

Abstract
Chronic traumatic encephalopathy (CTE) is a neurodegenerative disease characterized by a distinct pattern of hyperphosphorylated tau (p‐tau). Thought to be caused by repetitive concussive and subconcussive injuries, CTE is considered largely preventable. The majority of neuropathologically confirmed cases have occurred in professional contact sport athletes (eg, boxing, football). A recent post‐mortem case series has magnified concerns for the public's health following its identification in six high school level athletes. CTE is diagnosed with certainty only following a post‐mortem autopsy. Efforts to define the etiology and clinical progression during life are ongoing. The goal of this article is to characterize the clinical concepts associated with short‐ and long‐term effects of repetitive traumatic brain injury, with a special emphasis on new clinical diagnostic criteria for CTE. Utilizing these new diagnostic criteria, two cases of neuropathologically confirmed CTE, one in a professional football player and one in a professional boxer, are reported. Differences in cerebellar pathology in CTE confirmed cases in boxing and football are discussed.
Keywords: chronic traumatic encephalopathy, concussion, mild traumatic brain injury, football, boxing, neuropathology
Introduction
Participating in contact sports is thought to increase an individual's risk for later‐life impairments and neurodegeneration. Both active and retired athletes who report multiple concussions are significantly more likely to have problems with depression, emotional liability, executive function, attention and memory. In a study commissioned by the National Football League (NFL), it was reported that retired players were 20 times more likely than age‐matched controls to receive a diagnosis of dementia, Alzheimer's disease (AD), cognitive impairment or related memory impairment disorders 93. Lehman et al, for the National Institute for Occupational Safety and Health, found that neurodegeneration was listed as the cause of death three times more often in NFL players than in the general US population 46, 47. Mounting evidence suggests chronic traumatic encephalopathy (CTE) may be the major underlying etiology in these reports. In fact, a recent study with the National Alzheimer's Coordinating Center Uniform Data Set reported an atypical, tau predominant pathology in cases of suspected AD after grouping participants with significant traumatic brain injury (TBI) histories 73. Recently, the Chronic Traumatic Encephalopathy Center (CTEC) added 68 new cases of CTE to the literature 52, which doubled the number of cases reported in the world's literature. Of the 68 new cases, it is particularly concerning that six cases of neuropathologically confirmed CTE were identified in high school level athletes 17. As a result, the National Institutes of Health and the National Institute of Neurological Disorders and Stroke (NINDS) have teamed up with investigators to provide financial support for multicenter and multidisciplinary investigations into this condition. The purpose of this article is to review (i) the acute effects of sports‐related TBI, including clinical criteria for concussions, post‐concussion syndrome (PCS); second impact syndrome (SIS) (ii); the chronic effects of sports‐related TBI, including CTE (iii); review new clinical diagnostic criteria for CTE; and (iv) provide clinical and pathological details from two cases of neuropathologically confirmed CTE from a professional football player and a professional boxer.
TBI
Concussion
The word “concussion” derives from the Latin concutere, meaning “to shake violently.” Concussions are just that—a shaking of the brain inside the skull, which alters the alertness of the injured person or produces symptoms that fall into four major categories:
-
(i)
Somatic: headaches, nausea, vomiting, balance and/or visual problems, dizzy spells and issues such as sensitivity to light and noise.
-
(ii)
Emotional: sadness to the point of depression (even suicide), nervousness and irritability.
-
(iii)
Sleep disturbance: sleeping more or less than usual and trouble falling asleep.
-
(iv)
Cognitive: difficulty concentrating, troubles with memory, feeling mentally slow or as if in a fog that will not lift.
Changes in alertness can be relatively mild (slightly dazed) or profound (unconscious), yet both situations fall within the definition of concussion. Although concussion is often classified as a form of mild TBI (MTBI), when the profound potential effects are considered, many clinicians do not view a concussion as a necessarily mild injury. It is, however, generally agreed that:
-
(i)
Both direct and indirect head trauma produce linear and rotational forces on the brain, with rotational forces being the most injurious 14.
-
(ii)
Concussions do not typically cause structural changes seen on routine imaging studies, such as computed tomography (CT) and magnetic resonance imaging (MRI) scan, but rather exert their pathological changes at the microscopic and biomechanical levels from the brain being shaken within in the skull 13.
-
(iii) Following a concussive event, there is a destructive pathophysiological and biomechanical response that initiates a chain of neurometabolic and neurochemical reactions that include 79:
- Activation of inflammatory response.
- Imbalance of ionic concentrations.
- Increase in the excitatory amino acids.
- Dysregulation of neurotransmitter release and synthesis.
- Imbalance of mitochondrial functions and energy metabolism.
- Productions of free radicals.
-
(iv)
While an individual prognosis cannot be determined, all concussions are initially managed with both cognitive and physical rest 12, 15, 31.
-
(v) Following a concussive event, even after resolution of all symptoms, there may be long‐lasting, ultrastructural and functional brain alteration as shown by 79:
- Susceptibility weighted imaging MRI.
- Diffusion tensor imaging (DTI) MRI.
- Functional MRI.
- Magnetic resonance spectroscopy MRI.
- Positron emission tomography MRI.
-
(vi)
Because of the unique features of the maturing brain, young athletes are more vulnerable to the effects of a concussion than adults 32, 76.
PCS
While there are two well‐recognized definitions of PCS 2, 65 (Tables 1 and 2), most clinicians recognize PCS as the persistence of concussion symptoms lasting beyond a month.
Table 1.
International classification for diseases 10th revision clinical criteria for post‐concussion syndrome
| A. Head injury usually severe enough to cause loss of consciousness within 4 weeks of symptom onset |
| B. Preoccupation with symptoms and fear of brain damage with hypochondrial concern and adaptation of sick role |
| C. Three from below |
| Headache, dizziness, malaise, fatigue, noise intolerance |
| Irritability, depression, anxiety, emotional lability |
| Concentration, memory or intellectual deficit without neuropsychological evidence of deficit |
| Insomnia |
| Reduced alcohol intolerance |
Table 2.
Diagnostic and statistical manual of mental disorders fourth edition criteria for post‐concussion syndrome
|
Individuals at increased risk for this condition include athletes with multiple concussions, those with concussions in close proximity to each other and athletes subjected to a double hit such as a direct helmet‐to‐helmet hit and then the head hitting the ground as the athlete falls 18. At even higher risk is an athlete that experiences additional head trauma while they are symptomatic from a prior concussion through the course of the same game or match. PCS is usually very debilitating, but it typically clears up in a matter of months. Although rare, there are reports where post‐concussion symptoms take as long as 5 years to clear up after trauma 18. For those who are able to recover from PCS, especially those with shorter courses, many are able to safely return to competitive sports. For those who do not recover, it is presently not possible to rule out incipient CTE.
SIS
The most concerning concussion‐related problem is SIS. Autoregulation, the process by which our brain normally maintain a constant blood flow, is the basis of this serious condition. When the brain's blood pressure rises, there is a concurrent restriction in the diameter of arterioles. When the blood pressure falls, the opposite occurs: arterioles dilate, or relax, to maintain constant blood flow (Figure 1A–C).
Figure 1.
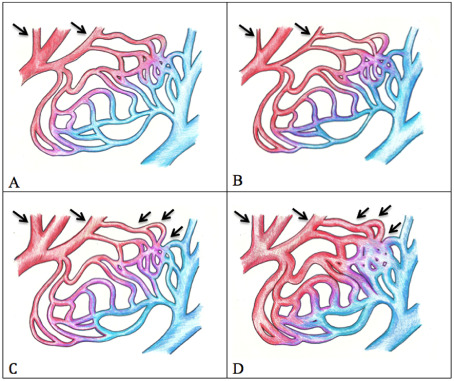
Autoregulation of brain arterioles and pathophysiology. Baseline (A): When blood pressure is normal, brain arteriole blood vessels are neither constricted nor dilated. Increased blood pressure (B): The brain seeks to maintain a constant blood flow. The brain arteriole blood vessels constrict. Decreased blood pressure (C): As pressure falls, brain arteriole dilation occurs. Blood flow to the brain remains unchanged. Dysautoregulation or second impact syndrome (D): A serious disruption occurs. The brain acts as if blood pressure is low when it is not low; it is normal or may be elevated. Brain arteriole blood vessels dilate and blood rushes into the brain and results in a massive rise in intracranial pressure. Within minutes, the brain can herniate, resulting in coma.
SIS disrupts autoregulation (Figure 1D). Instead of constricting when blood pressure is normal or elevated, arterioles dilate and allow blood to rush through. The result: a massive inflow of blood to the brain accompanied by an equally dramatic increase in intracranial pressure—a highly dangerous situation, which often leads to brain herniation and death. The patients who survive are almost always severely disabled.
Each year, a few athletes lose their lives to SIS, although the exact number is unclear. Since 1987, the senior author of this paper has been involved in a research study that tracks catastrophic injuries causing death, permanent brain injury or spinal cord damage among high school and college football players in the United States. During a 13‐year interval, 15 cases of SIS were identified out of 94 cases of catastrophic head injury 9. In that study, approximately 40% of athletes were kept on the field despite having concussion symptoms. With a greater commitment to keeping players with concussion symptoms off the field, deaths from SIS could be drastically reduced or eliminated.
