

Abstract
Olfactory dysfunction is common in multiple sclerosis (MS). Olfactory bulb and tract pathology in MS and other demyelinating diseases remain unexplored. A human autopsy cohort of pathologically confirmed cases encompassing the spectrum of demyelinating disease (MS; n = 17), neuromyelitis optica [(NMO); n = 3] and acute disseminated encephalomyelitis [(ADEM); n = 7] was compared to neuroinflammatory [herpes simplex virus encephalitis (HSE); n = 3], neurodegenerative [Alzheimer's disease (AD); n = 4] and non‐neurologic (n = 8) controls. For each case, olfactory bulbs and/or tracts were stained for myelin, axons and inflammation. Inferior frontal cortex and hippocampus were stained for myelin in a subset of MS and ADEM cases. Olfactory bulb/tract demyelination was frequent in all demyelinating diseases [MS 12/17 (70.6%); ADEM 3/7 (42.9%); NMO 2/3 (66.7%)] but was absent in HSE, AD and non‐neurologic controls. Inflammation was greater in the demyelinating diseases compared to non‐neurologic controls. Olfactory bulb/tract axonal loss was most severe in MS where it correlated significantly with the extent of demyelination (r = 0.610, P = 0.009) and parenchymal inflammation (r = 0.681, P = 0.003). The extent of olfactory bulb/tract demyelination correlated with that found in the adjacent inferior frontal cortex but not hippocampus. We provide unequivocal evidence that olfactory bulb/tract demyelination is frequent, can occur early and is highly inflammatory, and is specific to demyelinating disease.
Keywords: demyelinating disease, multiple sclerosis, neuroinflammation, olfaction, pathology
Introduction
Olfactory dysfunction is a common, but often overlooked, feature of multiple sclerosis (MS) 8, 12, 14, 15, 18, 19, 22, 24, 26, 31, 34. Even though MS is characterized by inflammatory demyelination disseminated throughout the central nervous system (CNS), evidence of olfactory bulb/tract demyelination is controversial. Cruveilhier 3, Carswell 1, Charcot 2 and Dawson 4 published among the most comprehensive surveys of MS pathology, but olfactory bulb/tract demyelination was never mentioned. Interestingly, pathologic involvement of the olfactory “nerve” was briefly mentioned by Gowers 23 at the end of the 19th century, but this has been largely ignored. This, in combination with Zimmerman and Netsky's failure to detect demyelination in the olfactory “nerve” in eight MS cases 33, has led to the conclusion that the olfactory bulb/tract is relatively unaffected in the disease. Consequently, the search for the pathologic substrate of olfactory loss in MS has shifted to the olfactory brain.
Radiographic studies have shed light onto the possible structural correlate of olfactory dysfunction in MS. Doty et al were the first to demonstrate a striking relationship between magnetic resonance imaging (MRI) measures of lesion burden of the olfactory brain (ie, inferior frontal and temporal lobes) and loss of smell using a validated test of olfaction 12. This landmark finding, subsequently corroborated by independent investigators, has further pointed attention away from possible olfactory bulb/tract pathology in MS 14, 15, 30, 34, 35.
Clinical tests of olfaction do not allow definitive distinction between dysfunction of the olfactory bulb/tract and olfactory brain. Further, MRI measures of lesion burden used in these studies did not have sufficient resolution to detect pathologic changes in the olfactory bulb/tract themselves. Therefore, olfactory bulb/tract involvement in MS should not be overlooked. Anatomically, the peripheral olfactory neuroepithelium lines the attic above the superior and anterior middle turbinates in the nasal cavity and consists of olfactory receptor cells that extend small unmyelinated axons that synapse in the glomerular apparatus of the olfactory bulb located in the CNS. With further relays within the olfactory bulb and tract, these central olfactory fibers pass via the olfactory tubercle and trigone to terminate in mesial temporal or inferior frontal cortex 10. Given that the olfactory bulb/tract are ensheathed by central oligodendrocyte‐derived myelin, are exposed to a multitude of pathogens, are immunologically active and provide a direct conduit into the CNS [reviewed in 10 ], they may be of particular relevance to MS where gene–environment interactions are thought to be central to disease pathogenesis.
We provide a descriptive account of the extent, distribution and specificity of inflammatory demyelination in the olfactory bulb/tract in a wide spectrum of demyelinating [MS, neuromyelitis optica (NMO), acute disseminated encephalomyelitis (ADEM)], neuroinflammatory [herpes simplex encephalitis (HSE)] and neurodegenerative [Alzheimer's disease (AD)] diseases. In this study, we unequivocally demonstrate that olfactory bulb/tract demyelination is frequent, can occur early and is highly inflammatory, and is specific to demyelinating disease.
Materials and Methods
Study population
A human autopsy cohort of pathologically confirmed cases encompassing the spectrum of demyelinating disease (MS, n = 17; NMO, n = 3; and ADEM, n = 7) with available olfactory bulb/tract tissue was used and compared to neuroinflammatory (HSE, n = 3), neurodegenerative (AD; Braak Stage V/VI, n = 4) and non‐neurologic (n = 8) controls derived from the Oxford Brain Bank with relevant ethics committee approval. A subset of the MS, ADEM and HSE cases have been the subject of previous reports on these diseases 6, 7, 16, 21. Aquaporin‐4 antibody serostatus was available in two of three NMO cases, both of which were positive. The selection of cases was based on the availability of olfactory bulbs and/or tracts for the respective diagnoses. No clinical information about olfactory function during life was available. Further details are provided in the Supporting Information.
