

Abstract
Amyotrophic Lateral Sclerosis (ALS) is recognized as a very complex disease. As we have learned in the past 20 years from studies in patients and in models based on the expression of mutant SOD1, ALS is not a purely motor neuron disease as previously thought. While undoubtedly motor neurons are lost in patients, a number of alterations in those cell‐types that interact functionally with motor neurons (astrocytes, microglia, muscle fibers, oligodendrocytes) take place even long before onset of symptoms. At the same time, disturbance of several, only partly inter‐related physiological functions play some role in the onset and progression of the disease. Traditionally, mitochondrial damage and oxidative stress, excitotoxicity, neuroinflammation, altered axonal transport, ER stress, protein aggregation and defective removal of toxic proteins have been considered as key factors in the pathogenesis of ALS, with the relatively recent addition of disturbances in RNA metabolism. This complexity makes the search for an effective treatment extremely difficult and prompts further studies to reveal other possible, previously unappreciated aspects of the pathogenesis of ALS. In this review, we focus on previous knowledge on ALS mechanisms as well as new facets emerging from studies on genetic ALS patients and models that may both provide precious information for a novel therapeutic approach.
Keywords: ALS, amyotrophic lateral sclerosis, motor neuron, protein aggregation, RNA metabolism
Introduction
An unquestionable set of genetic evidence indicates that disturbed RNA metabolism is a key player in ALS pathogenesis 106. Indeed, more and more genes with an established or probable role in RNA processing have been discovered as to be causally linked to familial as well as to apparently sporadic forms of the disease. In particular, the identification of mutations in TARDBP and FUS, both genes encoding RNA binding proteins, initially proposed a pivotal role for aberrant RNA processing, a topic that was previously not fully appreciated as a possible mechanism of the disease pathogenesis. However, it was the more recent identification of the hexanucleotide repeat expansion in the C9orf72 gene as the most frequent genetic cause for ALS that truly and completely shifted the focus of ALS research and placed RNA mis‐processing in the spotlight. Mutations in several other genes encoding proteins involved in RNA metabolism have been described: although they occur only in a small fraction of fALS cases, at the same time they strengthen the relevance of this pathway in the pathogenesis of ALS.
Yet, ALS is far from being a pure RNA‐dysfunction disease. A wealth of experimental data has been accumulated over the last 20 years indicating that dysfunctions in a number of cellular functions, that is, mitochondrial metabolism and regulation of oxidative balance, modulation of neuronal excitability, control of the inflammatory response, axonal transport, protein folding and degradation, do have a role in disease pathogenesis 26. As new information is rapidly getting available, it is becoming clear that previous knowledge can be integrated into the new one, thus helping in shedding light into a real intricate disease.
TDP‐43 and FUS: RNA DYS‐Metabolism as a Novel Player in ALS
Mutations in the gene TARDBP (Trans active response DNA binding protein), encoding the 43 KDa protein TDP‐43, have been identified in 2008 and account for about 3% of fALS and about 1.5% of sALS cases 46, 65, 78, 126.
Shortly after this discovery, mutations in the gene encoding for the fused in sarcoma/translocated in liposarcoma (FUS/TLS) protein were identified in about 4% of familial ALS patients and rare sporadic cases 74, 78, 133. Together, these discoveries caused a great enthusiasm and opened a new perspective in the field. Indeed, FUS and TDP‐43 share structural and functional homology, being both DNA/RNA binding proteins belonging to the family of heterogeneous ribonucleoproteins (hnRNPs) 78. These are proteins that are able to shuttle between the nucleus and the cytoplasm where they play various functions in RNA‐related pathways, including transcription, pre‐mRNA splicing, mRNA transport and local translation 78. At steady state, FUS and TDP‐43 are localized predominantly in the nucleus, while ALS mutations cause a relocation of the proteins from nucleus to cytoplasm and their accumulation into stress granules 10, transient cytoplasmic assemblies where nonessential mRNAs are translationally repressed in response to stress 3. The pathological significance of this process is unclear, although it might imply a role of both FUS and TDP‐43 in stress‐granule mediated translational repression, a central process in the control of gene expression which is emerging as a crucial issue in the field of neurodegenerative diseases, including ALS 10. Yet, as it is believed that stress granules are permissive sites for protein aggregation, the localization of mutant TDP‐43 and FUS (and even of their wild‐type forms under stress conditions) into stress granules might represent the initial step that eventually generates TDP‐43 or FUS‐positive inclusions that typically mark affected tissues from patients (see below) 31, 84. Overall, these findings imply both a loss of the normal protein function in the nucleus and a gain of toxic function of the aggregated forms of TDP‐43 and FUS in the cytoplasm as potential pathogenic mechanisms. However, they do not provide straightforward clues about the pathological mechanisms whereby TARDBP and FUS mutations cause neurodegeneration. So far, the approach that has been most extensively used to answer this question consisted on high‐throughput screenings of the RNAs that are bound to FUS and TDP‐43. To date, several thousands of RNA species targeted by TDP‐43 107, 120, 131, 138 and FUS 56, 59, 76, 112 have been identified, but which of these interactions have physiological and/or pathological implications is still really unclear. It has been proposed that a set of common transcripts that are targeted by both proteins might be the most relevant for the disease, but again experimental evidence showing that this is the case are still lacking. Interestingly, most of them are pre‐mRNAs with very long introns (>100 kb) and the expression of these types of mRNAs, which typically encode proteins involved in neuronal functionality, is enriched in brain, thus providing a potential explanation for the selective neuronal vulnerability associated to FUS or TDP‐43 dysfunction 76.
Considering the thousands of RNA targets and the multiple roles in RNA processing that both FUS and TDP‐43 might have, it is not surprising that alterations in subcellular distribution and/or deposition in insoluble inclusions that characterize ALS patients have potential deleterious consequences for each step of RNA‐related cellular pathways (Figure 1A).
Figure 1.
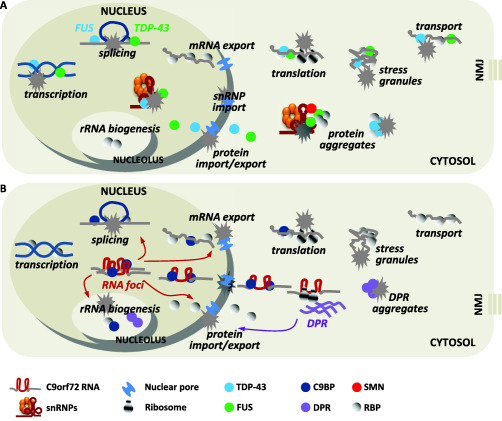
Converging pathogenic mechanisms triggered by RNA‐related mutant genes. A. FUS and TDP‐43 have a central role in different steps of RNA related pathway. While they are mainly localized in the nucleus, where they act as transcriptional and splicing regulators, both FUS and TDP‐43 are also involved in cytoplasmic mRNA transport and local translation. ALS mutations alter their normal cellular distribution, relocating both proteins in cytoplasmic protein aggregates and/or stress granules. This causes both a loss of their normal, and mainly nuclear functions, and a gain of toxic function by protein aggregates in the cytoplasm. In particular, mutated FUS and TDP‐43 affect the proper splicing regulation of thousands of RNA targets. This might also be a result of sequestration of snRNPs and SMN splicing complexes in the cytosol, which in turn leads to their depletion from nuclear gems. Further, mutated FUS and TDP‐43 alter stress granule dynamics, thus impairing stress granules‐mediated translational repression in condition of stress. Local translation into distal neuronal processes and neuromuscular junctions (MNJ) might also be affected, as TDP‐43 plays a role in the axonal transport of defined target mRNAs for local translation. Finally, ALS‐associated mutation in FUS and TDP‐43 affect nuclear protein import, as in the case of C9orf72. B. The transcription of mutant C9orf72 gene lead to the expression of both sense and antisense C9orf72 RNA transcripts containing expanded G4C2 (and C4G2) repeat, which can affect normal cellular pathway in numerous manners. First, the C9orf72 RNA transcripts accumulate in the nucleus as RNA foci which interact and sequester different RNA binding proteins, leading to alterations in their normal cellular functions. Interaction between G4C2 repeat expansion and nucleolin causes nucleolar stress and defects in rRNA biogenesis. Similarly, the hexanucleotide repeat binds several splicing factors, such as hnRNP H, hnRNP A3, SRSF2, thus impairing splicing regulation of hundreds of genes. Second, expanded repeats impact on the control of nucleo‐cytoplasmic trafficking of both mRNAs and proteins by targeting mRNA export adaptors and the nuclear pore complex machinery. As a consequence, mRNA translation is affected. Protein translation might also be impaired by sequestration of translational regulators, such as initiation and elongation factors, which in turn might lead to impairment in stress granules‐mediated translation repression. Finally, bypassing normal surveillance mechanisms in an obscure way, expanded RNAs translocate from nucleus to cytoplasm, where they are translated through an unconventional process independent from the presence of an upstream ATG (RAN translation). The ribosomal reading of the sense and antisense transcripts lead to the expression of five different poly‐dipeptide repeat proteins (DPRs). DPRs accumulate in cytosolic inclusions that affect cell viability by unknown mechanism, but they also target nuclear RNA processing and protein transport leading to a detrimental loop of RNA dysfunction.
