

Abstract
Neuroimaging has revealed a range of white matter abnormalities that are common in dementia, some that predict cognitive decline. The abnormalities may result from structural diseases of the cerebral vasculature, such as arteriolosclerosis and amyloid angiopathy, but can also be caused by nonstructural vascular abnormalities (eg, of vascular contractility or permeability), neurovascular instability or extracranial cardiac or vascular disease. Conventional histopathological assessment of the white matter has tended to conflate morphological vascular abnormalities with changes that reflect altered interstitial fluid dynamics or white matter ischemic damage, even though the latter may be of extracranial or nonstructural etiology. However, histopathology is being supplemented by biochemical approaches, including the measurement of proteins involved in the molecular responses to brain ischemia, myelin proteins differentially susceptible to ischemic damage, vessel‐associated proteins that allow rapid measurement of microvessel density, markers of blood–brain barrier dysfunction and axonal injury, and mediators of white matter damage. By combining neuroimaging with histopathology and biochemical analysis, we can provide reproducible, quantitative data on the severity of white matter damage, and information on its etiology and pathogenesis. Together these have the potential to inform and improve treatment, particularly in forms of dementia to which white matter hypoperfusion makes a significant contribution.
Keywords: Alzheimer's disease, cerebrovascular disease, ischemia, molecular markers, vascular dementia, white matter
Introduction
White matter abnormalities are demonstrable in vivo by computed X‐ray tomography (CT), magnetic resonance imaging (MRI) or positron emission tomography (PET) in most patients with dementia and many of those with mild cognitive impairment (MCI). The abnormalities include infarcts and ischemic leukoencephalopathy, white matter hyperintensities, enlarged perivascular spaces, microbleeds 13, 14, 15, 90, 101, 109, 111, changes in white matter “integrity” on brain diffusion tensor imaging (DTI) 81, 93, 101, 109 and reductions in white matter perfusion and glucose utilization 12, 40, 53. Some of the abnormalities are likely to be secondary to degenerative changes in the cerebral cortex and deep gray matter structures, but others reflect primary damage to the white matter. These include white matter infarcts and regions of ischemic damage; foci of hemorrhage; damage caused by impaired drainage of interstitial fluid, or leakage of neurotoxic molecules from the bloodstream as a result of blood–brain barrier damage; and in several types of dementia, primary degenerative changes in the white matter itself. Post‐mortem histopathological studies have confirmed the high prevalence of structural white matter abnormalities in dementia, the main types of lesion being microinfarcts, markedly enlarged perivascular spaces and regions of non‐cavitating ischemic damage (ischemic leukoencephalopathy) 16, 34, 36, 52. These abnormalities are particularly common and pronounced in vascular dementia (VaD) but are also seen significantly more often in Alzheimer's disease (AD) than control brains 34, 35, 52. In 60% of brains from patients with AD, the deep white matter was reported to show diffuse rarefaction and gliosis 15, thought to reflect ischemic damage 32, 33.
Clinical significance of white matter damage in dementia
White matter ischemic lesions are associated with impaired cognition 37, 96, 98 and are a significant contributor to cognitive decline in AD 37, 51, 96, 98, Parkinson's disease (PD) and dementia with Lewy bodies (DLB) 24, 25, 45, 60. Their contribution to cognitive decline in patients with AD is most significant in the early stages of disease 26; as the disease progresses, the contribution of ischemic lesions to cognitive decline is overwhelmed by the impact of neurodegenerative pathology 26, 34. White matter abnormalities are demonstrable by DTI even before detectable cognitive decline 42, 43, but the precise pathological substrate of these imaging abnormalities is unclear.
In early dementia, cognitive decline correlates with reduced white matter perfusion and glucose utilization 48, 89. In a prospective study of 7983 people aged 55 years and older, 1730 participants whose cognitive function had been monitored over a mean period of 6.5 years had blood flow velocity in the middle cerebral artery measured by transcranial Doppler ultrasonography 89. Those participants with higher blood flow velocity were significantly less likely to show cognitive decline, an association that persisted after adjustment for age, gender and MRI evidence of vascular brain disease (periventricular and subcortical white matter lesions and brain infarcts).
Causes of white matter damage in dementia
The white matter can be damaged by several processes and some of those processes may have multiple possible etiologies. In the context of dementia, the dominant pathogenic process is hypoperfusion. This is often caused by small vessel disease affecting the cerebral vasculature. Arteriolosclerosis and cerebral amyloid angiopathy (CAA) are the main contributors (Figure 1). However, intracranial (and even intracerebral) atherosclerosis, extracranial cardiovascular disease (eg, atrial fibrillation, aortic valve stenosis, atheromatous stenosis of the carotid artery), orthostatic hypotension and carotid sinus hypersensitivity (neurovascular instability) may all cause or contribute to white matter damage 5, 6, 55, 58, 59, 74, 88. Other factors may include impaired drainage of interstitial fluid (see accompanying paper by Weller et al in this mini‐symposium), systemic or local inflammatory processes 31, 84, 92, extravasation of red blood cells leading to the release of iron and possible oxidative damage, and breakdown of the blood–brain barrier as a result of pericyte loss 10 and increased bradykinin production 4 causing leakage of potentially toxic molecules such as thrombin 10, 21, 65 and plasmin 23. The anatomical distribution of white matter abnormalities that can be demonstrated by DTI in MCI and early dementia suggests that some of the abnormalities, for example, in the parahippocampal white matter 109 and fornices 81, are secondary to degenerative changes in the neurons from which the axons in the white matter originate and probably reflect axonal atrophy and degeneration. However, this presumptive pathogenic sequence is not necessarily correct. In their interpretation of DTI changes in the white matter in PD and DLB, Hattori et al 45 proposed the converse: that imaging abnormalities in the parietal and occipital white matter were caused by white matter hypoperfusion that was responsible for cognitive impairment in PD and led to the atrophy of cortical gray matter. In AD, DTI abnormalities are demonstrable at a preclinical stage. In a cohort of 139 people enriched for AD risk factors but not cognitively impaired, DTI revealed higher fractional anisotropy in the cingulum adjacent to the corpus callosum, hippocampal cingulum and lateral fornix in participants who were amyloid positive on PET with [C‐11] Pittsburgh Compound B than in those who were amyloid negative 83.
Figure 1.
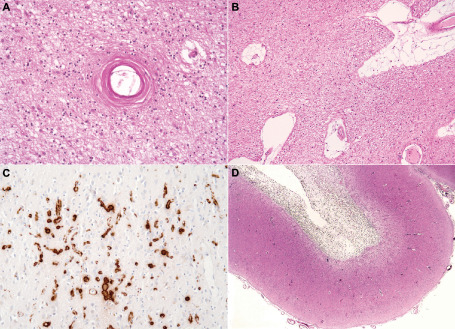
Structural disease of small cerebral blood vessels in dementia. A. Arteriolosclerosis in the parietal white matter. The white matter is rarefied and gliotic. B. In another brain with rarefied, gliotic white matter, perivascular spaces are markedly enlarged. C. The overlying cerebral cortex shows severe capillary cerebral amyloid angiopathy (CAA), demonstrated by immunohistochemistry for Aβ42. D. Old hematoma cavity in the subcortical white matter in association with arteriolar CAA.
