

Abstract
Cerebral white matter lesions (WML) are common in the aging brain and are associated with dementia and depression. They are associated with vascular risk factors and small vessel disease, suggesting an ischemic origin, but recent pathology studies suggest a more complex pathogenesis. Studies using samples from the population‐representative Medical Research Council Cognitive Function and Ageing Study neuropathology cohort used post‐mortem magnetic resonance imaging to identify WML for further study. Expression of hypoxia‐related molecules and other injury and protective cellular pathways in candidate immunohistochemical and gene expression microarray studies support a role for hypoxia/ischemia. However, these approaches also suggest that immune activation, blood–brain barrier dysfunction, altered cell metabolic pathways and glial cell injury contribute to pathogenesis. These abnormalities are not confined to WML, but are also found in apparently normal white matter in brains with lesions, suggesting a field effect of white matter abnormality within which lesions arise. WML are an active pathology with a complex pathogenesis that may potentially offer a number of primary and secondary intervention targets.
Keywords: aging, blood–brain barrier, dementia, small vessel disease, vascular, white matter lesions
Definitions and Clinical Significance
Cerebral white matter lesions (WML), sometimes referred to as white matter hyperintensities (WMH) are recognized in life as bright areas of high signal intensity in T2‐weighted and diffusion tensor magnetic resonance imaging (MRI) 56. They are very common in the aging brain, with an in‐life prevalence of over 90% in the over‐65 age group, the volume of lesions increasing with age group in the over‐60s 15, 42.
Although often an incidental finding, they are clinically significant. They are associated with dementia, reduced information processing speed, depression and impaired motor function, and they are associated with Alzheimer's disease 3, 8, 16, 35, 36, 61, 80, 85. The presence of WML may also be a risk factor for progression of mild cognitive impairment to dementia 11, 18. Volume and confluency of WML are important, so that the presence of confluent lesions is a predictor of their subsequent progression 56. WML may be subclassified by location into those within the white matter of the centrum semiovale (deep subcortical lesions, DSCL) and periventricular lesions (PVL). DSCL, with a tendency to become confluent, may extend into the periventricular zone, so that such subclassification may not be clear‐cut 65, but there is some evidence that DSCL and PVL may have different clinical associations 38.
WML in the Cognitive Function and Ageing Study (CFAS)
The Medical Research Council CFAS (for review see 89 ) is a longitudinal population‐based study of cognitive impairment and frailty in the elderly in England and Wales (http://www.cfas.ac.uk). Based around six urban and rural centers, the sample was recruited from individuals identified from family practitioner registers and aged over 65 years at study commencement. The study has a brain donation program and is one of a limited number of international studies combining neuropathology with selection criteria that can generate truly population‐representative analyses 93.
An initial CFAS study, based on the first 209 cases, combined WML with other forms of small vessel pathology 53. A further study, based on 456 consecutively donated cases, allowed assessment of the population prevalences of pathologies 50. This article reported PVL in 87% of non‐demented (at death) and 95% of demented donors, with an odds ratio for dementia of 4.3 [95% confidence interval (CI) 1.9–9.8]. DSCL were also frequent, although less common than PVL, being found in 60% of non‐demented and 73% of demented donors, with an odds ratio for dementia for severe deep lesions of 3.3 (95% CI 1.6–6.8). This study was sufficiently powered to allow calculation of attributable risk (AR) for specific pathologies. AR estimates the amount of dementia at death in the sample, determined by each of the factors in the model and is dependent on both the risk and prevalence of the factor. However, WML were not treated as separate single variables for AR analysis. Rather, the presence of confluent DSCL was included as a component of a global estimate of significant cerebral small vessel disease (SVD). Multivariate analysis estimated the AR for dementia associated with this index of SVD to be 12%. In combination with the presence of a macroscopic infarct the AR associated with SVD rises to 21% in this cohort.
WML are clinically identified by MRI, and a routine sampling approach for histology is likely to underestimate the burden of WML in pathology studies. The CFAS approach has been to use post‐mortem MRI to identify lesions and allow directed sampling for further histopathologic study 24. Formalin‐fixed 1‐cm thick coronal brain slices were examined from three levels of each brain, corresponding to Newcastle coronal brain map reference levels 10/12, 19/20 and 24/25. Slices were placed in a Perspex stack and imaged in a 1.0T machine to produce a single MRI image of each slice half way through its depth (Figure 1). Scans were then rated using a modification of the Sheltens’ semiquantitative scale 67, developed to score WMH in MRI in life. This approach identified PVL with a sensitivity of 95% and specificity of 71%, and DSCL with a sensitivity of 86% and specificity of 80%. The ability of post‐mortem MRI to identify subcortical vascular pathology has been confirmed in other studies by comparison with extensive histologic assessment, and is more sensitive than a routine histologic sampling approach 51. Post‐mortem MRI assessment in CFAS showed that 94% of brains scanned contained WML, with PVL and DSCL present in 81% and 64% of scanned brains, respectively. Severe DSCL assessed in this way are independent risk factors for cognitive decline and their inclusion in a multivariable model improved the incorrect prediction of dementia status from 25% to 17% 23. The population‐based neuropathologic approach in CFAS has therefore shown that WML are frequent, that they are independent risk factors for dementia and that post‐mortem MRI of brain slices can be used to identify and quantify lesions.
Figure 1.
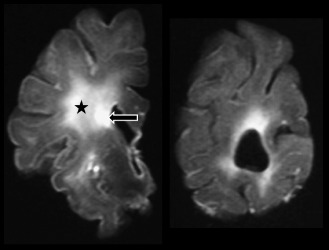
Magnetic resonance imaging scan of fixed brain slices. Left: high signal in the white matter identifies a periventricular lesion (arrow) and a deep white matter lesion (star). Right: slice at occipital level showing periventricular areas of high signal.