Neuropathological sampling
Coronally sliced brains were aligned so that the entire length of available olfactory structures (including olfactory bulb, anterior nucleus and tract) could be identified and sampled from each side (Figure 1). Inferior frontal cortex adjacent to the olfactory bulb/tract and hippocampus were sampled in MS and ADEM cases for comparative analyses of demyelination between brain regions, where available. Inferior frontal cortical and hippocampal blocks were taken in the coronal plane, the latter being sampled preferentially at the level of the lateral geniculate nucleus to enable comparable scoring between cases. Further details of the quantification of proportional lesional areas are outlined in Figure 4. Given that cortical and hippocampal demyelination are not established features of NMO, a similar sampling protocol for these regions was omitted 28.
Figure 1.
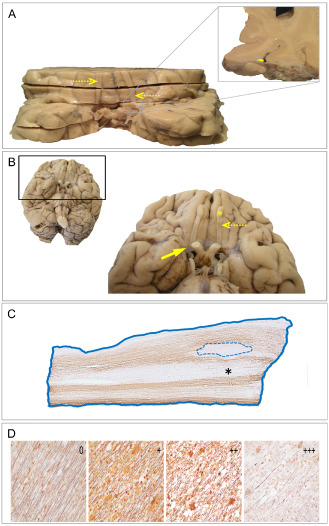
Olfactory bulb and tract anatomy, sampling and histopathological analysis strategies. A. Inferior view of coronally sliced brain from a multiple sclerosis (MS) case. Given that most brains had been sliced in the coronal plane for post‐mortem diagnostic evaluation, olfactory bulbs and tracts (dashed arrows) were interrupted. Accordingly, coronal slices were ordered (anterior to posterior) and realigned (left/right) anatomically so that the entire available length of olfactory structures could be identified and sampled from each side. Inset shows the anatomic relationship between the olfactory tract (yellow) and adjacent meninges and cortex. B. Inferior view of brain showing intact olfactory bulbs (yellow asterisk) and tracts (dashed arrow) from a non‐neurologic control case. The pia and arachnoid meningeal layers (solid arrow) form an intricate web covering both the olfactory bulbs/tracts and adjacent cortex. C. Myelin immunohistochemistry (proteolipid protein) of an olfactory tract derived from a neuromyelitis optica case. Areas of demyelination (dotted line) were related to the total sampled olfactory tissue area (solid line) to obtain proportional plaque load measures for each case. Regions that appear to have “myelin loss” (black asterisk) are, in fact, normal appearing gray matter. D. Palmgren‐stained sections of olfactory tracts demonstrating varying degrees of axonal loss. Semiquantitative scores ranging from 0 (no axonal loss) to +++ (severe axonal loss) were assigned to each olfactory bulb and tract section as illustrated, with a mean score obtained from each case for statistical analyses.
Figure 4.
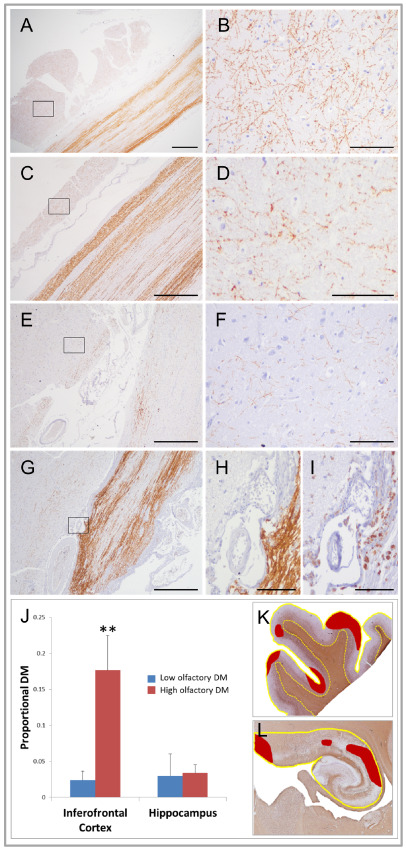
Relationship between cortical and olfactory bulb and tract demyelination in demyelinating diseases and non‐neurologic controls. A–H. Olfactory bulb and tract and adjacent cortex stained for myelin (proteolipid protein). Panels A, C, E and G show the close proximity of cortex (left) to olfactory structures (right) with shared meninges between. Panels B, D, F and H show high power views of cortical gray matter sampled in regions indicated by boxes. No demyelination is seen in control olfactory bulb and tract (A) or adjacent cortex (B). In multiple sclerosis (C–F), the extent of cortical demyelination mirrors the extent of olfactory bulb and tract demyelination with panels C and D showing minimal demyelination of these structures compared to panels E and F where demyelination is relatively severe. In acute disseminated encephalomyelitis (G–I), subpial cortical demyelination (H) adjacent to active olfactory tract demyelination marked by abundant macrophages (G). Panel I shows significant macrophage infiltration (immunostained by PG‐M1) of olfactory tract parenchyma and adjacent meninges and cortex in area depicted in panel H. J. Bar graphs of the extent of proportional demyelination in inferior frontal cortex and hippocampus in multiple sclerosis and acute disseminated encephalomyelitis cases with “low” (blue bars) and “high” (red bars) olfactory bulb/tract demyelination. Inferior frontal cortical (K) and hippocampal (L) demyelinated areas (red shading in K and L) were respectively related to total cortical and hippocampal area measures (yellow lines in K and L) to derive estimates of total proportional demyelination in these regions (NB: red shading represents hypothetical areas of demyelination for purposes of illustration). Inferior frontal cortical demyelination was significantly greater in cases with high levels of olfactory bulb and tract demyelination compared to cases with low levels. No such relationship was found between the extent of olfactory bulb/tract and hippocampal demyelination. Similar findings were found when multiple sclerosis and acute disseminated encephalomyelitis cases were analyzed separately. DM = demyelination. Low olfactory DM = cases with olfactory bulb/tract demyelination less than median value of olfactory bulb/tract demyelination. High olfactory DM = cases with olfactory bulb/tract demyelination greater than median value of olfactory bulb/tract demyelination. Scale bars represent 1 mm in panel A; 100 μm in panels B, D, F, H and I; and 500 μm in panels C, E and G. **P = 0.01.