Subconcussive brain trauma
A concussion is well recognized as an etiological factor for a spectrum of neurological conditions, including PCS and risk for developing CTE, a neurodegenerative disorder occurring later in life. The role of subconcussive head trauma, defined as head trauma that does not result in recognized concussion symptoms or signs, is not well known. In our published work from the Center for the Study of Traumatic Encephalopathy at Boston University Medical School, we have seen cases of CTE in deceased athletes who never had a recognized concussion. These athletes had, however, sustained many thousands, often more than 10 000 subconcussive blows during their athletic career 53. For athletes who have sustained concussions, our experience tells us that the risk of CTE occurrence correlates best with total head trauma, including both concussive and subconcussive blows 52.
Recent publications clearly point to the damaging effects of repetitive subconcussive trauma in male contact sport athletes when compared with age‐matched male noncontact sport athletes 16. Significant abnormal changes in DTI MRI, including fractional anisotropy (FA) and mean diffusivity, were observed in contact sport athletes. Abnormalities were most robust in the corpus callosum, external capsule and inferior fronto‐occipital fasciculus areas of the brain containing long myelinated axonal fiber tracts. These abnormal findings were not only seen preseason in the contact vs. noncontact groups, but increased when the groups were compared with preseason to postseason. An alarming finding, this preseason‐postseason difference suggests long‐term effects from the repeated head trauma associated with playing just one season of a contact sport. A study of 50 Division I football players compared with 25 matched controls found that the volume of the hippocampus (part of the brain important for memory) was reduced by 15%–25% in the football players 77. This diminished brain volume reflected neuronal loss and was correlated with decreased cognitive activity and reaction time. The study further found hippocampus size to be inversely correlated with an athlete's total years of play. These findings suggested that subconcussive hits have a harmful effect on young brains.
These data validate the idea that brain injury can occur from the repetitive impacts sustained in contact sports, even in the absence of clinically apparent TBI. These subconcussive hits still impart resultant linear and rotational accelerations on the brain as it violently shakes inside the skull.
What makes the author's findings so compelling and concerning is that in the last year alone, multiple reports in peer‐reviewed journals have shown significant differences in preseason vs. postseason values in contact sport athletes using a variety of tests. These findings have included metabolic brain function as measured by functional MRI 87, neurocognitive testing (ImPACT) 87 and structural breakdown of the blood–brain barrier as manifested by S100B protein in the blood 50 structural changes seen on DTI imaging 46. While the majority of these studies have involved American football players, some have included soccer and hockey athletes.
This demonstrates that all head trauma, even at the subconcussive level, can result in brain damage in susceptible individuals. It can be argued that the subconcussive group may have included some unrecognized concussions. It is necessary to carry out additional studies beyond the immediate postseason, up to 3, 6 or 12 months, to see in what percentage the abnormalities persist and result in permanent injury.
Cumulative exposure
In those exposed to repetitive head trauma, there are several possible long‐term outcomes that may occur: (i) none, that is, there is no appreciable neurological signs or symptoms; (ii) static deficits that are a result of the head trauma but do not progress; and (iii) a neurodegenerative process, with the repetitive head trauma as either a risk factor (eg, AD) or cause of, for example, CTE. To characterize the clinical features associated with repetitive head trauma, there may be no better groups to examine than football players and boxers. These athletes, particularly those who make it to the professional level, are exposed to thousands of blows to the head over the course of many years. Quantifying such exposure is among the major challenges in the field of TBI. Research efforts are underway to define “clinically practical” measurements of blows to the head among contact sport athletes. For now, the “gold standard” for determining a lifetime history of TBI is retrospective self‐reports or proxy reports obtained in a structured interview. These include the NINDS common data elements and recommendations from the Center for Disease Control and Prevention 23, 24, 34. The researchers at the CTEC developed novel questionnaires to obtain athletes' athletic and concussion histories and to provide meaningful and accurate estimates of overall repetitive TBI (RTBI) exposure 5, 6, 70. While there are inherent limitations of retrospective self‐reports, numerous studies have demonstrated their usefulness in evaluating the association between long‐term RTBI exposure and latent impairments with an acceptable level of reliability 23, 45. For example, the CTEC recently reported a link between previous football experience and executive dysfunction in older retired football players 74. In their study, 64 retired college and professional football players were compared with healthy adults. Subjects were administered the Behavior Rating Inventory of Executive Function, adult version, to evaluate nine areas of executive functioning with scores compared with published age‐corrected normative scores for healthy adults. Relative to healthy adults, the football players indicated significantly more problems overall, as well as on seven of the nine clinical scales, including inhibit, shift, emotional control, initiate, working memory, plan/organize and task monitor. These symptoms were greater in athletes aged 40 and older, indicating that although RTBI experienced by football players is associated with both short‐term and long‐term self‐reported executive dysfunction, these symptoms may develop or worsen in the fifth decade of life.
In the absence of a direct measure of a subject's cumulative trauma exposure, there are several potential surrogates, such as number of fights, fights per year, number of knockouts and years of fighting, that have been utilized in studies with boxers. Among football players, the total number of seasons, primary position and level achieved (high school, college, professional) have been utilized as well. However, each of these variables may actually have a slightly different influence on the development of long‐term impairments and underlying neuropathology, including CTE. Number of fights, for example, may act as a proxy for amount of training. Some have postulated that the effects of repeated blows to the head—even at a subconcussive level—that occur during sparring may play an important role in causing cumulative brain injury as the boxing match itself. Investigations using electroencephalography (EEG) and CT in professional boxers reported a stronger association between total number of years/bouts fought than to the total number of knockouts (KOs), implicating cumulative subconcussive effects 19, 72. Frequency of fighting may be a complementary variable that requires consideration; fighting more frequently may reduce the time the brain has to fully recover from prior trauma and be a risk factor that interacts with number of fights. On the other hand, when the period of unconsciousness exceeds 1 minute, KO may reflect the more severe end of the spectrum of MTBI. However, in the majority of cases where loss of consciousness (LOC) is only seconds, LOC is not correlated with a severe MTBI. While the number of KOs sustained in sanctioned professional fights can be tracked from commonly available records, KOs that may have occurred at other times are harder to trace.
Aside from the specific aspects of RTBI exposures (eg, severity, location and frequency), the timing of exposure in relation to brain maturation may also influence long‐term outcomes. Recently, Cantu and Hyman hypothesized that the age at which an individual is first exposed to RTBI could play a major role in the pathological cascade that leads to CTE 13. In the book “Concussions and Our Kids 13,” the authors provide a theoretical groundwork for why certain developmental ages are particularly vulnerable to the effects of RTBI. Experimental evidence to support this hypothesis first appeared in a study of amateur boxers in 1971 39. In this study, Jedlińksi et al demonstrated a stronger correlation between neurological presentations and pathological EEG (r = 0.47), and psychiatric findings (r = 0.60) in boxers who began their fighting careers at age 15, 16 or 17, showing a stepwise change in the association with each year 39. Since this publication, there has been little additional research into the subject of “age at first contact sport exposure.” Having identified this gap 13, a new study lead by Julie Stamm 83 demonstrated similar findings in American style football players. In this study, an association was made between participation in tackle football prior to age 12 and a greater cognitive impairment later in life; this was determined based on objective neuropsychological tests in a sample of 41 former NFL players (ages 40–69).
In a another recent study group of 730 National Collegiate Athletic Association Division I Football Championship Series athletes, it was demonstrated that while there were no significant differences between position groups in the number of diagnosed concussions, there were significant differences between position groups in the number of undiagnosed concussions (P = 0.008) and “dings” (P < 0.001), with offensive linemen reporting significantly greater numbers than any other positions 3. It is therefore reasonable to suggest that positions with greater risk of concussion have a greater likelihood for cumulative and latent neurodegeneration. Indeed, Lehman et al attempted to subgroup NFL players into “speed” and a “nonspeed” groups for analysis, but were limited by the available sample size 48. To date, no method has been shown to reliably predict which athletes are likely to develop late‐life impairments and CTE disease, other than to roughly classify risk as involvement in contact sports 47.
A contemporary study designed to better understand the effect of repetitive head trauma on clinical and subclinical outcomes is the Professional Fighters Brain Health Study (PFBHS). The PFBHS is a longitudinal study of active professional fighters (boxers and mixed martial arts), retired professional fighters and age‐/education‐matched controls 6. The main objective of the PFBHS is to determine the relationships between measures of head trauma exposure, along with other potential modifiers and changes in brain imaging and neurological/behavioral function over time. Initial results from the PFBHS indicate that increased exposure to head trauma, as measured either by number of professional fights or years of professional fighting, is associated with imaging and performance findings. Because of its ability to potentially reflect white matter integrity, MRI‐based DTI has been studied in many different groups exposed to repetitive head trauma. In the PFBHS, a relationship was found between number of KOs and DTI measures in several white matter and subcortical grey matter regions 75. Moreover, striking changes were seen in transcallosal motor pathways. These fibers transverse a long distance, and given the torsional movement of the brain that can occur with head trauma, may be particularly susceptible to injury. Specifically, in 17 active fighters—scanned two times with approximately 1‐year interval—transverse diffusivity (P = 0.055) and FA (P = 0.018) in a motor pathway, defined by DTI tracking from the left M1 seed, were significantly related to number of professional fights over a period of a year (see Figure 2).