WML are related to vascular risk factors 8, 43, 56, 62, 78, 86 and to insulin resistance in non‐diabetics 37. Lack of a relationship to carotid artery stenosis suggests that WML are not due to extracranial thromboembolic events 60. White matter attenuation is also recognized as a co‐pathology with Alzheimer's disease 10, 77. However, the pathogenesis of WML remains poorly defined. A difficulty in the neuropathologic study of WML is that, unless severe, they are difficult to recognize and therefore to sample at brain dissection 54. Post‐mortem MRI‐guided sampling has therefore been used in CFAS to identify WML (both DSCL and PVL) and non‐lesional control white matter for further histologic and pathogenic studies. Controls in these studies have been divided into those from cases with lesions (controls–lesional) and those without lesions (controls–non‐lesional). This has been important in assessing the wider white matter environment in which lesions exist, and as discussed later, implies a more widespread pathology of white matter in individuals with WML. It should be noted that, while the lesion frequency and dementia associations presented earlier are population‐based in their approach, the studies of lesion histopathology/pathogenesis have taken a more conventional case‐control design, nested within the CFAS population‐neuropathology cohort.
Glial Pathology of WML
WML may consist of infarct‐like areas of white matter loss, or areas of myelin attenuation and pallor. WML and Binswanger's disease where white matter pathology is severe and diffuse, have been associated with increased microglial and macro‐glial pathology, including astrogliosis, regressive changes in astrocytes and apoptosis of oligodendrocytes and astrocytes 1, 41, 68. Axonal loss is an area that still requires thorough investigation, ideally using rigorous stereological approaches.
Use of immunohistochemistry for myelin basic protein (MBP) in the CFAS cohort demonstrated loss of myelin in both DSCL and PVL, the sharply demarcated area of MBP loss in PVL being associated with a dense feltwork of subependymal glial fibrillary acidic protein (GFAP)‐positive processes 76. PVL are also characterized by loss of ependymal cells, leaving gaps in the ependymal lining of the ventricle. This breakdown of the ventricular lining has been postulated to play a role in white matter changes 68. With the dense band of myelin loss and gliosis in PVL, it is possible that exposure to back‐diffusion of cerebrospinal fluid (CSF) might contribute to lesion pathogenesis in this location.
Platelet‐derived growth factor α receptor (PDGFαR) and the +13 isoform of microtubule‐associated protein‐2 (MAP‐2+13) are both markers of oligodendrocyte precursor cells (OPC), although PDGFαR is not OPC‐specific, also being present in reactive astrocytes, endothelial cells and neurons 49, 70. Using these markers, OPC were identified in the CFAS cases (Figure 2A) 76. Some of the oligodendroglial cells showed a T‐shaped pattern of cytoplasmic labeling for MAP‐2+13, considered to be the morphology of pre‐myelinating oligodendrocytes. These cells were more frequent in PVL, particularly at lesion margins, suggesting that there may be some attempt at repair, although it may be surmised that the astrogliosis in PVL might inhibit the efficacy of this process, given that gliosis can impair remyelination 91. We also identified OPC at a low frequency in control white matter. OPC may be important in remyelination, repair and plasticity in white matter in adults, although repair mechanisms may be impaired by age and by pathology 6, 13, 52, 71. So, whether impaired repair is a significant contributor to age‐related white matter degeneration remains unclear at present. The paucity of OPC in DSCL is interesting and is reminiscent of findings in established multiple sclerosis (MS) plaques. Potentially, this difference may correlate with the greater prominence of hypoxia in the pathogenesis of DSCL (see later; and as postulated in late MS lesions) with a selective effect on oligodendroglial lineage components.
Figure 2.
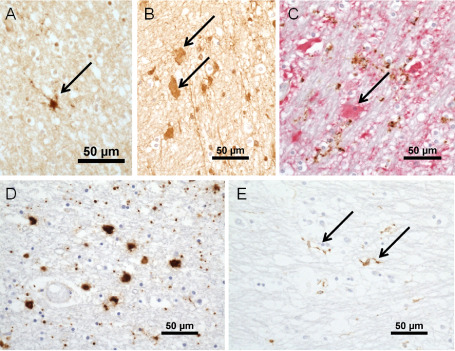
A. Immunohistochemistry for +13 isoform of microtubule‐associated protein‐2 identifies a process‐bearing oligodendrocyte precursor cells (arrow). B. Clasmatodendritic astrocytes labeled for fibrinogen (arrows). C. Double‐immunohistochemistry for glial fibrillary acidic protein (GFAP) (red) and CD68 (brown). The clasmatodendritic astrocyte labels for GFAP (arrow), but not CD68, antibody to which shows granular labeling of microglial processes. D. Amoeboid microglia in a deep subcortical white matter lesions. E. Ramified microglial labeled for major histocompatibility complex class II in a periventricular lesion (arrows). Magnification bars all 50 μm.
The role of the astroglial response remains to be determined and is likely to be complex 58, 59, 79. In MS white matter, for example, astrocytes have complex roles that can be either beneficial or harmful, or both, to white matter preservation and repair 91, so that it will be necessary to dissect their multifaceted responses. Astrocytes may be important for repair in white matter 95, and damage to the glia may impair their protective functions. Regressive changes and expression of apoptosis markers have been found in astrocytes in aging white matter pathology 41. Our recent studies in CFAS found expression of the DNA damage marker 8‐hydroxy deoxyguanosine in glia, particularly oligodendrocytes and microglia, and of senescence markers in astrocytes 96. In addition to myelination, remyelination and myelin maintenance (reviewed in 7), oligodendrocytes have important roles in the metabolic support of axons, and impairment of this support may be important in neurodegeneration 27, 44. The demonstration of damage to astrocytes and oligodendrocytes suggests that loss of glial function may be an important contributor to WML.
Evidence of a Hypoxic/Ischemic Environment in WML
Based on pathologic and biochemical findings, WML have been interpreted as ischemic in origin, representing areas of infarction or incomplete infarction 20, 21. As noted earlier, they share risk factors for vascular disease and in CFAS, epidemiologic results have been included as a component or manifestation of SVD. White matter is vulnerable to ischemia, particularly oligodendrocytes and myelinated axons 57. WML are associated with impaired cerebral vasomotor reactivity 2. Histologic studies demonstrate thickening of arterioles, perivascular widening 34, 84, capillary loss and an association with perivenous collagenosis 9, suggesting that there is vascular insufficiency and consequent hypoperfusion. Fernando et al sought direct evidence for a hypoxic environment in WML in a CFAS study 25. In addition to confirming thickened arterioles, the study examined the expression of hypoxia response molecules. Hypoxia inducible factor 1α, 2α, neuroglobin and matrix metalloproteinase‐7 were up‐regulated in WML, particularly in DSCL, providing evidence for a hypoxic environment in DSCL, with a difference from PVL where there is less evidence for hypoxia.
