

Abstract
Dendritic alteration of striatal medium spiny neurons is one of the earliest morphological abnormalities in Huntington's disease (HD). The main microtubule‐associated protein in dendrites is MAP2. The low‐molecular weight isoforms of MAP2 (LMW‐MAP2) are the juvenile forms resulting from exclusion of the sequence encoded by exons E7‐E9 and are downregulated after the early stages of neuronal development when E7‐E9 exon‐including high‐molecular weight isoforms (HMW‐MAP2) are favored. Splicing alteration has recently been proposed to contribute to HD in view of two pathogenic missplicing events resulting in a highly toxic N‐terminal version of mutant huntingtin and in a detrimental imbalance in MAP Tau isoforms with three or four tubulin‐binding repeats. Both splicing events are postulated targets of the SR splicing factor SRSF6 which has recently been reported to be dramatically altered in HD. SR proteins often regulate functionally related sets of genes and SRSF6 targets are enriched in genes involved in brain organogenesis including several actin‐and tubulin‐binding proteins. Here we hypothesized that MAP2 might be target of SRSF6 and altered in HD. By SRSF6 knockdown in neuroblastoma cells, we demonstrate that splicing of MAP2 E7‐E9 exons is affected by SRSF6. We then show a disbalance in LMW and HMW MAP2 mRNA isoforms in HD striatum in favor of the juvenile LMW forms together with a decrease in total MAP2 mRNA. This is accompanied by a global decrease in total MAP2 protein due to almost total disappearance of HMW‐MAP2 isoforms with preservation of LMW‐MAP2 isoforms. Accordingly, the predominant dendritic MAP2 staining in striatal neuropil of control subjects is absent in HD cases. In these, MAP2‐immunoreactivity is faint and restricted to neuronal cell bodies often showing a sharp boundary at the base of dendrites. Together, our results highlight the importance of splicing alteration in HD and suggest that MAP2 alteration contributes to dendritic atrophy.
Keywords: dendrite, MAP2, Huntington's disease, splicing, SRSF6
Introduction
Huntington's disease (HD) is an autosomal dominant inherited neurodegenerative disorder 25. Neuropathological changes in HD are strikingly selective, with prominent cell loss and atrophy of the caudate and putamen in the corpus striatum. Other brain areas are also affected but less severely. The causing mutation is a CAG repeat expansion in the Huntingtin (HTT) gene (HDRCG, 1993) encoding a toxic expanded polyglutamine (polyQ) tract. HD thus belongs to the group of CAG trinucleotide repeat/PolyQ disorders 22. It has been suggested that the expanded CAG mRNA itself might also be a toxic entity by affecting the splicing machinery, as has been described for other trinucleotide repeat expansion disorders like myotonic dystrophy 1 that is caused by a CUG repeat 22, 23. In line with this, two splicing alterations have been described that may be important for the pathogenesis of HD; one in the HTT gene itself giving rise to a highly toxic N‐terminal form of mutant HTT 27 and more recently, we and others have demonstrated missplicing of the MAP Tau exon 10 in HD patients 7, 31. Both splicing events altered in HD are likely to be regulated by the splicing factor SRSF6 7, 27. Besides, we have reported profound alterations of SRSF6 (also known as SRp55) in the brain of HD patients as it accumulates into HTT‐inclusion bodies, its total level in the striatum is increased and its phosphorylation is highly increased 7. This robust alteration of SRSF6 in HD brains, made us hypothesize that many other alternatively spliced genes apart from HTT and MAP Tau will be altered and might also contribute to HD pathogenesis.
An emerging concept in splicing factor physiology is the possibility that these proteins regulate multiple functionally related genes as entire gene expression programs 1, 10. The closest homolog to MAP Tau is the microtubule‐binding protein MAP2 5. MAP2 is a neuronal cytoskeleton regulator that can bind both microtubules and F‐actin participating in nucleation and stabilization of microtubules and microfilaments. Particularly, it has been hypothesized to be a critical stabilizer of microtubules in mature dendrites and it may also participate in organelle transport and scaffold functions within this neuronal compartment. (5, 26). MAP2 has multiple isoforms divided in two groups: High Molecular Weight MAP2 isoforms (HMW MAP2) and Low Molecular Weight MAP2 isoforms (LMW MAP2) 26. HMW and LMW MAP2 isoforms share the N‐ and C‐terminal regions, however exons 7, 8 and 9 (according to Ensembl isoform MAP2‐001; Figure 1A) are skipped in LMW and, as a result of this alternative splicing, LMW isoforms lack the central domain (CD). Alternative splicing of MAP2 is regulated during neuronal development and is critical for its subcellular distribution. HMW isoforms are restricted to dendrites and cell bodies and are highly expressed during adulthood 28, whereas LMW isoforms are widely distributed in every neuronal compartment during development, and their expression in the adult brain is low except in the cerebellum 6. The subcellular localization of MAP2 in dendrites and cell bodies suggests a complementary function with MAP Tau, as Tau is the main microtubule‐associated protein expressed specifically in axons 5, 26.
Figure 1.