Most of the scientific literature on abnormalities of the cerebral vasculature in dementia has focused on structural changes to vessel walls, for example, collagenous thickening, deposition of amyloid, loss of smooth muscle cells or loss of pericytes. However, blood flow and blood–brain barrier function are also affected by alterations in vascular contractility and permeability that are not caused by structural changes but are nonetheless likely to contribute to ischemic damage and edema. Aβ peptides were shown to reduce cerebral blood flow, enhance vasoconstriction and impair both functional hyperemia and cerebral autoregulation in mice transgenic for mutant human amyloid‐β precursor protein (hAPP) 71, 72, 73. The level of the vasoconstrictor endothelin‐1 (ET‐1) is significantly elevated in the cerebral cortex in AD 76, in keeping with the increased synthesis of endothelin‐converting enzyme 2 (ECE‐2) 75 and increased activity of ECE‐1 77. Aβ is, like big‐ET‐1 (the biologically inactive precursor of ET‐1), a substrate of ECE‐1 and ‐2 28, 29, 30, both of which can be upregulated in vitro by Aβ peptides: ECE‐1 by exposure of endothelial cells to Aβ40, and ECE‐2 by exposure of neuroblastoma cells to Aβ42 75, 77. Upregulation of ECEs and elevated ET‐1 production in the cerebral cortex in AD may be unfortunate side effects of the accumulation of excessive substrate in the form of Aβ.
The production of another vasoconstrictor is also increased in the cerebral cortex in AD: angiotensin II, which is converted from its inactive precursor angiotensin I by the action of angiotensin‐converting enzyme (ACE) 68. The level and activity of ACE were significantly higher in frontal cortex in AD than control brains and increased with progression of disease as marked by Braak tangle stage 68. ACE level and activity were also upregulated in vitro in neuroblastoma cells when they were exposed to aggregated Aβ42. The upregulation of ACE may also counteract the upregulation of plasma kallikrein in AD 4; plasma kallikrein catalyses the production of bradykinin, which causes arterial and venous dilatation and increased permeability but is cleaved and inactivated by ACE 103. Although neither ET‐1 nor ACE is increased in the white matter in AD 7, reduced perfusion of the white matter in AD may result in part from angiotensin II‐ and ET‐1‐mediated constriction of the perforating arterioles that traverse the cortex but supply blood to the cerebral white matter.
Cerebral hypoperfusion and neurodegeneration
A range of experimental evidence suggests that cerebral hypoperfusion is likely to exacerbate some of the neurodegenerative disease processes in patients with dementia. Most of the evidence relates to AD, a disease in which the initiating abnormality is thought to be the cerebral accumulation of Aβ peptide (particularly Aβ1–42) produced by the actions of β‐ and γ‐secretases on amyloid‐β precursor protein (APP). Transient focal cerebral ischemia caused overexpression of APP mRNA 94, and hypoperfusion and neuronal hypoxia upregulated BACE1 mRNA and protein, increased β‐secretase activity and the production of Aβ peptide 44, 61, 112, 121, 122. Ischemia also induced expression of PSEN1 mRNA in the gerbil hippocampus 100 and hypoxia enhanced the expression of APH‐1a, another component of γ‐secretase complex 61. Conversely, exposure of neuroblastoma cells and primary neuronal cultures to hypoxia and oxidative stress reduced the expression and activity of neprilysin 41, a major Aβ‐degrading enzyme 18, 66, 67, 104, 105. Other studies showed that oxidative modification of neprilysin, leading to the formation of 4‐hydroxynonenal adducts, reduced Aβ catalytic activity and caused Aβ accumulation 107, 108. Oxidative modification also inactivated insulin‐degrading enzyme, another contributor to Aβ degradation 95. Lastly, transient cerebral ischemia caused hyperphosphorylation of tau 113, 114; this was significantly reduced when cyclin‐dependent kinases were inhibited with roscovitine.
In studies of potential effects of cerebral hypoperfusion on the development of Lewy body pathology, transient cerebral ischemia increased the amount of α‐synuclein that could be detected immunohistochemically in the gerbil hippocampus 49, 117. Acute hypoxia increased the level of α‐synuclein in mouse cerebral cortex 118, and glucose deprivation caused the formation of α‐synuclein inclusions in neuroblastoma cells in vitro 11. Unal‐Cevik et al 106 found that transient middle cerebral artery occlusion induced oligomerization of wild‐type α‐synuclein in mouse brain. When we exposed neuroblastoma cells overexpressing human wild‐type α‐synuclein to either glucose deprivation or combined oxygen and glucose deprivation (simulating ischemia), there were significant increases in the levels of total α‐synuclein and α‐synuclein phosphorylated at serine 129 [a modification reported to promote α‐synuclein aggregation and neurotoxicity 20, 22 ] 69.
There is some evidence from studies in patients and from post‐mortem examination of human brain tissue that cerebral hypoperfusion increases AD pathology. White matter hyperinsensities were significantly associated with cortical atrophy in patients with clinically probable AD 17. Hypoxia due to cardiac arrest was found to increase serum amyloid β levels in humans 120. Yuan et al 119 reported that patients with probable severe AD (ie, having a clinical dementia rating score of 3) had a significantly greater prevalence of severe atheromatous stenosis of intracranial or extracranial arteries than did patients with “early” or mild AD (clinical dementia rating score of 0.5 or 1), and Hofman et al 46 found that severe atherosclerosis increased the odds ratio for AD by about threefold (95% confidence interval 1.5–6.0), although this was partly attributable to an association between APOE ε4 and both AD and atherosclerosis. In several post‐mortem studies, the severity of cerebrovascular atherosclerosis correlated with the burden of AD pathology 9, 47, 56, 86, 87, 88, 116. However, other post‐mortem studies did not find an association between the severity of AD pathology and the presence of vascular clinical risk factors, cerebral infarcts or other vascular pathological abnormalities 27, 85, 91.
The association between cerebral hypoperfusion and neurodegenerative pathology in patients with dementia is likely to be bidirectional. As noted above (see Causes of white matter damage in dementia), Aβ upregulates the production of several vasoconstrictors within the brain. It also has anti‐angiogenic activity 79, binds to (and possibly sequesters) vascular endothelial growth factor (VEGF) 115 and interacts with the extracellular domain of VEGF receptor 2 and interferes with VEGF‐mediated signaling 78. Less information is available in relation to other neurodegenerative diseases that cause dementia. However, in post‐mortem samples of occipital cortex from patients DLB, we showed that von Willebrand factor (factor VIII‐related antigen), an excellent surrogate marker of capillary density 7, was reduced and that this was associated with a lower concentration of VEGF 69. VEGF was also reduced in SH‐SY5Y neuroblastoma cells overexpressing wild‐type human α‐synuclein, the extent of reduction correlating closely with the level of α‐synuclein, suggesting that VEGF deficiency secondary to the accumulation of α‐synuclein may lead to reduced microvessel density in this disease.