Figure 2.
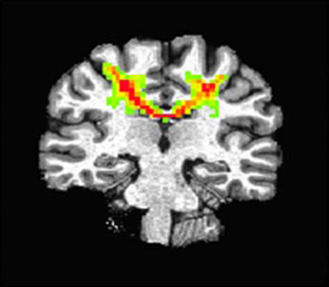
A representative diffusion tensor imaging tracking in a motor pathway. Tracking was conducted from the left M1 seed in 17 paired fighters.
While findings have been reported in a variety of sports and military settings, there is no uniform method for DTI analysis that can be applied at an individual level on differing MRI equipment. Measurement of MRI volumes may be a more practical tool, as methods for automated volumetrics are commercially available. Cross‐sectional analysis of over 200 active fighters have shown significant correlations between higher number of fights or years of fighting and lower volumes of the thalamus and caudate (see Figure 3) 5.
Figure 3.
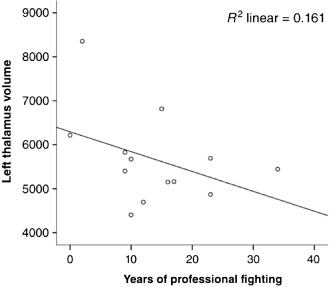
Left thalamus volume and years of professional fighting in retired boxers.
Overall, these findings cannot be interpreted as being indicative of risk for any specific long‐term outcome (ie, CTE). However, additional research examining the relationship between different potential RTBI exposure variables (ie, age of first exposure, subconcussions, years of fighting) and neuropathologically confirmed CTE lesions is warranted.
CTE
Historical context
As a result of its early discovery in boxers, CTE has been variously referred to as “cumulative encephalopathy of the boxer 20,” “chronic progressive traumatic encephalopathy 27” and “chronic traumatic encephalopathy 26, 62” to reflect its trauma etiology and clinical presentation. In the earlier half of the 20th century, it was suggested that the punch‐drunk condition was unique to the sport of boxing. The first neuropathologically characterized case of CTE was reported by Brandenberg and Hallervorden in 1954 10. As additional cases appeared 21, 33, an association formed between the blows endured from boxing and the subsequent development of neurofibrillary tangle predominant dementia. Augustus Thorndike MD, a Massachusetts General Hospital with the Harvard University Athletic Department (1952) declared in the New England Journal of Medicine: “The college health authorities are conscious of the pathology of the ‘punch‐drunk’ boxer. Just how much one should permit recurrence of cerebral concussion in college athletes is a matter of opinion (p. 556) 88.” New research shows that CTE is more common in former contact sport athletes than previously realized. Interest in this condition has grown considerably since 2005, following the first post‐mortem autopsy report characterizing the pathological hallmarks of CTE in a professional football player 64. Additional neuropathological evidence of CTE has been reported in many sports other than professional boxing and football, including professional soccer, mixed martial arts, rugby, ice hockey, wrestling and baseball 52, 53, 84.
CTE with motor neuron disease (MND)
MND has been reported in a subset of cases with CTE 54, 57. It is not yet clear whether the pathology in these cases represents a variant of CTE or CTE with overlapping comorbid disease. Recent data suggest that professional American football players have more than four times the risk of dying from amyotrophic lateral sclerosis (ALS) MND than age‐ and gender‐matched controls 47. Our recent review of the world's literature 61 uncovered the first case of CTE MND ever reported 57 with pathological evidence to support the link between sport‐related RTBI and atypical ALS. Meyers et al reported clinical and neuropathological findings from a 41‐year‐old retired professional boxer who presented clinically with signs of progressive motor weakness 57. The subject had a significant boxing history, with a career that began at age 14 and included only two known KOs, which were verified by the Pennsylvania Boxing Commission. This case is historically important because the findings reported by Meyers et al corroborate the findings reported in our more recent case series 52, 54. In 2010, the members of the CTEC reported the first case series with neuropathologically confirmed CTE and ALS in two football players and one boxer 54. Among our recent series of 68 cases, approximately 11% demonstrated pathological evidence of MND 52. The predominant presentation (63%) involved motor weakness, atrophy and fasciculations in addition to cognitive and behavioral symptoms.
Preclinical CTE
Analogous to other neurodegenerative diseases, there were neuropathologically confirmed CTE cases in our sample that lacked overt clinical symptoms or impairment 52, 81. As is the case in preclinical AD, developing methods to identify persons at this early stage of CTE disease is crucial for investigating preventative methods and treatments. Additionally, preclinical cases could provide invaluable information about the etiology and development of symptoms in living individuals. Several studies in boxers and football players support the interpretation that asymptomatic head trauma can cause long‐term brain damage that may only become apparent once the normal aging process has contributed to neuronal degeneration 89.
Clinical symptoms of CTE
The symptoms associated with CTE pathology typically manifest in one of four clinical domains: (i) cognitive, (ii) behavior, (iii) mood and (iv) motor 61, 86.
Table 3 summarizes the clinical symptoms identified in our recent systematic review of 202 previously published cases of male athletes with histories of RTBI that met review criteria for possible, probable and neuropathologically confirmed CTE 61. The sample included 141 boxers, 54 American football players, 5 ice hockey players and 2 professional wrestlers, making this the largest pooled case review of the clinical features in CTE to date. Progression was identified in 137 cases (68%), most often reported in cognitive symptoms, resulting in dementia. The cases described as “stable” were notably younger in age. Symptoms typically manifest 8–10 years after initial RTBI. The clinical course of CTE is slow, much slower then AD or Frontotemporal dementia, with a progression rate estimated at 11–14 years between pathological stages 52.
Table 3.
Symptoms of chronic traumatic encephalopathy
| Cognitive features | Behavioral features | Mood features | Motor features |
|---|---|---|---|
|
aMemory impairment aExecutive dysfunction aImpaired attention bDysgraphia Lack of insight Perseveration Language difficulties Dementia Alogia Visuospatial difficulties Cognitive impairment Reduced intelligence |
aPhysical violence aVerbal violence aExplosivity aLoss of control aShort fuse bImpulsivity bParanoid delusions Aggression Rage Inappropriate speech Boastfulness Childish behavior Socially inappropriate Disinhibited behavior Personality changes Psychosis Social isolation |
aDepression aHopelessness bSuicidality bAnxiety bFearfulness bIrritability bApathy bLoss of interest Labile emotions Fatigue Flat affect Insomnia Mania Euphoria Mood swings Prolix |
bAtaxia bDysarthria bParkinsonism bGait bTremor bMasked facies bRigidity Weakness Spasticity Clonus |
Core diagnostic clinical feature, defined as any feature that appeared in 70% or more of the neuropathologically confirmed chronic traumatic encephalopathy (CTE) cases without comorbid disease.
Supportive diagnostic feature, defined as any feature that appeared in neuropathologically confirmed CTE cases without comorbid disease.
Table adapted from Montenigro et al 61 with permission.
Clinical subtypes of CTE
Similar to other neurodegenerative conditions, the clinical features in CTE are heterogeneous. Stern et al, identified two relatively distinct clinical presentations: one consisting of behavioral and mood symptoms with an earlier age at onset [mean age at onset 34.5 standard deviation (SD) = 11.6] and another consisting of cognitive impairment with a later age at onset (mean age at onset 58.5, SD = 17.7) 86. Among cases with initial behavioral and mood symptoms, 86% progressed to include cognitive symptoms whereas only 46% of cognitive cases developed behavior and mood symptoms. In addition to the two subtypes described in Stern et al, Montenigro et al identified an additional “mixed subtype” (mean age at onset 43.0, SD = 14.0) in neuropathologically confirmed cases where the predominant presentation was neither behavioral‐mood or cognitive, but rather a combination of the two 61. Consistent with recent subtype descriptions 61, 86, earlier studies in boxers also reported having identified recurring subtypes in the presentation of CTE. Classifications identified in the earlier literature 61 were based on various clinical features, including initial presentation, progression, age at onset and occurrence of dementia. For example, Ernst Jokl (founder of the American College of Sports Medicine) distinguished between the two types of chronic impairment in punch‐drunk boxers, namely a “behavioral‐psychopathic” type and a “neurological‐psychiatric” one 60. The former involved cases with presentations involving “viciousness,” “murder committed from jealousy” and “delinquency.” Research involving these subtypes represents an opportunity to refine risk‐factor definitions, develop targeted prevention strategies and someday assess treatment responsiveness.
Clinical diagnostic criteria
To date, the only definitive means of diagnosing CTE is through post‐mortem autopsy. However, the ability to diagnose CTE during life is critical to conduct epidemiologic studies on CTE and to eventually plan treatment trials. To address this gap, Montenigro et al proposed new clinical research diagnostic criteria for CTE 61 that overcome the limitations identified 49, 58 in the previous criteria 40, 43, 44, 92. The new criteria are based on a systematic review of the previous literature, as well as on the clinical features reported in neuropathologically confirmed cases of CTE without comorbid disease 52, 53, 86. The proposed diagnostic criteria include five general criteria, three core clinical features and nine supportive features to identify the “traumatic encephalopathy syndrome” (TES). The term TES is used to describe the “syndrome” of clinical features that comprise this condition when the underlying pathology is speculative (Table 4). Criteria for the behavioral/mood variant, cognitive variant, mixed variant and TES dementia phenotypes are also provided (Table 5).