WML and Acute vs. Chronic Ischemia/Hypoxia
The implication of studies that associate WML with vascular risk factors, local arterial disease and evidence of hypoxia‐related markers in lesional tissue challenges current understanding of cerebral hypoxic/ischemic injury. Fundamentally there is a disconnection between the concepts of chronic ischemia now invoked in human clinical practice and research and the canon of published work into the pathogenesis and consequences of hypoxic/ischemic injury based on experimental neuroscience 46. This literature has predominantly focused on neuronal vulnerability, has used relatively acute experimental models, and is heavily dominated by research in species with a very high ratio of grey : white matter compared with the human brain. The introduction of the term “incomplete infarction” 20 into the literature was necessitated by this problem despite the term having no real pathogenic validity or definition. Canonically, brain ischemia is perceived to be an acute phenomenon that preferentially affects neurons (more precisely neuronal perikarya) and initiates an injury cascade that either kills cells or is sublethal such that they are able to fully recover. In WMLs, the tissue is not infarcted, oligodendroglia may be the major ischemic target, and the severity of tissue attenuation falls along a gradient from normality to severe loss of tissue elements. The concept of selective oligodendroglial vulnerability is established in pediatric neuroscience 39 and may be relevant to WML in older people; however, as discussed earlier, the relative proportion of oligodendroglial vs. axonal damage is not yet well‐characterized. The anatomy of human white matter is likely to contribute to its susceptibility to formation of WML based on one or both of its potential to be a watershed region for arterial perfusion and through local arterial and arteriolar pathology associated with systemic hypertension in older people. There remains, however, no established experimental model that provides validation of the concept that varying degrees of chronic misery perfusion, remaining above the threshold that would lead to tissue infarction, account for the patchy distribution, tendency to progression and variable severity of tissue attenuation that characterize this human pathology.
Blood–Brain Barrier (BBB) Leakage
There is evidence that the BBB, which resides at the level of the endothelium, is impaired in brain aging, neurodegeneration and vascular dementias, including white matter disease, although studies have produced varying findings 22, 64, 87, 94. BBB disruption has a number of potential adverse effects on tissue. Leakage of plasma proteins such as fibrinogen, thrombin and plasmin may promote neuroinflammation, cell and vascular injury, affect the extracellular matrix and cause tissue edema, while red cell extravasation may lead to hemoglobin and iron release and consequent oxidative stress through Fenton chemistry 92. Studies in CFAS have shown increased iron in WML; however, this is associated with alterations in iron metabolism‐related genes and proteins 29. Polymorphism of the hemochromatosis gene may also be a risk factor for WML 28. These results in CFAS suggest that altered iron metabolism can be a mechanism for increased tissue iron, with consequent oxidative stress.
The presence of clasmatodendritic astrocytes is histopathologic evidence for BBB leakage in WML 25, 76, 81. Clasmatodendritic astrocytes are rounded, weakly eosinophilic cells that show immunoreactivity for plasma proteins (Figure 2B). They have therefore been considered to reflect BBB leakage, perhaps representing a homeostatic response 17. Their morphology is not typical of a reactive astrocyte. Indeed, they rather resemble amoeboid microglia, but they label for GFAP and not with microglial markers (Figure 2C). Studies in CFAS also demonstrated extravasation of albumin that was widespread in the aged white matter, but enhanced in WML 74.
This report also described the expression of the proteins claudin‐V, zona‐occludin‐1 and occludin, which form the tight junction complexes that are part of the structural basis of the BBB 74. Tight junction protein reduction has been observed in a number of disorders, and is thought to be associated with BBB dysfunction 14, 40, 55, 66, 87. Staining for these proteins demonstrates linear profiles along vessels, representing the intercellular junctions. Quantification of gaps in staining was used to assess tight junction disruption. Changes in BBB permeability in white matter in the CFAS cohort were not associated with altered tight junction protein expression. Such an approach has detected changes in tight junctions in human autopsy‐derived material in MS 40, 45, but whether the lack of tight junction protein changes in CFAS white matter reflects technical insensitivity or a real pathophysiologic difference from MS is presently unclear. BBB alterations can occur without tight junction protein alterations 32 and junctional proteins are subject to molecular alterations such as phosphorylation 87. These more subtle regulatory aspects of BBB remain to be investigated.
In addition to BBB leakage, there may be other disruptions to tissue fluid homeostasis and movement in WML. Fernando et al 25 demonstrated widened perivascular spaces. It is possible that this reflects increased retention of tissue fluid related to impaired perivascular fluid drainage, a subject discussed in depth in the accompanying review by Weller et al 88.
Inflammation in WML
The presence of microglia in WML, and increased intercellular adhesion molecule 1 (ICAM‐1) expression in endothelium, suggest immune activation in WML 25, 76, and BBB disruption may also suggest an inflammatory response 64. The commonly used marker CD68 can identify white matter microglia in their ramified and more activated states, through to the amoeboid macrophages that are seen in DSCL (Figure 2D). Microglia, however, vary in their activation pattern with potentially different functions, for example, adopting more of an antigen‐presenting cell or innate immune phenotype 69. Microglia can express major histocompatibility complex (MHC) class II (Figure 2E), and can also express co‐stimulatory molecules, such as integrin B7 and CD40 63, which are necessary for antigen presentation and signalling in an immune response. In further characterizing the microglial response in WML, Simpson et al examined these markers in the CFAS cohort 72. While the number of CD68‐positive microglia is higher in the DSCL, the number of MHC class II‐positive cells is greater in the PVL, suggesting that the nature of the microglial response differs between DSCL and PVL. MHC class II expression in PVL, supported by expression of the accessory molecules B7 and CD40 suggests that the microglial phenotype differs between PVL and DSCL, with more immune activation in the former and a more innate pattern in the latter. In PVL, higher levels of minichromosome maintenance protein 2, which is a marker of cells that are licensed to replicate (although not proof of replication) 90, further suggested that PVL provide a more proliferation‐permissive environment. The pathogenesis of immune activation in PVL is uncertain. BBB breakdown may be a contributor. Clasmatodendritic astrocytes are more frequent in PVL than DSCL, suggesting higher levels of BBB disruption in PVL 76. Leakage of plasma immunoglobulins and other elements in serum can activate microglia 47. Other factors, such as denudation of the ependyma with consequent CSF exposure may also be relevant.