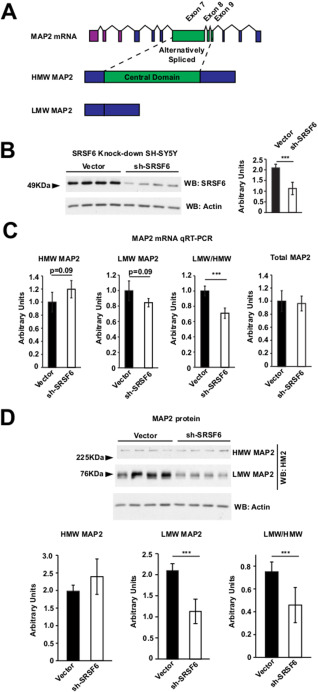
SRSF6 knockdown alters LMW/HMW MAP2 alternative splicing. A. Upper: scheme of human MAP2 exons using Ensembl human MAP2‐001 isoform as reference. Purple exons are 5’ UTR, blue exons are common to all MAP2 isoforms and green exons are alternatively spliced. Middle: HMW MAP2 isoform. Regions in blue are common to LMW MAP2 isoforms; regions in green are specific of HMW MAP2 isoforms. Lower: LMW MAP2 isoform. B. Western‐blot analysis of SRSF6 in SH‐SY5Y SRSF6 knocked‐down cells. Graphs represent the quantification of western‐blots. n = 3 with four technical replicates each condition. Experiment shown is representative of the three experiments performed. Student's t‐test, *** P < 0.001. C. Graphs show relative RT‐qPCRs performed to detect (from left) HMW MAP2, LMW MAP2 the ratio LMW/HMW and Total MAP2, in control or sh‐SRSF6 knockdown cells. n = 4 in every experiment with three technical replicates each condition. Experiment shown is representative of the two experiments performed. Student's t‐test, *** P < 0.001. D. Western‐blot analysis of MAP2 isoforms in SH‐SY5Y SRSF6 knocked‐down cells. Graphs represent the quantification of western‐blots. Last graph below right correspond to the ratio LMW/HMW. n = 3 with four technical replicates each condition. Experiment shown is representative of the three experiments performed. Student's t‐test, *** P < 0.001.
Dendrites of striatal medium‐size spiny neurons are altered in HD patients 9. In moderate grades of HD dendrites show recurving of distal segments, short‐segment branching and increased numbers and size of dendritic spines. In severe grade, truncated dendritic arborizations, occasional focal dendritic swellings and marked spine loss have been reported 9. These changes in dendritic morphology made us hypothesize that splicing of MAP2 isoforms might be deregulated in HD possibly as a consequence of SRSF6 alteration.
Here we show that SRSF6 knockdown alters the MAP2 alternative splicing event that originates HMW and LMW forms. We then investigated the status of MAP2 mRNA isoforms in HD brains, finding an imbalance in the MAP2 isoforms in the striatum of HD patients. More precisely, we found decreased HMW MAP2 mRNA and a tendency to increased LMW MAP2 mRNA, despite decreased total MAP2 mRNA. Accordingly, protein levels of MAP2 isoforms in the striatum of HD patients show a similar disbalance, with a clear reduction of the HMW MAP2 with a mild reduction of LMW MAP2 isoforms. This mRNA and protein disbalance correlates with abnormal distribution of MAP2 in HD striatal neurons. In the striatum of control subjects MAP2 immunoreactivity stains dendritic arbors and cell bodies excluding the nucleus. In good agreement with the biochemical data, HD striatal MAP2 immunoreactivity is decreased and restricted to neuronal cell bodies, as expected given the predominance of LMW MAP2 isoforms.
Materials and Methods
Human brain tissue samples
Brain specimens used in this study from HD patients and controls (frontal cortex and striatum) were provided by the Neurological Tissue Bank of the IDIBAPS Biobank (Barcelona, Spain), the Banco de Tejidos Fundación Cien (BT‐CIEN, Madrid, Spain) and the Netherlands Brain Bank (Amsterdam, The Netherlands). Written informed consent for brain removal after death for diagnostic and research purposes was obtained from brain donors and/or next of kin. Procedures, information and consent forms have been approved by the Medical Ethics Committee of the VU University Medical Center (Amsterdam, The Netherlands) and the Bioethics Subcommittee of Centro Superior de Investigaciones Científicas (Madrid, Spain).
Quantitative real‐time reverse transcriptase‐PCR
Total tissue RNA was extracted from striatum and cortex of HD and control subjects using the Maxwell® 16 LEV simplyRNA Tissue Kit (Promega, Madison, WI). The resulting total RNA (750 ng) was used for cDNA synthesis with a Super Script III First‐Strand Synthesis SuperMix kit from Invitrogene Invitrogen, Carlsbad (CA) (PN 11752250) with the amplification protocol 30” at 95°C + (5” at 95°C + 5” at 60°C) x 40 + (5” at 60°C + 5” at 95°C). Quantification was performed by real‐time PCR using a CFX 384 System (Bio‐Rad, Hercules (CA)) in combination with SsoFast Eva Green (Bio‐Rad), as per manufacturer's protocol and 1 µL of primer pair was used. Data were analyzed by GenEx 5.3.7 software (Multid AnaLyses AB). The mRNA levels were normalized first relative to total RNA and then relative to the mean of 18S ribosome subunit, GADPH, β‐Actin and β‐Tubulin gene expression in each sample. The PCR primers used for human were: Total MAP2 (Exon 4) forward: 5′ GGAAGGACTTGTCCGAAGC 3′, Total MAP2 (Exon 4) reverse: 5′ GTGACCCATGCTCTCCAAA 3′; HMW MAP2 (Exon 7) forward: 5′ ATGACCCCCTCATCCAAAG 3′; HMW MAP2 (Exon 7) reverse: 5′ CATGTGGCCAGACTCAACAC 3′; LMW MAP2 (Junction Exons 6‐10) forward: 5′ TGCCTCCTTCTCCACCCC 3′ and LMW MAP2 (Junction Exons 6‐10) reverse: 5′ TTCCCCACCTGCTGCTTC 3′.