Neuropathological evaluation
Olfactory bulb and tract
Adjacent formalin‐fixed, paraffin‐embedded (FFPE) transverse sections of the olfactory bulbs and/or tracts were immunostained with primary antibodies to demonstrate myelin, inflammation, glial cells and aquaporin‐4 and impregnated with Palmgren silver to demonstrate axons (Supporting Information Table S1) 5. The extent of demyelination, inflammation and axonal loss was quantified by two independent observers blinded to disease category (see Figure 1 for details).
Areas of demyelination were related to the total sampled olfactory tissue area to obtain proportional plaque load measures for each case as outlined in Figure 1. Stage of demyelination (ie, acute, border active or chronic inactive) was determined using established criteria based on the intensity and distribution of microglial infiltrate in demyelinated regions 9. Semiquantitative scores of axonal loss with scores ranging from 0 (no axonal loss) to +++ (severe axonal loss) were assigned to each olfactory bulb and tract section, with a mean score obtained from each case for statistical analyses (Figure 1). A similar semiquantative scoring method was used to estimate the extent of microglial/macrophage (from PG‐M1 immunolabeled sections), T‐cell (from CD3+ immunolabeled sections) and B‐cell (from CD20+ immunolabeled sections) inflammation for each compartment (ie, parenchyma, perivascular, meninges) as follows: 0 = no inflammatory cells; + = average of 1 positively labeled cell per 200× field (≈7.1 cells/mm2); ++ = average of 2–4 positively labeled cells per 200× field (≈14.3–28.5 cells/mm2); +++ = average > 4 positively labeled cells per 200× field (>28.5 cells/mm2).
Inferior frontal cortical and hippocampal demyelination
Similar to the olfactory bulb/tract, inferior frontal cortical and hippocampal sections were stained for myelin to quantify areas of demyelination in each of these regions. These lesional areas were related to the total sampled tissue area of the region analyzed to obtain proportional plaque load measures for each case as outlined in Figure 4.
Statistical analysis
Statistical methods used to analyze the data were parametric where data are normally distributed and nonparametric otherwise, to compare findings in between disease groups and controls. Additional details are provided in the Supporting Information.
Results
Demographics
Age and duration of disease differed significantly between disease groups reflecting the wide spectrum of diseases selected for study. Clinical details are provided in Table 1 and Supporting Information Table S2. Post‐mortem interval did not differ significantly between groups (data not shown).
Table 1.
Demographic and clinical details. Abbreviations: F = female; M = male; N/A = not applicable
| Demographics | Disease | |||||
|---|---|---|---|---|---|---|
| Control | Multiple sclerosis | Neuromyelitis optica | Acute disseminated encephalomyelitis | Herpes simplex encephalitis | Alzheimer's disease | |
| Sex | F: 3, M: 5 | F: 11, M: 6 | F: 3, M: 0 | F: 3, M: 4 | F: 0, M :3 | F: 1, M: 3 |
| Age (years) | 63.0 (52–77) | 53.4 (25–76) | 39.3 (18–64) | 25.4 (10–39) | 36.0 (18–47) | 76.3 (73–80) |
| Duration of disease | N/A | 8.2 years (4 months–32 years) | 8.0 years (1–15 years) | 7.1 days (2–14 days) | 5.0 days | 10.8 years (8–15 years) |
Mean values with range in parentheses are given for age and duration of disease.
Demyelination
The topography of myelination in the olfactory bulb/tract was complex. In non‐neurologic controls, olfactory gray matter structures typically demonstrated a fine reticulated meshwork of myelin save for olfactory glomeruli, which consistently lacked myelin. In contrast, myelin in olfactory white matter structures was more densely packed and linearly organized (Figure 2). Only areas completely devoid of myelin (not including olfactory glomeruli that are not myelinated) were classified as demyelinated.
Figure 2.
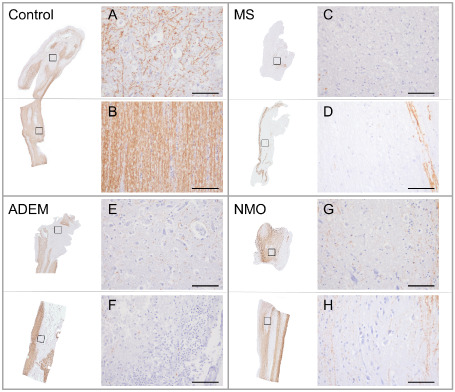
Olfactory bulb and tract demyelination in demyelinating diseases compared to non‐neurologic controls. A–H. Olfactory bulbs and tracts stained for myelin (proteolipid protein). High power views of boxed areas represent gray matter (A, C, E, G) and white matter (B, D, F, H) regions for each disease category. A. In controls, olfactory gray matter structures (external and internal plexiform layers, mitral and granule cell layers, anterior olfactory nuclei) typically demonstrated a fine reticulated meshwork of myelin save for olfactory glomeruli, which consistently lacked myelin (not shown). B. In contrast, myelin in olfactory white matter structures (lateral, medial and intermediate striae) was more densely packed and linearly organized. In each of the demyelinating diseases (multiple sclerosis (MS) (C, D), acute disseminated encephalomyelitis (ADEM) (E, F) and neuromyelitis optica (NMO) (G, H), olfactory bulb and tract demyelination affected both gray and white matter regions, the extent of which was consistently most severe in MS. Scale bars represent 100 μm.