The work on WML to date has not taken into account more refined phenotyping of microglia, for example into classical (M1) and alternative (M2) activation states 5, nor have interactions with glia been investigated. However, these studies suggest that immune mechanisms are important in the pathogenesis of WML, and that mechanisms may differ according to location.
A Microarray Approach to WML
The application of genome expression studies provides an opportunity to move beyond candidate approaches to pathogenesis, with the opportunity to identify novel cellular pathways. Whole genome microarray technology combined with pathway analysis has found application in autopsy tissue from a wide range of neurologic disorders, including neurodegenerative diseases and MS, with the potential to identify novel mechanisms and biomarkers 12, 19. It has also been used, in conjunction with laser capture microdissection, to investigate the changes in the transcriptome of astrocytes with progression of Alzheimer‐type pathology 75. Gene expression analysis of WML and controls, using RNA isolated from whole‐tissue, revealed alterations in 502 genes in DSCL compared with controls, with 331 up‐regulated and 171 down‐regulated 73. Pathway analysis showed that immune‐regulatory genes were a key pathway to show differential expression. The genes affected included those involved with antigen presentation (including up‐regulation of MHC class II), complement, lymphocyte activation, proinflammatory cytokine signaling and phagocytosis. Increased expression of hypoxia‐related genes was also noted. These pathway changes were consistent with evidence of immune mechanisms and hypoxia from previous candidate histologic studies. However, the study also revealed significant alterations in a number of other cellular pathways, including cell cycle, proteolysis and ion transport. These alterations, supported by validation studies, suggest that hitherto uninvestigated cellular mechanisms may be operating in the pathogenesis of WML, providing novel targets for investigation.
Evidence for a Field Effect of Abnormal White Matter
Although WML might be regarded as ischemic foci, there is evidence from the CFAS studies that they are present in a background of abnormal white matter. This has some parallels with MS where apparently normal white matter that does not show abnormality on standard MRI can have subtle abnormalities, including increased microglial reactivity, and is different from control tissue from normal subjects. These changes may be relevant to the dynamic nature of MS and to new lesion formation 30, 48, 82, 83. In the CFAS WML studies, microglial activation, identified by MHC class II up‐regulation, was increased in control white matter from cases with lesions compared with control white matter from individuals without WML 72. More recently, we have shown that oxidative DNA damage, identified by immunohistochemistry for 8‐hydroxydeoxyguanosine, is also increased not only in DSCL but also in control white matter from lesional cases (manuscript in submission). Gene expression microarrays also show that “control” white matter from cases with lesions differs from that in cases without lesions. Thus, 419 genes were differentially expressed in lesional control vs. non‐lesional control white matter. The pathways affected are similar to those altered in the lesions themselves 73. The abnormal state of apparently normal white matter in brains that have lesions is supported by imaging abnormalities revealed by more sophisticated techniques, such as diffusion tensor imaging and magnetic resonance spectroscopy 26. In a number of respects therefore, normal‐appearing white matter from cases with lesions more closely resembles the lesions themselves than it does white matter from non‐lesional cases, and this suggests that WML exist or arise in white matter that shows a field effect of diffuse abnormality. This abnormality includes immune activation and alterations of a variety of cellular pathways, as detailed in the microarray section earlier. At present, it is unclear whether this is secondary to the effects of lesions or whether the lesions develop in the context of pathologic white matter creating a pre‐lesional environment. Indeed, this might predict the presence of small, subclinical early lesions not detectable on routine MRI. In either case, these findings indicate that lesions are present within a background of abnormal white matter, and suggests that abnormal white matter function may be widespread in individuals with lesions and that there are dynamic pathologic processes at work.
Model of Pathogenesis and Future Perspectives
While ischemia and hypoperfusion are important, these studies suggest that other factors also contribute to the pathogenesis of WML, including inflammation, BBB alterations and possibly age‐related alterations to glial function (Figure 3). In addition to loss of myelin and axons, altered functions of astrocytes, oligodendroglia and microglia have emerged. Thus, WML are not simply the tombstone markers of an episode of ischemia, but reflect an active and complex pathogenesis, while the “field effect” of changes in surrounding white matter indicate that active pathologic process are widespread in white matter in those with lesions. The active nature of this pathology has translational implications and opportunities: (i) targeting WML pathogenesis is of value for ameliorating dementia in a population setting; (ii) WML have potentially modifiable risk factors; and (iii) the pathogenesis presents a number of potential targets for intervention, such as myelin support, glial support and modulation of neuroinflammation.
Figure 3.
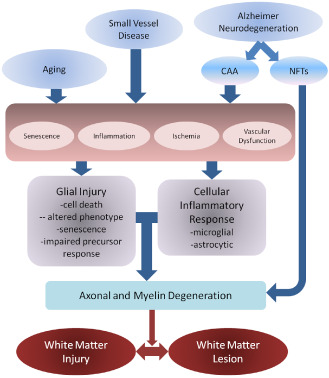
Possible pathogenic mechanisms for white matter damage in the context of vascular and Alzheimer‐related pathologies. Factors such as aging and small vessel disease may result in glial injury and inflammation, acting via a number of pathogenic mechanisms, including cell damage and senescence, inflammation, ischemia and vascular dysfunction, which may include blood–brain barrier changes and altered drainage. Glial injury and inflammation may then damage myelin and axons, resulting in the myelin pallor observed histologically. Alzheimer's may also act through small vessel ischemia related to cerebral amyloid angiopathy (CAA), while neurodegeneration related to neurofibrillary tangle (NFT) formation may directly cause axonal damage and secondary myelin loss. Although shown with arrows, mechanisms are interactive and processes may go in both directions and produce feed‐forward cycles. Further, different antecedents may cause white matter pallor and dysfunction via different routes and predilection to affect either myelin or axon primarily. These differing routes to white matter injury and lesion formation require definition.