Western blot
The human brain striatums that were stored at −80°C were ground with a mortar in a frozen environment with liquid nitrogen to prevent thaw of the samples. The resulting powder was homogenized. For SH‐SY5Y cells plates were washed twice and placed on an ice‐cold plate. Both extracts were prepared by homogenizing the brain areas in ice‐cold extraction buffer consisting of 20 mM HEPES pH 7.4, 100 mM NaCl, 20 mM NaF, 1% Triton X‐100, 1 mM sodium orthovanadate, 1μM okadaic acid, 5mM sodium pyrophosphate, 30mM β‐glycerophosphate, 5 mM EDTA, and protease inhibitors (2 mM PMSF, 10 µg/mL aprotinin, 10 µg/mL leupeptin and 10 µg/mL pepstatin). Samples were homogenized and centrifuged at 15 000 g for 15 minute at 4°C. The resulting supernatant was collected, and protein content determined by Bradford assay. Fifteen micrograms of total protein were electrophoresed on 10% SDS‐polyacrylamide gel and transferred to a nitrocellulose membrane and blocked in TBS‐T (150 mM NaCl, 20 mM Tris–HCl, pH 7.5, 0.05% Tween 20) with 5% nonfat dry milk. The experiments were performed using the following primary antibodies: anti‐MAP2 clone HM2 (M4403 from Sigma‐Aldrich, Saint Louis (MO)), anti‐SRSF6/SRP55 clone 9‐1‐56 (MABE152 from Merck Millipore, Darmstadt, Germany), anti‐Vinculin (sc‐5573 from Santa Cruz, CA) and β‐actin clone AC‐74 (A2228 from Sigma‐Aldrich). The membranes were incubated with antibody overnight at 4°C in 5% nonfat dried milk washed in TBS‐T, and next incubated with secondary anti‐mouse IgG HRP‐conjugated (P0447 from DAKO Cytomation) and developed using the ECL detection kit (Perkin Elmer).
Immunohistochemistry and immunofluorescence
Formalin fixed (4%, 24 h) paraffin embedded tissue from striatum were used. Sections (5 μm thick) were mounted on superfrost‐plus tissue slides (Menzel‐Gläser) and deparaffinized. Subsequently, sections were immersed in 0.3% H2O2 in methanol for 30 minutes to quench endogenous peroxidase activity, and treated in 10 mM pH 6.0 citrate buffer heated by microwave during 15 minutes for antigen retrieval. For immunohistochemical staining, slides were blocked for 1 h in PBS containing 0.5% Normal Serum, 0.3% Triton X‐100 and 1% bovine serum albumin. (Sigma‐Aldrich). Sections were incubated overnight at 4°C in phosphate buffered saline. containing 0.3% Triton X‐100 and 1% BSA with anti‐MAP2 clone HM2 (M4403 from Sigma‐Aldrich). Finally, brain sections were incubated in avidin‐biotin complex using the Elite Vectastain kit (Vector Laboratories). Chromogen reactions were performed with diaminobenzidine (SIGMAFASTTM DAB, Sigma) for 10 minutes. Sections were mounted on glass slides and coverslipped with Mowiol (Calbiochem). Images were captured using an Olympus BX41 microscope with an Olympus camera DP‐70 (Olympus Denmark A/S). For immunofluorescence, after making the antigen retrieval, slides were pretreated with 0.1% Triton X‐100 for 15 minutes, 1M Glycine for 30 minutes and Cu2SO4 in ammonium acetate for 15 minutes, and blocking solution (1% BSA and 0.1% Triton X‐100) for 1 h. Sections were then incubated overnight at 4°C° with primary antibodies in blocking solution at the following concentrations: anti‐MAP2 (ab32454 Abcam, Cambridge, UK) 1:100 and anti Tau (HT‐7, MN1000, Thermo Fisher, Waltham, MA, USA) 1:100. The following day, sections were washed in PBS. To‐Pro‐3 and secondary antibodies used were from Life Technologies (Life Technologies, Thermo Fisher Scientific, Carlsbad, CA, USA). Sections were mounted in Mowiol (Merk Millipore, Darmstadt, Germany). Confocal analysis was performed with LSM 510 Confocal Laser Scanning Microscope from Carl Zeiss (Oberkochen, Germany). Images were taken with same settings. Analysis and treatment of images was performed using LSM Image Browser, Fiji and Adobe Photoshop.
Cell culture
SH‐SY5Y neuroblastoma cell line was grown on DMEM, 10% serum and antibiotics. Cells were nucleofected with Amaxa Nucleofector kit V (Lonza, Basel, Switzerland) according to manufacturer's instructions. pKLO.1 and shRNA MISSION for SRSF6 were obtained from Sigma.
Statistical analyses
Statistical analysis was performed with SPSS 19.0 (SPSS Statistic IBM). Data are represented as means ± standard deviation. The normality of the data was analyzed by Shapiro–Wilk test. For two‐group comparison, two‐tailed Student's t‐test (data with normal distribution) or Mann–Whitney U‐test (data with non‐normal distribution) was performed. A critical value for significance of P < 0.05 was used throughout the study. No statistical method was used to predetermine sample size. The investigators were blinded to genotype during experiments in the immunohistochemical counting of the TNRs and behavioral tests. The experiments were not randomized.
Results
SRSF6 knockdown alters MAP2 alternative splicing
We hypothesized that SRSF6 might regulate the alternative splicing event that results in LMW and HMW isoforms of MAP2 (Figure 1A). In line with this, by using ESEfinder 3.0 29, we were able to find two high‐score SRSF6 binding sites at the 5′ portion of exon 7 (data not shown). Then, to analyze SRSF6 involvement in the MAP2 splicing event originating LMW‐HMW isoforms, we used the human neuroblastoma cell line SHSY‐5Y which expresses high levels of SRSF6 compared with other neuroblastoma cell lines such as mouse cell line N2A (data not shown). We nucleofected SHSY‐5Y cells with an empty vector or an SRSF6 sh‐RNA vector to analyze the effect of knocking‐down SRSF6 on the ratio of HMW MAP2 and LMW MAP2 mRNAs (Figure 1B). First, we confirmed by Western‐blot the efficiency of SRSF6 silencing (Figure 1B). Then we analyzed HMW MAP2 and LMW MAP2 mRNA levels as well as total MAP2 mRNA level (Figure 1C). HMW MAP2 mRNA showed a tendency to increase while LMW MAP2 mRNA levels showed a tendency to decrease in cells nucleofected with sh‐SRSF6, (Figure 1C). This resulted in a significant decrease in the LMW/HMW isoform ratio of MAP2 mRNA in cells with reduced SRSF6. Total MAP2 mRNA level was not altered by SRSF6 knockdown. We then analyzed the impact of SRSF6 knockdown and altered MAP2 mRNA isoforms ratio in MAP2 isoforms protein level (Figure 1D). Western‐blot analysis revealed a mild tendency to increase of HMW MAP2 isoforms with a strong reduction of the LMW MAP2 isoforms in SRSF6 knocked‐down cells (Figure 1D). This disbalance resulted in a significant decrease in the ratio LMW/HMW MAP2 isoforms. These results demonstrate, as postulated, that SRSF6 is able to regulate the alternative splicing of MAP2 by controlling the equilibrium between the LMW and the HMW isoforms of MAP2 mRNA.