Table 4.
General diagnostic criteria for traumatic encephalopathy 61
| All five criterion (1–5) must be met for diagnosis | |
| 1. History of multiple impacts | |
| Types of injuries | Concussion or mild traumatic brain injury. If no other repetitive traumatic brain injury then minimum of 4. |
| Moderate/severe traumatic brain injury. If no other repetitive traumatic brain injury then minimum of 2. | |
| Subconcussive trauma. | |
| Source of exposures | Contact sports. Minimum of 6 years. |
| Military service. | |
| Other repetitive traumatic brain injury exposures (eg, domestic abuse) | |
| 2. Other neurological disorder that likely accounts for all clinical features | |
| Exclude if | A single traumatic brain injury. |
| Or persistent post‐concussion syndrome. | |
| Can be present | Substance abuse. |
| Post‐traumatic stress disorder. | |
| Mood/anxiety disorders. | |
| Other neurodegenerative diseases. | |
| 3. Clinical features must be present for a minimum of 12 months | |
| 4. “Core clinical features” of traumatic encephalopathy syndrome | |
| At least one must be present | Cognitive. Difficulties identified by standardized mental status or cognitive neuropsychological test at least 1.5. standard deviation below normal |
| Behavioral. Described as explosive, short fuse, out of control, physically and/or verbally violent. Or intermittent explosive disorder. | |
| Mood. Feeling overly sad, depressed or hopeless. Or diagnosis of major depressive disorder or persistent depressive disorder. | |
| 5. “Supportive features” of traumatic encephalopathy syndrome | |
| At least two must be present | Documented decline (1 year), delayed onset, impulsivity, anxiety, apathy, paranoia, suicidality, headache, motor. |
Table 5.
Criteria for diagnostic subtypes with modifiers 61
| A. Traumatic encephalopathy syndrome diagnostic variants | ||
| Select one | “Cognitive” | Cognitive core features without behavioral/mood. |
| “Behavioral/mood” | Behavioral/mood core features without cognitive. | |
| “Mixed” | Both cognitive and behavioral/mood core features. | |
| “Dementia” | Progressive cognitive core and functional impairment. | |
| B. “With motor features” modifier | ||
| “With motor features” | Dysarthria, dysgraphia, bradykinesia, tremor, rigidity, gait change, falls and/or other features of parkinsonism. | |
| C. Clinical course modifier | ||
| Select one | “Stable” | History or tests indicate little if any change. |
| “Progressive” | Clear indication of progression over 2 years. | |
| “Unknown/inconsistent” | Unknown or inconsistent information | |
Additional biomarker evidence is required to indicate the likelihood that the etiology underlying TES is caused by the CTE pathology. Several potential biomarkers for “probable CTE,” “possible CTE” and “unlikely CTE” are proposed based on recent and ongoing biomarker research ( Table 6) 4, 59, 80. Additional research is needed to validate the usefulness of the proposed biomarkers for CTE. Efforts to validate the utility of the proposed clinical criteria 61 are currently underway (Table 4, 5, 6).
Table 6.
Chronic traumatic encephalopathy (CTE) likelihood criteria 61
| “Probable CTE” | Does not satisfy criteria for another disorder more consistently | |
| Meets classification for any TES variant. | ||
| Progressive course. | ||
| At least one positive “potential biomarker” | Positive PET tau imaging. | |
| Negative PET amyloid imaging. | ||
| Normal beta‐amyloid CSF levels. | ||
| Elevated CSF p‐tau/tau ratio. | ||
| Cavum septum pellucidum. | ||
| Cortical thinning or atrophy. | ||
| “Possible CTE” | May satisfy diagnostic criteria another disorder. | |
| Meets classification for any TES variant. | ||
| Progressive course. | ||
| No testing or one negative biomarker except for PET tau. | ||
| “Unlikely CTE” | Does not meet general criteria (1–5) for TES. | |
| Or has had negative PET tau imaging. | ||
Abbreviations: CSF = cerebrospinal fluid; PET = positron emission tomography; TES = traumatic encephalopathy syndrome.
Treatment and disease management
The treatment of CTE is currently theoretical and remains to be validated with prospective treatment trials. Treatment and management are likely to vary from case to case. Once deficits related to CTE appear, rehabilitation and medications to treat specific symptoms may still be useful 49. For behavioral and/or mood issues (ie, aggression, violence) potentially useful treatments may include antipsychotics, lithium, antidepressants, sedatives, anxiolytics, anticonvulsants, opiate antagonists and beta blockers 22, 56. To reduce drug‐induced extrapyramidal symptoms, risperidone and other neuroleptics drugs can be useful to treat behavioral issues (ie, psychotic behavior) 56. In a limited number of cases treated for psychotic symptoms, medications such as trifluoperazine were effective 22, 36. Methylphenidate may be used to treat apathy, as well as cognitive symptoms 35, 49. Anti‐parkinsonian medications have had mixed results in case reports 41, 56. One case report documented successful stereotactic surgical treatment of parkinsonian features 7. Cholinergic dysfunction is thought to underlie cognitive impairments in CTE 90, however, the use of anti‐cholinergic treatments (ie, tacrine, donepezil) for CTE remains speculative and further investigation is required 42, 66. In another case report, verbal memory, but not visual memory, improved following treatment with physostigmine and lecithin 42. Additionally, there are no known preventative interventions for CTE, although it has been suggested that treatments demonstrating effectiveness for AD and TBI might also show promise for CTE 49. For example, one candidate is amantadine, which is considered to be a safe and effective treatment for severe TBI 68. Recent preliminary investigations have suggested that nonpharmacological interventions may benefit contact sport athletes. Studies of professional football players report statistically significant improvements in neurocognitive function for up to 6 months with dietary supplementation (eg, omega‐3 fatty acids) 1, 78. Additional research is needed to determine whether or not reported improvements were maintained in the long term 49. Because of the severe behavioral mood manifestations of this disease, counseling and cognitive behavioral therapy may also help to mitigate the aberrant behavioral and mood manifestations of this disease 49.
CTE then and now
Recently, certain authors have made a distinction between “classic” and “modern” descriptions of CTE 30, 51. The “classic” entity, proposed by McCrory et al, is defined by the cases reported by Roberts and Corsellis et al in their boxing subjects 25, 71. This particular description highlighted early cases that had prominent motor features, including dysarthria, difficulties with gait and pyramidal problems. It was noted that early reports in boxers identified progression in “the physical signs and problems, but not the cognitive deficits” (p. 2) 30, which is the distinguishing factor from what are considered to be “modern” CTE cases. Alternatively, “modern” cases (ie, cases published after 2004) are characterized by prominent mood, behavior and progressive cognitive features, but with a reduced frequency of motor symptoms. Our assessment of the evidence suggests that this distinction between “classic” and “modern” CTE presentations is largely an artifact of review bias. The first source of this bias involves the “classic CTE” article by Roberts, who writes: “more attention has been paid, intentionally, to the clinical signs which indicate lesions of cerebellar, pyramidal and extra‐pyramidal systems, than to the evidence of dementia or personality change … (for) lesions in these systems are readily comparable … Leaving aside for later consideration the question of dementia and psychiatric disturbance, which undoubtedly occurs” (p. 47) 71. This methodological limitation prevents any reasonable inferences about the frequency of behavior, mood and cognitive symptoms in classic cases. Although, “in the first case described,” Roberts emphasized that in addition to motor features “dementia had clearly progressed over the years” with “development of a paranoid illness” (p. 44) 71. This evidence does not support the definition of “classic” CTE, rather it suggests that some of the perceived differences in symptoms reported in earlier cases were caused by the methodological limitations and biased review. For instance, the distinction between “classic” and “modern” presentations also does not account for possible group effects related to different sport exposures, that is, the “classic” presentation is derived from boxers while the “modern” presentation is predominantly derived from American football players 61. It is our hypothesis that sport‐specific differences in exposure alter the course and severity of certain clinical manifestations in CTE. In the sections that follow, we explore our hypothesis by re‐examining the CTE case evidence in boxers and football players previously reported by McKee et al 52, 53 and provide two detailed clinical and pathological case reports from two professional level athletes, a former US professional lightweight boxing champion and an American NFL player.
Boxing and American Football
Trauma risk factors for CTE
All reported neuropathologically confirmed CTE cases have a significant history of brain trauma, usually repetitive, which suggests that RTBI is a necessary factor in acquirement of CTE degenerative pathology. Not every case of CTE has a history of concussions, leading to the belief that subconcussive impacts may be sufficient to induce neuronal degeneration and subsequent neurodegeneration. Alternately, not every individual that is exposed to RTBI, either concussive or subconcussive, necessarily develops CTE 37. It is not known what specific aspect of exposure (sport, age, level, position, severity, frequency and mechanics) influences the risk of acquiring CTE. The threshold of damage required for induction and progression of tau pathology is likely multifactorial and may incorporate genetic, environmental and/or nutritional factors 85. Most of what we know about CTE comes from limited information provided in post‐mortem case series. Investigations with confirmed cases have identified factors that influence the severity 52 and phenotype 86 of CTE pathology. In American football players with neuropathologically confirmed CTE, there is a positive correlation with the severity of pathology and the total number of years played (Spearman's test, r = 0.805, P < 0.0001), as well as years since retirement (Spearman's test, r = 0.753, P < 0.0001) and age at death (Spearman's test, r = 0.806, P < 0.0001) 52. Conversely, informant‐reported number of concussions (Spearman's test, r = 0.259, P = 0.184), years of education (Spearman's test, r = 0.258, P = 0.134) and lifetime steroid use (Wilcoxon–Mann–Whitney test, P = 0.731) were not significantly correlated. In boxers with neuropathologically confirmed CTE, the severity of the tau pathology appears to correlate with the total number of years exposed 53.