The majority of TDP‐43 and FUS binding sites in RNAs are introns, suggesting that a major function of these proteins is to control the splicing of hundreds of gene transcripts. Based on the possibility that ALS mutations might induce neurodegeneration as a consequence of both loss‐of‐function and gain‐of‐function splicing defects, different transcriptomic studies have been performed after protein knockdown or overexpression of wild‐type and mutant FUS and TDP‐43 4, 55, 76, 107, 110. Not surprisingly, an altered expression of thousands of aberrantly spliced mRNA molecules emerged in all of these studies, strengthening the link between the two proteins with splicing regulation. In addition to a direct alteration on this process, ALS‐related mutations may affect also the network of protein–protein interaction of FUS and TDP‐43 with other splicing factors. Indeed, FUS interacts with both RNA polymerase II and the small nuclear ribonucleoprotein complex snRNP U1, thus suggesting that it might have a role in coupling transcription to splicing 143. Moreover, FUS and TDP‐43 interact with the SMN complex, which is essential for spliceosome assembly 132, 140 and mutant FUS sequesters both SMN and snRNPs in protein aggregates in the cytoplasm, leading to alteration in the alternative splicing process 45, 142. Further, ALS mutations disrupt the interactions between TDP‐43 and different hnRNPs (A1, A2/B1, C1/C2 and A3) 12. Finally, both FUS and TDP‐43 are required for the formation of nuclear particles called gems, which are involved in the final stages of snRNP modification and assembly and are typically lost in spinal motor neurons from ALS patients 60, 132. Interestingly, mutant SOD1, a major determinant of familial ALS, impairs the recruitment of SMN into nuclear gems, which are significantly reduced in spinal motor neurons of mutant SOD1 mice 69. Considering that splicing alterations have been clearly reported in cell and mouse models of SOD1‐ALS 6, 81, one interesting possibility is that motor neurons are particularly vulnerable to splicing defects that might well represent a converging mechanism of different types of genetic ALS. This conclusion is further supported by the observation that mutations in SMN1 gene coding for SMN are responsible for spinal muscular atrophy (SMA), the most common form of juvenile motor neuron degeneration, and that SMN is a significant modifier of ALS pathogenesis in mouse models and in human patients 1.
TDP‐43 and FUS also play a critical role in the biogenesis of noncoding regulatory RNAs such as microRNA (miRNA) 43, interacting with the Drosha miRNA processing complex 51 and, at least for TDP‐43, with Dicer complexes 70. Moreover, TDP43 binds directly to some miRNAs and silencing of TDP43 significantly alters the abundance of several miRNAs in human cultured cells 13, 70. Importantly, the expression levels of these microRNAs were found up‐ or down‐regulated in some ALS patients, although the results were somehow inconsistent, which might depend on the different tissues analyzed 38.
FUS and TDP‐43 are key component of neuronal RNA transport granules, which contain mRNAs that must be transported from the cell body to axons and dendrites and locally translated, and that are, therefore, essential for the normal functionality of highly polarized cells as neurons in general, and motor neurons in particular 2, 39, 40, 86, 135. Interestingly, disease‐associated mutations in TARDBP impair this process 2, 86.
In addition to their function in RNA trafficking, cytoplasmic FUS and TDP‐43 might be involved in the regulation of mRNA translation. Although a direct role of these proteins in the control of translation machinery is still lacking, it is clear that sequestration of mRNA transcripts into stress granules in response to various cellular insults, such as oxidative, heat or endoplasmic reticulum stress, might impact on the overall control of stress granules‐mediated translational repression. As a matter of fact, stress granules dynamics is strongly affected by ALS mutations 84, and both genetic and pharmacological modulation of this process provide beneficial effects on animal models of the disease 32, 72.
C9orf72: Solid Foundations for RNA DYS‐Metabolism in ALS
A mutation in the C9orf72 gene is the most frequent cause of ALS, accounting for about 40% of familial and about 7% of sporadic ALS cases 89, and consists of an expansion of a GGGGCC (G4C2) hexanucleotide repeat between the noncoding exons 1a and 1b 28, 111. In healthy individuals, the hexanucleotide sequences have a median length of two repeats (range 0–20), whereas most of C9orf72‐associated ALS patients have several hundreds or even thousands of repeats. However, the minimal pathogenic repeat size remains undefined 113.
Although the mechanisms whereby the G4C2 repeat expansion leads to neurodegeneration are currently unknown, the nature of the mutation and its analogies with other noncoding repeat expansion disorders suggest two potential pathogenic mechanisms, including a loss of function of the protein encoded by C9orf72 gene, and a gain of toxic function of RNA transcripts containing the expanded repeat. While haploinsufficiency of C9orf72 protein, whose function is only scarcely characterized 35, 82, 145, does not seem to cause neurodegeneration 73, the simple overexpression of expanded G4C2 repeats causes reduced viability in cellular models and motor deficits in Drosophila melanogaster and mice 18, 37, 80, 139, 148. In these cases, cell toxicity could be mediated by the accumulation of pre‐mRNAs containing the expanded repeat and/or the formation of poly‐dipeptide proteins, which are unconventionally produced by the so called repeat‐associated non‐ATG (RAN) translation.
Intracellular accumulation of pre‐mRNAs containing repeat expansions as RNA foci is a clinical feature of C9orf72‐associated patients: both sense and antisense RNA foci have been indeed detected in cortical, hippocampal, cerebellar and spinal motor neurons of ALS patients 44, 77, 94. RNA foci may exert a deleterious gain‐of‐function effect through the sequestration of RNA binding proteins, whose decreased availability might lead to alterations, once again, in various steps of RNA processing. To date, different groups have reported several candidate binding partners, including splicing and translational factors, and identified the presence of various of these proteins in RNA foci 21, 33, 52, 80, 97, 115, 139. Although a pathogenic role of specific repeat binding proteins in ALS neurodegeneration has not yet been definitively proved, emerging data have started to shed light on the possible toxic mechanisms induced by expanded RNAs (Figure 1B). We have recently observed that the expression of a (G4C2)31 repeat expansion in cultured cells induces stress granules‐associated translational repression, which is accompanied by a marked accumulation of poly(A) mRNAs in cell nuclei, thus suggesting that defective trafficking of mRNA, as a consequence of impaired nuclear mRNA export, might contribute to the pathogenesis of C9orf72 ALS 115. Nuclear retention of mRNAs was later confirmed by Freibaum et al in the nuclei of Drosophila cells expressing expanded G4C2 repeats and in induced pluripotent stem‐cell‐derived neurons from C9orf72‐associated patients 37. Importantly, at least 18 proteins involved in the nucleo‐cytoplasmic transport act as genetic modifiers (enhancers or suppressors) of eye degeneration induced by expanded G4C2 in Drosophila 37, further suggesting the involvement of this pathway in the pathogenesis of C9orf72 ALS. A similar conclusion was obtained by Zhang et al, who picked up the protein RanGAP1, a central regulator of protein trafficking between nucleus and cytoplasm, as a key target of the repeat expansion 148. In particular, the overexpression of RanGAP (orthologue of human RanGAP1) is a potent suppressor of eye degeneration and locomotor defects observed in Drosophila model expressing G4C2 repeats. RanGAP is able to bind the hexanucleotide repeat, can colocalize with repeat RNA foci and is mislocalized in repeat‐expressing flies, iPSC‐derived neurons and in C9orf72 ALS patient brain tissue. Altered localization of RanGAP is associated to a loss of function mechanism, as demonstrated by the impairment of import of nuclear proteins in Drosophila cells and in iPSC neurons. Interestingly, pharmacological targeting of nuclear export machinery rescues the neurodegenerative phenotype, suggesting a potential therapeutic approach 148.