MAP2 splicing isoforms are altered in the striatum of HD patients
We have recently reported an alteration of SRSF6 in the striatum of HD patients 7. This fact, together with results in Figure 1, would suggest that alteration of MAP2 alternative splicing might take place in the striatum of HD patients. First, we confirmed the alteration of SRSF6 in striatum of HD patients and explored the status of SRSF6 in cortex (Figure 2A). In agreement with our previous results, SRSF6 level was increased in the striatum of HD patients (Figure 2A, left panels, 42% increase, P < 0.01). Also, as previously described, SRSF6 in striatum shows a pattern of multiple bands that may be the result of multiple phosphorylation. Then, by using the phospho‐SR antibody we also observed higher levels of the 55kDa band that would correspond to phosphorylated‐SRSF6 (Figure 2A). However levels of SRSF6 in the cortex of HD patients were similar to levels in control subjects (Figure 2A) and only an increase in the band at 55kDa was observed using the phospho‐SR antibody (Figure 2A). Then, we analyzed the levels of total MAP2, HMW MAP2 and LMW MAP2 mRNAs in the striatum and cortex of HD patients and control subjects. Levels of total MAP2 mRNA were severely decreased in the striatum of HD patients (Figure 2B, left panels). In good agreement, levels of HMW MAP2 isoforms were also highly reduced in the striatum of HD patients (Figure 2B, left panels). Strikingly, levels of LMW MAP2 mRNA were not decreased, and instead showed a tendency to increase in the striatum of HD patients, which resulted in a significant increase in the LMW/HMW MAP2 isoform ratio (Figure 2B, left panels). This was specific for the LMW/HMW‐generating alternative splicing event as no change was observed for the other alternative splicing events of MAP2 (data not shown). In cortex we did not observe changes in the total levels of MAP2 mRNA in HD compared to controls nor in the levels of HMW MAP2 mRNA (Figure 2B, right panels). We only observed a mild tendency to increase in the levels of the LMW MAP2 isoforms that resulted in an almost significantly increased LMW/HMW ratio (Figure 2B, right panels). These results confirm the previously described alteration of SRSF6 in the striatum of HD patients and in parallel show alteration of the SRSF6‐dependent splicing of MAP2 in favor of the LMW MAP2 isoform, as well as a decrease in MAP2 total mRNA levels.
Figure 2.
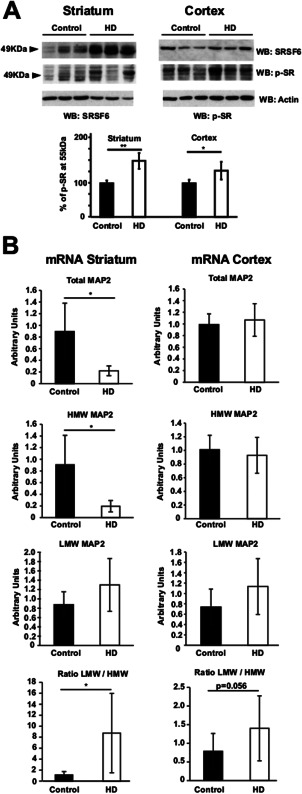
MAP2 splicing is altered in the striatum of HD patients. A. Western‐blot analysis of SRSF6 and p‐SR in controls and HD patients (striatum in left panels, n = 3/7 and cortex in right panels n = 5/6). Graph represents the quantification of p‐SR western‐blots at 55kDa (SRSF6). Student's t‐test, * P < 0.05, **P < 0.01. B. Graphs show relative RT‐qPCRs performed to detect (from above) Total MAP2, HMW MAP2 and LMW MAP2 in controls and HD patients. Last graph below correspond to the ratio LMW/HMW. Striatum in left panels n = 5/7. Cortex in right panels n = 6/7. Mann‐Whitney U‐test,* P < 0.05.
MAP2 isoform imbalance at the protein level in striatum of HD patients
We then tested whether the described alteration of MAP2 isoforms at the mRNA level in striatum of HD patients would have an impact on the protein level of the corresponding MAP2 isoforms. We performed Western blot on striatal protein extracts from HD patients and control subjects with a mouse monoclonal antibody that recognizes all MAP2 isoforms 18. HMW MAP2 isoforms with molecular weight higher than 225kDa were clearly observed in controls while they were barely detectable in HD patients (Figure 3A, upper panel). On the contrary, levels of LMW MAP2 isoforms with molecular weight between 70 and 75 kDas, showed a mild reduction in HD patients together with an altered pattern of bands (Figure 3A, middle panel). In good agreement with the mRNA data, the LMW/HMW ratio was highly increased in HD patients (Figure 3B) as the HMW MAP2 isoforms dropped and LMW MAP2 isoforms became predominant.
Figure 3.