While studies to date have begun to define the cellular and molecular pathologies of WML and its surrounding white matter environment, the mechanisms by which they lead finally to axonal dysfunction and disconnection remain to be defined. This uncertainty includes the significance of the field changes in surrounding white matter for the generation of new lesions and for white matter dysfunction. Interactions of ischemic drivers of pathology with the effects of cortical neurodegeneration on white matter axons and with the effects of age‐associated changes are further areas to be defined.
A particular area of interest will be interactions among cell types, for example astrocytes and microglia in the development of neuroinflammation. The ability of reactive astrocytes to promote endothelial repair in white matter injury provides evidence for the importance of interactions in the gliovascular unit 33, 95. The role of genetic polymorphisms as risk factors is also an area of interest. In addition to their roles as risk factors for vascular disease and neurodegeneration, polymorphisms may affect cellular mechanisms. APOE genotype, for example, may affect BBB integrity and neurovascular unit function 4, 31.
These questions suggest a need for further studies, including better models to address pathogenic mechanisms experimentally, but also further longitudinal aging population‐based studies, ideally combining functional imaging in life with post‐mortem investigation.
Conclusion
In a population/community setting, as opposed to tertiary clinic practice, dementia may involve combinations of pathologies each of which may offer therapeutic and preventative opportunities. WML are important, independent contributors to dementia, and involve active processes that can potentially be targeted therapeutically.
Acknowledgments
This work has been funded by grants from the UK Medical Research Council. JES is currently funded by the MRC (MR/J004308/1). SBW is also supported by grants from Alzheimer's Research UK (ARUK) and Biotechnology and Biological Sciences Research Council (BBSRC). The CFAS study is supported by the Department of Health and the Medical Research Council [grants MRC/G9901400, MRC U.1052.00.0013 and Medical Research Council (MRC/G0900582/1)]; the UKNIHR Biomedical Research Centre for Ageing and Age‐related Disease Award to the Newcastle upon Tyne Hospitals Foundation Trust; the Cambridge Brain Bank is supported by the NIHR Cambridge Biomedical Research Centre; The Cambridgeshire and Peterborough NIHR CLAHRC; Nottingham University Hospitals NHS Trust; University of Sheffield and the Sheffield Teaching Hospitals NHS Foundation Trust; The Thomas Willis Oxford Brain Collection, supported by the Oxford Biomedical Research Centre; The Walton Centre NHS Foundation Trust, Liverpool. We would like to acknowledge the essential contribution of the liaison officers, the general practitioners, their staff, and nursing and residential home staff. We are grateful to our respondents and their families for their generous gift to medical research, which has made this study possible.
References
- 1. Akiguchi I, Tomimoto H, Suenaga T, Wakita H, Budka H (1997) Alterations in glia and axons in the brains of Binswangers disease patients. Stroke 28:1423–1429. [DOI] [PubMed] [Google Scholar]
- 2. Bakker S, de Leeuw F, de Groot C, Hofman A, Koudstaal P, Breteler M (1999) Cerebral vasomotor reactivity and cerebral white matter lesions in the elderly. Neurology 52:578–583. [DOI] [PubMed] [Google Scholar]
- 3. Barber R, Scheltens P, Gholkar A, Ballard C, McKeith I, Ince P et al (1999) White matter lesions on magnetic resonance imaging in dementia with Lewy bodies, Alzheimer's disease, vascular dementia, and normal aging. J Neurol Neurosurg Psychiatr 67:66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bell R, Winkler E, Singh I, Sagare A, Deane R, Wu Z et al (2012) Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485:512–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boche D, Perry V, Nicoll J (2013) Activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol 39:3–18. [DOI] [PubMed] [Google Scholar]
- 6. Boulanger J, Messier C (2014) From precursors to myelinating oligodendrocytes: contribution of intrinsic and extrinsic factors to white matter plasticity in the adult brain. Neuroscience 269:343–355. [DOI] [PubMed] [Google Scholar]
- 7. Bradl M, Lassmann H (2010) Oligodendrocytes: biology and pathology. Acta Neuropathol 119:37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Breteler M, Van Swieten J, Bots M, Grobbee D, Claus J, Van Den Hout J et al (1994) Cerebral white matter lesions, vascular risk factors, and cognitive function in a population‐based study: the Rotterdam Study. Neurology 44:1246–1252. [DOI] [PubMed] [Google Scholar]
- 9. Brown W, Moody D, Thore C, Anstrom J, Challa V (2009) Microvascular changes in the white matter in dementia. J Neurol Sci 283:28–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brun A, Englund E (1986) A white matter disorder in dementia of the Alzheimer type: a pathoanatomical study. Ann Neurol 19:253–262. [DOI] [PubMed] [Google Scholar]
- 11. Clerici F, Caracciolo B, Cova I, Fusari I, Maggiore L, Galimberti D et al (2012) Does vascular burden contribute to the progression of mild cognitive impairment to dementia? Dement Geriatr Cogn Disord 34:235–243. [DOI] [PubMed] [Google Scholar]
- 12. Cooper‐Knock J, Kirby J, Ferraiuolo L, Heath P, Rattray M, Shaw P (2012) Gene expression profiling in human neurodegenerative disease. Nat Rev Neurol 8:518–530. [DOI] [PubMed] [Google Scholar]
- 13. Crawford A, Chambers C, Franklin R (2013) Remyelination: the true regeneration of the central nervous system. J Comp Pathol 149:242–254. [DOI] [PubMed] [Google Scholar]