Current approaches to assessment of white matter damage (and their limitations)
As noted in the Introduction, CT and MRI have proven invaluable in demonstrating ischemic and other types of white matter abnormality in vivo. Newer MRI techniques such as DTI are highly sensitive to cellular and subcellular changes in the white matter that alter the separation and orientation of membranes and the diffusivity of water molecules in different planes 3, 50, 64. These techniques allow the identification of distinct patterns of white matter damage in different types of dementia, even in preclinical stages 80, 81, 93, 101, 109. Neuroimaging is less useful in determining the process responsible for the damage (eg, ischemia, leakage of neurotoxic molecules from the bloodstream, impaired drainage of interstitial fluid) or the underlying etiology (eg, whether ischemic changes result from arteriolosclerosis, CAA, cardiac dysrhythmias or neurovascular instability and postural hypotension), although intravenous administration of gadolinium can be used to assess the integrity of the blood–brain barrier 110. Imaging methods for measuring cerebral blood flow and glucose utilization may add information, particularly if there are regional reductions in blood flow 12, 40, 53, 57, but do not generally enable a pathogenic reduction in blood flow leading to tissue hypoxia to be distinguished from a reduction secondary to tissue damage and lower metabolic demand.
Conventional histopathological approaches to assessing white matter damage have focused mainly on ischemic changes and foci of hemorrhage and have tended to conflate changes that affect vessel walls (atherosclerosis, arteriosclerosis, arteriolosclerosis, CAA) with changes that reflect altered interstitial fluid dynamics or white matter ischemic damage, even though the latter may sometimes be of extracranial etiology (Figure 2) or a reflection of nonstructural vascular abnormalities (eg, of vascular contractility or permeability). In histopathological studies, the severity of white matter damage or of vascular abnormalities has generally been specified (rather than measured) with reference to arbitrary, semi‐quantitative or categorical scales. These studies have provided some insights into the pathogenesis of the damage [eg, its relationship to arteriolosclerosis, severe CAA or to Aβ parenchymal load 19, 26, 62 ] but have been limited by subjectivity, lack of specificity and relative insensitivity. Abnormalities in the subcortical and periventricular white matter can also be demonstrated in formalin‐fixed post‐mortem brain tissue by T2‐weighted MRI, although post‐mortem MRI was reported to be generally less sensitive than histopathological examination for detecting abnormalities 38, 63.
Figure 2.
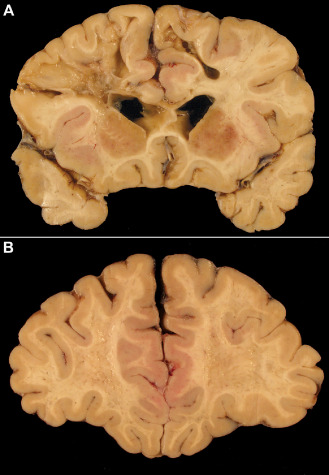
Ischemic white matter damage in a dementia patient with cardiac valvular disease and recurrent episodes of bradyarrhythmia. A. A coronal slice through the frontal and temporal lobes reveals multiple cavitated infarcts and foci of softening and gray discoloration. There is only mild atherosclerosis. B. In a more anterior slice through the same brain, the white matter appears pitted, with numerous confluent foci of gray discoloration.
Molecular pathological approaches
Another approach to assessing ischemic white matter damage in post‐mortem brain tissue is the measurement of gene transcripts and proteins involved in the molecular responses to brain ischemia. Hypoxia‐inducible factor (HIF) α subunits are the principal sensors of tissue hypoxia 2, 54. Under normal conditions these constitutively expressed proteins undergo proline hydroxylation, which initiates their interaction with E3 ubiquitin ligase and consequent degradation. Hypoxia inhibits hydroxylation of the proline residues and results in persistence of the HIF α subunits, which associate with HIF β to form transcriptionally active HIF‐1 and HIF‐2 heterodimers. In vitro studies suggest that Aβ can also activate HIF‐1 directly 97. Under conditions of chronic or intermittent hypoxia, there is an increase in HIF‐1α transcription 70, 82. Tissue hypoxia also causes upregulation of a large number of other genes. The transcription of some of these, such as VEGF and heme‐oxygenase 1, is upregulated by HIFs. Others, such as neuroglobin 99, are upregulated by hypoxia independently of HIFs. Fernando et al 39 demonstrated an increase in immunohistochemical labeling of white matter for HIF‐1α and HIF‐2α and neuroglobin in deep subcortical white matter lesions in the elderly. In more recent studies, we demonstrated the post‐mortem stability of VEGF in human brain tissue, for up to 72 h at 4°C or even room temperature 69, and used a sandwich enzyme‐linked immunosorbent assay (ELISA) to measure the concentration of the protein in both gray and white matter 7, 69, 102. As noted below, there was a highly significant correlation between VEGF concentration in the white matter and other measures of white matter ischemia.
A further index of the adequacy of ante‐mortem perfusion of the white matter is the ratio of the “distal” periaxonal oligodendrocyte protein, myelin‐associated glycoprotein (MAG) that is highly susceptible to white matter ischaemia, to more “proximal” myelin proteins such as proteolipid protein 1 (PLP1) and myelin basic protein (MBP), which are relatively resistant 1, 7, 8. Unfortunately, over a period of several hours, MBP undergoes post‐mortem degradation, even when stored at 4°C 8. However, both PLP1 and MAG are relatively stable for at least 72 h, and comparison of the level of these two proteins is a useful means of detecting and quantifying the severity of ante‐mortem hypoperfusion. We found that both MAG concentration and the MAG : PLP1 ratio showed highly significantly negative correlation with semi‐quantitative scores of the severity of small vessel disease, in a Bristol cohort (n = 99) and an independently scored Oxford cohort (n = 77) 8 (Figure 3). In a subsequent study, we demonstrated that as the MAG : PLP1 ratio declined, the concentration of VEGF in the white matter increased 7 (Figure 4). The highly significant correlation between these entirely distinct measures of white matter hypoperfusion provides reassurance as to their validity, particularly as the findings were reproduced in two independent cohorts. A further indicator of white matter hypoperfusion was a decline in the concentration of the vasoconstrictor ET‐1, presumably as a protective response to reduce vasoconstriction, increase blood flow and minimize the risk of ischemic damage. In VaD, however, there was a trend toward increased ET‐1, raising the possibility that ET1‐mediated vasoconstriction may contribute to white matter hypoperfusion in this disease 7.
Figure 3.
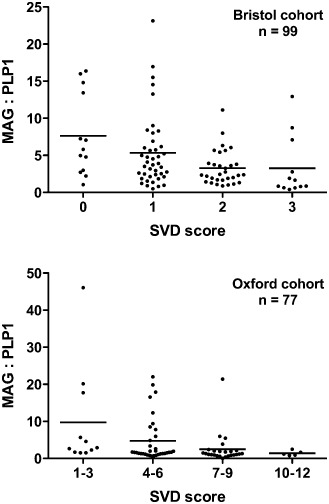
Myelin‐associated glycoprotein (MAG) : proteolipid protein 1 (PLP1) ratio as a measure of the severity of chronic hypoperfusion of the cerebral white matter. In a study of two independent post‐mortem cohorts from Bristol and Oxford, MAG : PLP1 declined significantly with increasing severity of arteriolosclerotic small vessel disease (SVD) (Spearman's test: Bristol P = 0.0003, Oxford P = 0.0009). Note that severity of SVD was scored on a 4‐point scale in the Bristol cohort and on a 12‐point scale 96 in the Oxford cohort. Reproduced with permission from 8.
