ORF72‐ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84:1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Whitwell JL, Weigand SD, Boeve BF, Senjem ML, Gunter JL, DeJesus‐Hernandez M et al (2012) Neuroimaging signatures of frontotemporal dementia genetics: C9orf72, tau, progranulin and sporadics. Brain 135:794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM (2008) Disturbance of nuclear and cytoplasmic TAR DNA‐binding protein (TDP‐43) induces disease‐like redistribution, sequestration, and aggregate formation. J Biol Chem 283:13302–13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Yu C‐E, Bird TD, Bekris LM, Montine TJ, Leverenz JB, Steinbart E et al (2010) The spectrum of mutations in progranulin: A collaborative study screening 545 cases of neurodegeneration. Arch Neurol 67:161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Zhang YJ, Jansen‐West K, Xu YF, Gendron TF, Bieniek KF, Lin WL et al (2014) Aggregation‐prone c9FTD/ALS poly(GA) RAN‐translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol 128:505–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM et al (2002) Conversion of proepithelin to epithelins: Roles of SLPI and elastase in host defense and wound repair. Cell 111:867–878. [DOI] [PubMed] [Google Scholar]
- 181. Zu T, Liu Y, Banez‐Coronel M, Reid T, Pletnikova O, Lewis J et al (2013) RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci USA 110:E4968–E4977. [DOI] [PMC free article] [PubMed] [Google Scholar]