A direct effect of repeat expansion on RNA processing has also emerged from gene expression profiling, which revealed a number of differentially expressed genes and exons in purified motor neurons and other tissues from C9orf72‐associated patients compared to normal controls 22, 109, 117. Notably, among the genes found aberrantly spliced, enrichment in genes encoding post‐transcriptional regulatory factors was observed, suggesting a potentially detrimental vicious cycle for proper RNA metabolism.
It is therefore clear from this short survey of the available experimental evidence, that RNA processing is a crucial issue in ALS pathogenesis, a concept that is further strengthened by a number of mutations in other genes encoding for proteins involved in RNA metabolism that were found to be associated, although rarely, with ALS.
Mutations in the gene encoding senataxin (SETX), a DNA/RNA helicase predicted to be involved in several steps of gene expression and the maintenance of genomic integrity, have been identified in rare cases of autosomal dominant juvenile ALS 9, 17. Mutations in the gene encoding angiogenin (ANG), a tRNA specific RNase involved in the transcription of ribosomal RNA, were found in adult onset forms of familial and sporadic ALS 50. Mutations in elongation protein 3 (ELP3), the histone H3/H4 acetyl transferase which is the catalytic subunit of the elongator complex, regulator of transcriptional elongation and post‐transcriptional processing of tRNA, were also associated to ALS 123. Moreover, mutations in genes encoding Ewing sarcoma breakpoint region 1 (EWSR1) and TATA box binding protein‐associated factor 15 (TAF15) also cause ALS 23, 24, 130. Both these proteins are RNA binding proteins structurally and functionally related to the other disease‐associated protein FUS, that form together the FET family, and play a regulatory role in transcription and alternative splicing. Additional genetic mutations were more recently identified in different hnRNPs, such as hnRNP A1, hnRNP A2/B1 and Matrin3 63, 71. Altogether, these findings reinforce the notion that RNA metabolism is a vulnerable point for motor neurons, although the pathogenic mechanisms triggered by mutations in all these genes remain to be defined.
Dipeptide Repeat Aggregation: A New Player in An Old Scenario
The discovery that expanded nucleotide repeats can be translated into polypeptides (RAN peptides) with mechanisms that bypass the conventional rules of mRNA translation, such as the presence of an ATG‐initiated open reading frame, not only represents one of the most exciting and important findings in basic research over the last years, but it also promises to deeply impact on our knowledge of the mechanisms underlying different disease of the nervous and muscular tissues. To date, spinocerebellar ataxia type 8 (SCA8), myotonic dystrophy type 1 (DM1) and Fragile‐X tremor ataxia syndrome (FXTAS) have been shown to be associated to this process 19. Most importantly, growing experimental evidence points to RAN peptides as major determinants of C9orf72 ALS pathogenesis.
Both sense and antisense C9orf72 transcripts containing G4C2 repeat expansion undergo RAN translation, resulting in the expression of five dipeptide repeat proteins (DPR): Glycine‐Proline (GP), Glycine‐Arginine (GR), Glycine‐Alanine (GA), Proline‐Alanine (PA) and Proline‐Arginine (PR) 5, 44, 96, 98, 150. Analysis of postmortem tissues from C9orf72‐associated patients has shown that these dipeptide repeat proteins form cytoplasmic p62 positive and TDP‐43 negative inclusions and, less frequently, intranuclear inclusions in neuronal cells, which might both contribute to the disease pathogenesis 88, 90. The translational products of C9orf72 repeat expansion seem to be sufficient per se to cause ALS neurodegeneration, as expression of synthetic constructs lacking G4C2 repeats and producing DPRs are toxic in cellular models, as well as in Drosophila 93, 95, 129, 137, 141, 147. However, these evidence are inconsistent with the observations that the amount of DPR inclusions does not seem to correlate with the clinical severity in ALS patients 88, and that the DPR protein aggregates are mainly distributed in brain regions such as cerebellum, hippocampus, neocortex, while are rare in the brainstem and in the spinal cord and, strikingly, almost absent in motor neurons 27, 48. Yet, the presence of soluble DPRs cannot be excluded in these tissues, where they might interfere with the normal cellular pathways independently from their accumulation into detectable inclusions.
How DPRs cause neurotoxicity is completely unknown. Interestingly, even DPR proteins seem to be capable of affecting RNA metabolism. Different groups have indeed shown that Arginine‐rich DPRs, in particular PR, translocate to the nucleus, where they localize and aggregate in the nucleoli, leading to nucleolar stress response and alteration in RNA biogenesis 75, 129, 137. Notably, nucleolar stress is also triggered by the interaction of expanded RNA transcript with nucleolin, an essential nucleolar protein 52. Moreover, several proteins involved in nucleocytoplasmic transport have been identified as potent modifiers of toxicity induced by PR dipeptides in Saccharomyces cerevisiae 64. Finally, among the recently identified GR and PR interacting proteins, many are RNA binding proteins and ribosomal proteins, further suggesting a crucial role of this pathway in the disease pathogenesis 129. Interestingly the analysis of poly‐GA coaggregating proteins revealed an interactome network enriched for proteins involved in the ubiquitin‐proteasome system regulation 93, thus pointing to protein aggregation as a major player in ALS, a concept that has been repeatedly invoked to explain the action of other aggregated proteins in ALS.
Protein Aggregation: A New Scenario for An Old Player
Protein aggregates in motor neurons are a pathological hallmark of ALS, which may be regarded as a classical proteinopathy. Motor neuron degeneration is often preceded by the formation of inclusions containing ALS associated proteins that are usually also ubiquitin‐positive. At variance with mutant SOD1, which is found aggregated exclusively in patients carrying SOD1 mutations, and mutant FUS, which is aggregated mainly in patients carrying FUS mutations and only in a few juvenile and sporadic forms of the disease 29, TDP‐43 is aggregated in most patients with sporadic and familial ALS including those carrying C9orf72 expansion 87 or other unrelated mutations (ie, OPTN, UBQLN2, VCP, ANG, ATXN1, PFN1) 29. A complex interplay between the aggregated ALS proteins and the mis‐functioning of specific mechanisms that are involved in their proper folding/disposal seems to be involved in ALS (Figure 2).
Figure 2.
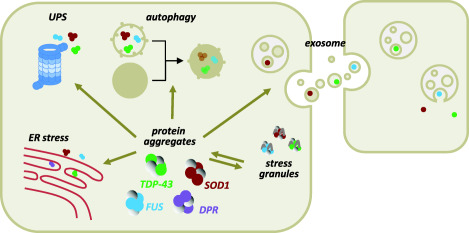
Protein aggregation‐related toxicity in ALS. SOD1, TDP‐43 and FUS may share toxicity mediated by protein aggregates that form both as a consequence of gene mutations and/or as a result of oxidative or ER stress. In the case of FUS and TDP‐43, this process can be enhanced by their localization into stress granules. Correct protein handling by the Ubiquitin Proteasome System (UPS), as well as proper aggregate removal by the autophagic machinery can be hampered by the same aggregated proteins, but also by dysfunction/mutations in proteins that regulate these processes. As a result, aggregates accumulate over time and can propagate into neighboring cells following their extracellular release, which is due to discharge of seeds of aggregates by dying cells, or mediated by exosomes. Inclusions of Dipeptide Repeat Proteins (DPRs) that are formed by an aberrant translation of the C9orf72 expanded repeat, are thought to share the same toxic mechanisms as the other aggregation‐prone ALS factors.