Figure 4.
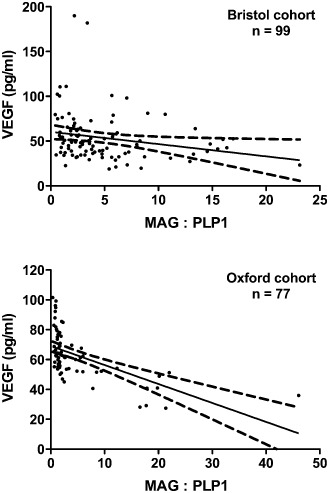
Correlation between two independent measures of white matter hypoperfusion. In both the Bristol and Oxford cohorts, vascular endothelial growth factor (VEGF) concentration in the white matter increased significantly as the myelin‐associated glycoprotein (MAG) : proteolipid protein 1 (PLP1) ratio declined (Spearman's test: Bristol P = 0.0015, Oxford P < 0.0001). Reproduced with permission from 7.
A biochemical approach can also be used to assess the microvascular density in brain tissue. We found good correlation between computer‐assisted morphometric measurements of vessel density (assessed immunohistochemically in paraffin sections) and the concentration of the endothelial marker, von Willebrand factor, in both cerebral cortex and white matter 7, 69. Computer‐assisted morphometry of microvessel density in immunolabeled paraffin sections is excellent for identification of structural abnormalities of the blood vessels but has several limitations. It is relatively labor intensive and time consuming, more affected by sampling variation than are measurements on tissue homogenates (which yield mean values for a much larger volume of tissue), and does not allow direct comparison of microvessel density and biochemical measurements such as VEGF or MAG : PLP1 in the same samples. We found a significant positive correlation between von Willebrand factor level and VEGF concentration in the white matter 7, as might be expected given the pro‐angiogenic actions of VEGF: that is, part of the response to hypoperfusion includes a HIF‐mediated increase in VEGF that leads to an increase in vessel density. Other potential biochemical approaches to the post‐mortem assessment of white matter damage and its pathogenesis include measurement of serum proteins such as albumin to quantify blood–brain barrier dysfunction; of phosphorylated and non‐phosphorylated neurofilament proteins to assess axonal injury; and of a range of potential mediators of damage to oligodendrocytes, myelin, axons and blood vessels.
Conclusions
An ideal method for assessing white matter hypoperfusion and post‐mortem damage would be sensitive and specific, and provide reproducible, quantitative data on severity, as well as information on etiology and pathogenesis. No single method meets all of these objectives. However, by combining in vivo neuroimaging data with post‐mortem histopathology and biochemical analysis of a range of molecular markers, we can now meet most of these objectives and have the potential to make detailed assessment of white matter damage in dementia, to gain insight into physiological and pathological processes involved in the regulation of cerebral perfusion in the human brain and to identify an expanded range of pathogenic processes that contribute to cognitive decline.
Acknowledgments
This work was supported by grants from Alzheimer's Research UK (ARUK) and the Medical Research Council (MRC). The South West Dementia Brain Bank is funded by the MRC, BRACE (Bristol Research into Alzheimer's and Care of the Elderly) and ABBUK (Alzheimer's Brain Bank UK, supporting Brains for Dementia Research).
References
- 1. Aboul‐Enein F, Rauschka H, Kornek B, Stadelmann C, Stefferl A, Bruck W et al (2003) Preferential loss of myelin‐associated glycoprotein reflects hypoxia‐like white matter damage in stroke and inflammatory brain diseases. J Neuropathol Exp Neurol 62:25–33. [DOI] [PubMed] [Google Scholar]
- 2. Acker T, Acker H (2004) Cellular oxygen sensing need in CNS function: physiological and pathological implications. J Exp Biol 207:3171–3188. [DOI] [PubMed] [Google Scholar]
- 3. Alexander AL, Lee JE, Lazar M, Field AS (2007) Diffusion tensor imaging of the brain. Neurotherapeutics 4:316–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ashby EL, Love S, Kehoe PG (2012) Assessment of activation of the plasma kallikrein‐kinin system in frontal and temporal cortex in Alzheimer's disease and vascular dementia. Neurobiol Aging 33:1345–1355. [DOI] [PubMed] [Google Scholar]
- 5. Ballard C, O'Brien J, Barber B, Scheltens P, Shaw F, McKeith I, Kenny RA (2000) Neurocardiovascular instability, hypotensive episodes, and MRI lesions in neurodegenerative dementia. Ann N Y Acad Sci 903:442–445. [DOI] [PubMed] [Google Scholar]
- 6. Ballard C, Shaw F, McKeith I, Kenny R (1998) High prevalence of neurovascular instability in neurodegenerative dementias. Neurology 51:1760–1762. [DOI] [PubMed] [Google Scholar]
- 7. Barker R, Ashby EL, Wellington D, Barrow VM, Palmer JC, Kehoe PG et al (2014) Pathophysiology of white matter perfusion in Alzheimer's disease and vascular dementia. Brain 137:1524–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barker R, Wellington D, Esiri MM, Love S (2013) Assessing white matter ischemic damage in dementia patients by measurement of myelin proteins. J Cereb Blood Flow Metab 33:1050–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Beach TG, Wilson JR, Sue LI, Newell A, Poston M, Cisneros R et al (2007) Circle of Willis atherosclerosis: association with Alzheimer's disease, neuritic plaques and neurofibrillary tangles. Acta Neuropathol 113:13–21. [DOI] [PubMed] [Google Scholar]
- 10. Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV (2010) Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68:409–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bellucci A, Collo G, Sarnico I, Battistin L, Missale C, Spano P (2008) α‐synuclein aggregation and cell death triggered by energy deprivation and dopamine overload are counteracted by D2/D3 receptor activation. J Neurochem 106:560–577. [DOI] [PubMed] [Google Scholar]
- 12. Borroni B, Perani D, Broli M, Colciaghi F, Garibotto V, Paghera B et al (2005) Pre‐clinical diagnosis of Alzheimer disease combining platelet amyloid precursor protein ratio and rCBF spect analysis. J Neurol 252:1359–1362. [DOI] [PubMed] [Google Scholar]
- 13. Bozzali M, Falini A, Franceschi M, Cercignani M, Zuffi M, Scotti G et al (2002) White matter damage in Alzheimer's disease assessed in vivo using diffusion tensor magnetic resonance imaging. J Neurol Neurosurg Psychiatry 72:742–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brun A, Englund E (1986) Brain changes in dementia of Alzheimer's type relevant to new imaging diagnostic methods. Prog Neuropsychopharmacol Biol Psychiatry 10:297–308. [DOI] [PubMed] [Google Scholar]
