

Abstract
The blood–brain barrier (BBB) consists of dense contacts between endothelial cells, the tight junctions, which are complemented by membrane‐bound transporters belonging to the ATP‐binding cassette (ABC) transporter family. Liver X receptors (LXR) have previously been shown to stabilize the integrity of atherosclerotic noncerebral arteries. Their effects on ischemic cerebral vessels are still unknown. By delivering LXR agonists, T0901317 and GW3965, to mice submitted to 30 minutes intraluminal middle cerebral artery occlusion, we show that LXR activation reduces brain swelling and decreases BBB permeability by upregulating LXR's target calpastatin that deactivates calpain‐1/2, stabilizing p120 catenin. p120 catenin specifically interacts with RhoA and Cdc42, inactivating the former and overactivating the latter, thus restoring the postischemic expression, phosphorylation and interaction of the tight junction proteins occludin and zona occludens‐1. Moreover, LXR activation deactivates matrix metalloproteases‐2/9 and inhibits microvascular apoptosis by deactivating JNK1/2 and caspase‐3. In addition to the cholesterol transporters ABCA1 and ABCG1, which have previously been shown to be upregulated by LXR in noncerebral vessels, LXR activation increases the abundance of the drug transporters ABCB1 and ABCC1 on ischemic brain capillaries, as we further show. That LXR activation promotes endothelial integrity in different ways makes this receptor attractive as target for stroke therapies.
Keywords: ATP‐binding cassette transporter, blood‐brain barrier, brain edema, calpain‐1/2, focal cerebral ischemia, middle cerebral artery occlusion, Rho GTPase, tight junction
INTRODUCTION
The blood–brain barrier (BBB) is characterized by high‐electrical resistance contacts between endothelial cells, the tight junctions, which prevent the diffusion of blood–borne molecules into the brain tissue 15, 29. The BBB is complemented by active efflux transporters located on endothelial membranes, among which ATP‐binding cassette (ABC) transporters have received particular interest as they eliminate a large number of lipophilic and amphipathic xenobiotics, including pharmacological compounds, from the brain tissue 16, 24. Ischemic stroke challenges BBB function, resulting in the loss of expression of tight junction proteins, such as occludin and zona occludens (ZO)‐1 (20), in the altered phosphorylation of these proteins and their disassembly (38), and the deregulation of ABC transporters (17).
A variety of events compromises the BBB integrity after stroke. Endothelial Ca2+ influx activates calpain proteases (4) and matrix metalloproteinases (MMPs) 14, 43. Calpains are calcium‐dependent nonlysosomal cysteine proteases that are ubiquitously expressed. Calpains consist in two isoforms, calpain‐1 and ‐2, representing heterodimers of a common 28‐kDa subunit with two different 80‐kDa subunits (13). Calpain‐1/2 activity is tightly controlled by calpastatin, its endogenous specific inhibitor. Calpastatin expression has been shown to decrease after stroke, increasing calpain‐1/2 activity (34). MMP‐2 (gelatinase A) and MMP‐9 (gelatinase B) are zinc‐dependent endopeptidases, promoting extracellular matrix degradation (31). MMP‐2 and MMP‐9 are activated upon cerebral hypoxia and ischemia, increasing brain edema and exacerbating neuronal injury 32, 42. MMP‐9 activity is negatively regulated by calpain inhibitors (39), suggesting that calpain mediates matrix protease activation. However, the mechanisms, how calpains and MMPs mediate BBB permeability, are unknown.
It has been suggested that Rho GTPases induce the BBB permeability changes after stroke 7, 33. Rho GTPases are small G‐proteins transducing signals by binding and activating downstream effectors (11). The most studied are RhoA, Rac1 and Cdc42, which are active in their GTP bound state and inactive in their GDP bound state. Their activities are controlled by (i) guanine exchange factors (GEF) that promote exchanging GDP with GTP; (ii) GTPase activating proteins (GAP) that catalyze GTP hydrolysis; and (iii) GDP dissociation inhibitors (GDIs) that sequester the inactive GDP bound forms into the cytosol (11). In endothelial cells, RhoA activation induces stress fiber formation, thus destabilizing tight junction complexes and increasing BBB leakage 18, 33. Cdc42 activation controls cell polarity and promotes BBB integrity by enabling tight junction proteins assembly, such as occludin and ZO‐1 5, 12.