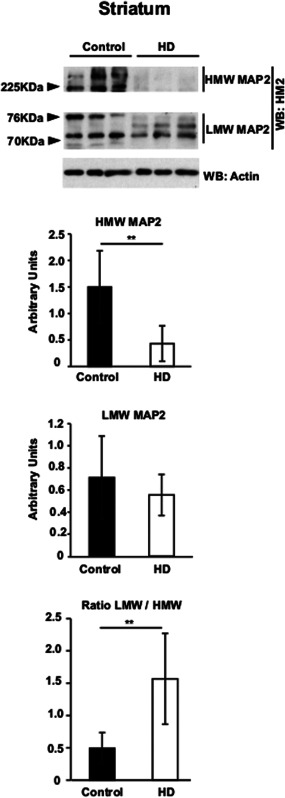
MAP2 isoforms are disbalanced at the protein level in the striatum of HD patients. Western‐blot analysis of MAP2 isoforms in striatum of Controls and HD patients. Graphs represent the quantification of western‐blots. Last graph below correspond to the ratio LMW/HMW. n = 7/8. Student's t‐test, ** P < 0.01.
Altered MAP2 histopathology in striatum of HD patients
HMW and LMW MAP2 isoforms have differential distribution in neurons. More precisely, HMW MAP2 isoforms, which are predominant in mature neurons, are preferentially located in cell bodies and dendrites. On the contrary LMW MAP2 isoforms can be found at any compartment of the neurons during developmental stages (19, 20, 26). As we have observed a decrease in total MAP2 levels with LMW MAP2 isoforms being predominant, we hypothesized MAP2 histopathological changes by MAP2 immunoreactivity in HD. We performed MAP2 immunohistochemical analysis in striatum of HD patients and control subjects with the HM2 mouse monoclonal antibody that detects all MAP2 isoforms. In the neuropil of controls we observed profuse staining of dendritic arbors (Figure 4A, left panels). Neuronal cell bodies were also detected with dendrites clearly stained from their base to more distal parts (Figure 4A, left panels). On the contrary, MAP2 immunoreactivity in the striatum of HD patients showed a different pattern; dendritic arbors were barely detected in the neuropil and, although cell body staining was preserved (Figure 4A, right panels), in many neurons it did not extend to dendrites as it was truncated at their base (Figure 4A, right panels, arrowhead). Analysis also revealed sparse neurons showing MAP2 staining across the nucleus, resembling recently described Tau Nuclear Rods 7, 31 and we observed this deposits both in control and in HD subjects (Figure 4B). Together, the changes observed in MAP2 immunoreactivity in HD, especially the loss of MAP2 dendritic staining, fit with splicing alteration observed at the mRNA level.
Figure 4.
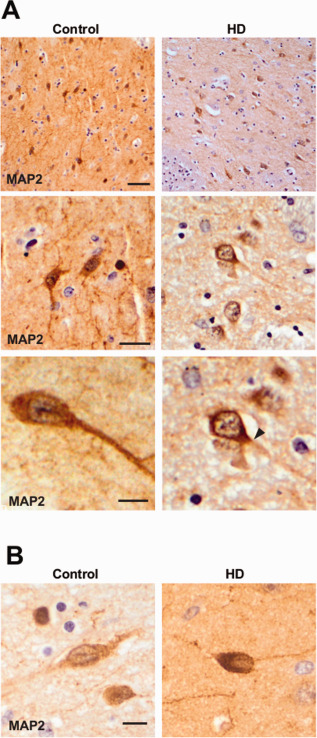
MAP2 immunostaining is altered in the striatum of HD patients. Images show MAP2 immunohistochemical analysis of striatum of controls and HD patients. Images are representative of all cases analyzed n = 4/5. A. Upper panels: Loss of MAP2 in dendritic arbors in HD neurons. Scale bar 20 µm. Middle and lower panels: MAP2 is stacked at the base of the dendrite in striatal HD neurons (solid arrowhead). Scale bars 10 µm (middle), 5 µm (lower). Lower panel right is a magnification of middle panel right. B. MAP2 nuclear deposit in Control and HD striatal neurons. Scale bar 5 µm.
MAP2 is stacked at the base of the dendrite in HD striatal neurons
In order to confirm our findings, we performed immunofluorescence with a rabbit polyclonal N‐terminal MAP2 antibody that recognizes all MAP2 isoforms, together with To‐Pro‐3 nuclear counterstaining, and analyzed it under confocal microscopy. First, we confirmed that MAP2 staining in dendritic arbors of HD patients was severely reduced (Figure 5A, 25x) while staining within the cell bodies remained. Then, we analyzed cell bodies and the base of the dendrites in detail. We observed that HD neurons showed MAP2 staining with a pattern similar to that in than control neurons but fainter. In agreement with the immunohistochemistry data, MAP2 staining in HD neurons was truncated at the base of dendrites (Figure 5A, 100x). Finally, we were able to find scattered neurons, either in control or HD subjects, with nuclear membrane indentations [initially described by others 24 and also recently by our group 7]. These indentations were filled with mild MAP2 staining and colocalizes with MAP Tau deposits recently described by us denominated Tau nuclear rods (Figure 5B).
Figure 5.
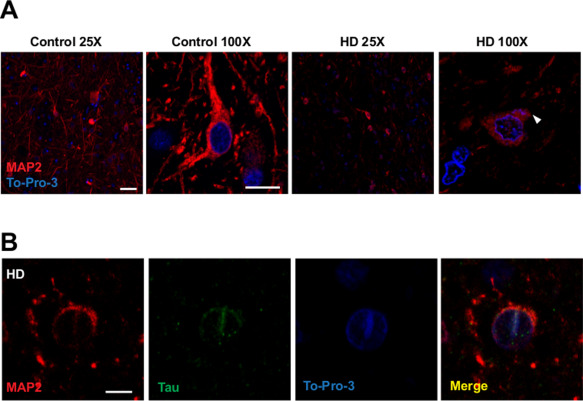
Dendritic loss of MAP2 in striatal HD neurons and detection of MAP2 nuclear deposits in striatal neurons. A. Images show MAP2 immunofluorescence (red) with a To‐Pro‐3 counterstaining (blue) of striatum from controls and HD patients. Images are a single stack representative of all cases analyzed n = 4/5. Loss of MAP2 in dendritic arbors in HD neurons. Scale bar 50 µm in 25X images, 7,5 µm in 100X images. White arrowhead shows MAP2 stacked at the base of the dendrite in striatal HD neurons. B. MAP2 and MAP Tau nuclear deposit in HD striatal neuron. Scale bar 5 µm.