Impact type and biomechanics
The types of impacts athletes endure differ by sport. However, each impact is composed of both linear and rotational forces 13, 14.
Rotational acceleration (Figure 4A,B) occurs when a force is eccentric or tangential to the center of gravity of the head. In boxing, when a punch is directed toward the lateral side of an opponent's face (ie, “hook punch”) or chin (ie, “upper cut”), the force of the impact will cause the head to twist and rotate outward along the fixed spinal axis (Figure 4A) 13, 38. The sudden rotational acceleration of the skull and hyperextension of the neck causes brain deformation at mechanically rigid inflection points, such as the cerebellopontine angle (Figure 4B). Regions of the brain that are composed of multiple tissue types with different tensile strengths near rigid boney structures, such as the midbrain, are particularly vulnerable to shearing forces that stretch and injure vessels, axons and glia. The greatest risk a boxer faces for concussion is the result of impacts that generate rotational accelerations, such as the hook punch 8. Linear acceleration (Figure 4C,D) occurs when a force is applied directly through the center of gravity of the head, such as in the anterior to posterior direction 14, 67. Investigations with accelerometers placed in helmets have shown that the majority of impacts in American‐style football occur from helmet‐to‐helmet impacts at the top‐front of the helmet (Figure 4C). This is true of all levels (ie, youth and professional) and positions, with the exception being the quarterback position. The forces generated in this type of impact are absorbed at the point of origin, which is nearest the frontal lobes (anterior), and transmitted through the brain, down to the brainstem and cerebellum (‐posterior) (Figure 4D). The helmet‐to‐helmet impacts in American football generate a larger net linear acceleration experienced by the frontal lobes, when compared with the net rotational acceleration generated by the hook punch in boxing 91. The significance of impact differences between sports on long‐term consequences and neurodegeneration is not yet known. Cumulative exposure to different impact types has the potential to influence the onset, type, location, severity and progression of underlying neuropathology.
Figure 4.
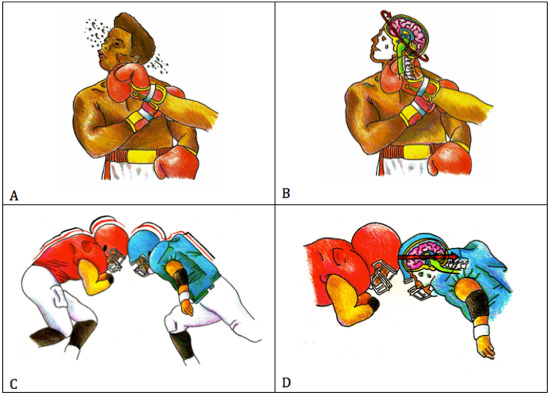
Impact mechanics in boxing and football. Hook punch (A). Primarily rotational acceleration (B). Helmet‐to‐helmet impact (C). Primarily linear acceleration (D). Figures A and C each depict the impact type that typically leads to injury, including concussions, for each sport, respectively. Figures B and D depict the biomechanics of each impact, including the predominant acceleration involved and its transmission into brain.
CTE in boxers and American football players
As the types of impact and predominant forces differ between sports (Figure 4), it was hypothesized 61 that the phenotype of CTE would also differ with the type of sport exposure. Some authors question whether the underlying pathology between CTE in American football players and boxers is really the same 8, 30, 51, 63. To explore this issue and to test our hypothesis, we compared the frequency and severity of motor symptoms in addition to cerebellar pathology in professional boxers and professional American football players with neuropathologically confirmed CTE previously described by McKee et al 52. Because of the spectrum of pathology in CTE stages I through IV, cases were grouped into early CTE (stages I and II) and late CTE (stages III and IV). In CTE, neurofibrillary tangle (NFT) lesions progress from a multifocal state (stage II) to a widespread state (stage III) 55. This grouping allowed us to be reasonably confident that any significant difference between groups was caused by the differences in sport exposure and not the natural progression of CTE disease (Table 7).
Table 7.
Comparison of chronic traumatic encephalopathy (CTE) clinicopathological features in boxers vs. football players
| Novel new (secondary) analysis of previously reported cases in McKee et al 52 | Pro Football | Pro Boxing |
|---|---|---|
| Late stage CTE (III–IV) | 25 | 7 |
| Mean decade age at symptom onset | 50–60 | 50–60 |
| Mean decade age at time of death | 70–80 | 80–90 |
| aLate stage CTE “with motor features” | 18.8% (3/16)b | 83% (5/6)b |
| Late stage CTE with cerebellar dentate neurofibrillary tangles | 57% (12/21) | 71% (5/7) |
| Late stage CTE with severe (++/+++) dentate neurofibrillary tangles | 17% (2/12)b | 80% (4/5)b |
Motor symptoms include parkinsonism, gait changes and dysarthria, unrelated to motor neuron disease.
Statistically significant difference in the proportions between groups (P < 0.05, Fisher's exact test).
Our analysis found that the proportion of boxers with motor symptoms (parkinsonism, gait changes and dysarthria) was significantly greater (Table 7 , P < 0.05, Fisher's exact test) than the proportion in American football players. The proportion of boxers identified with NFT pathology in their cerebellum dentate (71%) was also greater than American football players (57%), although this trend did not quite reach significance. However, we also compared the proportion of cases that had severe (++/+++) NFT pathology in the cerebellum dentate and found that boxers had significantly more severe NFT deposition (Table 7, P < 0.05, Fisher's exact test) than American football players. The lack of significance in frequency of cerebellar involvement between the two sports suggests either one of two possibilities: (i) our sample size was too small to detect the difference or (ii) the severe NFT deposition in the cerebellum dentate of boxers reached clinical thresholds for motor symptoms, whereas in football players it did not. In summary, the results of our secondary analysis (Table 7) of previously reported CTE cases 52 supports the hypothesis that impacts in boxing cause greater strain to the midbrain and cerebellum (Figure 4B) than and therefore having a greater frequency of motor symptoms in boxers with confirmed CTE than football players. This was also evidenced by the severity of CTE NFT pathology in the cerebellum dentate. Although preliminary, these results suggest that different biomechanic exposure profiles influence the risk for specific CTE phenotypes and alter the underlying pathological severity.
Illustrative case studies
The history and clinical presentation of the following cases were obtained through post‐mortem telephone interviews with family members while the interviewer remained blind to neuropathological diagnosis and apolipoprotein E status. Semi‐structured interviews and well‐characterized informant questionnaires were utilized in combination with medical records. For a detailed description of the methods used, see Stern et al 86. Post‐mortem pathological analysis and diagnosis methods are described in McKee et al 52.
NFL player, case 1
Clinical history: 56‐year‐old Caucasian male with a history of high blood pressure and a basal ganglia cerebral vascular accident at 54 and death from a myocardial infarction at age 56. He participated in American football for a total of 17 years, beginning at age 12, and played for 7 years in the NFL at the running back and full back positions. He endured approximately 20 concussions, 10 in the NFL level and five in college. Two concussions resulted in loss of consciousness. At 54, he awoke with symptoms of leg weakness and an MRI demonstrated “a small lacunar infarct to the basal ganglia.” Post‐stroke changes were mild and stable, with slowness in movement and complaints of fatigue. After 3 months of rest, he returned to his work as a bank vice president and did well. Around the time that he retired from the NFL, he began to experience recurring posterior headaches, with onset at age 32. Family informants reported that his mood changed in his mid‐30s and indicated that he struggled with feelings of sadness and depression. His depression was further supported with the informant short version of the geriatric depression scale (GDS) 11 (total GDS = 6), as well as having been prescribed with Wellbutrin. In the year prior to death, he developed a sense of worthlessness and hopelessness. His behavior was characterized as emotionally explosive and easily frustrated. He would often have sudden angry reactions and “little things would set him off.” Overall, he had a “short fuse” and was easily frustrated. Difficulties with rage did not worsen; however, in the year prior to his death, he began acting out of character, becoming somewhat detached and started listening to old music. Post‐stroke neurology follow‐up visits indicated improvement in motor symptoms and family informants reported no new symptoms in the time leading up to his death. A change in his cognitive function was observed by family informants during the 5 years leading up to his death. The family version of the cognitive difficulties scale identified mild to moderate cognitive difficulties in short‐term memory, attention and concentration and language 28, 82. Several of these changes were reported to have occurred prior to his stroke and with progression. In contrast, results on the functional activities questionnaire (total score = 0) and the modified AD8 informant interview for dementia (total score = 1) did not indicate significant functional impairment or dementia 29, 69. He did not serve in the military nor did he receive a diagnosis related to dementia, parkinsonism or AD.