ABC transporters exhibit coordinated changes in abundance on the luminal and abluminal endothelial membrane in the ischemic brain. As such, the transporter ABCB1, which is predominantly found on the luminal membrane of brain capillary cells, is upregulated (37), whereas ABCC1, which is present mainly on the abluminal endothelial membrane, is downregulated in response to stroke (22). The regulation of ABC transporters has profound effects on the brain accumulation of neuroprotective compounds, as we further showed, influencing drug accumulation in the brain by up to an order of magnitude or even more 22, 37. We recently reported that apolipoprotein E (ApoE) controls the balance between the luminal transporter ABCB1 and the abluminal transporter ABCC1 via its receptor ApoER2 (10). It was noteworthy that ApoE neither influenced the integrity of tight junctions nor brain edema, suggesting that ABC transporters and tight junctions may differentially be modulated after focal cerebral ischemia.
Liver X receptors (LXRs) are members of the orphan nuclear receptor family acting as transcription factors. They exist in two isoforms, LXRα and LXRβ, which are activated by their endogenous ligands, oxysterols (34). Initially, LXRs have been described as sterol sensors protecting cells from cholesterol overload by regulating the lipid transporters ABCA1 and ABCG1, which also belong to the ABC transporter family 3, 9. Recently, LXRs have been shown to protect against glucose intolerance and diabetes (9) and to provide anti‐inflammatory effects under conditions of atherosclerosis and ischemic stroke 3, 26. Thus, LXRs have been recognized as promising targets in metabolic diseases. Synthetic agonists such as T0901317 and GW3965 have been developed in the past that induce LXR activity more potently than endogenous LXR ligands.
In view of the profound effects of LXR on the vascular system, considering that LXR control the expression of cholesterol transporters belonging to the ABC transporter family, we were interested how LXR activation influences BBB permeability, tight junction expression and functionality similarly as drug transporter expression on brain capillary cells. For this reason, we delivered T0901317 and GW3965 to mice subjected to 30 minutes of middle cerebral artery (MCA) occlusion. Our study shows that LXR activation enhances BBB integrity via inhibition of the proteases calpain‐1/2, affecting Rho GTPase balance that in turn modulates the expression, phosphorylation and assembly of tight junction proteins. At the same time, Jun N‐terminal kinase (JNK)‐1/2 and caspase‐3 activation was inhibited and the expression of ABCB1 and ABCC1 was increased on brain capillaries. Our data show that LXR activation stabilizes the integrity of ischemic brain capillaries in several ways.
MATERIALS AND METHODS
Animal groups
Experiments were done according to the National Institutes of Health guidelines for the care and use of laboratory animals. A first set of C57BL6/j mice (20–25 g) were divided into two groups that were intraperitoneally treated once daily over 1 week prior to stroke with (i) 5% ethanol in 0.1 M phosphate‐buffered saline (PBS; vehicle) and (ii) the LXR agonist T0901317 (1.25 mg/animal/day; 270–309‐M050; Alexis Biochemicals, San Diego, CA, USA) dissolved in 5% ethanol in 0.1 M PBS (n = 8 animals/group).
To elucidate whether the observations made could be reproduced by postischemic T0901317 delivery and to elucidate the role of calpain‐1/2 in the regulation of BBB integrity, additional C57BL6/j mice were divided into three groups that were intraperitoneally treated 1 h after MCA occlusion with (i) 10% dimethyl sulfoxide (DMSO) in 0.1 M PBS (vehicle); (ii) the LXR agonist T0901317 (1.25 mg/animal, single dose), diluted in 10% DMSO in 0.1 M PBS; and (iii) the calpain‐1/2 inhibitor MDL28170 (0.75 mg/animal; Sigma, Deisenhofen, Germany), diluted in 10% DMSO in 0.1 M PBS (n = 5 animals/group).