Discussion
MAP2 is a neuronal specific microtubule associated protein that stabilizes microtubules in cell bodies and particularly in dendrites 5, 26. Alteration of MAP2 has been associated to neurodegenerative and physichiatric disorders. A decrease of MAP2 staining has been reported in the subiculum and enthorthinal cortex of subjects with schizophrenia 2. Also, it has been reported a strong depletion of MAP2 in temporal and medial lobes in patients of Lewy body variant of Alzheimer's disease 21. In Parkinson's disease, some neurons of the substania nigra show abnormal MAP2 immunolabeling, colocalizing with alpha synuclein and ubiquitin in Lewy bodies 4. Curiously, in this report MAP2 is also found altered in HD cases and colocalizing with Tau Nuclear Rods both in control and in HD subjects.
We show that both MAP2 mRNA and protein levels are decreased in striatum of HD patients. A previous study reported a modest decrease in cortical layer V area 9 of the HD patients 30, but here, for the first time to our knowledge, we present MAP2 alteration in the striatum, the most affected brain region in this disease. Our data show that MAP2 decrease in the striatum of HD does not occur evenly across its isoforms: HMW MAP2 isoforms, which are preferentially located in dendrites and are the predominant isoforms in adult brain, are highly reduced at mRNA and protein levels in HD while LMW MAP2 mRNA levels are slightly increased and protein levels remained constant. We also observed changes in the electrophoretic mobility of the LMW MAP2 isoforms in HD. However, this may be caused by changes in phosphorylation as MAP2 is highly phosphorylated 26 and both kinases and phosphatases that regulate phosphorylation of various MAPs are known to be altered in HD 8, 11, 33.
We also present evidence of changes in MAP2 immunolabeling in the striatum of HD patients. We observe a loss of the MAP2 staining in the dendritic arbors of HD striatal neurons that fits with dropped levels of HMW MAP2 isoforms, which are the dendrite associated MAP2 isoforms. MAP2 stabilizes microtubules within the dendrite (5, 26). One of the first neuropathological events in HD is the loss of synapses in corticostriatal pathway 25. Striatal dendrites receive an immense amount of connections from cortical axons. In the striatum of moderate grades of HD recurving of distal dendritic segments, short‐segment branching along dendrites, and increased numbers and size of dendritic spines have been observed while in the striatum of severe grades of HD truncated dendritic arborizations, occasional focal dendritic swellings, and marked spine loss have been reported 9. Loss of HMW MAP2 isoforms would be either cause or consequence of the pathophysiological changes observed in the dendrites of striatal neurons, however we can hypothesize that there might be a connection between both observations.
Cell bodies of HD striatal neurons show staining with total MAP2 antibodies, as expected given the remaining levels of LMW MAP2 isoforms. Detailed observation of cell bodies suggests that MAP2 is stacked at the base of the dendrites in HD striatal neurons. This may be consequence of the nonuniform polarity of microtubules in the dendrite 3, 13 and suggests that dendrite itself is compartmentalized. Another observation from the analysis of the MAP2 staining within the cell bodies of control and HD subjects is the presence on scattered neurons of filamentous MAP2 structures that appear to span along the nucleus, similar to those observed in Parkinson's disease 4. However confocal sections show that these MAP2 positive structures fill nuclear indentations previously described in HD 24 and colocalizes with the new histopathological hallmark denominated Tau Nuclear Rods 7. MAP2 deposits appear in control and in HD patients in a similar number, suggesting that both MAP2 isoforms, HMW and LMW, may be filling nuclear indentations, however as the total levels of MAP2 are severely decreased in the striatum of HD, it is difficult to interpret this result.
The MAP2 alterations observed in HD originate from changes at the mRNA levels. In the striatum of HD patients the levels of Total MAP2 mRNA drop as a consequence of the decreased levels of the predominant HMW MAP2 mRNA isoform, while levels of LMW MAP2 isoforms remain constant or even show a tendency to increase. In the cortex of HD patients we observed a similar tendency to increase in the LMW MAP2 isoforms mRNA. This MAP2 isoform disbalance in HD suggests a splicing alteration rather than a transcriptional defect, and we hypothesized that may be caused by alterations of the splicing factor SRSF6. Our data show that interference of SRSF6 in SHSY‐5Y neuroblastoma cell line, which expresses high levels of SRSF6, alters the HMW/LMW MAP2 ratio at mRNA level and consequently at protein level. Also, it has been suggested that SRSF6 binds preferentially CAG expanded Huntingtin mRNA 27 and the predicted consensus sequence for SRSF6 binding is CCAGCAGCA 14; curiously, MAP2 contains a CAG repeat motif in its exon 1 15. SRSF6 is severely altered in the striatum of HD patients with increased protein levels and increased phosphorylation 7. Our present data thus confirm our previous findings regarding HD striatum and also show increased phosphorylation of SRSF6 in cortex of HD patients, which may explain why MAP2 mRNA is more altered in striatum than in cortex of HD patients, and may suggest that increase of total levels of SRSF6 would have a higher impact on MAP2 splicing than changes in SRSF6 phosphorylation.