The mechanisms of mutant SOD1 aggregation are quite well understood as it involves cysteine‐mediated polymerization and amyloid fibrils formation 15; intracellular accumulation of aggregates is favored by impairment of protein degradation systems in neurons 8 and, to a lesser extent, in muscle cells 42. It is also quite clear that these aggregates are linked to induction of mitochondrial damage, another recognized facet of ALS pathology 25, 36, 57. Conversely, the mechanisms ruling aggregation of wild‐type and mutant TDP‐43 are still largely debated, with evidence attributing a main role in the cysteine‐mediated aggregation process to the C‐terminal RNA‐recognition motifs (RRMs) of the protein, as well as to a glycine‐rich sequence containing a prion‐like domain 105, 122, 136 or to the N‐terminal domain 11, 114, 146.
Oxidative stress promotes TDP‐43 cysteine oxidation and disulfide crosslinking 20, 122, a fact that may explain aggregation of the wild‐type protein in most ALS patients. It also promotes TDP‐43 acetylation on two specific lysine residues (K145 and K192) within the RNA‐binding domains; in turn, acetylation may prompt aggregation and loss of function of the protein 20. A direct physical interaction between histone deacetylase 6 (HDAC6) and TDP‐43 exists in vivo 54 and, thus, it is also possible that under exposure to oxidative stimuli TDP‐43 aggregation prevents effective HDAC6 binding.
Oxidative stress also prompts hyperphosphorylation of aggregated TDP‐43 on C‐terminal serine residues 58 and this may contribute to TDP‐43 aggregation in patients 53, although other studies suggest that phosphorylation prevents rather than promotes TDP‐43 aggregation 83.
Aggregation of wild‐type and mutant FUS has also been investigated, although most studies were focused on FUS mis‐localization in specific protein aggregates such as stress granules (see above) 10 rather than in large unfunctional aggregates. FUS is also able to form RNA‐dependent aggregates which are distinct from stress granules 121, with both the low‐complexity domain and the R/G rich domain of FUS contributing to assembly of fibrous β zipper structures 118. As for SOD1 and TDP‐43, oxidative stress accelerates the formation of FUS granules 127. Most important, as other neurodegeneration‐linked proteins including TDP‐43, FUS contains disordered, prion‐like regions that render it very prone to aggregation. In particular, as demonstrated in a recent elegant study by Patel et al, FUS function requires its existence in the form of “liquid droplets” that convert with time to an aggregated state, and this transition is accelerated by ALS mutations 104.
Why motor neurons should be more sensitive to protein aggregates than other cell‐types is still not clear. However, as discussed in two recent reviews 42, 61 glia and muscle cells may be better equipped than neurons to handle misfolded proteins and counteract toxicity due to aggregates, possibly because they are able to better activate molecular chaperones and protein degradation systems including the immunoproteasome.
Correct protein folding and inhibition of protein aggregation is facilitated by a quality control system that involves a network of proteins including molecular chaperones and the ubiquitin proteasome system (UPS). A number of studies reported defects in this system in models for mutant SOD1 toxicity (reviewed in [8]) and in postmortem tissue from patients 66, and a recent study in two strains of G93A‐SOD1 mice correlates severity of phenotype in terms of early onset and fast progression with low levels of soluble chaperones and with malfunction of the proteasome degradation machinery 91. Thus, increasing the level of chaperones or even inducing autophagy may constitute an approach to removal of SOD1 aggregates as suggested from studies in models 67.
However, the relation between ALS pathogenesis and autophagy is still not clear. For instance, rapamycin, an MTOR‐dependent autophagic activator, accelerates disease progression in the SOD1(G93A) mouse model of ALS 144. Conversely, treatment with trehalose (an MTOR‐independent autophagic inducer) was reported to have a number of beneficial effects in these mice, including delay of onset, prolonged life span, reduction of motor neuron loss, decreased SOD1 aggregation and ubiquitinated protein accumulation in one study 149, while it became less effective at delaying further disease progression as the disease progressed and it failed to extend the survival of the same mice in another study 85. Interestingly, BECN1 levels are upregulated in mutant SOD1 mice but life span is increased in these mice if they are made haploinsufficient for Becn1 compared with littermate control animals. Furthermore, an altered equilibrium between monomeric and oligomeric forms of mutant SOD1 is observed in the spinal cord of these mice, suggesting an abnormal interaction of the mutant protein with the BECN1‐BCL2L1 complex 100.
TDP‐43 proteostasis is also maintained by the coordinated action of the UPS and autophagy, which may be particularly important for clearing TDP‐43 oligomers and aggregates as these are found in most ALS patients. Activators of the UPS or autophagy promote TDP‐43 clearance and/or mitigate toxicity in models overexpressing TDP‐43 119. Interestingly, autophagy activation with rapamycin reduces the accumulation of FUS‐positive SGs and also reduces neurite fragmentation and cell death in neurons expressing mutant FUS under oxidative stress 116.
In this context, it is worth recalling that mutations in Ubiquilin 2, a member of the ubiquilin family, which regulates the degradation of ubiquitinated proteins, are linked to abnormal protein aggregation and neurodegeneration in a small subset of familial ALS and ALS/FTLD patients and that Ubiquilin 2 dysfunction is found also in patients without UBQLN2 mutations 30. Mice expressing mutant forms of UBQLN2 variably develop a motor phenotype at 3–4 months accompanied by large neuronal cytoplasmic inclusions and ubiquilin‐2‐positive inclusions that colocalize with ubiquitin, p62/SQSTM, optineurin, and occasionally TDP‐43, but never with FUS 14.
Protein misfolding may also be a consequence of ER stress. Dysfunction in protein handling in the ER and the following stress are typically associated with neuronal damage in SOD1 models and in patients 68, 92, 128 including those carrying FUS mutation 34, 134, with a controversial role of protein disulphide isomerase (PDI), a disulphide bond‐modulating ER chaperone that also facilitates the ER‐associated degradation of misfolded proteins 62.
ER stress is activated by the expression of poly(GA) DPRs in cultured cells and primary neurons 147, although the relevance of this phenomenon in patients has not been documented. ER stress is seen also in skeletal muscle across the lifespan of G93A‐SOD1 mice; activation begins before onset of symptoms and increases with disease progression and most probably contributes to muscle atrophy and weakness in ALS 16. Interestingly, it has been recently reported that ALS‐typical mutant SOD1, TDP‐43 and FUS perturb protein transport in the early secretory pathway between ER and Golgi compartments by distinct mechanisms, but each process is dependent on Rab1, which colocalizes with the mutant proteins and is probably misfolded itself 125.
Prion Propagation: A New Mechanism for the Toxicity of ALS Protein Aggregates
Whether TDP‐43 and FUS are really toxic inside neurons, and by which mechanism beside protein and RNA sequestration (if any), is also still not entirely clear.
What is now emerging as an interesting facet of the toxicity of protein aggregates in ALS is the possibility that these aggregates do not act simply through a loss‐of‐function due to sequestration of the proteins (or RNAs?), but rather actively diffuse damage to neighbor cells via a prion‐like mechanism that might contribute to the noncell autonomous nature of the disease.
ALS neurodegeneration typically begins focally and then spreads in an orderly propagating process 108 that is reminiscent of the seeding and self‐propagation seen in prion disease 79. As mentioned above, both TDP‐43 and FUS contain prion‐like domains that render them quite prone to spontaneous aggregation. Thus, it is tempting to assume that these domains allow diffusion of toxic aggregates to neighboring cells as prions do. However, to consider ALS a prion‐like disease, several conditions must be fulfilled 124.
First, ALS misfolded proteins must have “seeding” properties and form self‐aggregates. This condition seems to be verified for SOD1 103, for TDP‐43 41 and FUS 101.