To test whether the observations made were indeed specific for LXR activation, other C57BL6/j mice were intraperitoneally treated 1 h after MCA occlusion with (i) 10% DMSO in 0.1 M PBS (vehicle) or (ii) the LXR‐specific agonist GW3965 (0.5 mg/animal, single dose; Sigma), diluted in 10% DMSO in 0.1 M PBS (n = 5 animals/group).
Induction of focal cerebral ischemia
Thirty minutes of MCA occlusion was induced using an intraluminal filament technique in mice anesthetized with 1% isoflurane (30% O2, remainder N2O). In all animals, cerebral laser Doppler flow (LDF) was monitored during and after ischemia using flexible probes attached to the intact skull overlying the core of the MCA territory 22, 37. Twenty‐four hours after MCA occlusion, animals were sacrificed by transcardiac perfusion with normal saline. Brains were removed and cut on a cryostat into coronal 20 µm sections. In addition, tissue samples were collected from the ischemic and contralateral nonischemic MCA territory (striatum and overlying cortex) for Western blotting, immunoprecipitation studies and protease activity assays.
Measurement of infarct volume and brain edema and IgG extravasation studies
Representative brain sections were stained with 0.5% cresyl violet. On these sections, the infarct area and brain swelling were evaluated, as previously described 22, 37. Briefly, stained sections were digitized and infarct borders between infarcted and noninfarcted tissue were outlined using an image analysis system (Image J, National Institutes of Health, Bethesda, MD, USA). Infarct size was measured by subtracting the area of the nonlesioned ipsilateral hemisphere from that of the contralateral hemisphere, and brain swelling size was measured by subtracting the area of ipsilateral hemisphere from that of contralateral hemisphere. In a number of studies, infarct areas and edema areas were determined at the rostrocaudal level of the bregma, which is the level of the maximum extension of the MCA territory, at which brain infarcts are most reproducible. In other studies, infarct volumes and edema volumes were assessed. For that purpose, infarct areas and edema areas were determined on five equidistant sections 1 mm apart throughout the forebrain. Areas were integrated, resulting in the assessment of infarct volumes corrected for brain edema and edema volumes, respectively. Adjacent brain sections obtained from the level of the mid‐striatum were processed by immunohistochemistry for extravasated IgG (41). These sections were digitized and evaluated for areas exhibiting IgG extravasation.
Microvessel isolation and protein extraction
For molecular biological studies, tissue samples from animals belonging to the same group were pooled (Western blots, pull‐down assays, immunoprecipitation and coprecipitation studies, gelatin zymography) or individually processed (microplate assays, G‐LISA™, Cytoskeleton Inc, Denver, CO, USA), thus allowing group by group and animal‐wise comparisons. Tissue samples were gently homogenized in a glass Teflon® homogenizer in ice‐cold microvessel isolation buffer (MIB; 15 mM HEPES, 147 mM NaCl, 4 mM KCl, 3 mM CaCl2 and 12 mM MgCl2), supplemented with 5% protease inhibitor cocktail (P8340, Sigma) and 1% phosphatase inhibitor cocktail‐2 (P5726, Sigma). Homogenates were centrifuged at 3200 rpm for 10 minutes at 4°C. The resulting pellets were resuspended in 20% dextran (MW 64 000–76 000; D4751, Sigma) in MIB. Suspensions were centrifuged at 6500 rpm for 20 minutes at 4°C. The resulting crude microvessel‐rich pellets were resuspended in MIB and filtered through two nylon filters of 100 µm and 30 µm mesh size (Millipore, Schwalbach, Germany). The quality of trapped microvessels in 30‐µm filters was checked. Microvessels were stored at −80°C until further use. Isolated microvessels were homogenized in appropriate lysis buffers (see below) supplemented with 5% protease inhibitor cocktail and 1% phosphatase inhibitor cocktail‐2. Lysate samples were sonicated over two cycles, lasting 20 s each at 4°C. Protein concentrations were measured using Bradford assay kit with an iMark microplate reader (Bio‐Rad, Hercules, CA, USA).