Clinical diagnosis 61: TES mixed variant; progressive course; possible CTE.
Pathological findings: brain weight was 1550 g. Gross findings include a corpus callosum that is thinned throughout its extent, ventricular enlargement, pallor of the substantia nigra and pallor of the locus coeruleus. Microscopic (Figure 5A–D) findings were diagnostic of CTE 52 and included numerous tau‐immunoreactive NFTs, neurites and astrocytic tangles in the perivascular, sulcal depth, superficial cortical, subpial and glial distribution. No beta‐amyloid protein was found.
Figure 5.
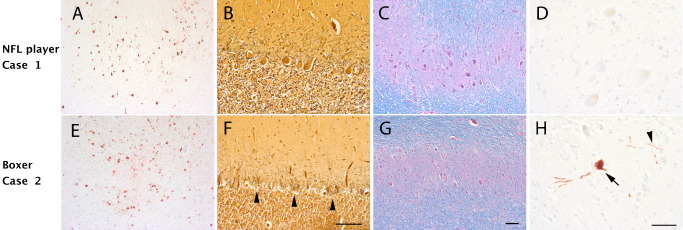
Neuropathology and cerebellar degeneration in a former professional boxer and football player. Case 1 (A–D): A 56‐year‐old former National Football League (NFL) player had chronic traumatic encephalopathy (CTE) characterized by phosphorylated tau‐positive neurofibrillary tangles present at the sulcal depths (A, AT8 immunostain). His cerebellum appeared intact with a well‐populated Purkinje cell layer (B, Bielschowsky silver stain) and dentate nucleus (C, Luxol hematoxylin and eosin). There was no phosphorylated tau accumulation present within the dentate nucleus (D). Case 2 (E–H): A 61‐year‐old former professional boxer had a similar degree of CTE‐related tauopathy (E, AT8 immunostain). In fact, both the NFL player and the boxer met neuropathological criteria 49 for CTE stage III tauopathy. However, in contrast to the NFL player, there is marked degeneration within the cerebellum with marked Purkinje cell loss (F, Bielschowsky silver stain; arrowhead, Basket cell processes without Purkinje cells “empty Baskets”). There is also loss of neurons within the dentate nucleus (G), which contains scattered phosphorylated tau‐positive neurons (H, arrow) and processes (H, arrowhead). These two cases illustrate the need for careful clinical phenotyping and demonstrate the utility of recently published criteria 61. Scale bars, A, C, E and G, 100 μm; B and F, 100 μm; D and H, 50 μm.
Pathology diagnoses 52: CTE stage III/IV; vascular disease.
Boxer, case 2
Clinical history: 61‐year‐old Caucasian male with a history of hepatitis C virus and significant liver cirrhosis that led to a transplant at age 58. The subject died from liver failure and complications of pneumonia. He was a former professional boxer and a US lightweight boxing champion. His boxing career began when he was 8, lasting a total of 26 years. He went on to win the Golden Gloves championship and was ranked the top US lightweight boxing champion for several years, with a total of 36 professional bouts in his 12 years at the professional level. He retired at age 34. Over the course of his career, he reported that he had suffered multiple concussions with only one with loss of consciousness, although the specific number of concussions he experienced is not known. He was also involved in one motor vehicle accident at the age of 56, at which time he experienced headaches, backache and signs of a concussion. A noncontrast brain CT scan found no acute intracranial process and therefore he “did not require medical care.” His brain CT did, however, discover that his ventricles and sulcal spaces showed prominent “cerebral atrophy,” including decreased attenuation within the periventricular white matter suggestive of small vessel disease, both of “uncertain significance.” There was also evidence of chronic fractures in the nasal and orbital bones. He did not serve in the military nor did he receive a diagnosis related to dementia, parkinsonism or AD. Family informants described him as being violent and hot‐headed since childhood, but noted that his behavior deteriorated significantly starting at age 30, with onset of impulsivity, spousal abuse, intermittent explosivity and generally being out of control. In his early 50s, his mood was depressed and he developed feelings of hopelessness and worthlessness. At age 55, cognitive symptoms related to executive dysfunction became apparent to the family. At age 56, an altercation with his daughter lead to a formal psychiatric consultation, which noted he had “increased memory problems (frequently forgets placement of personal items and previous conversations),” as well as worsening dysphoria, irritability and paranoid delusions that his girlfriend was attempting to kill him. He also suggested that the boxing association had officially diagnosed him with “pugilistic dementia.” In his late 50s he reported falling at home from “feeling unsteady and weak.” Motor impairments were obvious in the year prior to his death; he was never diagnosed with parkinsonism or ALS, however, he lost ability to speak, developed dysphagia for food and liquids and his handwriting deteriorated.
Clinical diagnosis 61: TES mixed variant; with motor features; progressive course; probable CTE.
Pathological findings: brain weight was 1230 g. Gross findings include generalized atrophy of the cerebral cortex, atrophy, atrophy of the fornix, cavum septum (1.0 cm), multiple septal fenestrations posteriorly and atrophy of the thalamus and mammillary bodies. There was pallor in the substantia nigra and locus coeruleus. Microscopic (Figure 5E–H) findings were diagnostic of CTE 52 and included numerous tau‐immunoreactive NFTs, neurites and astrocytic tangles in the perivascular, sulcal depth, superficial cortical, subpial and glial distribution. No beta‐amyloid protein was found. The tau pathology in the substantia nigra, basis pontis and cerebellum was particularly severe. There was mild proliferation of protoplasmic astrocytes consistent with hepatic disease.
Pathological diagnosis 52: CTE with classic microscopic of stage III/IV with mild transactive response DNA binding protein (TDP‐43) proteinopathy.
Conclusions and Future Directions
Participating in a contact sport is now thought to increase an individual's risk for later‐life impairment and possibly developing CTE. CTE has been diagnosed in a wide range of individuals with a history of head trauma, including American football players, soccer and hockey players, boxers, wrestlers and soldiers who have received battlefield injuries. It has even been reported in athletes as young as 17, who only played sports in high school or college. Given the wide range of people that are diagnosed and the recent increase in reported cases, CTE is likely more prevalent than previously thought. To date, the only definitive means to diagnose CTE is through post‐mortem autopsy. The neuropathological features of CTE are increasingly well‐characterized, yet the clinical aspects of CTE require further elucidation. There is an urgent need for methods that can reliably diagnose CTE during life. To address this gap, new clinical research diagnostic criteria for CTE were proposed and studies to validate its utility are ongoing.
Disclosure Statement
P. Montenigro reports no disclosures. C. Bernick receives research support for the Professional Fighters Brain Health study from Top Rank Promotions, Golden Boy Promotions, Bellator/Spike TV, Zuffa. R. Cantu receives compensation from the National Football League as Senior Advisor to the Head Neck and Spine Committee, from the National Operating Committee on Safety of Athletic Equipment as Chairman of the Scientific Advisory Committee and from Sports Legacy Institute as co‐founder and Medical Director for some talks given and research conducted. He receives royalties from Houghton Mifflin Harcourt and compensation from expert legal opinion.
Acknowledgments
The authors thank and gratefully acknowledge the extraordinary contribution of neuropathologist Dr. Thor D. Stein, Dr. Ann C. McKee and all other members of the VA Boston Brain Bank, as well as the individuals and families whose participation and donation made this work possible. Additionally, we gratefully acknowledge the work of Dr. Robert A. Stern, Christopher Nowinski, Lisa McHale and the members of the Chronic Traumatic Encephalopathy Center at Boston University. Medical illustrations by Danielle Eble (dje3@bu.edu).
References
- 1. Amen DG, Wu JC, Taylor D, Willeumier K (2011) Reversing brain damage in former NFL players: implications for traumatic brain injury and substance abuse rehabilitation. J Psychoactive Drugs 43:1–5. [DOI] [PubMed] [Google Scholar]
- 2. American Psychiatric Association (1994) Diagnostic and statistical manual of mental disorders. DSM‐IV, Washington, DC.