Furthermore, splicing alteration of MAP2 may also explain reduced Total MAP2 mRNA levels. The juvenile LMW MAP2 isoform appears to be favored in adult HD brains, and as a consequence of this, exons 7, 8 and 9 are processed as introns. There are many miRNA generated from intronic sequences 16 and most of them are silencing the expression of their host genes in an autoregulatory loop 12. Then, it would be possible than an excess of LMW MAP2 mRNA in adult brain may result in a decreased levels of total MAP2. Finally, there is an emerging concept suggesting that splicing factors may regulate entire gene expression programs 1. It is well described that SRSF6 regulates the splicing of MAP Tau 17, 32, and now we present evidence suggesting SRSF6 may regulate MAP2 splicing. Curiously, our preliminary results suggest the other member of this family of microtubule associated proteins, MAP4 5, may be also altered at the mRNA level (not shown). If confirmed, these results would indicate mRNA alteration of this entire family of MAPs. It should be noted though that MAP4 expression is glial specific and further studies will be required for full characterization. All together this suggests that SRSF6 may act as a master regulator of neuronal cytoarchitecture by regulating the splicing of two critical microtubule associated proteins MAP Tau and MAP2, and its misregulation in HD may be critical for the axonal and dendritic alteration of striatal neurons.
Acknowledgments
This work was supported by CiberNed‐ISCIII collaborative grant PI2013/09‐2, CoEN grant (NEURO‐MIR) and by grants from Ministerio de Ciencia (MICINN, MINECO) SAF2012‐34177, SAF2015‐65271 and from Fundación BBVA. JRC is supported by CIBERNED. CBMSO is recipient of an institutional grant from Fundacion Ramón Areces. We thank M. Lucas and Dr. M. Santos for excellent technical support. We also thank the support of the confocal (SMOC) and the Genomic and Massive Sequencing services at Centro de Biología Molecular Severo Ochoa. Authors have no conflict of interest to declareThis work was supported by CiberNed‐ISCIII collaborative grant PI2013/09‐2, CoEN grant (NEURO‐MIR) and by grants from Ministerio de Ciencia (MICINN, MINECO) and from Fundación BBVA. JRC is supported by CIBERNED. CBMSO is recipient of an institutional grant from Fundacion Ramón Areces. We thank M. Lucas and Dr. M. Santos for excellent technical support. We also thank the support of the confocal (SMOC) and the Genomic and Massive Sequencing services at Centro de Biología Molecular Severo Ochoa. Authors have no conflict of interest to declare.
References
- 1. Ankö M‐L (2014) Regulation of gene expression programs by serine‐arginine rich splicing factors. Semin Cell Dev Biol 32: 1–11. doi: 10.1016/j.semcdb.2014.03.011 [DOI] [PubMed] [Google Scholar]
- 2. Arnold SE, Lee VM, Gur RE, Trojanowski JQ (1991) Abnormal expression of two microtubule‐associated proteins (MAP2 and MAP5) in specific subfields of the hippocampal formation in schizophrenia. Proc Natl Acad Sci USA 88:10850–10854. doi: 10.1073/pnas.88.23.10850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baas PW, Deitch JS, Black MM, Banker GA (1988) Polarity orientation of microtubules in hippocampal neurons: uniformity in the axon and nonuniformity in the dendrite. Proc Natl Acad Sci USA 85:8335–8339. doi: 10.1073/pnas.85.21.8335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. D'Andrea MR, Ilyin S, Plata‐Salaman CR (2001) Abnormal patterns of microtubule‐associated protein‐2 (MAP‐2) immunolabeling in neuronal nuclei and Lewy bodies in Parkinson's disease substantia nigra brain tissues. Neurosci Lett 306:137–140. doi: 10.1016/S0304-3940(01)01811-0 [DOI] [PubMed] [Google Scholar]
- 5. Dehmelt L, Halpain S (2005) The MAP2/Tau family of microtubule‐associated proteins. Genome Biol 6:204. doi: 10.1186/gb-2004-6-1-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ferhat L, Represa A, Ferhat W, Ben‐Ari Y, Khrestchatisky M (1998) MAP2d mRNA is expressed in identified neuronal populations in the developing and adult rat brain and its subcellular distribution differs from that of MAP2b in hippocampal neurones. Eur J Neurosci 10:161–171. [DOI] [PubMed] [Google Scholar]
- 7. Fernández‐Nogales M, Cabrera JR, Santos‐Galindo M, Hoozemans JJM, Ferrer I, Rozemuller AJM et al (2014) Huntington's disease is a four‐repeat tauopathy with tau nuclear rods. Nat Med 20:881–885. doi: 10.1038/nm.3617 [DOI] [PubMed] [Google Scholar]
- 8. Fernández‐Nogales M, Hernández F, Miguez A, Alberch J, Ginés S, Pérez‐Navarro E, Lucas JJ (2015) Decreased glycogen synthase kinase‐3 levels and activity contribute to Huntington's disease. Hum Mol Genet 24:5040–5052. doi: 10.1093/hmg/ddv224 [DOI] [PubMed] [Google Scholar]
- 9. Ferrante RJ, Kowall NW, Richardson EP (1991) Proliferative and degenerative changes in striatal spiny neurons in Huntington's disease: a combined study using the section‐Golgi method and calbindin D28k immunocytochemistry. J Neurosci 11:3877–3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Germann S, Gratadou L, Dutertre M, Auboeuf D (2012) Splicing programs and cancer. J Nucleic Acids 2012:269570. doi: 10.1155/2012/269570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gratuze M, Noel A, Julien C, Cisbani G, Milot‐Rousseau P, Morin F et al (2014) Tau hyperphosphorylation and deregulation of calcineurin in mouse models of Huntington's disease. Hum Mol Genet 24:86–99. doi: 10.1093/hmg/ddu456 [DOI] [PubMed] [Google Scholar]