- 14. Dallasta L, Pisarov L, Esplen J, Werley J, Moses A, Nelson J et al (1999) Blood–brain barrier tight junction disruption in human immunodeficiency virus‐1 encephalitis. Am J Pathol 155:1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. De Leeuw F‐E, De Groot J, Achten E, Oudkerk M, Ramos L, Heijboer R et al (2001) Prevalence of cerebral white matter lesions in elderly people: a population based magnetic resonance imaging study. The Rotterdam Scan Study. J Neurol Neurosurg Psychiatr 70:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Debette S, Markus H (2010) The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systematic review and meta‐analysis. Br Med J 341:c3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Del Bigio M, Deck J, Davidson G (2000) Glial swelling with eosinophilia in human post‐mortem brains: a change indicative of plasma extravasation. Acta Neuropathol 100:688–694. [DOI] [PubMed] [Google Scholar]
- 18. Devine M, Fonseca J, Walker Z (2013) Do cerebral white matter lesions influence the rate of progression from mild cognitive impairment to dementia? Int Psychogeriatr 25:120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dutta R, Trapp B (2012) Gene expression profiling in multiple sclerosis brain. Neurobiol Dis 45:108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Englund E, Brun A (1990) White matter changes in dementia of Alzheimer's type: the difference in vulnerability between cell compartments. Histopathology 16:433–439. [DOI] [PubMed] [Google Scholar]
- 21. Englund E, Brun A, Alling C (1988) White matter changes in dementia of Alzheimer's type. Brain 111:1425–1439. [DOI] [PubMed] [Google Scholar]
- 22. Farrall A, Wardlaw J (2009) Blood brain barrier: ageing and microvascular disease—systematic review and meta‐analysis. Neurobiol Aging 30:337–352. [DOI] [PubMed] [Google Scholar]
- 23. Fernando M, Ince P (2004) Vascular pathologies and cognition in a population‐based cohort of elderly people. J Neurol Sci 226:13–17. [DOI] [PubMed] [Google Scholar]
- 24. Fernando M, O'Brien J, Perry R, English P, Forster G, McMeekin W et al (2004) Comparison of the pathology of cerebral white matter with post‐mortem magnetic resonance imaging (MRI) in the elderly brain. Neuropathol Appl Neurobiol 30:385–395. [DOI] [PubMed] [Google Scholar]
- 25. Fernando M, Simpson J, Matthews F, Brayne C, Lewis C, Barber R et al (2006) White matter lesions in an unselected cohort of the elderly: molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 37:1391–1398. [DOI] [PubMed] [Google Scholar]
- 26. Firbank M, Minett T, O'Brien J (2003) Changes in DWI and MRS associated with white matter hyperintensities in elderly subjects. Neurology 61:95–954. [DOI] [PubMed] [Google Scholar]
- 27. Funfschilling U, Supplie L, Mahad D, Boretius S, Saab A, Edgar J et al (2012) Glycolytic oligodendrocytes maintain myelin and long‐term axonal integrity. Nature 485:517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gebril O, Kirby J, Savva G, Brayne C, Ince P (2011) HFE H63D, C282Y and AGTR1 A1166C polymorphisms and brain white matter lesions in the aging brain. J Neurogenet 25:7–14. [DOI] [PubMed] [Google Scholar]
- 29. Gebril O, Simpson J, Kirby J, Brayne C, Ince P (2011) Brain iron dysregulation and the risk of ageing white matter lesions. Neuromolecular Med 13:289–299. [DOI] [PubMed] [Google Scholar]
- 30. Gobin S, Montagne L, Van Zutphen M, Van Der Valk P, Van Den Elsen P, De Groot C (2001) Upregulation of transcription factors controlling MHC expression in multiple sclerosis lesions. Glia 36:68–77. [DOI] [PubMed] [Google Scholar]
- 31. Halliday M, Pomara N, Sagare A, Mack W, Frangione B, Zlokovic B (2013) Relationship between cyclophilin A levels and matrix metalloproteinase 9 activity in cerebrospinal fluid of cognitively normal apolipoprotein E4 carriers and blood–brain barrier breakdown. JAMA Neurol 70:1198–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hamm S, Dehouck B, Kraus J, Wolburg‐Buchholz K, Wolburg H, Risau W et al (2004) Astrocyte mediated modulation of blood–brain barrier permeability does not correlate with a loss of tight junction proteins from the cellular contacts. Cell Tissue Res 315:157–166. [DOI] [PubMed] [Google Scholar]
- 33. Hayakawa K, Miyamoto N, Seo J, Pham L, Kim K, Lo E et al (2013) High‐mobility group box 1 from reactive astrocytes enhances the accumulation of endothelial progenitor cells in damaged white matter. J Neurochem 125:273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang Y, Zhang W, Lin L, Feng J, Zhao X, Guo W et al (2010) Could changes in arterioles impede the perivascular drainage of interstitial fluid from the cerebral white matter in leukoaraiosis? Neuropathol Appl Neurobiol 36:237–247. [DOI] [PubMed] [Google Scholar]
- 35. Ikram M, Luijendijk H, Vernooij M, Horfman A, Niessen W, van der Lugt A et al (2010) Vascular brain disease and depression in the elderly. Epidemiology 21:78–81. [DOI] [PubMed] [Google Scholar]
- 36. Inaba M, White L, Bell C, Chen R, Petrovich H, Launer L et al (2011) White matter lesions on brain magnetic resonance imaging scan and 5‐year cognitive decline: the Honolulu‐Asia aging study. J Am Geriatr Soc 59:1484–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Katsumata T, Otori T, Nishiyama Y, Okubo S, Nishiyama Y, Nagayama H et al (2010) Correlation between insulin resistance and white matter lesions among non‐diabetic patients with ischaemic stroke. Neurol Res 32:743–747. [DOI] [PubMed] [Google Scholar]
- 38. Kee H, Lee J, Na D, Kim S, Cheong H, Moon S et al (2011) Different associations of periventricular and deep white matter lesions with cognition, neuropsychiatric symptoms, and daily activities in dementia. J Geriatr Psychiatry Neurol 24:84–90. [DOI] [PubMed] [Google Scholar]
- 39. Kinney H, Back S (1998) Human oligodendroglial development: relationship to periventricular leukomalacia. Semin Pediatr Neurol 5:180–189. [DOI] [PubMed] [Google Scholar]