Second, protein aggregates must be able to spread and propagate to neighboring cells. This is the case, at least in cultured cells, for TDP‐43 102. Aggregated mutant SOD1 is able to enter cells efficiently by macropinocytosis and to nucleate aggregation of the cytosolic protein with a self‐perpetuating mechanism 99. This process does not require cell‐to‐cell contact, but obviously depends on the extracellular release of aggregates, which can be due to discharge of seeds by dying cells or mediated by exosomes 49. Mutant SOD1 is secreted by a mechanism involving exosomes in NSC34 cells 47 and astrocyte‐derived exosomes efficiently transfer mutant SOD1 to spinal neurons and induce selective motor neuron death 7, which is the third and final property to be possessed by a protein to be considered “prion‐like.” TDP‐43 is also secreted via exosomes at least in part and it is also able to induce neurotoxicity in vitro 102.
Whether FUS possesses all the required prion characteristics and whether C9orf72 dipeptide repeat proteins are capable of similar spreading mechanisms is currently not known.
Conclusions
Studies sprouting from the individuation of the main genetic causes of ALS have greatly increased our knowledge of the processes playing a role in the pathogenesis of this complex disorder. Most importantly, they have substantially enlarged and modified our view of the molecular factors that are relevant for the development of the disease. Although the real relevance of some of these factors is still to be conclusively ascertained, precious suggestions for therapy may hopefully soon derive and be translated into clinical trials.
Acknowledgments
This work was supported by Ministero della Salute [Project RF‐2010‐2309849 to M.C.]. M.T.C. is funded by Fondazione Italiana di Ricerca per la Sclerosi Laterale Amiotrofica (ARiSLA), project ‘OligoALS’, M.C. is funded by ARiSLA, project ‘FUSMALS’.
The authors declare no conflict of interest.
References
- 1. Achsel T, Barabino S, Cozzolino M, Carri MT (2013) The intriguing case of motor neuron disease: ALS and SMA come closer. Biochem Soc Trans 41:1593–1597. [DOI] [PubMed] [Google Scholar]
- 2. Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SS et al (2014) Axonal transport of TDP‐43 mRNA granules is impaired by ALS‐causing mutations. Neuron 81:536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anderson P, Kedersha N (2009) Stress granules. Curr biol 19:R397–R398. [DOI] [PubMed] [Google Scholar]
- 4. Arnold ES, Ling SC, Huelga SC, Lagier‐Tourenne C, Polymenidou M, Ditsworth D et al (2013) ALS‐linked TDP‐43 mutations produce aberrant RNA splicing and adult‐onset motor neuron disease without aggregation or loss of nuclear TDP‐43. Proc Natl Acad Sci USA 110:E736–E745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus‐Hernandez M et al (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77:639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bandyopadhyay U, Cotney J, Nagy M, Oh S, Leng J, Mahajan M et al (2013) RNA‐Seq profiling of spinal cord motor neurons from a presymptomatic SOD1 ALS mouse. PloS One 8:e53575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Basso M, Pozzi S, Tortarolo M, Fiordaliso F, Bisighini C, Pasetto L et al (2013) Mutant copper‐zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis. J biol Chem 288:15699–15711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bendotti C, Marino M, Cheroni C, Fontana E, Crippa V, Poletti A, De Biasi S (2012) Dysfunction of constitutive and inducible ubiquitin‐proteasome system in amyotrophic lateral sclerosis: implication for protein aggregation and immune response. Prog Neurobiol 97:101–126. [DOI] [PubMed] [Google Scholar]
- 9. Bennett CL, La Spada AR (2015) Unwinding the role of senataxin in neurodegeneration. Discov Med 19:127–136. [PubMed] [Google Scholar]
- 10. Bentmann E, Haass C, Dormann D (2013) Stress granules in neurodegeneration–lessons learnt from TAR DNA binding protein of 43 kDa and fused in sarcoma. FEBS J 280:4348–4370. [DOI] [PubMed] [Google Scholar]
- 11. Budini M, Romano V, Quadri Z, Buratti E, Baralle FE (2015) TDP‐43 loss of cellular function through aggregation requires additional structural determinants beyond its C‐terminal Q/N prion‐like domain. Hum Mol Genet 24:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE (2005) TDP‐43 binds heterogeneous nuclear ribonucleoprotein A/B through its C‐terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J biol Chem 280:37572–37584. [DOI] [PubMed] [Google Scholar]
- 13. Buratti E, De Conti L, Stuani C, Romano M, Baralle M, Baralle F (2010) Nuclear factor TDP‐43 can affect selected microRNA levels. FEBS J 277:2268–2281. [DOI] [PubMed] [Google Scholar]
- 14. Ceballos‐Diaz C, Rosario AM, Park HJ, Chakrabarty P, Sacino A, Cruz PE et al (2015) Viral expression of ALS‐linked ubiquilin‐2 mutants causes inclusion pathology and behavioral deficits in mice. Mol Neurodegener 10:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chattopadhyay M, Valentine JS (2009) Aggregation of copper‐zinc superoxide dismutase in familial and sporadic ALS. Antioxid Redox Signal 11:1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen D, Wang Y, Chin ER (2015) Activation of the endoplasmic reticulum stress response in skeletal muscle of G93A*SOD1 amyotrophic lateral sclerosis mice. Front Cell Neurosci 9:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J et al (2004) DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet 74:1128–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes‐Casey M et al (2015) Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP‐43 pathology, neuronal loss, and behavioral deficits. Science 348:1151–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cleary JD, Ranum LP (2014) Repeat associated non‐ATG (RAN) translation: new starts in microsatellite expansion disorders. Curr Opin Genet Dev 26C:6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cohen TJ, Hwang AW, Unger T, Trojanowski JQ, Lee VM (2012) Redox signalling directly regulates TDP‐43 via cysteine oxidation and disulphide cross‐linking. EMBO J 31:1241–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cooper‐Knock J, Walsh MJ, Higginbottom A, Robin Highley J, Dickman MJ, Edbauer D et al (2014) Sequestration of multiple RNA recognition motif‐containing proteins by C9orf72 repeat expansions. Brain 137(Pt 7):2040–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cooper‐Knock J, Bury JJ, Heath PR, Wyles M, Higginbottom A, Gelsthorpe C et al (2015) C9ORF72 GGGGCC expanded repeats produce splicing dysregulation which correlates with disease severity in amyotrophic lateral sclerosis. PloS One 10:e0127376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Couthouis J, Hart MP, Shorter J, DeJesus‐Hernandez M, Erion R, Oristano R et al (2011) A yeast functional screen predicts new candidate ALS disease genes. Proc Nat Acad Sci USA 108:20881–20890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Couthouis J, Hart MP, Erion R, King OD, Diaz Z, Nakaya T et al (2012) Evaluating the role of the FUS/TLS‐related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet 21:2899–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cozzolino M, Pesaresi MG, Amori I, Crosio C, Ferri A, Nencini M, Carri MT (2009) Oligomerization of mutant SOD1 in mitochondria of motoneuronal cells drives mitochondrial damage and cell toxicity. Antioxid Redox Signal 11:1547–1558. [DOI] [PubMed] [Google Scholar]
- 26. Cozzolino M, Pesaresi MG, Gerbino V, Grosskreutz J, Carri MT (2012) Amyotrophic lateral sclerosis: new insights into underlying molecular mechanisms and opportunities for therapeutic intervention. Antioxid Redox Signal 17:1277–1330. [DOI] [PubMed] [Google Scholar]
- 27. Davidson YS, Barker H, Robinson AC, Thompson JC, Harris J, Troakes C et al (2014) Brain distribution of dipeptide repeat proteins in frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun 2:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron 72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deng H, Gao K, Jankovic J (2014) The role of FUS gene variants in neurodegenerative diseases. Nat Rev Neurol 10:337–48. [DOI] [PubMed] [Google Scholar]