- 12. Heyn P, Kalinka AT, Tomancak P, Neugebauer KM (2015) Introns and gene expression: cellular constraints, transcriptional regulation, and evolutionary consequences. Bioessays 37:148–154. doi: 10.1002/bies.201400138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hirokawa N, Takemura R (2005) Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci 6:201–214. doi: 10.1038/nrn1624 [DOI] [PubMed] [Google Scholar]
- 14. Jensen MA, Wilkinson JE, Krainer AR (2014) Splicing factor SRSF6 promotes hyperplasia of sensitized skin. Nat Struct Mol Biol 21:189–197. doi: 10.1038/nsmb.2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kalcheva N, Lachman HM, Shafit‐Zagardo B (1999) Survey for CAG repeat polymorphisms in the human MAP‐2 gene [In Process Citation]. Psychiatr Genet 9:43–46. [DOI] [PubMed] [Google Scholar]
- 16. Kim VN, Han J, Siomi MC (2009) Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10:126–139. doi: 10.1038/nrm2632 [DOI] [PubMed] [Google Scholar]
- 17. Liu F, Gong C‐X (2008) Tau exon 10 alternative splicing and tauopathies. Mol Neurodegener 3:8. doi: 10.1186/1750-1326-3-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Loveland KL, Herszfeld D, Chu B, Rames E, Christy E, Briggs LJ et al (1999) Novel low molecular weight microtubule‐associated protein‐2 isoforms contain a functional nuclear localization sequence. J Biol Chem 274:19261–19268. [DOI] [PubMed] [Google Scholar]
- 19. Matus A (1988) Microtubule‐associated proteins: their potential role in determining neuronal morphology. Annu Rev Neurosci 29–44. doi: 10.1146/annurev.ne.11.030188.000333 [DOI] [PubMed] [Google Scholar]
- 20. Meichsner M, Doll T, Reddy D, Weisshaar B, Matus A (1993) The low molecular weight form of microtubule‐associated protein 2 is transported into both axons and dendrites. Neuroscience 54:873–880. doi: 0306‐4522(93)90581‐Y [pii] [DOI] [PubMed] [Google Scholar]
- 21. Mukaetova‐Ladinska EB, Xuereb JH, Garcia‐Sierra F, Hurt J, Gertz H‐J, Hills R et al (2009) Lewy body variant of Alzheimer's disease: selective neocortical loss of t‐SNARE proteins and loss of MAP2 and alpha‐synuclein in medial temporal lobe. ScientificWorldJournal 9:1463–1475. doi: 10.1100/tsw.2009.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Orr HT, Zoghbi HY (2007) Trinucleotide repeat disorders. Annu Rev Neurosci 30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042 [DOI] [PubMed] [Google Scholar]
- 23. Ranum LPW, Cooper TA (2006) Rna‐mediated neuromuscular disorders. Annu Rev Neurosci 29:259–277. doi: 10.1146/annurev.neuro.29.051605.113014 [DOI] [PubMed] [Google Scholar]
- 24. Roos RA, Bots GT (1983) Nuclear membrane indentations in Huntington's chorea. J Neurol Sci 61:37–47. [DOI] [PubMed] [Google Scholar]
- 25. Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH et al (2014) Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol 10:204–216. doi: 10.1038/nrneurol.2014.24 [DOI] [PubMed] [Google Scholar]
- 26. Sánchez C, Díaz‐Nido J, Avila J (2000) Phosphorylation of microtubule‐associated protein 2 (MAP2) and its relevance for the regulation of the neuronal cytoskeleton function. Prog Neurobiol 61:133–168. doi: 10.1016/S0301-0082(99)00046-5 [DOI] [PubMed] [Google Scholar]
- 27. Sathasivam K, Neueder A, Gipson TA, Landles C, Benjamin AC, Bondulich MK et al (2013) Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci USA 110:2366–2370. doi: 10.1073/pnas.1221891110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shafit‐Zagardo B, Kalcheva N, Dickson D, Davies P, Kress Y (1997) Distribution and subcellular localization of high‐molecular‐weight microtubule‐associated protein‐2 expressing exon 8 in brain and spinal cord. J Neurochem 68:862–873. [DOI] [PubMed] [Google Scholar]
- 29. Smith PJ, Zhang C, Wang J, Chew SL, Zhang MQ, Krainer AR (2006) An increased specificity score matrix for the prediction of SF2/ASF‐specific exonic splicing enhancers. Hum Mol Genet 15:2490–2508. doi: 10.1093/hmg/ddl171 [DOI] [PubMed] [Google Scholar]
- 30. Somenarain L, Jones LB (2010) A comparative study of MAP2 immunostaining in areas 9 and 17 in schizophrenia and Huntington chorea. J Psychiatr Res 44:694–699. doi: 10.1016/j.jpsychires.2009.12.006 [DOI] [PubMed] [Google Scholar]
- 31. Vuono R, Winder‐Rhodes S, de Silva R, Cisbani G, Drouin‐Ouellet J, Spillantini MG et al (2015) The role of tau in the pathological process and clinical expression of Huntington's disease. Brain 138:1907–1918. doi: 10.1093/brain/awv107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang J, Gao QS, Wang Y, Lafyatis R, Stamm S, Andreadis A (2004) Tau exon 10, whose missplicing causes frontotemporal dementia, is regulated by an intricate interplay of cis elements and trans factors. J Neurochem 88:1078–1090. doi: 10.1046/j.1471-4159.2003.02232.x [DOI] [PubMed] [Google Scholar]
- 33. Xifró X, García‐Martínez JM, Del Toro D, Alberch J, Pérez‐Navarro E (2008) Calcineurin is involved in the early activation of NMDA‐mediated cell death in mutant huntingtin knock‐in striatal cells. J Neurochem 105:1596–1612. doi: 10.1111/j.1471-4159.2008.05252.x [DOI] [PubMed] [Google Scholar]