Demyelination was found exclusively in cases with demyelinating disease. Demyelinating lesions were found in 12/17 (70.6%) of MS, 2/3 (66.7%) of NMO and 3/7 (42.9%) of ADEM cases. In patients with demyelination, the global mean of demyelination relative to the total sampled olfactory bulb/tract area was 18.7% in MS, 3.0% in NMO and 2.9% in ADEM (Supporting Information Table S3). Demyelination involved both gray and white matter areas. Of the cases with demyelination, active (ie, acute and border active) lesions were found in 0/12 (0%) of MS, 0/2 (0%) of NMO and 3/3 (100%) of ADEM cases. Chronic inactive lesions in olfactory bulb/tract demyelination were found even in MS and NMO cases who died within months of disease onset. In MS, no significant difference in disease duration was observed in cases with or without evidence of olfactory bulb/tract demyelination [with olfactory demyelination: mean 8.5 years, range 4 months–32 years; without olfactory demyelination: (mean 7.0 years, range 2–14 years)]. In contrast, ADEM lesions were significantly more inflammatory with myelin inclusions within macrophages occasionally seen (Figure 3). Aquaporin‐4 was absent within NMO lesions compared to MS and ADEM where it was relatively spared (Supporting Information Figure S1). HSE, AD and non‐neurologic control cases showed no demyelination or aquaporin‐4 loss.
Figure 3.
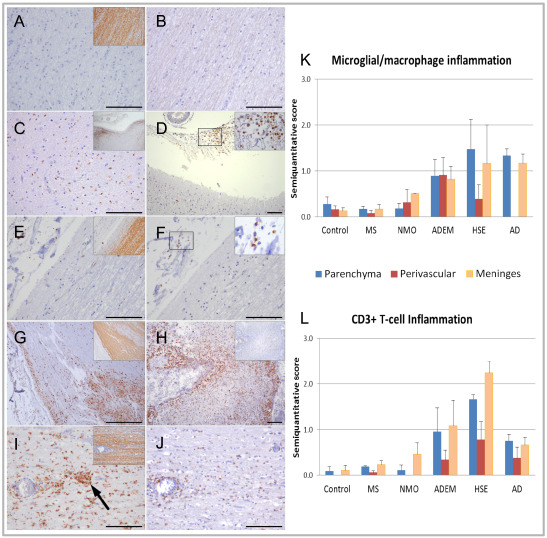
Olfactory bulb and tract inflammation in demyelinating diseases compared to non‐neurologic, neuroinflammatory and neurodegenerative controls. Olfactory structures from control (A, B); multiple sclerosis (MS) (C, D); neuromyelitis optica (NMO) (E, F); acute disseminated encephalomyelitis (ADEM) (G, H); and herpes simplex encephalitis (HSE) (I, J). Panels A, C, E, G and I show immunostaining for microglia/macrophages (PG‐M1) with inset showing myelin‐stained region (proteolipid protein) from which the inflammation‐stained sections are derived. Panels B, D, F, H and J show immunostaining for CD3+ T‐cells in the same regions unless otherwise indicated, with inset in D and F showing high power views of CD3+ T‐cells in region indicated by black box. Control cases demonstrate negligible microglial/macrophage (A) or T‐cell (B) inflammation. Moderately severe microglial/macrophage and T‐cell inflammation is evenly distributed throughout the parenchyma and meninges in MS (C, D) and mild inflammation in NMO (E, F). In ADEM, fulminant microglial/macrophage (G) and T‐cell inflammation (H) is concentrated in the subpial margins of the olfactory bulb and tract parenchyma and meninges, particularly surrounding vessels (panel H—inset showing myelin‐stained section for corresponding region). HSE cases demonstrate severe microglial/macrophage (I) and T‐cell infiltration (J) throughout the parenchyma and meninges, with occasional microglial clusters observed in the former (arrow in panel I), but no demyelination (inset panel I). (K, L) Mean semiquantitative scores of microglial/macrophage (K) and CD3+ T‐cell (L) inflammation of olfactory bulb and tract parenchyma (blue), perivascular area (red) and meninges (orange) in non‐neurologic controls, MS, NMO, ADEM, HSE and Alzheimer's disease (AD). Each of the demyelinating diseases demonstrated increased olfactory bulb and tract inflammation compared to non‐neurologic controls; however, this only reached statistical significance in meningeal macrophage inflammation in ADEM (P = 0.03) and NMO (P = 0.02). HSE and AD showed statistically significantly higher levels of microglial/macrophage and T‐cell inflammation compared to controls (HSE vs. controls): (i) Microglial/macrophage inflammation—parenchyma (P = 0.04), meninges (P = 0.05); (ii) T‐cell inflammation—parenchyma (P = 0.009), perivascular (P = 0.01), meninges (P = 0.01); AD vs. controls: (i) Microglial/macrophage inflammation—parenchyma (P = 0.008), meninges (P = 0.006); (ii) T‐cell inflammation—parenchyma (P = 0.008), perivascular (P = 0.037), meninges (P = 0.028). In the demyelinating diseases, ADEM and NMO showed significantly greater macrophage inflammation compared to MS in the perivascular region (ADEM vs. MS—P = 0.01; NMO vs. MS—P = 0.04) and meninges (ADEM vs. MS—P = 0.004; NMO vs. MS—P = 0.01). HSE and AD showed significantly greater levels of microglial/macrophage and T‐cell inflammation in all compartments compared to MS (data not shown). Error bars represent standard errors of the mean. Significance was measured by Mann–Whitney U‐tests. Scale bars represent 100 μm in all panels except in panel G where the scale bar represents 500 μm.