- 3. Baugh CM, Kiernan PT, Kroshus E, Daneshvar DH, Montenigro PH, McKee AC, Stern R (2014) Frequency of head impact related outcomes by position in NCAA Division I collegiate football players. J Neurotrauma 32(5):314–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baugh CM, Robbins CA, Stern RA, McKee AC (2014) Current understanding of chronic traumatic encephalopathy. Curr Treat Options Neurol 16:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bernick C, Banks S (2013) What boxing tells us about repetitive head trauma and the brain. Alzheimers Res Ther 5:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bernick C, Banks S, Phillips M, Lowe M, Shin W, Obuchowski N et al (2013) Professional fighters brain health study: rationale and methods. Am J Epidemiol 178:280–286. [DOI] [PubMed] [Google Scholar]
- 7. Betti O, Ottino C (1969) Pugilistic encephalopathy. Acta Neurol Latinoam 15:47–51. [PubMed] [Google Scholar]
- 8. Blennow K, Hardy J, Zetterberg H (2012) The neuropathology and neurobiology of traumatic brain injury. Neuron 76:886–899. [DOI] [PubMed] [Google Scholar]
- 9. Boden BP, Tacchetti RL, Cantu RC, Knowles SB, Mueller FO (2007) Catastrophic head injuries in high school and college football players. Am J Sports Med 35:1075–1081. [DOI] [PubMed] [Google Scholar]
- 10. Brandenburg W, Hallervorden J (1954) Dementia pugilistica mit anatomischem Befund. Virchows Arch 325:680–709. [DOI] [PubMed] [Google Scholar]
- 11. Brown LM, Schinka JA (2005) Development and initial validation of a 15‐item informant version of the Geriatric Depression Scale. Int J Geriatr Psychiatry 20:911–918. [DOI] [PubMed] [Google Scholar]
- 12. Cantu R (1986) Guidelines for return to contact sports after a cerebral concussion. Physician Sportsmed 14:75. [DOI] [PubMed] [Google Scholar]
- 13. Cantu R, Hyman M (2012) Concussions and Our Kids. Houghton Mifflin: New York, New York. [Google Scholar]
- 14. Cantu RC (2000) Biomechanics of head injury. In: Neurologic Athletic Head and Spine Injuries. Cantu RC (ed.), pp. 2–5. WB Saunders Company: Philadelphia. [Google Scholar]
- 15. Cantu RC (2006) An overview of concussion consensus statements since 2000. Neurosurg Focus 21:1–6. [DOI] [PubMed] [Google Scholar]
- 16. Cantu RC (2013) Role of diffusion tensor imaging MRI in detecting brain injury in asymptomatic contact athletes. World Neurosurg 80:792–793. [DOI] [PubMed] [Google Scholar]
- 17. Cantu RC (2013) The role of the neurologist in concussions: when to tell your patient to stop. JAMA Neurol 70:1481–1482. [DOI] [PubMed] [Google Scholar]
- 18. Cantu RC, Guskiewicz K, Register‐Mihalik JK (2010) A retrospective clinical analysis of moderate to severe athletic concussions. PM R 2:1088–1093. [DOI] [PubMed] [Google Scholar]
- 19. Casson IR, Sham R, Campbell EA, Tarlau M, Didomenico A (1982) Neurological and CT evaluation of knocked‐out boxers. J Neurol Neurosurg Psychiatr 45:170–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cava L (1952) The injuries of boxing. Deutsch Sportéirzte Kongress 79:817. [Google Scholar]
- 21. Constantinides J, Tissot R (1967) [Generalized Alzheimer's neurofibrillary lesions without senile plaques. (Presentation of one anatomo‐clinical case)]. Schweiz Arch Neurol Neurochir Psychiatr 100:117–130. [PubMed] [Google Scholar]
- 22. Cordeiro Júnior Q, Oliveira AMD (2001) Parkinsonian, cerebellar, psychotic and demential symptoms in ex‐boxer: case report. Arq Neuropsiquiatr 59 (2A):283–285. [PubMed] [Google Scholar]
- 23. Corrigan JD, Bogner J (2007) Initial reliability and validity of the Ohio State University TBI identification method. J Head Trauma Rehabil 22:318–329. [DOI] [PubMed] [Google Scholar]
- 24. Corrigan JD, Bogner J (2007) Screening and identification of TBI. J Head Trauma Rehabil 22:315–317. [DOI] [PubMed] [Google Scholar]
- 25. Corsellis J, Bruton C, Freeman‐Browne D (1973) The aftermath of boxing. Psychol Med 3:270–303. [DOI] [PubMed] [Google Scholar]
- 26. Critchley M (1949) Punch‐drunk syndromes: the chronic traumatic encephalopathy of boxers. In: Hommage a Clovis Vincent. pp. 131–141. Maloine: Paris. [Google Scholar]
- 27. Critchley M (1957) Medical aspects of boxing, particularly from a neurological standpoint. Br Med J 1:357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Crook T, Ferris S, Bartus R (1983) Assessment in geriatric Psychopharmacology. Mark Powley Associates: New Canaan, Conn. [Google Scholar]
- 29. Galvin J, Roe C, Powlishta K, Coats M, Muich S, Grant E et al (2005) The AD8: a brief informant interview to detect dementia. Neurology 65:559–564. [DOI] [PubMed] [Google Scholar]
- 30. Gardner A, Iverson GL, McCrory P (2014) Chronic traumatic encephalopathy in sport: a systematic review. Br J Sports Med 48:84–90. [DOI] [PubMed] [Google Scholar]
- 31. Giza C, Kutcher J, Ashwal S (2013) Guideline Development Subcommittee of the American Academy of Neurology. Summary of evidence‐based guideline update: evaluation and management of concussion in sports. Neurology 80:2250–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Giza CC, Griesbach GS, Hovda DA (2005) Experience‐dependent behavioral plasticity is disturbed following traumatic injury to the immature brain. Behav Brain Res 157:11–22. [DOI] [PubMed] [Google Scholar]
- 33. Grahmann H, Ule G (1957) [Diagnosis of chronic cerebral symptoms in boxers (dementia pugilistica & traumatic encephalopathy of boxers).]. Psychiatr Neurol (Basel) 134:261–283. [PubMed] [Google Scholar]
- 34. Grinnon ST, Miller K, Marler JR, Lu Y, Stout A, Odenkirchen J, Kunitz S (2012) National institute of neurological disorders and stroke common data element project–approach and methods. Clin Trials 9:322–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Handratta V, Hsu E, Vento J, Yang C, Tanev K (2010) Neuroimaging findings and brain‐behavioral correlates in a former boxer with chronic traumatic brain injury. Neurocase 16:125–134. [DOI] [PubMed] [Google Scholar]
- 36. Harvey P, Newsom Davis J (1974) Traumatic encephalopathy in a young boxer. Lancet 304:928–929. [DOI] [PubMed] [Google Scholar]
- 37. Hazrati LN, Tartaglia MC, Diamandis P, Davis KD, Green RE, Wennberg R et al (2013) Absence of chronic traumatic encephalopathy in retired football players with multiple concussions and neurological symptomatology. Front Hum Neurosci 7:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jayarao M, Chin LS, Cantu RC (2010) Boxing‐related head injuries. Physician Sportsmed 38:18–26. [DOI] [PubMed] [Google Scholar]
- 39. Jedliński J, Gatarski J, Szymusik A (1971) Encephalopathia pugilistica (punch drunkeness). Acta Med Pol 12:443–451. [PubMed] [Google Scholar]
- 40. Jordan B (1987) Neurologic aspects of boxing. Arch Neurol 44:453–459. [DOI] [PubMed] [Google Scholar]
- 41. Jordan BD (1992) Neurologic injuries in boxing. In: Medical Aspects of Boxing. Jordan Barry (ed.), pp. 150–152. CRC Press: Boca Raton, Florida. [Google Scholar]
- 42. Jordan BD (1998) Dementia pugilistica. In: Neurobiology of Primary Dementia. Folstein MD Marshal F. (ed.), pp. 191–203, American Psychiatric Press: Washington, DC. [Google Scholar]
- 43. Jordan BD (2000) Chronic traumatic brain injury associated with boxing. Semin Neurol 20:179–185. [DOI] [PubMed] [Google Scholar]
- 44. Jordan BD (2013) The clinical spectrum of sport‐related traumatic brain injury. Nat Rev Neurol 9:222–230. [DOI] [PubMed] [Google Scholar]
- 45. Kerr ZY, Marshall SW, Guskiewicz KM (2012) Reliability of concussion history in former professional football players. Med Sci Sports Exerc 44:377–382. [DOI] [PubMed] [Google Scholar]
- 46. Koerte IK, Kaufmann D, Hartl E, Bouix S, Pasternak O, Kubicki M et al (2012) A prospective study of physician‐observed concussion during a varsity university hockey season: white matter integrity in ice hockey players. Part 3 of 4. Neurosurg Focus 33:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lehman EJ (2013) Epidemiology of neurodegeneration in American‐style professional football players. Alzheimer Res Ther 5:34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lehman EJ, Hein MJ, Baron SL, Gersic CM (2012) Neurodegenerative causes of death among retired National Football League players. Neurology 79:1970–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Levin B, Bhardwaj A (2014) Chronic traumatic encephalopathy: a critical appraisal. Neurocrit Care 20:334–344. [DOI] [PubMed] [Google Scholar]
- 50. Marchi N, Bazarian JJ, Puvenna V, Janigro M, Ghosh C, Zhong J et al (2013) Consequences of repeated blood‐brain barrier disruption in football players. PLoS ONE 8:e56805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McCrory P, Meeuwisse WH, Kutcher JS, Jordan BD, Gardner A (2013) What is the evidence for chronic concussion‐related changes in retired athletes: behavioural, pathological and clinical outcomes? Br J Sports Med 47:327–330. [DOI] [PubMed] [Google Scholar]