- 40. Kirk J, Plumb J, Mirakhur M, McQuaid S (2003) Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood–brain barrier leakage and active demyelination. J Pathol 201:319–327. [DOI] [PubMed] [Google Scholar]
- 41. Kobayashi K, Hayashi M, Nakano H, Fukutani Y, Sasaki K, Shimazaki M et al (2002) Apoptosis of astrocytes with enhanced lysosomal activity and oligodendrocytes in white matter lesions in Alzheimer's disease. Neuropathol Appl Neurobiol 28:238–251. [DOI] [PubMed] [Google Scholar]
- 42. Launer L, Berger K, Breteler M, Dufouil C, Fuhrer R, Giampaoli S et al (2006) Regional variability in the prevalence of cerebral white matter lesions: an MRI study in 9 European countries (CASCADE). Neuroepidemiology 26:23–29. [DOI] [PubMed] [Google Scholar]
- 43. Lee S, Kim J, Chung S, Kim B, Ahn K, Lee K (2011) White matter hyperintensities (WMH) are associated with intracranial atherosclerosis rather than extracranial atherosclerosis. Arch Gerontol Geriatr 53:e129–e132. [DOI] [PubMed] [Google Scholar]
- 44. Lee Y, Morrison B, Li Y, Lengacher S, Farah M, Hoffman P et al (2012) Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487:443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Leech S, Kirk J, Plumb J, McQuaid S (2007) Persistent endothelial abnormalities and blood–brain barrier leak in primary and secondary progressive multiple sclerosis. Neuropathol Appl Neurobiol 33:86–98. [DOI] [PubMed] [Google Scholar]
- 46. Lipton P (1999) Ischemic cell death in brain neurons. Physiol Rev 79:1431–1568. [DOI] [PubMed] [Google Scholar]
- 47. Lu J, Moochhala S, Kaur C, Ling E (2001) Cellular inflammatory response associated with breakdown of the blood–brain barrier after closed head injury in rats. J Neurotrauma 18:399–408. [DOI] [PubMed] [Google Scholar]
- 48. Lund H, Krakauer M, Skimminge A, Sellebjerg F, Garde E, Siebner H et al (2013) Blood–brain barrier permeability of normal appearing white matter in relapsing‐remitting multiple sclerosis. PLoS ONE 8:e56375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maeda Y, Solanky M, Menonna J, Chapin J, Li W, Dowling P (2001) Platelet‐derived growth factor‐α receptor positive oligodendroglia are frequent in multiple sclerosis lesions. Ann Neurol 49:776–785. [DOI] [PubMed] [Google Scholar]
- 50. Matthews F, Brayne C, Lowe J, McKeith I, Wharton S, Ince P (2009) Epidemiological pathology of dementia: attributable‐risks at death in the MRC Cognitive Function and Ageing Study. PLoS Med 6:e1000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McAleese K, Firbank M, Hunter D, Sun L, Hall R, Neal J et al (2013) Magnetic resonance imaging of fixed post mortem brains reliably reflects subcortical vascular pathology of frontal, parietal and occipital white matter. Neuropathol Appl Neurobiol 39:485–497. [DOI] [PubMed] [Google Scholar]
- 52. Miyamoto N, Pham L, Hayakawa K, Matsuzaki T, Seo J, Magnain C et al (2013) Age‐related decline in oligodendrogliosis retards white matter repair in mice. Stroke 44:2573–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. MRC‐CFAS (2001) Pathological correlates of late‐onset dementia in a multicentre, community‐based population in England and Wales. Lancet 357:169–175. [DOI] [PubMed] [Google Scholar]
- 54. Munoz D (2006) Leukoaraiosis and ischaemia. Beyond the myth. Stroke 37:1348–1349. [DOI] [PubMed] [Google Scholar]
- 55. Nag S, Kapadia A, Stewart D (2011) Review: molecular pathogenesis of blood–brain barrier breakdown in acute brain injury. Neuropathol Appl Neurobiol 37:3–23. [DOI] [PubMed] [Google Scholar]
- 56. Ovbiagele B, Saver J (2006) Cerebral white matter hyperintensities on MRI: current concepts and therapeutic implications. Cerebrovasc Dis 22:83–90. [DOI] [PubMed] [Google Scholar]
- 57. Pantoni L, Garcia J, Gutierrez J (1996) Cerebral white matter is highly vulnerable to ischaemia. Stroke 27:1641–1647. [DOI] [PubMed] [Google Scholar]
- 58. Parpura V, Heneka M, Montana V, Oliet S, Schousboe A, Haydon P et al (2012) Glial cells in (patho)physiology. J Neurochem 121:4–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pekny M, Nilsson M (2005) Astrocyte activation and reactive gliosis. Glia 50:427–434. [DOI] [PubMed] [Google Scholar]
- 60. Potter G, Doubal F, Jackson C, Sudlow C, Dennis M, Wardlaw J (2012) Lack of association of white matter lesions with ipsilateral carotid artery stenosis. Cerebrovasc Dis 33:378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Prins N, van Dijk E, den Heijer T, Vermeer S, Jolles J, Koudstaal P et al (2005) Cerebral small‐vessel disease and decline in information processing speed, executive function and memory. Brain 128:2034–2041. [DOI] [PubMed] [Google Scholar]
- 62. Raiha I, Tarvonen S, Kurki T, Rajala T, Sourander L (1993) Relationship between vascular factors and white matter low attenuation of the brain. Acta Neurol Scand 87:286–289. [DOI] [PubMed] [Google Scholar]
- 63. Raivich G, Banati R (2004) Brain microglia and blood‐derived macrophages: molecular profiles and functional roles in multiple sclerosis and animal models of autoimmune demyelinating disease. Brain Res Rev 46:261–281. [DOI] [PubMed] [Google Scholar]
- 64. Rosenberg G (2009) Inflammation and white matter damage in vascular cognitive impairment. Stroke 40(Suppl. 1):S20–S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rossor M (2006) Is the whole brain periventricular? J Neurol Neurosurg Psychiatr 77:143–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sandoval K, Witt K (2008) Blood–brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis 32:200–219. [DOI] [PubMed] [Google Scholar]
- 67. Scheltens P, Barkhof F, Leys D, Pruvo J, Nauta J, Vermersch P et al (1993) A semiquantitative rating scale for the assessment of signal hyperintensities on magnetic resonance imaging. J Neurol Sci 114:7–12. [DOI] [PubMed] [Google Scholar]
- 68. Scheltens P, Barkhof F, Leys D, Wolters E, Ravid R, Kamphorst W (1995) Histopathologic correlates of white matter changes on MRI in Alzheimer's disease and normal aging. Neurology 45:883–888. [DOI] [PubMed] [Google Scholar]