On evaluating the influence of demographic factors on demyelination, there was a trend for disease duration to correlate with extent of demyelination in ADEM cases only (r = 0.783, P = 0.06). No relationship was observed between age, sex and extent of demyelination in MS, NMO and ADEM.
Inflammation
CD3+ T‐cell inflammation was increased in all disease groups compared to non‐neurologic controls, being most severe in HSE. In all cases, the majority of T‐cells were CD8+. In the demyelinating diseases, T‐cell inflammation was variable between cases but was most extensive in ADEM. Microglial/macrophage infiltration was increased in all disease groups compared to non‐neurologic controls, with HSE cases being most severely affected (Figure 3). In comparison to the demyelinating diseases, AD cases showed higher levels of microglial/macrophage activation. B‐cells were occasionally detected in demyelinating and neuroinflammatory disease groups but were virtually absent in AD and non‐neurologic controls.
The pattern of T‐cell and microglia/macrophage infiltration differed between demyelinating disease groups. In comparison to controls, MS and NMO cases showed only a slightly increased level of T‐cell and microglial/macrophage infiltration. In these cases, inflammation was variable and, where present, was diffuse and evenly distributed throughout the olfactory bulb/tract parenchyma and overlying meninges. Mild perivascular T‐cell inflammation was observed in MS and NMO in levels above controls. More striking inflammation was observed in ADEM where the T‐cell and microglia/macrophage response was moderately severe and primarily concentrated in subpial margins of the parenchyma and adjacent meninges, particularly surrounding vessels as is well recognized (Figure 3) 21.
In HSE, T‐cell and microglia/macrophage inflammation was consistently diffuse and its extent fulminant throughout the parenchyma, with occasional microglial clusters being observed (Figure 3). Striking inflammation was also found within the meninges and to a lesser extent within the perivascular space in HSE. In contrast, AD cases showed moderate levels of diffuse T‐cell and microglial/macrophage inflammation with a paucity of inflammation associated with vessels.
Evaluation of the influence of demographic factors on inflammation showed relationships only in ADEM cases where duration of disease correlated significantly with the extent of CD3+ T‐cell infiltration in the parenchyma (r = 0.938, P = 0.006).
Axonal loss
Axonal loss was demonstrated in all cases (mean semiquantitative axonal loss scores: MS = 1.0; NMO = 0.3; ADEM = 0.7; HSE = 0.8; AD = 1.7; non‐neurologic controls = 0.4) (Supporting Information Table S3). In demyelinating disease, axonal loss was most pronounced in MS and to a lesser extent in ADEM. The extent of axonal loss in NMO was similar to that seen in non‐neurologic controls. HSE showed heightened axonal loss compared to controls despite being significantly younger. AD showed the most striking degree of axonal loss compared to all groups studied. No correlations were found between axonal loss and age, sex and duration of disease in controls and disease groups.
Correlations between demyelination, inflammation and axonal loss in the olfactory bulb/tract
Evaluation of the relationships between demyelination, inflammation and axonal loss was restricted to MS and ADEM cases given the small sample sizes of the other cohorts studied. In MS, the extent of demyelination correlated significantly with CD3+ T‐cell inflammation (parenchyma, r = 0.592, P = 0.01; perivascular, r = 0.566, P = 0.01). CD3+ T‐cell inflammation correlated significantly with microglial/macrophage inflammation (data not shown). The magnitude of axonal loss correlated significantly with the extent of demyelination (r = 0.610, P = 0.009) and CD3+ T‐cell parenchymal inflammation (r = 0.681, P = 0.003). In ADEM, similar relationships between demyelination and CD3+ T‐cell (parenchyma: r = 0.900, P = 0.006; perivascular: r = 0.957, P = 0.001; meningeal: r = 0.935, P = 0.006) and microglial/macrophage inflammation (parenchyma: r = 0.795, P = 0.03; perivascular: r = 0.818, P = 0.02) were found. However, unlike in MS, these did not correlate with axonal loss (data not shown).
Association between olfactory pathology and adjacent cortical pathology
In a limited number of cases, cortical structures of the gyrus rectus adjacent to olfactory bulb/tract were sampled in situ (MS, n = 4; ADEM, n = 2). Pia/arachnoid meningeal layers appeared to be shared between cortex and olfactory bulb/tract. The extent of olfactory bulb/tract demyelination and inflammation mirrored that found in superficial cortical layers where subpial demyelination and inflammation were observed in both MS and ADEM (Figure 4).
The relationship between olfactory bulb/tract and adjacent cortical demyelination was further evaluated in MS and ADEM. A total of 180 lesions from 73 inferior frontal cortical blocks juxtaposed to the analyzed olfactory bulb/tract tissue from 22 cases (MS, n = 15; ADEM, n = 7) and 23 lesions from 27 hippocampal blocks from 12 cases (MS, n = 7; ADEM, n = 5) were studied. Cortical demyelination was significantly more severe in cases demonstrating high levels of olfactory bulb/tract demyelination compared to those with low levels (P = 0.01). Cortical demyelination was not limited to the straight gyrus and medial orbital gyrus; however, given our sampling strategy, these cortical areas were intentionally overrepresented in our dataset given their proximity to the olfactory sulcus. The extent of hippocampal demyelination did not differ between groups with low and high levels of olfactory bulb/tract demyelination (Figure 4).