- 15. Brun A, Englund E (1986) A white matter disorder in dementia of the Alzheimer type: a pathoanatomical study. Ann Neurol 19:253–262. [DOI] [PubMed] [Google Scholar]
- 16. Brundel M, de Bresser J, van Dillen JJ, Kappelle LJ, Biessels GJ (2012) Cerebral microinfarcts: a systematic review of neuropathological studies. J Cereb Blood Flow Metab 32:425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Capizzano AA, Acion L, Bekinschtein T, Furman M, Gomila H, Martinez A et al (2004) White matter hyperintensities are significantly associated with cortical atrophy in Alzheimer's disease. J Neurol Neurosurg Psychiatry 75:822–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carson JA, Turner AJ (2002) β‐amyloid catabolism: roles for neprilysin (NEP) and other metallopeptidases? J Neurochem 81:1–8. [DOI] [PubMed] [Google Scholar]
- 19. Chalmers K, Wilcock G, Love S (2005) Contributors to white matter damage in the frontal lobe in Alzheimer's disease. Neuropathol Appl Neurobiol 31:623–631. [DOI] [PubMed] [Google Scholar]
- 20. Chau KY, Ching HL, Schapira AH, Cooper JM (2009) Relationship between α synuclein phosphorylation, proteasomal inhibition and cell death: relevance to Parkinson's disease pathogenesis. J Neurochem 110:1005–1013. [DOI] [PubMed] [Google Scholar]
- 21. Chen B, Cheng Q, Yang K, Lyden PD (2010) Thrombin mediates severe neurovascular injury during ischemia. Stroke 41:2348–2352. [DOI] [PubMed] [Google Scholar]
- 22. Chen L, Feany MB (2005) α‐synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci 8:657–663. [DOI] [PubMed] [Google Scholar]
- 23. Chen ZL, Strickland S (1997) Neuronal death in the hippocampus is promoted by plasmin‐catalyzed degradation of laminin. Cell 91:917–925. [DOI] [PubMed] [Google Scholar]
- 24. Choi SA, Evidente VG, Caviness JN (2010) Comparing cerebral white matter lesion burdens between Parkinson's disease with and without dementia. J Mov Disord 3:6–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Choi SA, Evidente VG, Caviness JN, Shill HA, Sabbagh MN, Connor DJ et al (2010) Are there differences in cerebral white matter lesion burdens between Parkinson's disease patients with or without dementia? Acta Neuropathol 119:147–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chui HC, Zarow C, Mack WJ, Ellis WG, Zheng L, Jagust WJ et al (2006) Cognitive impact of subcortical vascular and Alzheimer's disease pathology. Ann Neurol 60:677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chui HC, Zheng L, Reed BR, Vinters HV, Mack WJ (2012) Vascular risk factors and Alzheimer's disease: are these risk factors for plaques and tangles or for concomitant vascular pathology that increases the likelihood of dementia? An evidence‐based review. Alzheimers Res Ther 4:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eckman EA, Adams SK, Troendle FJ, Stodola BA, Kahn MA, Fauq AH et al (2006) Regulation of steady‐state β‐amyloid levels in the brain by neprilysin and endothelin‐converting enzyme but not angiotensin‐converting enzyme. J Biol Chem 281:30471–30478. [DOI] [PubMed] [Google Scholar]
- 29. Eckman EA, Reed DK, Eckman CB (2001) Degradation of the Alzheimer's amyloid β peptide by endothelin‐converting enzyme. J Biol Chem 276:24540–24548. [DOI] [PubMed] [Google Scholar]
- 30. Eckman EA, Watson M, Marlow L, Sambamurti K, Eckman CB (2003) Alzheimer's disease β‐amyloid peptide is increased in mice deficient in endothelin‐converting enzyme. J Biol Chem 278:2081–2084. [DOI] [PubMed] [Google Scholar]
- 31. Eng JA, Frosch MP, Choi K, Rebeck GW, Greenberg SM (2004) Clinical manifestations of cerebral amyloid angiopathy‐related inflammation. Ann Neurol 55:250–256. [DOI] [PubMed] [Google Scholar]
- 32. Englund E, Brun A (1990) White matter changes in dementia of Alzheimer's type: the difference in vulnerability between cell compartments. Histopathology 16:433–439. [DOI] [PubMed] [Google Scholar]
- 33. Englund E, Brun A, Alling C (1988) White matter changes in dementia of Alzheimer's type. Biochemical and neuropathological correlates. Brain 111:1425–1439. [DOI] [PubMed] [Google Scholar]
- 34. Esiri MM, Joachim C, Sloan C, Christie S, Agacinski G, Bridges LR et al (2014) Cerebral subcortical small vessel disease in subjects with pathologically confirmed Alzheimer disease: a clinicopathologic study in the Oxford Project to Investigate Memory and Ageing (OPTIMA). Alzheimer Dis Assoc Disord 28:30–35. [DOI] [PubMed] [Google Scholar]
- 35. Esiri MM, Nagy Z, Smith MZ, Barnetson L, Smith AD (1999) Cerebrovascular disease and threshold for dementia in the early stages of Alzheimer's disease. Lancet 354:919–920. [DOI] [PubMed] [Google Scholar]
- 36. Esiri MM, Wilcock GK, Morris JH (1997) Neuropathological assessment of the lesions of significance in vascular dementia. J Neurol Neurosurg Psychiatry 63:749–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fernando MS, Ince PG, Function MRCC, Ageing Neuropathology Study Group (2004) Vascular pathologies and cognition in a population‐based cohort of elderly people. J Neurol Sci 226:13–17. [DOI] [PubMed] [Google Scholar]
- 38. Fernando MS, O'Brien JT, Perry RH, English P, Forster G, McMeekin W et al (2004) Comparison of the pathology of cerebral white matter with post‐mortem magnetic resonance imaging (MRI) in the elderly brain. Neuropathol Appl Neurobiol 30:385–395. [DOI] [PubMed] [Google Scholar]
- 39. Fernando MS, Simpson JE, Matthews F, Brayne C, Lewis CE, Barber R et al (2006) White matter lesions in an unselected cohort of the elderly: molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 37:1391–1398. [DOI] [PubMed] [Google Scholar]
- 40. Firbank MJ, Colloby SJ, Burn DJ, McKeith IG, O'Brien JT (2003) Regional cerebral blood flow in Parkinson's disease with and without dementia. Neuroimage 20:1309–1319. [DOI] [PubMed] [Google Scholar]
- 41. Fisk L, Nalivaeva NN, Boyle JP, Peers CS, Turner AJ (2007) Effects of hypoxia and oxidative stress on expression of neprilysin in human neuroblastoma cells and rat cortical neurones and astrocytes. Neurochem Res 32:1741–1748. [DOI] [PubMed] [Google Scholar]