- 30. Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N et al (2011) Mutations in UBQLN2 cause dominant X‐linked juvenile and adult‐onset ALS and ALS/dementia. Nature 477:211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dewey CM, Cenik B, Sephton CF, Johnson BA, Herz J, Yu G (2012) TDP‐43 aggregation in neurodegeneration: are stress granules the key? Brain Res 1462:16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Di Salvio M, Piccinni V, Gerbino V, Mantoni F, Camerini S, Lenzi J et al (2015) Pur‐alpha functionally interacts with FUS carrying ALS‐associated mutations. Cell Death Dis 6:e1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, Vidensky S et al (2013) RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80:415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Farg MA, Soo KY, Walker AK, Pham H, Orian J, Horne MK et al (2012) Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide‐isomerase. Neurobiol Aging 33:2855–2868. [DOI] [PubMed] [Google Scholar]
- 35. Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V et al (2014) C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet 23:3579–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ferri A, Cozzolino M, Crosio C, Nencini M, Casciati A, Gralla EB et al (2006) Familial ALS‐superoxide dismutases associate with mitochondria and shift their redox potentials. Proc Nat Acad Sci USA 103:13860–13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Freibaum BD, Lu Y, Lopez‐Gonzalez R, Kim NC, Almeida S, Lee KH et al (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525:129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Freischmidt A, Muller K, Ludolph AC, Weishaupt JH (2013) Systemic dysregulation of TDP‐43 binding microRNAs in amyotrophic lateral sclerosis. Acta Neuropathol Commun 1:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fujii R, Okabe S, Urushido T, Inoue K, Yoshimura A, Tachibana T et al (2005) The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Current Biol 15:587–593. [DOI] [PubMed] [Google Scholar]
- 40. Fujii R, Takumi T (2005) TLS facilitates transport of mRNA encoding an actin‐stabilizing protein to dendritic spines. J Cell Sci 118(Pt 24):5755–5765. [DOI] [PubMed] [Google Scholar]
- 41. Furukawa Y, Kaneko K, Watanabe S, Yamanaka K, Nukina N (2011) A seeding reaction recapitulates intracellular formation of Sarkosyl‐insoluble transactivation response element (TAR) DNA‐binding protein‐43 inclusions. J Biol Chem 286:18664–18672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Galbiati M, Crippa V, Rusmini P, Cristofani R, Cicardi ME, Giorgetti E et al (2014) ALS‐related misfolded protein management in motor neurons and muscle cells. Neurochem Int 79:70–78. [DOI] [PubMed] [Google Scholar]
- 43. Gascon E, Gao FB (2014) The emerging roles of microRNAs in the pathogenesis of frontotemporal dementia‐amyotrophic lateral sclerosis (FTD‐ALS) spectrum disorders. J Neurogenet 28:30–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gendron TF, Bieniek KF, Zhang YJ, Jansen‐West K, Ash PE, Caulfield T et al (2013) Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat‐associated non‐ATG translation in c9FTD/ALS. Acta Neuropathol 126:829–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gerbino V, Carri MT, Cozzolino M, Achsel T (2013) Mislocalised FUS mutants stall spliceosomal snRNPs in the cytoplasm. Neurobiol Dis 55:120–128. [DOI] [PubMed] [Google Scholar]
- 46. Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D et al (2008) TDP‐43 A315T mutation in familial motor neuron disease. Ann Neurol 63:535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gomes C, Keller S, Altevogt P, Costa J (2007) Evidence for secretion of Cu,Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neurosci Lett 428:43–46. [DOI] [PubMed] [Google Scholar]
- 48. Gomez‐Deza J, Lee YB, Troakes C, Nolan M, Al‐Sarraj S, Gallo JM, Shaw CE (2015) Dipeptide repeat protein inclusions are rare in the spinal cord and almost absent from motor neurons in C9ORF72 mutant amyotrophic lateral sclerosis and are unlikely to cause their degeneration. Acta Neuropathol Commun 3:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Grad LI, Yerbury JJ, Turner BJ, Guest WC, Pokrishevsky E, O'Neill MA et al (2014) Intercellular propagated misfolding of wild‐type Cu/Zn superoxide dismutase occurs via exosome‐dependent and ‐independent mechanisms. Proc Nat Acad Sci USA 111:3620–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C et al (2006) ANG mutations segregate with familial and ‘sporadic' amyotrophic lateral sclerosis. Nat Genet 38:411–413. [DOI] [PubMed] [Google Scholar]
- 51. Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R (2004) The Microprocessor complex mediates the genesis of microRNAs. Nature 432:235–240. [DOI] [PubMed] [Google Scholar]
- 52. Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS et al (2014) C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507:195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y et al (2008) Phosphorylated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol 64:60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hebron ML, Lonskaya I, Sharpe K, Weerasinghe PP, Algarzae NK, Shekoyan AR, Moussa CE (2013) Parkin ubiquitinates Tar‐DNA binding protein‐43 (TDP‐43) and promotes its cytosolic accumulation via interaction with histone deacetylase 6 (HDAC6). J Biol Chem 288:4103–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Highley JR, Kirby J, Jansweijer JA, Webb PS, Hewamadduma CA, Heath PR et al (2014) Loss of nuclear TDP‐43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones. Neuropathol Appl Neurobiol 40:670–685. [DOI] [PubMed] [Google Scholar]
- 56. Hoell JI, Larsson E, Runge S, Nusbaum JD, Duggimpudi S, Farazi TA et al (2011) RNA targets of wild‐type and mutant FET family proteins. Nat Struct Mol Biol 18:1428–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Igoudjil A, Magrane J, Fischer LR, Kim HJ, Hervias I, Dumont M et al (2011) In vivo pathogenic role of mutant SOD1 localized in the mitochondrial intermembrane space. J Neurosci 31:15826–15837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Iguchi Y, Katsuno M, Takagi S, Ishigaki S, Niwa J, Hasegawa M et al (2012) Oxidative stress induced by glutathione depletion reproduces pathological modifications of TDP‐43 linked to TDP‐43 proteinopathies. Neurobiol Dis 45:862–870. [DOI] [PubMed] [Google Scholar]
- 59. Ishigaki S, Masuda A, Fujioka Y, Iguchi Y, Katsuno M, Shibata A et al (2012) Position‐dependent FUS‐RNA interactions regulate alternative splicing events and transcriptions. Sci Rep 2:529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ishihara T, Ariizumi Y, Shiga A, Kato T, Tan CF, Sato T et al (2013) Decreased number of Gemini of coiled bodies and U12 snRNA level in amyotrophic lateral sclerosis. Hum Mol Genet 22:4136–4147. [DOI] [PubMed] [Google Scholar]
- 61. Jansen AH, Reits EA, Hol EM (2014) The ubiquitin proteasome system in glia and its role in neurodegenerative diseases. Front Mol Neurosci 7:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jaronen M, Goldsteins G, Koistinaho J (2014) ER stress and unfolded protein response in amyotrophic lateral sclerosis‐a controversial role of protein disulphide isomerase. Front Cell Neurosci 8:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE et al (2014) Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci 17:664–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB et al (2015) Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci 18:1226–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C et al (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572–574. [DOI] [PubMed] [Google Scholar]
- 66. Kabashi E, Agar JN, Strong MJ, Durham HD (2012) Impaired proteasome function in sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler 13:367–371. [DOI] [PubMed] [Google Scholar]
- 67. Kalmar B, Lu CH, Greensmith L (2014) The role of heat shock proteins in Amyotrophic Lateral Sclerosis: The therapeutic potential of Arimoclomol. Pharmacol Ther 141(1):40–54. [DOI] [PubMed] [Google Scholar]
- 68. Karademir B, Corek C, Ozer NK (2015) Endoplasmic reticulum stress and proteasomal system in amyotrophic lateral sclerosis. Free Radic Biol Med 88(Pt A):42–50. [DOI] [PubMed] [Google Scholar]