Discussion
We provide unequivocal evidence that olfactory bulb/tract demyelination in MS is frequent and extensive and report, for the first time, that it commonly occurs in other demyelinating diseases such as NMO and ADEM. Olfactory bulb and tract demyelination can occur early and is highly inflammatory, relates to adjacent meningeal and cortical pathology, and is specific to demyelinating disease.
In our cohort, demyelination of olfactory bulb/tract was common and specific to demyelinating disease. Olfactory bulb/tract demyelination was most extensive in MS where it affected approximately 18.7% of the olfactory structures sampled. The frequency of olfactory bulb/tract demyelination in MS (ie, 70.6%) is similar to other areas of the CNS known to be preferentially affected in the disease such as the optic nerve, corpus callosum, periventricular white matter and cervical spinal cord 7, 25. In NMO, olfactory bulb/tract lesions demonstrated selective loss of aquaporin‐4 reflecting the established role of antibodies directed against this water channel in NMO pathogenesis 29. Unlike in MS and NMO, lesions in the olfactory bulb/tract in ADEM were concentrated around blood vessels and showed signs of active, ongoing demyelination in a pattern similar to what is seen in brain lesions in this disorder (Supporting Information Figure S2) 21. All of our ADEM cases died within days to weeks of onset of neurologic symptoms, indicating that olfactory bulb/tract demyelination is an early feature in this disease. Despite being highly inflammatory, HSE and AD cases were without olfactory bulb/tract demyelination highlighting how olfactory demyelination is not merely a bystander effect of inflammation or neurodegeneration. These findings support the concept that molecular targets central to demyelinating disease pathogenesis are shared between olfactory bulb/tract and other CNS structures, making them vulnerable to similar patterns of tissue injury.
Olfactory bulb/tract inflammation was a common feature in each of the demyelinating diseases evaluated but varied considerably between them. MS and NMO demonstrated low grade, diffuse parenchymal and meningeal inflammation above that seen in non‐neurologic controls, suggesting propagation of an aberrant inflammatory response in the olfactory bulb/tract. Lesions were mostly chronic and devoid of heightened inflammatory activity, likely reflecting long‐standing disease duration. Given that chronic inactive plaques were observed in MS and NMO cases who died within months of disease onset suggests possible early olfactory involvement. In our ADEM cohort, the clinical course was monophasic and time to death was short. ADEM cases showed fulminant macrophage and T‐cell infiltration along the subpial rim of the olfactory bulb/tract parenchyma and adjacent meninges, most notably around blood vessels. In both MS and ADEM, the significant relationship between T‐cell inflammation and demyelination implicates an important role of a cellular‐mediated immune response in the olfactory bulb/tract pathology and, more broadly, CNS demyelinating disease. While the inflammation in ADEM was more severe compared to MS, the extent of demyelination was significantly less than that seen in MS, likely reflecting the typically restricted perivenular distribution of pathology, shorter disease duration and monophasic disease course. The pronounced meningeal inflammation in the acute phase of ADEM and its intimate juxtaposition to both olfactory bulb/tract and cortex suggests that meningeal pathology may affect both structures. This is supported by the fact that meningeal inflammation correlated significantly with the extent of demyelination in the olfactory bulb/tract, which, in turn, appeared to be related to the extent of demyelination in the adjacent inferior frontal cortex but not in more remote hippocampal structures (Figure 4). While the small number of cases with available hippocampal tissue for study may have confounded this latter observation, it is plausible that focal factors (ie, blood—brain/CSF barrier integrity, cerebrospinal fluid (CSF) humoral inflammation) influence hippocampal pathology independent of that seen in more distant olfactory structures. It is intriguing that inferior frontal (eg, rectus gyrus) and medial temporal (eg, inferior temporal gyrus) cortical structures are among the most consistently affected in MS 25, with the inferior frontal lobe being particularly closely related to the olfactory bulb/tract. Our findings support the possible role of meningeal/CSF inflammation in the pathogenesis and propagation of olfactory bulb/tract and adjacent cortical pathology.
Axonal loss is a recognized feature of demyelinating diseases 6, 9, 21. In our series, we observed that axonal loss in the olfactory bulb/tract was most significant in MS and ADEM compared to non‐neurologic controls. It is difficult to compare the extent of axonal loss based on axonal density measures in MS and ADEM. In ADEM, the commonly observed fulminant inflammatory and edematous response both within and outside of lesions could lead to a spurious reduction in axonal density measures compared to MS where inflammation was more chronic and less severe. Chronic olfactory disturbances are an established feature of MS with several studies describing impaired olfactory threshold, discrimination and identification even at disease onset 13, 15, 26. Our findings of substantial axonal loss in the olfactory bulb/tract of MS patients align with this clinical observation. Based on our pathological findings of demyelination and an intense inflammatory reaction in ADEM, we would predict that patients with ADEM might also suffer olfactory loss, at least during the peak of their disease. We did not observe striking axonal loss in NMO despite evidence of demyelination and inflammation above that seen in non‐neurologic controls. The reason for this is not clear and may stem from the small sample size and/or the relatively smaller burden of olfactory bulb/tract pathology. Interestingly, HSE cases showed levels of axonal loss similar to MS despite short disease durations. While this may be secondary to the fulminant inflammatory response against the herpes simplex viral antigen in the olfactory nerve, it may also be contributed to by lysis of infected neurons 16. As expected, AD patients showed the most striking axonal loss compared to the demyelinating diseases possibly reflecting the older age of the AD patient group and the established presence of amyloid and tau pathology in the olfactory bulb, anterior olfactory nucleus and tract 17.