- 69. Schwartz M, Butovsky O, Bruck W, Hanisch U‐K (2006) Microglial phenotype: is the commitment reversible? Trends Neurosci 29:68–74. [DOI] [PubMed] [Google Scholar]
- 70. Shafit‐Zagardo B, Kress Y, Zhao M, Lee S (1999) A novel microtubule‐associated protein‐2 expressed in oligodendrocytes in multiple sclerosis lesions. J Neurochem 73:2531–2537. [DOI] [PubMed] [Google Scholar]
- 71. Sim F, Zhao C, Penderis J, Franklin R (2002) The age‐related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J Neurosci 22:2451–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Simpson J, Ince P, Higham C, Gelsthorpe C, Fernando M, Matthews F et al (2007) Microglial activation in white matter lesions and nonlesional white matter of ageing brains. Neuropathol Appl Neurobiol 33:670–683. [DOI] [PubMed] [Google Scholar]
- 73. Simpson J, El‐Sayad O, Wharton S, Heath P, Holden H, Fernando M et al (2009) Microarray RNA expression analysis of cerebral white matter lesions reveals changes in multiple functional pathways. Stroke 40:369–375. [DOI] [PubMed] [Google Scholar]
- 74. Simpson J, Wharton S, Cooper J, Gelsthorpe C, Baxter L, Forster G et al (2010) Alterations of the blood–brain barrier in cerebral white matter lesions in the ageing brain. Neurosci Lett 486:246–251. [DOI] [PubMed] [Google Scholar]
- 75. Simpson J, Ince P, Shaw P, Heath P, Raman R, Garwood C et al (2011) Microarray analysis of the astrocyte transcriptome in the ageing brain: relationship to Alzheimer's pathology and APOE genotype. Neurobiol Aging 32:1795–1807. [DOI] [PubMed] [Google Scholar]
- 76. Simpson JE, Fernando M, Clark L, Ince P, Matthews F, Forster G et al (2007) White matter lesions in an unselected cohort of the elderly: astrocytic, microglial and oligodendrocyte precursor cell responses. Neuropathol Appl Neurobiol 33:410–419. [DOI] [PubMed] [Google Scholar]
- 77. Sjobeck M, Haglund M, Englund E (2006) White matter mapping in Alzheimer's disease: a neuropathological study. Neurobiol Aging 27:673–680. [DOI] [PubMed] [Google Scholar]
- 78. Skoog I, Lernfelt B, Landahl S, Palmertz B, Andreasson L‐A, Nilsson L et al (1996) 15‐year longitudinal study of blood pressure and dementia. Lancet 347:1141–1145. [DOI] [PubMed] [Google Scholar]
- 79. Sofroniew M, Vinters H (2010) Astrocytes: biology and pathology. Acta Neuropathol 119:7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Srikanth V, Beare R, Blizzard L, Phan T, Stapleton J, Chen J et al (2009) Cerebral white matter lesions, gait, and the risk of incident falls. A prospective population‐based study. Stroke 40:175–180. [DOI] [PubMed] [Google Scholar]
- 81. Tomimoto H, Akiguchi I, Suenaga T, Nishimura M, Wakita H, Nakamura S et al (1996) Alterations of the blood–brain barrier and glial cells in white‐matter lesions in cerebrovascular and Alzheimer's disease patients. Stroke 27:2069–2074. [DOI] [PubMed] [Google Scholar]
- 82. van der Valk P, Amor S (2009) Preactive lesions in multiple sclerosis. Curr Opinion Neurol 22:207–213. [DOI] [PubMed] [Google Scholar]
- 83. van der Valk P, De Groot C (2000) Staging of multiple sclerosis (MS) lesions: pathology of the time frame of MS. Neuropathol Appl Neurobiol 26:2–10. [DOI] [PubMed] [Google Scholar]
- 84. van Swieten J, van den Hout J, van Ketel B, Hijdra A, Wokke J, van Gijn J (1991) Periventricular lesions in the white matter on magnetic resonance imaging in the elderly. A morphometric correlation with arteriolosclerosis and dilated perivascular spaces. Brain 114:761–774. [DOI] [PubMed] [Google Scholar]
- 85. Viana‐Baptista M, Bugalho P, Jordao C, Ribeiro O, Esperanca‐Pina J, Ferro J (2011) Motor dysfunction correlates with frontal white matter ischemic changes in patients with leukoaraiosis. J Aging Res 2011:950341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wardlaw J (2005) What causes lacunar stroke? J Neurol Neurosurg Psychiatr 76:617–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Weiss N, Miller F, Cazaubon S, Couraud P‐O (2009) The blood–brain barrier in brain homeostasis and neurological diseases. Biochim Biophys Acta 1788:842–857. [DOI] [PubMed] [Google Scholar]
- 88. Weller RO, Hawkes CA, Kalaria RN, Werring DJ, Carare RO (2015) White matter changes in dementia: role of impaired drainage of interstitial fluid. Brain Pathol 25:63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wharton S, Brayne C, Savva G, Matthews F, Forster G, Simpson J et al (2011) Epidemiological neuropathology: the MRC Cognitive Function and Ageing Study experience. J Alzheimer Dis 25:359–372. [DOI] [PubMed] [Google Scholar]
- 90. Willams G, Stoeber K (2012) The cell cycle and cancer. J Pathol 226:352–364. [DOI] [PubMed] [Google Scholar]
- 91. Williams A, Piaton G, Lubetzki C (2007) Astrocytes—friends or foes in multiple sclerosis? Glia 55:1300–1312. [DOI] [PubMed] [Google Scholar]
- 92. Winkler E, Sagare A, Zlokovic B (2014) The pericyte: a forgotten cell type with important implications for Alzheimer's disease? Brain Pathol 24:371–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zaccai J, Ince P, Brayne C (2006) Population‐based neuropathological studies of dementia: design, methods and areas of investigation—a systematic review. BMC Neurol 6:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Zlokovic B (2008) The blood–brain barrier in health and chronic neurodegenerative disorders. Neuron 57:178–201. [DOI] [PubMed] [Google Scholar]
- 95. Zlokovic B (2013) A gliovascular idea for the white matter repair? J Neurochem 125:172–174. [DOI] [PubMed] [Google Scholar]
- 96. Al‐Mashhadi S, Simpson JE, Heath PR, Dickman M, Forster G, Matthews FE, Brayne C, Ince PG, Wharton SB (2014) Oxidative glial cell damage associated with white matter lesions in the ageing human brain. Brain Pathol doi: 10.1111/bpa.12216. [DOI] [PMC free article] [PubMed] [Google Scholar]