- 42. Gold BT, Jiang Y, Powell DK, Smith CD (2012) Multimodal imaging evidence for axonal and myelin deterioration in amnestic mild cognitive impairment. J Alzheimers Dis 31(Suppl. 3):S19–S31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gold BT, Johnson NF, Powell DK, Smith CD (2012) White matter integrity and vulnerability to Alzheimer's disease: preliminary findings and future directions. Biochim Biophys Acta 1822:416–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guglielmotto M, Aragno M, Autelli R, Giliberto L, Novo E, Colombatto S et al (2009) The up‐regulation of BACE1 mediated by hypoxia and ischemic injury: role of oxidative stress and HIF1α. J Neurochem 108:1045–1056. [DOI] [PubMed] [Google Scholar]
- 45. Hattori T, Orimo S, Aoki S, Ito K, Abe O, Amano A et al (2012) Cognitive status correlates with white matter alteration in Parkinson's disease. Hum Brain Mapp 33:727–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hofman A, Ott A, Breteler MM, Bots ML, Slooter AJ, van Harskamp F et al (1997) Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer's disease in the Rotterdam Study. Lancet 349:151–154. [DOI] [PubMed] [Google Scholar]
- 47. Honig LS, Kukull W, Mayeux R (2005) Atherosclerosis and AD: analysis of data from the US National Alzheimer's Coordinating Center. Neurology 64:494–500. [DOI] [PubMed] [Google Scholar]
- 48. Imran MB, Kawashima R, Awata S, Sato K, Kinomura S, Ono S et al (1999) Tc‐99m HMPAO SPECT in the evaluation of Alzheimer's disease: correlation between neuropsychiatric evaluation and CBF images. J Neurol Neurosurg Psychiatry 66:228–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ishimaru H, Ueda K, Takahashi A, Maruyama Y (1998) Changes in presynaptic protein NACP/α‐synuclein in an ischemic gerbil hippocampus. Brain Res 788:311–314. [DOI] [PubMed] [Google Scholar]
- 50. Ito R, Mori S, Melhem ER (2002) Diffusion tensor brain imaging and tractography. Neuroimaging Clin N Am 12:1–19. [DOI] [PubMed] [Google Scholar]
- 51. Jacobs HI, Visser PJ, Van Boxtel MP, Frisoni GB, Tsolaki M, Papapostolou P et al (2012) Association between white matter hyperintensities and executive decline in mild cognitive impairment is network dependent. Neurobiol Aging 33:e1–e8. [DOI] [PubMed] [Google Scholar]
- 52. Jellinger KA (2008) Morphologic diagnosis of “vascular dementia”—a critical update. J Neurol Sci 270:1–12. [DOI] [PubMed] [Google Scholar]
- 53. Johnson NA, Jahng GH, Weiner MW, Miller BL, Chui HC, Jagust WJ et al (2005) Pattern of cerebral hypoperfusion in Alzheimer disease and mild cognitive impairment measured with arterial spin‐labeling MR imaging: initial experience. Radiology 234:851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kaelin WG Jr, Ratcliffe PJ (2008) Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 30:393–402. [DOI] [PubMed] [Google Scholar]
- 55. Kalaria RN, Perry RH, O'Brien J, Jaros E (2012) Atheromatous disease in small intracerebral vessels, microinfarcts and dementia. Neuropathol Appl Neurobiol 38:505–508. [DOI] [PubMed] [Google Scholar]
- 56. Kalback W, Esh C, Castano EM, Rahman A, Kokjohn T, Luehrs DC et al (2004) Atherosclerosis, vascular amyloidosis and brain hypoperfusion in the pathogenesis of sporadic Alzheimer's disease. Neurol Res 26:525–539. [DOI] [PubMed] [Google Scholar]
- 57. Kaneko K, Kuwabara Y, Sasaki M, Ogomori K, Ichimiya A, Koga H et al (2004) Posterior cingulate hypoperfusion in Alzheimer's disease, senile dementia of Alzheimer type, and other dementias evaluated by three‐dimensional stereotactic surface projections using Tc‐99m HMPAO SPECT. Clin Nucl Med 29:362–366. [DOI] [PubMed] [Google Scholar]
- 58. Kenny RA, Kalaria R, Ballard C (2002) Neurocardiovascular instability in cognitive impairment and dementia. Ann N Y Acad Sci 977:183–195. [DOI] [PubMed] [Google Scholar]
- 59. Kenny RA, Shaw FE, O'Brien JT, Scheltens PH, Kalaria R, Ballard C (2004) Carotid sinus syndrome is common in dementia with Lewy bodies and correlates with deep white matter lesions. J Neurol Neurosurg Psychiatry 75:966–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lee SJ, Kim JS, Yoo JY, Song IU, Kim BS, Jung SL et al (2010) Influence of white matter hyperintensities on the cognition of patients with Parkinson disease. Alzheimer Dis Assoc Disord 24:227–233. [DOI] [PubMed] [Google Scholar]
- 61. Li L, Zhang X, Yang D, Luo G, Chen S, Le W (2009) Hypoxia increases Aβ generation by altering β‐ and γ‐cleavage of APP. Neurobiol Aging 30:1091–1098. [DOI] [PubMed] [Google Scholar]
- 62. Love S, Miners S, Palmer J, Chalmers K, Kehoe P (2009) Insights into the pathogenesis and pathogenicity of cerebral amyloid angiopathy. Front Biosci (Landmark Ed) 14:4778–4792. [DOI] [PubMed] [Google Scholar]
- 63. McAleese KE, Firbank M, Hunter D, Sun L, Hall R, Neal JW et al (2013) Magnetic resonance imaging of fixed post mortem brains reliably reflects subcortical vascular pathology of frontal, parietal and occipital white matter. Neuropathol Appl Neurobiol 39:485–497. [DOI] [PubMed] [Google Scholar]
- 64. Melhem ER, Mori S, Mukundan G, Kraut MA, Pomper MG, van Zijl PC (2002) Diffusion tensor MR imaging of the brain and white matter tractography. AJR Am J Roentgenol 178:3–16. [DOI] [PubMed] [Google Scholar]
- 65. Mhatre M, Nguyen A, Kashani S, Pham T, Adesina A, Grammas P (2004) Thrombin, a mediator of neurotoxicity and memory impairment. Neurobiol Aging 25:783–793. [DOI] [PubMed] [Google Scholar]
- 66. Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S (2008) Aβ‐degrading enzymes in Alzheimer's disease. Brain Pathol 18:240–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Miners JS, Barua N, Kehoe PG, Gill S, Love S (2011) Aβ‐degrading enzymes: potential for treatment of Alzheimer disease. J Neuropathol Exp Neurol 70:944–959. [DOI] [PubMed] [Google Scholar]
- 68. Miners S, Ashby E, Baig S, Harrison R, Tayler H, Speedy E et al (2009) Angiotensin‐converting enzyme levels and activity in Alzheimer's disease: differences in brain and CSF ACE and association with ACE1 genotypes. Am J Transl Res 1:163–177. [PMC free article] [PubMed] [Google Scholar]