Caseinase microplate assay for calpain‐1/2
For measurement of calpain‐1/2 activity, we developed a caseinase microplate assay that evaluates caseinase activity in the presence of calpain‐1/2's endogenous inhibitors. Twenty micrograms of protein extracts from each sample, obtained using NP‐40 lysis buffer containing 50 mM Tris‐HCl, 150 mM NaCl and 1% NP‐40 (pH 7.4), was added into 96 well microplate and complemented with calibrating buffer containing 20 mM Tris‐HCl, 1 mM EDTA, 100 mM KCl and 0.1% 2‐mercaptoethanol (pH 7.4), resulting in volumes of 60 µL in each well.
Two solutions were prepared containing casein at 20 µg/mL:
-
(i)
Solution A (= activation) consisted of 20 mM Tris‐HCl, 1 mM EDTA, 10 mM Ca2+, 100 mM KCl and 0.1% 2‐mercaptoethanol (pH 7.4), allowing to study the enzymatic activity of caplain‐1/2 owing to the fact that samples contained Ca2+ that is required for calpain‐1/2 to exert its protease activity.
-
(ii)
Solution I (= inhibition) contained 20 mM Tris‐HCl, 50 mM EDTA, 100 mM KCl and 0.1% mercaptoethanol (pH 7.4). In this solution calpain‐1/2 was inactive as the sample lacked Ca2+ ions. Experiments were conducted in triplicate for each sample, 100 µL of solution A being added to the first set of wells containing protein samples, and 100 µL of solution I being added to the next set, increasing the total volume in each well to 160 µL (60 µL + 100 µL). As such, the assay was run four times for statistical comparisons.
Each microplate was incubated for 2 h at room temperature. Then, 120 µL of 1X G‐250 dye (Quick Start Bradford Protein Assay, Biorad) was added simultaneously to each well and incubated for 5 minutes at room temperature, the G‐250 dye forming a stable complex upon binding proteins shifting light absorbance from 470 nm to 595 nm. The dye does not bind proteolytic fragments of digested proteins like small peptides or amino acids, assessing the enzymatic activity of calpain‐1/2, the higher the activity the less absorbance values are. The absorbance was read at 595 nm using the iMark microplate reader and calpain‐1/2 caseinase activity was calculated for each sample by subtracting absorbances between solution I and solution A. Calpain‐1/2 protease activity was expressed as absorbance difference at 595 nm units (AD 595 nm).
Gelatin zymography
To examine MMP‐2/9 activity, we used a gelatin zymography method that was previously described (41), as well as a gelatinase microplate assay, which we established in analogy to the caseinase microplate assay. For gelatin zymography, 25 µg proteins were mixed with 5× nonreducing loading buffer for 15 minutes at room temperature and subjected to sodium dodecylsulfate (SDS)‐polyacrylamide gel electrophoresis (PAGE) using 9% acrylamide‐bis gel containing 0.1% gelatin (Sigma). Gels were removed and washed, incubated for 1 h at room temperature with slight shaking in modified enzymatic activation buffer (50 mM Tris‐HCl, 6 mM CaCl2, 1.5 µM ZnCl2, pH 7.4) containing 2.5% Triton‐X100 to remove SDS and restore gelatinase activity. Gels were incubated for 24 h at 37°C in modified enzymatic activation buffer. Gels were stained in Coomassie Brilliant Blue R‐250 (Bio‐Rad). A total of four independent experiments were run, which were digitized and densitometrically analyzed.
Gelatinase microplate assay for MMP‐2/9
To study MMP‐2/9's activity in the presence of their endogenous inhibitors, the caseinase microplate assay was adopted with the following changes: 5 µg of proteins was added to a 96 well microplate and complemented with a calibrating buffer containing 50 mM Tris‐HCl and 6 mM CaCl2 (pH 7.4) to 60 µL volumes. Two solutions were prepared containing gelatin at a concentration of 20 µg/mL: (i) Solution A (= activation) (50 mM Tris‐HCl, 6 mM CaCl2, 50 mM EDTA, 1.5 µM ZnCl2, pH 7.4) and (ii) solution I (= inhibition) (50 mM Tris‐HCl, 6 mM CaCl2, 50 mM EDTA, pH 7.4), the two samples differing in terms of the presence and absence of ZnCl2, which is a cofactor needed for gelatinase to exert its enzymatic activity. After incubation for 2 h at 37°C, the G‐250 dye was added, the absorbance read, and MMP‐2/9 gelatinase activity was calculated by subtracting absorbance between solution I and solution A. MMP‐2/9 protease activity was again expressed as AD 595 nm.