Does pathological involvement of the olfactory bulb/tract in MS, ADEM and NMO have implications for the pathogenesis of these diseases? Several observations suggest that this may be so. The olfactory system connects the external world (olfactory receptor cells in the nasal mucosa) to the CNS (olfactory bulb/tract and adjacent meninges and cortex). Animal models have shown that olfactory structures are vulnerable to invasion by diverse pathogens, some of which can bypass the blood–brain barrier to penetrate deep regions of the brain and induce an inflammatory response to cause disease 27, 32. This has led some to hypothesize that environmental agents enter the brain via this route to cause and/or propagate diseases where anosmia is an early feature, such as Parkinson's and Alzheimer's (ie, nose to brain hypothesis) 10, 11. As anosmia can similarly occur early in MS, our olfactory bulb/tract findings may not only explain this symptom but also may spin a different light onto observations which link chronic sinusitis to MS susceptibility, relapse risk and its demography 20. Whether the olfactory system plays a role in the pathogenesis of CNS demyelinating diseases remains uncertain, but our findings support its further investigation.
In summary, we provide definitive evidence that olfactory bulb and tract demyelination is frequent, can occur early and is highly inflammatory, relates to adjacent cortical pathology and is specific to demyelinating disease.
Authors Contributions
The specific authors were involved in the following aspects of the study: study design (GD, AJ, MME), data gathering (GD, AJ, JG, RY, MHo, MH), data analysis (GD, AJ, JG, RY), vouch for the data and the analysis (GD, AJ, JG, RY, MH, MHo, MME), wrote the manuscript (GD, AJ, MME), decided to publish the paper (GD, AJ, MME).
Supporting information
Figure S1. Aquaporin‐4 staining in non‐neurologic controls and demyelinated lesions.
Figure S2. Characteristic lesions from demyelinating diseases cohort.
Table S1. Antibodies for immunohistochemistry.
Table S2. Clinical and demographic features of cohort.
Table S3. Olfactory bulb and tract demyelination and axonal loss in demyelinating diseases and non‐neurologic, neuroinflammatory, and neurodegenerative controls.
Acknowledgments
GD is supported by the AANF/CMSC John F. Kurtzke Clinician‐Scientist Development Award and a Goodger Scholarship (University of Oxford). GD and MME receive support from the NIHR Biomedical Research Centre, Oxford. RY was supported by a Medical Research Council PhD Studentship. The authors acknowledge the support of the Oxford Brain Bank for supplying tissue samples and clinical information presented in this study.
References
- 1. Carswell R (1838) Pathological Anatomy; Illustrations of the Elementary Forms of Disease. Longman, Orme, Brown, Green and Longman: London. [Google Scholar]
- 2. Charcot J Histologie de la sclerose en plaques. Gaz Hop (Paris) 1868.
- 3. Cruveilhier J (1835) Anatomie pathologique du corps humain; descriptions avec figures lithographiees et coloriees; des diverses alterations morbides dont le corps humain est susceptible. JB Bailliere: Paris. pp. 1835–1842. [Google Scholar]
- 4. Dawson J (1916) The histology of disseminated sclerosis. Trans R Soc Edinburgh 50:517–740. [Google Scholar]
- 5. DeLuca GC, Alterman R, Martin JL, Mittal A, Blundell S, Bird S et al (2013) Casting light on multiple sclerosis heterogeneity: the role of HLA‐DRB1 on spinal cord pathology. Brain 136(Pt 4):1025–1034. [DOI] [PubMed] [Google Scholar]
- 6. DeLuca GC, Ebers GC, Esiri MM (2004) Axonal loss in multiple sclerosis: a pathological survey of the corticospinal and sensory tracts. Brain 127(Pt 5):1009–1018. [DOI] [PubMed] [Google Scholar]
- 7. DeLuca GC, Williams K, Evangelou N, Ebers GC, Esiri MM (2006) The contribution of demyelination to axonal loss in multiple sclerosis. Brain 129(Pt 6):1507–1516. [DOI] [PubMed] [Google Scholar]
- 8. Demarquay G, Ryvlin P, Royet JP (2007) [Olfaction and neurological diseases: a review of the literature]. Rev Neurol (Paris) 163:155–167. [DOI] [PubMed] [Google Scholar]
- 9. Diaz‐Sanchez M, Williams K, DeLuca GC, Esiri MM (2006) Protein co‐expression with axonal injury in multiple sclerosis plaques. Acta Neuropathol 111:289–299. [DOI] [PubMed] [Google Scholar]
- 10. Doty RL (2008) The olfactory vector hypothesis of neurodegenerative disease: is it viable? Ann Neurol 63:7–15. [DOI] [PubMed] [Google Scholar]
- 11. Doty RL (2012) Olfactory dysfunction in Parkinson disease. Nat Rev Neurol 8:329–339. [DOI] [PubMed] [Google Scholar]
- 12. Doty RL, Li C, Mannon LJ, Yousem DM (1997) Olfactory dysfunction in multiple sclerosis. N Engl J Med 336:1918–1919. [DOI] [PubMed] [Google Scholar]
- 13. Doty RL, Li C, Mannon LJ, Yousem DM (1998) Olfactory dysfunction in multiple sclerosis. Relation to plaque load in inferior frontal and temporal lobes. Ann N Y Acad Sci 855:781–786. [DOI] [PubMed] [Google Scholar]