- 69. Miners S, Moulding H, de Silva R, Love S (2014) Reduced vascular endothelial growth factor and capillary density in the occipital cortex in dementia with Lewy bodies. Brain Pathol 24:334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nanduri J, Yuan G, Kumar GK, Semenza GL, Prabhakar NR (2008) Transcriptional responses to intermittent hypoxia. Respir Physiol Neurobiol 164:277–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Niwa K, Kazama K, Younkin L, Younkin SG, Carlson GA, Iadecola C (2002) Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am J Physiol Heart Circ Physiol 283:H315–H323. [DOI] [PubMed] [Google Scholar]
- 72. Niwa K, Kazama K, Younkin SG, Carlson GA, Iadecola C (2002) Alterations in cerebral blood flow and glucose utilization in mice overexpressing the amyloid precursor protein. Neurobiol Dis 9:61–68. [DOI] [PubMed] [Google Scholar]
- 73. Niwa K, Porter VA, Kazama K, Cornfield D, Carlson GA, Iadecola C (2001) Aβ‐peptides enhance vasoconstriction in cerebral circulation. Am J Physiol Heart Circ Physiol 281:H2417–H2424. [DOI] [PubMed] [Google Scholar]
- 74. Ott A, Breteler MM, de Bruyne MC, van Harskamp F, Grobbee DE, Hofman A (1997) Atrial fibrillation and dementia in a population‐based study. The Rotterdam Study. Stroke 28:316–321. [DOI] [PubMed] [Google Scholar]
- 75. Palmer JC, Baig S, Kehoe PG, Love S (2009) Endothelin‐converting enzyme‐2 is increased in Alzheimer's disease and up‐regulated by Aβ. Am J Pathol 175:262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Palmer JC, Barker R, Kehoe PG, Love S (2012) Endothelin‐1 is elevated in Alzheimer's disease and upregulated by amyloid‐β. J Alzheimers Dis 29:853–861. [DOI] [PubMed] [Google Scholar]
- 77. Palmer JC, Tayler HM, Love S (2013) Endothelin‐converting enzyme‐1 activity, endothelin‐1 production, and free radical‐dependent vasoconstriction in Alzheimer's disease. J Alzheimers Dis 36:577–587. [DOI] [PubMed] [Google Scholar]
- 78. Patel NS, Mathura VS, Bachmeier C, Beaulieu‐Abdelahad D, Laporte V, Weeks O et al (2009) Alzheimer's β‐amyloid peptide blocks vascular endothelial growth factor mediated signaling via direct interaction with VEGFR‐2. J Neurochem 112:66–76. [DOI] [PubMed] [Google Scholar]
- 79. Patel NS, Quadros A, Brem S, Wotoczek‐Obadia M, Mathura VS, Laporte V et al (2008) Potent anti‐angiogenic motifs within the Alzheimer β‐amyloid peptide. Amyloid 15:5–19. [DOI] [PubMed] [Google Scholar]
- 80. Perea RD, Rada RC, Wilson J, Vidoni ED, Morris JK, Lyons KE et al (2013) A comparative white matter study with Parkinson's disease, Parkinson's disease with dementia and Alzheimer's disease. J Alzheimers Dis Parkinsonism 3:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pievani M, Agosta F, Pagani E, Canu E, Sala S, Absinta M et al (2010) Assessment of white matter tract damage in mild cognitive impairment and Alzheimer's disease. Hum Brain Mapp 31:1862–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Powell FL, Fu Z (2008) HIF‐1 and ventilatory acclimatization to chronic hypoxia. Respir Physiol Neurobiol 164:282–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Racine AM, Adluru N, Alexander AL, Christian BT, Okonkwo OC, Oh J et al (2014) Associations between white matter microstructure and amyloid burden in preclinical Alzheimer's disease: a multimodal imaging investigation. Neuroimage Clin 4:604–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ravaglia G, Forti P, Maioli F, Chiappelli M, Montesi F, Tumini E et al (2007) Blood inflammatory markers and risk of dementia: the Conselice Study of Brain Aging. Neurobiol Aging 28:1810–1820. [DOI] [PubMed] [Google Scholar]
- 85. Richardson K, Stephan BC, Ince PG, Brayne C, Matthews FE, Esiri MM (2012) The neuropathology of vascular disease in the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Curr Alzheimer Res 9:687–696. [DOI] [PubMed] [Google Scholar]
- 86. Roher AE, Esh C, Kokjohn TA, Kalback W, Luehrs DC, Seward JD et al (2003) Circle of Willis atherosclerosis is a risk factor for sporadic Alzheimer's disease. Arterioscler Thromb Vasc Biol 23:2055–2062. [DOI] [PubMed] [Google Scholar]
- 87. Roher AE, Esh C, Rahman A, Kokjohn TA, Beach TG (2004) Atherosclerosis of cerebral arteries in Alzheimer disease. Stroke 35:2623–2627. [DOI] [PubMed] [Google Scholar]
- 88. Roher AE, Tyas SL, Maarouf CL, Daugs ID, Kokjohn TA, Emmerling MR et al (2011) Intracranial atherosclerosis as a contributing factor to Alzheimer's disease dementia. Alzheimers Dement 7:436–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ruitenberg A, den Heijer T, Bakker SL, van Swieten JC, Koudstaal PJ, Hofman A, Breteler MM (2005) Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam Study. Ann Neurol 57:789–794. [DOI] [PubMed] [Google Scholar]
- 90. Scheltens P, Barkhof F, Valk J, Algra PR, van der Hoop RG, Nauta J, Wolters EC (1992) White matter lesions on magnetic resonance imaging in clinically diagnosed Alzheimer's disease. Evidence for heterogeneity. Brain 115(Pt 3):735–748. [DOI] [PubMed] [Google Scholar]
- 91. Schneider JA, Wilson RS, Bienias JL, Evans DA, Bennett DA (2004) Cerebral infarctions and the likelihood of dementia from Alzheimer disease pathology. Neurology 62:1148–1155. [DOI] [PubMed] [Google Scholar]
- 92. Scolding NJ, Joseph F, Kirby PA, Mazanti I, Gray F, Mikol J et al (2005) Aβ‐related angiitis: primary angiitis of the central nervous system associated with cerebral amyloid angiopathy. Brain 128:500–515. [DOI] [PubMed] [Google Scholar]
- 93. Sexton CE, Kalu UG, Filippini N, Mackay CE, Ebmeier KP (2011) A meta‐analysis of diffusion tensor imaging in mild cognitive impairment and Alzheimer's disease. Neurobiol Aging 32:e5–e18. [DOI] [PubMed] [Google Scholar]
- 94. Shi J, Yang SH, Stubley L, Day AL, Simpkins JW (2000) Hypoperfusion induces overexpression of beta‐amyloid precursor protein mRNA in a focal ischemic rodent model. Brain Res 853:1–4. [DOI] [PubMed] [Google Scholar]
- 95. Shinall H, Song ES, Hersh LB (2005) Susceptibility of amyloid β peptide degrading enzymes to oxidative damage: a potential Alzheimer's disease spiral. Biochemistry 44:15345–15350. [DOI] [PubMed] [Google Scholar]