- 52. McKee A, Stern R, Nowinski C, Stein T, Alvarez V, Daneshvar D et al (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain 136:43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McKee AC, Cantu RC, Nowinski CJ, Hedley‐Whyte ET, Gavett BE, Budson AE et al (2009) Chronic traumatic encephalopathy in athletes: progressive tauopathy following repetitive head injury. J Neuropathol Exp Neurol 68:709–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW et al (2010) TDP‐43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 69:918–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. McKee AC, Daneshvar DH, Alvarez VE, Stein TD (2014) The neuropathology of sport. Acta Neuropathol 127:29–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mendez MF (1995) The neuropsychiatric aspects of boxing. Int J Psychiatr Med 25:249–262. [DOI] [PubMed] [Google Scholar]
- 57. Meyers KR, Dorencamp DG, Suzuki K (1974) Amyotrophic lateral sclerosis with diffuse neurofibrillary changes: report of a case. Arch Neurol 30:84–89. [DOI] [PubMed] [Google Scholar]
- 58. Mez J, Stern RA, McKee AC (2013) Chronic traumatic encephalopathy: where are we and where are we going? Curr Neurol Neurosci Rep 13:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mitsis E, Riggio S, Kostakoglu L, Dickstein D, Machac J, Delman B et al (2014) Tauopathy PET and amyloid PET in the diagnosis of chronic traumatic encephalopathies: studies of a retired NFL player and of a man with FTD and a severe head injury. Transl Psychiatr 4:e441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Montenigro P, Stern R (2014) Author response clinical presentation of chronic traumatic encephalopathy. Neurology 83:1992–1993. [PubMed] [Google Scholar]
- 61. Montenigro P, Baugh C, Daneshvar D, Mez J, Budson A, Au R et al (2014) Clinical subtypes of chronic traumatic encephalopathy: literature review and proposed research diagnostic criteria for traumatic encephalopathy syndrome. Alzheimers Res Ther 6:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Montenigro P, Corp D, Stein T, Cantu R, Stern R (2015) Chronic traumatic encephalopathy: historical origins and current perspective. Annu Rev Clin Psychol doi: 10.1146/annurev-clinpsy-032814-112814 [DOI] [PubMed] [Google Scholar]
- 63. Ng TS, Lin AP, Koerte IK, Pasternak O, Liao H, Merugumala S et al (2014) Neuroimaging in repetitive brain trauma. Alzheimers Res Ther 6:10–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH (2005) Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 57:128–134, discussion ‐34. [DOI] [PubMed] [Google Scholar]
- 65. World Health Organization (1992) The ICD‐10 classification of mental and behavioural disorders: clinical descriptions and diagnostic guidelines http://www.who.int/classifications/icd/en/bluebook.pdf
- 66. Otero Siliceo E, Padilla Rubio J (2004) Dementia pugilistica 1a. parte. Arch Neurocien (México, DF) 9:114–119. [Google Scholar]
- 67. Pellman EJ, Viano DC, Tucker AM, Casson IR, Waeckerle JF (2003) Concussion in professional football: reconstruction of game impacts and injuries. Neurosurgery 53:799–814. [DOI] [PubMed] [Google Scholar]
- 68. Petraglia AL, Maroon JC, Bailes JE (2012) From the field of play to the field of combat: a review of the pharmacological management of concussion. Neurosurgery 70:1520–1533. [DOI] [PubMed] [Google Scholar]
- 69. Pfeffer R, Kurosaki T, Harrah C, Chance J, Filos S (1982) Measurement of functional activities in older adults in the community. J Gerontol 37:323–329. [DOI] [PubMed] [Google Scholar]
- 70. Robbins CA, Daneshvar DH, Picano JD, Gavett BE, Baugh CM, Riley DO et al (2014) Self‐reported concussion history: impact of providing a definition of concussion. Open Access J Sports Med 5:99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Roberts A (1969) Brain Damage in Boxers: A Study of the Prevalence of Traumatic Encephalopathy among ex‐Professional Boxers. Pitman Medical & Scientific Publishing Co: London. [Google Scholar]
- 72. Ross RJ, Cole M, Thompson JS, Kim KH (1983) Boxers—computed tomography, EEG, and neurological evaluation. JAMA 249:211–213. [DOI] [PubMed] [Google Scholar]
- 73. Sayed N, Culver C, Dams‐O'Connor K, Hammond F, Diaz‐Arrastia R (2013) Clinical phenotype of dementia after traumatic brain injury. J Neurotrauma 30:1117–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Seichepine DR, Stamm JM, Daneshvar DH, Riley DO, Baugh CM, Gavett BE et al (2013) Profile of self‐reported problems with executive functioning in college and professional football players. J Neurotrauma 30:1299–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Shin W, Mahmoud S, Sakaie K, Banks S, Lowe M, Phillips M et al (2014) Diffusion measures indicate fight exposure–related damage to cerebral white matter in boxers and mixed martial arts fighters. AJNR Am J Neuroradiol 35:285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sim A, Terryberry‐Spohr L, Wilson KR (2008) Prolonged recovery of memory functioning after mild traumatic brain injury in adolescent athletes. J Neurosurg 108:511–516. [DOI] [PubMed] [Google Scholar]
- 77. Singh R, Meier TB, Kuplicki R, Savitz J, Mukai I, Cavanagh L et al (2014) Relationship of collegiate football experience and concussion with hippocampal volume and cognitive outcomes. JAMA 311:1883–1888. [DOI] [PubMed] [Google Scholar]
- 78. Sinnott RA, Maddela RL, Bae S, Best T (2012) Dietary supplementation and the quality of life of retired football players. J Int Soc Sports Nutr 9(Suppl. 1):P28. [Google Scholar]
- 79. Slobounov S, Bazarian J, Bigler E, Cantu R, Hallett M, Harbaugh R et al (2013) Sports‐related concussion: ongoing debate. Br J Sports Med 48:75–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Small GW, Kepe V, Siddarth P, Ercoli LM, Merrill DA, Donoghue N et al (2013) PET scanning of brain tau in retired national football league players: preliminary findings. Am J Geriatr Psychiatr 21:138–144. [DOI] [PubMed] [Google Scholar]
- 81. Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM et al (2011) Toward defining the preclinical stages of Alzheimer's disease: Recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 7:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Spitznagel MB, Tremont G (2005) Cognitive reserve and anosognosia in questionable and mild dementia. Arch Clin Neuropsychol 20:505–515. [DOI] [PubMed] [Google Scholar]
- 83. Stamm JM, Bourlas AP, Baugh CM, Fritts NG, Daneshvar DH, Martin BM et al (2015) Age of first exposure to football and later‐life cognitive impairment in former NFL players. Neurology 84:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Stein TD, Alvarez VE, McKee AC (2014) Chronic traumatic encephalopathy: a spectrum of neuropathological changes following repetitive brain trauma in athletes and military personnel. Alzheimers Res Ther 6:4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Stern RA, Riley DO, Daneshvar DH, Nowinski CJ, Cantu RC, McKee AC (2011) Long‐term consequences of repetitive brain trauma: chronic traumatic encephalopathy. PM R 3:S460–S467. [DOI] [PubMed] [Google Scholar]
- 86. Stern RA, Daneshvar DH, Baugh CM, Seichepine DR, Montenigro PH, Riley DO et al (2013) Clinical presentation of chronic traumatic encephalopathy. Neurology 81:1122–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Talavage TM, Nauman EA, Breedlove EL, Yoruk U, Dye AE, Morigaki KE et al (2014) Functionally‐detected cognitive impairment in high school football players without clinically‐diagnosed concussion. J Neurotrauma 31:327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Thorndike A (1952) Serious recurrent injuries of athletes contraindications to further competitive participation. N Engl J Med 247:554–556. [DOI] [PubMed] [Google Scholar]
- 89. Tremblay S, Henry LC, Bedetti C, Larson‐Dupuis C, Gagnon J‐F, Evans AC et al (2014) Diffuse white matter tract abnormalities in clinically normal ageing retired athletes with a history of sports‐related concussions. Brain 137:2997–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Uhl G, McKinney M, Hedreen J, White C, Coyle J, Whitehouse P, Price D (1982) Dementia pugilistica‐loss of basal forebrain cholinergic neurons and cortical cholinergic markers. Ann Neurol 12:99. [Google Scholar]
- 91. Viano DC, Casson IR, Pellman EJ, Bir CA, Zhang L, Sherman DC, Boitano MA (2005) Concussion in professional football: comparison with boxing head impacts—part 10. Neurosurgery 57:1154–1172, discussion ‐72. [DOI] [PubMed] [Google Scholar]
- 92. Victoroff J (2013) Traumatic encephalopathy: review and provisional research diagnostic criteria. Neurorehabilitation 32:211–224. [DOI] [PubMed] [Google Scholar]
- 93. Weir DR, Jackson JS, Sonnega A (2009) National football league player care foundation study of retired NFL players. Ann Arbor: University of Michigan Institute for Social Research.
- 94. Margulies SS, Kilbaugh T, Sullivan S, Smith C, Propert K, Byro M et al (2015) Establishing a clinically relevant large animal model platform for TBI therapy development: using cyclosporin a as a case study. Brain Pathology 25:289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Koerte IK, Lin AP, Willems A, Muehlmann M, Hufschmidt J, Coleman MJ et al (2015) A review of neuroimaging findings in repetitive brain trauma. Brain Pathology 25:304–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. McKee AC, Stein T, Kiernan P, Alvarez V (2015) The neuropathology of chronic traumatic encephalopathy. Brain Pathology 25:336–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Smith C (2015) The study and consequences of repetitive traumatic brain injury. Brain Pathology 25:287–288. [DOI] [PMC free article] [PubMed] [Google Scholar]