- 69. Kariya S, Re DB, Jacquier A, Nelson K, Przedborski S, Monani UR (2012) Mutant superoxide dismutase 1 (SOD1), a cause of amyotrophic lateral sclerosis, disrupts the recruitment of SMN, the spinal muscular atrophy protein to nuclear Cajal bodies. Hum Mol Genet 21:3421–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kawahara Y, Mieda‐Sato A (2012) TDP‐43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc Nat Acad Sci USA 109:3347–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z et al (2013) Mutations in prion‐like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495:467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kim HJ, Raphael AR, LaDow ES, McGurk L, Weber RA, Trojanowski JQ et al (2014) Therapeutic modulation of eIF2alpha phosphorylation rescues TDP‐43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet 46:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Koppers M, Blokhuis AM, Westeneng HJ, Terpstra ML, Zundel CA, Vieira de Sa R et al (2015) C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol 78:426–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208. [DOI] [PubMed] [Google Scholar]
- 75. Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T et al (2014) Poly‐dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 345:1139–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lagier‐Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC et al (2012) Divergent roles of ALS‐linked proteins FUS/TLS and TDP‐43 intersect in processing long pre‐mRNAs. Nat Neurosci 15:1488–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lagier‐Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR et al (2013) Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Nat Acad Sci USA 110:E4530–E4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lattante S, Rouleau GA, Kabashi E (2013) TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat 34:812–826. [DOI] [PubMed] [Google Scholar]
- 79. Lee S, Kim HJ (2015) Prion‐like Mechanism in Amyotrophic Lateral Sclerosis: are Protein Aggregates the Key? Exp Neurobiol 24:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lee YB, Chen HJ, Peres JN, Gomez‐Deza J, Attig J, Stalekar M et al (2013) Hexanucleotide repeats in ALS/FTD form length‐dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep 5:1178–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lenzken SC, Romeo V, Zolezzi F, Cordero F, Lamorte G, Bonanno D et al (2011) Mutant SOD1 and mitochondrial damage alter expression and splicing of genes controlling neuritogenesis in models of neurodegeneration. Hum Mutat 32:168–182. [DOI] [PubMed] [Google Scholar]
- 82. Levine TP, Daniels RD, Gatta AT, Wong LH, Hayes MJ (2013) The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab‐GEFs. Bioinformatics 29:499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Li HY, Yeh PA, Chiu HC, Tang CY, Tu BP (2011) Hyperphosphorylation as a defense mechanism to reduce TDP‐43 aggregation. PloS One 6:e23075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Li YR, King OD, Shorter J, Gitler AD (2013) Stress granules as crucibles of ALS pathogenesis. J Cell biol 201:361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Li Y, Guo Y, Wang X, Yu X, Duan W, Hong K, Wang J, Han H, Li C (2015) Trehalose decreases mutant SOD1 expression and alleviates motor deficiency in early but not end‐stage amyotrophic lateral sclerosis in a SOD1‐G93A mouse model. Neuroscience 298:12–25. [DOI] [PubMed] [Google Scholar]
- 86. Liu‐Yesucevitz L, Lin AY, Ebata A, Boon JY, Reid W, Xu YF et al (2014) ALS‐linked mutations enlarge TDP‐43‐enriched neuronal RNA granules in the dendritic arbor. J Neurosci 34:4167–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mackenzie IR, Frick P, Neumann M (2014) The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol 127:347–357. [DOI] [PubMed] [Google Scholar]
- 88. Mackenzie IR, Arzberger T, Kremmer E, Troost D, Lorenzl S, Mori K et al (2013) Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico‐pathological correlations. Acta Neuropathol 126:859–879. [DOI] [PubMed] [Google Scholar]
- 89. Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S et al (2012) Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross‐sectional study. Lancet Neurol 11:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mann DM, Rollinson S, Robinson A, Bennion CJ, Thompson JC, Snowden JS et al (2013) Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun 1:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Marino M, Papa S, Crippa V, Nardo G, Peviani M, Cheroni C et al (2015) Differences in protein quality control correlate with phenotype variability in 2 mouse models of familial amyotrophic lateral sclerosis. Neurobiol Aging 36:492–504. [DOI] [PubMed] [Google Scholar]
- 92. Matus S, Valenzuela V, Medinas DB, Hetz C (2013) ER Dysfunction and Protein Folding Stress in ALS. Int J Cell Biol 2013:674751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM et al (2014) C9orf72 FTLD/ALS‐associated Gly‐Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta neuropathol 128:485–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mizielinska S, Lashley T, Norona FE, Clayton EL, Ridler CE, Fratta P, Isaacs AM (2013) C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta neuropathol 126:845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Mizielinska S, Gronke S, Niccoli T, Ridler CE, Clayton EL, Devoy A et al (2014) C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine‐rich proteins. Science 345:1192–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mori K, Arzberger T, Grasser FA, Gijselinck I, May S, Rentzsch K et al (2013) Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol 126:881–893. [DOI] [PubMed] [Google Scholar]
- 97. Mori K, Lammich S, Mackenzie IR, Forne I, Zilow S, Kretzschmar H et al (2013) hnRNP A3 binds to GGGGCC repeats and is a constituent of p62‐positive/TDP43‐negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol 125:413–423. [DOI] [PubMed] [Google Scholar]
- 98. Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E et al (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide‐repeat proteins in FTLD/ALS. Science 339:1335–1338. [DOI] [PubMed] [Google Scholar]
- 99. Munch C, O'Brien J, Bertolotti A (2011) Prion‐like propagation of mutant superoxide dismutase‐1 misfolding in neuronal cells. Proc Nat Acad Sci USA 108:3548–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Nassif M, Valenzuela V, Rojas‐Rivera D, Vidal R, Matus S, Castillo K et al (2014) Pathogenic role of BECN1/Beclin 1 in the development of amyotrophic lateral sclerosis. Autophagy 10:1256–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Nomura T, Watanabe S, Kaneko K, Yamanaka K, Nukina N, Furukawa Y (2014) Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J Biol Chem 289:1192–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Nonaka T, Masuda‐Suzukake M, Arai T, Hasegawa Y, Akatsu H, Obi T et al (2013) Prion‐like properties of pathological TDP‐43 aggregates from diseased brains. Cell Rep 4:124–134. [DOI] [PubMed] [Google Scholar]
- 103. Ogawa M, Furukawa Y (2014) A seeded propagation of Cu, Zn‐superoxide dismutase aggregates in amyotrophic lateral sclerosis. Front Cell Neurosci 8:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY et al (2015) A liquid‐to‐solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162:1066–1077. [DOI] [PubMed] [Google Scholar]
- 105. Pesiridis GS, Tripathy K, Tanik S, Trojanowski JQ, Lee VM (2011) A “two‐hit” hypothesis for inclusion formation by carboxyl‐terminal fragments of TDP‐43 protein linked to RNA depletion and impaired microtubule‐dependent transport. J Biol Chem 286:18845–18855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Peters OM, Ghasemi M, Brown RH Jr (2015) Emerging mechanisms of molecular pathology in ALS. J Clin Invest 125:1767–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Polymenidou M, Lagier‐Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY et al (2011) Long pre‐mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP‐43. Nat Neurosci 14:459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Pradat PF, Kabashi E, Desnuelle C (2015) Deciphering spreading mechanisms in amyotrophic lateral sclerosis: clinical evidence and potential molecular processes. Curr Opin Neurol 28:455–461. [DOI] [PubMed] [Google Scholar]
- 109. Prudencio M, Belzil VV, Batra R, Ross CA, Gendron TF, Pregent LJ et al (2015) Distinct brain transcriptome profiles in C9orf72‐associated and sporadic ALS. Nat Neurosci 18:1175–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Qiu H, Lee S, Shang Y, Wang WY, Au KF, Kamiya S et al (2014) ALS‐associated mutation FUS‐R521C causes DNA damage and RNA splicing defects. J Clin Invest 124:981–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Renton AE, Majounie E, Waite A, Simon‐Sanchez J, Rollinson S, Gibbs JR et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron 72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Rogelj B, Easton LE, Bogu GK, Stanton LW, Rot G, Curk T et al (2012) Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci Rep 2:603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Rohrer JD, Isaacs AM, Mizielinska S, Mead S, Lashley T, Wray S et al (2015) C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. Lancet Neurol 14:291–301. [DOI] [PubMed] [Google Scholar]