- 14. Doty RL, Li C, Mannon LJ, Yousem DM (1999) Olfactory dysfunction in multiple sclerosis: relation to longitudinal changes in plaque numbers in central olfactory structures. Neurology 53:880–882. [DOI] [PubMed] [Google Scholar]
- 15. Erb K, Bohner G, Harms L, Goektas O, Fleiner F, Dommes E et al (2012) Olfactory function in patients with multiple sclerosis: a diffusion tensor imaging study. J Neurol Sci 316:56–60. [DOI] [PubMed] [Google Scholar]
- 16. Esiri MM (1982) Herpes simplex encephalitis. An immunohistological study of the distribution of viral antigen within the brain. J Neurol Sci 54:209–226. [DOI] [PubMed] [Google Scholar]
- 17. Esiri MM, Wilcock GK (1984) The olfactory bulbs in Alzheimer's disease. J Neurol Neurosurg Psychiatry 47:56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fleiner F, Dahlslett SB, Schmidt F, Harms L, Goektas O (2010) Olfactory and gustatory function in patients with multiple sclerosis. Am J Rhinol Allergy 24:e93–e97. [DOI] [PubMed] [Google Scholar]
- 19. Garcia‐Gonzalez D, Murcia‐Belmonte V, Clemente D, De Castro F (2013) Olfactory system and demyelination. Anat Rec (Hoboken) 296:1424–1434. [DOI] [PubMed] [Google Scholar]
- 20. Gay D, Dick G, Upton G (1986) Multiple sclerosis associated with sinusitis: case‐controlled study in general practice. Lancet 1:815–819. [DOI] [PubMed] [Google Scholar]
- 21. Ghosh N, DeLuca GC, Esiri MM (2004) Evidence of axonal damage in human acute demyelinating diseases. J Neurol Sci 222:29–34. [DOI] [PubMed] [Google Scholar]
- 22. Goektas O, Schmidt F, Bohner G, Erb K, Ludemann L, Dahlslett B et al (2011) Olfactory bulb volume and olfactory function in patients with multiple sclerosis. Rhinology 49:221–226. [DOI] [PubMed] [Google Scholar]
- 23. Gowers WA (1893) Manual of Diseases of the Nervous System, Vol. 2, 2nd edn. J & A Churchill: London. [Google Scholar]
- 24. Hawkes CH, Shephard BC, Kobal G (1997) Assessment of olfaction in multiple sclerosis: evidence of dysfunction by olfactory evoked response and identification tests. J Neurol Neurosurg Psychiatry 63:145–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lassmann H (2011) Review: the architecture of inflammatory demyelinating lesions: implications for studies on pathogenesis. Neuropathol Appl Neurobiol 37:698–710. [DOI] [PubMed] [Google Scholar]
- 26. Lutterotti A, Vedovello M, Reindl M, Ehling R, DiPauli F, Kuenz B et al (2011) Olfactory threshold is impaired in early, active multiple sclerosis. Mult Scler 17:964–969. [DOI] [PubMed] [Google Scholar]
- 27. Okun E, Griffioen KJ, Mattson MP (2011) Toll‐like receptor signaling in neural plasticity and disease. Trends Neurosci 34:269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Popescu BF, Parisi JE, Cabrera‐Gomez JA, Newell K, Mandler RN, Pittock SJ et al (2010) Absence of cortical demyelination in neuromyelitis optica. Neurology 75:2103–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Roemer SF, Parisi JE, Lennon VA, Benarroch EE, Lassmann H, Bruck W et al (2007) Pattern‐specific loss of aquaporin‐4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain 130(Pt 5):1194–1205. [DOI] [PubMed] [Google Scholar]
- 30. Rolet A, Magnin E, Millot JL, Berger E, Vidal C, Sileman G, Rumbach L (2013) Olfactory dysfunction in multiple sclerosis: evidence of a decrease in different aspects of olfactory function. Eur Neurol 69:166–170. [DOI] [PubMed] [Google Scholar]
- 31. Silva AM, Santos E, Moreira I, Bettencourt A, Coutinho E, Goncalves A et al (2012) Olfactory dysfunction in multiple sclerosis: association with secondary progression. Mult Scler 18:616–621. [DOI] [PubMed] [Google Scholar]
- 32. Wada Y, Fujinami RS (1993) Viral infection and dissemination through the olfactory pathway and the limbic system by Theiler's virus. Am J Pathol 143:221–229. [PMC free article] [PubMed] [Google Scholar]
- 33. Zimmerman HM, Netsky MG (1950) The pathology of multiple sclerosis. Res Publ Assoc Res Nerv Ment Dis 28:271–312. [PubMed] [Google Scholar]
- 34. Zivadinov R, Zorzon M, Monti Bragadin L, Pagliaro G, Cazzato G (1999) Olfactory loss in multiple sclerosis. J Neurol Sci 168:127–130. [DOI] [PubMed] [Google Scholar]
- 35. Zorzon M, Ukmar M, Bragadin LM, Zanier F, Antonello RM, Cazzato G, Zivadinov R (2000) Olfactory dysfunction and extent of white matter abnormalities in multiple sclerosis: a clinical and MR study. Mult Scler 6:386–390. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Aquaporin‐4 staining in non‐neurologic controls and demyelinated lesions.
Figure S2. Characteristic lesions from demyelinating diseases cohort.
Table S1. Antibodies for immunohistochemistry.
Table S2. Clinical and demographic features of cohort.
Table S3. Olfactory bulb and tract demyelination and axonal loss in demyelinating diseases and non‐neurologic, neuroinflammatory, and neurodegenerative controls.