- 96. Smallwood A, Oulhaj A, Joachim C, Christie S, Sloan C, Smith AD, Esiri M (2012) Cerebral subcortical small vessel disease and its relation to cognition in elderly subjects: a pathological study in the Oxford Project to Investigate Memory and Ageing (OPTIMA) cohort. Neuropathol Appl Neurobiol 38:337–343. [DOI] [PubMed] [Google Scholar]
- 97. Soucek T, Cumming R, Dargusch R, Maher P, Schubert D (2003) The regulation of glucose metabolism by HIF‐1 mediates a neuroprotective response to amyloid β peptide. Neuron 39:43–56. [DOI] [PubMed] [Google Scholar]
- 98. Strozyk D, Dickson DW, Lipton RB, Katz M, Derby CA, Lee S et al (2010) Contribution of vascular pathology to the clinical expression of dementia. Neurobiol Aging 31:1710–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Sun Y, Jin K, Mao XO, Zhu Y, Greenberg DA (2001) Neuroglobin is up‐regulated by and protects neurons from hypoxic‐ischemic injury. Proc Natl Acad Sci U S A 98:15306–15311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Tanimukai H, Imaizumi K, Kudo T, Katayama T, Tsuda M, Takagi T et al (1998) Alzheimer‐associated presenilin‐1 gene is induced in gerbil hippocampus after transient ischemia. Brain Res Mol Brain Res 54:212–218. [DOI] [PubMed] [Google Scholar]
- 101. Teipel SJ, Meindl T, Wagner M, Stieltjes B, Reuter S, Hauenstein KH et al (2010) Longitudinal changes in fiber tract integrity in healthy aging and mild cognitive impairment: a DTI follow‐up study. J Alzheimers Dis 22:507–522. [DOI] [PubMed] [Google Scholar]
- 102. Thomas TL, Kehoe PG, Love S (2013) Elevated VEGF in the cerebral cortex in Alzheimer's disease is not attributable to arteriosclerotic small vessel disease or cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 39:45.23339288 [Google Scholar]
- 103. Tschope C, Schultheiss HP, Walther T (2002) Multiple interactions between the renin‐angiotensin and the kallikrein‐kinin systems: role of ACE inhibition and AT1 receptor blockade. J Cardiovasc Pharmacol 39:478–487. [DOI] [PubMed] [Google Scholar]
- 104. Turner AJ, Isaac RE, Coates D (2001) The neprilysin (NEP) family of zinc metalloendopeptidases: genomics and function. Bioessays 23:261–269. [DOI] [PubMed] [Google Scholar]
- 105. Turner AJ, Tanzawa K (1997) Mammalian membrane metallopeptidases: NEP, ECE, KELL, and PEX. FASEB J 11:355–364. [DOI] [PubMed] [Google Scholar]
- 106. Unal‐Cevik I, Gursoy‐Ozdemir Y, Yemisci M, Lule S, Gurer G, Can A et al (2011) α‐Synuclein aggregation induced by brief ischemia negatively impacts neuronal survival in vivo: a study in [A30P]α‐synuclein transgenic mouse. J Cereb Blood Flow Metab 31:913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Wang DS, Iwata N, Hama E, Saido TC, Dickson DW (2003) Oxidized neprilysin in aging and Alzheimer's disease brains. Biochem Biophys Res Commun 310:236–241. [DOI] [PubMed] [Google Scholar]
- 108. Wang R, Wang S, Malter JS, Wang DS (2009) Effects of HNE‐modification induced by Abeta on neprilysin expression and activity in SH‐SY5Y cells. J Neurochem 108:1072–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Wang Y, West JD, Flashman LA, Wishart HA, Santulli RB, Rabin LA et al (2012) Selective changes in white matter integrity in MCI and older adults with cognitive complaints. Biochim Biophys Acta 1822:423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wardlaw JM, Doubal FN, Valdes‐Hernandez M, Wang X, Chappell FM, Shuler K et al (2013) Blood‐brain barrier permeability and long‐term clinical and imaging outcomes in cerebral small vessel disease. Stroke 44:525–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R et al (2013) Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 12:822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wen Y, Onyewuchi O, Yang S, Liu R, Simpkins JW (2004) Increased β‐secretase activity and expression in rats following transient cerebral ischemia. Brain Res 1009:1–8. [DOI] [PubMed] [Google Scholar]
- 113. Wen Y, Yang S, Liu R, Brun‐Zinkernagel AM, Koulen P, Simpkins JW (2004) Transient cerebral ischemia induces aberrant neuronal cell cycle re‐entry and Alzheimer's disease‐like tauopathy in female rats. J Biol Chem 279:22684–22692. [DOI] [PubMed] [Google Scholar]
- 114. Wen Y, Yang S, Liu R, Simpkins JW (2004) Transient cerebral ischemia induces site‐specific hyperphosphorylation of tau protein. Brain Res 1022:30–38. [DOI] [PubMed] [Google Scholar]
- 115. Yang S‐P, Bae D‐G, Kang HJ, Gwag BJ, Gho YS, Chae C‐B (2004) Co‐accumulation of vascular endothelial growth factor with β‐amyloid in the brain of patients with Alzheimer's disease. Neurobiol Aging 25:283–290. [DOI] [PubMed] [Google Scholar]
- 116. Yarchoan M, Xie SX, Kling MA, Toledo JB, Wolk DA, Lee EB et al (2012) Cerebrovascular atherosclerosis correlates with Alzheimer pathology in neurodegenerative dementias. Brain 135:3749–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Yoon DK, Hwang IK, Yoo KY, Lee YB, Lee JJ, Kim JH et al (2006) Comparison of α‐synuclein immunoreactivity and protein levels in ischemic hippocampal CA1 region between adult and aged gerbils and correlation with Cu,Zn‐superoxide dismutase. Neurosci Res 55:434–441. [DOI] [PubMed] [Google Scholar]
- 118. Yu S, Liu XM, Li YH, Lu GW, Chen B (2004) Effect of repeated acute hypoxic treatment on the expression of α‐synuclein in the mouse brain cortex [Chinese]. Sheng Li Xue Bao 56:263–268. [PubMed] [Google Scholar]
- 119. Yuan J, Wen G, Li Y, Liu C (2013) The occurrence of cerebrovascular atherosclerosis in Alzheimer's disease patients. Clin Interv Aging 8:581–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Zetterberg H, Mortberg E, Song L, Chang L, Provuncher GK, Patel PP et al (2011) Hypoxia due to cardiac arrest induces a time‐dependent increase in serum amyloid β levels in humans. PLoS ONE 6:e28263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Zhang X, Zhou K, Wang R, Cui J, Lipton SA, Liao FF et al (2007) Hypoxia‐inducible factor 1α (HIF‐1α)‐mediated hypoxia increases BACE1 expression and β‐amyloid generation. J Biol Chem 282:10873–10880. [DOI] [PubMed] [Google Scholar]
- 122. Zhiyou C, Yong Y, Shanquan S, Jun Z, Liangguo H, Ling Y, Jieying L (2009) Upregulation of BACE1 and β‐amyloid protein mediated by chronic cerebral hypoperfusion contributes to cognitive impairment and pathogenesis of Alzheimer's disease. Neurochem Res 34:1226–1235. [DOI] [PubMed] [Google Scholar]