- 114. Romano V, Quadri Z, Baralle FE, Buratti E (2015) The structural integrity of TDP‐43 N‐terminus is required for efficient aggregate entrapment and consequent loss of protein function. Prion 9:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Rossi S, Serrano A, Gerbino V, Giorgi A, Di Francesco L, Nencini M et al (2015) Nuclear accumulation of mRNAs underlies G4C2‐repeat‐induced translational repression in a cellular model of C9orf72 ALS. J Cell Sci 128:1787–1799. [DOI] [PubMed] [Google Scholar]
- 116. Ryu HH, Jun MH, Min KJ, Jang DJ, Lee YS, Kim HK, Lee JA (2014) Autophagy regulates amyotrophic lateral sclerosis‐linked fused in sarcoma‐positive stress granules in neurons. Neurobiol Aging 35:2822–2831. [DOI] [PubMed] [Google Scholar]
- 117. Satoh J, Yamamoto Y, Kitano S, Takitani M, Asahina N, Kino Y (2014) Molecular network analysis suggests a logical hypothesis for the pathological role of c9orf72 in amyotrophic lateral sclerosis/frontotemporal dementia. J Cent Nerv Syst Dis 6:69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Schwartz JC, Wang X, Podell ER, Cech TR (2013) RNA seeds higher‐order assembly of FUS protein. Cell Rep 5:918–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Scotter EL, Chen HJ, Shaw CE (2015) TDP‐43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics 12:352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Sephton CF, Cenik C, Kucukural A, Dammer EB, Cenik B, Han Y et al (2011) Identification of neuronal RNA targets of TDP‐43‐containing ribonucleoprotein complexes. J Biol Chem 286:1204–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Shelkovnikova TA, Robinson HK, Southcombe JA, Ninkina N, Buchman VL (2014) Multistep process of FUS aggregation in the cell cytoplasm involves RNA‐dependent and RNA‐independent mechanisms. Hum Mol Genet 23:5211–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Shodai A, Morimura T, Ido A, Uchida T, Ayaki T, Takahashi R et al (2013) Aberrant assembly of RNA recognition motif 1 links to pathogenic conversion of TAR DNA‐binding protein of 43 kDa (TDP‐43). J Biol Chem 288:14886–14905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Simpson CL, Lemmens R, Miskiewicz K, Broom WJ, Hansen VK, van Vught PW et al (2009) Variants of the elongator protein 3 (ELP3) gene are associated with motor neuron degeneration. Hum Mol Genet 18:472–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Smethurst P, Sidle KC, Hardy J (2015) Review: Prion‐like mechanisms of transactive response DNA binding protein of 43 kDa (TDP‐43) in amyotrophic lateral sclerosis (ALS). Neuropathol Appl Neurobiol 41:578–597. [DOI] [PubMed] [Google Scholar]
- 125. Soo KY, Halloran M, Sundaramoorthy V, Parakh S, Toth RP, Southam KA et al (2015) Rab1‐dependent ER‐Golgi transport dysfunction is a common pathogenic mechanism in SOD1, TDP‐43 and FUS‐associated ALS. Acta neuropathol 130:679–697. [DOI] [PubMed] [Google Scholar]
- 126. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B et al (2008) TDP‐43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Svetoni F, Caporossi D, Paronetto MP (2014) Oxidative stress affects FET proteins localization and alternative pre‐mRNA processing in cellular models of ALS. Free Radic Biol Med 75(Suppl. 1):S51. [DOI] [PubMed] [Google Scholar]
- 128. Tadic V, Prell T, Lautenschlaeger J, Grosskreutz J (2014) The ER mitochondria calcium cycle and ER stress response as therapeutic targets in amyotrophic lateral sclerosis. Front Cell Neurosci 8:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Tao Z, Wang H, Xia Q, Li K, Jiang X, Xu G, Wang G, Ying Z (2015) Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation‐induced cytotoxicity. Hum Mol Genet 24:2426–2441. [DOI] [PubMed] [Google Scholar]
- 130. Ticozzi N, Vance C, Leclerc AL, Keagle P, Glass JD, McKenna‐Yasek D et al (2011) Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. Am J Med Genet B Neuropsychiatr Genet 156B:285–290. [DOI] [PubMed] [Google Scholar]
- 131. Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M et al (2011) Characterizing the RNA targets and position‐dependent splicing regulation by TDP‐43. Nat Neurosci 14:452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Tsuiji H, Iguchi Y, Furuya A, Kataoka A, Hatsuta H, Atsuta N et al (2013) Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol Med 5:221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Walker AK, Farg MA, Bye CR, McLean CA, Horne MK, Atkin JD (2010) Protein disulphide isomerase protects against protein aggregation and is S‐nitrosylated in amyotrophic lateral sclerosis. Brain 133(Pt 1):105–116. [DOI] [PubMed] [Google Scholar]
- 135. Wang IF, Wu LS, Chang HY, Shen CK (2008) TDP‐43, the signature protein of FTLD‐U, is a neuronal activity‐responsive factor. J Neurochem 105:797–806. [DOI] [PubMed] [Google Scholar]
- 136. Wang YT, Kuo PH, Chiang CH, Liang JR, Chen YR, Wang S et al (2013) The truncated C‐terminal RNA recognition motif of TDP‐43 protein plays a key role in forming proteinaceous aggregates. J Biol Chem 288:9049–9057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y et al (2014) Antisense proline‐arginine RAN dipeptides linked to C9ORF72‐ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84:1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Xiao S, Sanelli T, Dib S, Sheps D, Findlater J, Bilbao J et al (2011) RNA targets of TDP‐43 identified by UV‐CLIP are deregulated in ALS. Mol Cell Neurosci 47:167–180. [DOI] [PubMed] [Google Scholar]
- 139. Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL et al (2013) Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Nat Acad Sci USA 110:7778–7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Yamazaki T, Chen S, Yu Y, Yan B, Haertlein TC, Carrasco MA et al (2012) FUS‐SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep 2:799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Yang D, Abdallah A, Li Z, Lu Y, Almeida S, Gao FB (2015) FTD/ALS‐associated poly(GR) protein impairs the Notch pathway and is recruited by poly(GA) into cytoplasmic inclusions. Acta Neuropathol 130:525–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Yu Y, Chi B, Xia W, Gangopadhyay J, Yamazaki T, Winkelbauer‐Hurt ME et al (2015) U1 snRNP is mislocalized in ALS patient fibroblasts bearing NLS mutations in FUS and is required for motor neuron outgrowth in zebrafish. Nucleic Acids Res 43:3208–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Yu Y, Reed R (2015) FUS functions in coupling transcription to splicing by mediating an interaction between RNAP II and U1 snRNP. Proc Nat Acad Sci USA 112:8608–8613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Zhang X, Li L, Chen S, Yang D, Wang Y, Wang Z, Le W (2011) Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy 7:412–425. [DOI] [PubMed] [Google Scholar]
- 145. Zhang D, Iyer LM, He F, Aravind L (2012) Discovery of novel DENN proteins: implications for the evolution of eukaryotic intracellular membrane structures and human disease. Front Genet 3:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Zhang YJ, Caulfield T, Xu YF, Gendron TF, Hubbard J, Stetler C, Sasaguri H, Whitelaw EC, Cai S, Lee WC, Petrucelli L (2013) The dual functions of the extreme N‐terminus of TDP‐43 in regulating its biological activity and inclusion formation. Hum Mol Genet 22:3112–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Zhang YJ, Jansen‐West K, Xu YF, Gendron TF, Bieniek KF, Lin WL et al (2014) Aggregation‐prone c9FTD/ALS poly(GA) RAN‐translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol 128:505–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P et al (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525:56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Zhang X, Chen S, Song L, Tang Y, Shen Y, Jia L, Le W (2014) MTOR‐independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy 10:588–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Zu T, Liu Y, Banez‐Coronel M, Reid T, Pletnikova O, Lewis J et al (2013) RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Nat Acad Sci USA 110:E4968–E4977. [DOI] [PMC free article] [PubMed] [Google Scholar]