Western blot analysis
For Western blots, lysates containing 20 µg protein were complemented with 5× SDS loading buffer. Samples were pretreated by heating except for ABC transporter blots, for which nonheated samples were loaded to avoid aggregation of these highly glycosylated membrane proteins. Samples were subjected to SDS‐PAGE followed by Western blot analysis using primary antibodies diluted 1:100 for ABCC1 and 1:1000 for all other proteins in 5% skim milk and 0.1 M tris buffered saline containing Tween 20% (TBS‐T) 10, 22. Blots were digitized, densitometrically analyzed and corrected for protein loading by means of the β‐actin blots.
The following antibodies and recombinant proteins were used: anti‐LXRαβ (sc‐1000), anti‐LXRβ (sc‐1203), anti‐calpain‐1/2 (sc‐58326), anti‐calpastatin (sc‐20779), anti‐ABCB1 (sc‐8313), anti‐ABCG1 (sc‐11130), anti‐occludin (sc‐27151), anti‐RhoA monoclonal (sc‐418), anti‐RhoA polyclonal (sc‐179), anti‐Cdc42 (sc‐6083), anti‐CD31 (sc‐8306), anti‐Vav2 (sc‐20803), anti‐p‐Tyr(PY20) (sc‐508), anti‐Thr(1E11) (sc‐81527), and protein A/G plus‐agarose (sc‐2003) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rhotekin (RTK)—Rho binding domain (RBD)—glutathione S‐transferase (GST) beads (RT02), Wiskott‐Aldrich syndrome protein (WASP)—Cdc42 binding domain of WASP (CDB)—GST beads (WS03), constitutively inactive Cdc42‐(T17N)—GST beads (C17G01) and constitutively active Cdc42‐(Q61L)—GST beads (C61G01) were obtained from Cytoskeleton Inc. (Frankfurt, Germany). Anti‐p120 catenin (4989), anti‐cleaved caspase‐3 (Asp175) (9661), anti‐total‐JNK1/2 (9252), anti‐phosphorylated JNK1/2Thr183/Tyr185 (9255), anti‐total MMP‐2 (4022), anti‐total MMP‐9 (3852) and anti‐β‐actin (4967) were from Cell Signaling (Allschwil, Switzerland); anti‐ABCA1 (NB400‐105) was from Novus Biologicals (Cambridge, UK); anti‐ZO‐1 (40‐2300) was from Invitrogen (Karlsruhe, Germany); anti‐LXRα (ab28478) was from Abcam (Cambridge, UK); and anti‐ABCC1 (ALX‐801‐007‐C250) was from Alexis Biochemicals.
Rho GTPase affinity binding (= pull‐down) assays
Equal protein quantities (800 µg) of lysates obtained using a MgCl2 lysis buffer containing 50 mM Tris base, 100 mM NaCl, 2 mM MgCl2 and 1% NP‐40 (pH 7.4) were added in Eppendorf tubes and complemented to a total volume of 1 mL by adding MgCl2 lysis buffer. For Cdc42 affinity binding assay, 20 µL (20 µg) of recombinant GST‐WASP‐CDB prefixed on agarose beads was loaded and incubated under slight rotation overnight at 4°C. The next day, samples were centrifuged for 30 s at 15 000 rpm, supernatants were removed and pellets were washed three times in ice‐cold MgCl2 lysis buffer. A volume of 20 µL of 2× SDS plus 5% 2‐mercaptoethanol loading buffer was added to each sample and boiled for 5 minutes (Heater Plate, Eppendorf, Germany), followed by a short centrifugation at 4000 rpm to precipitate beads. Supernatants from each sample were subjected to SDS‐PAGE using 12.5% acrylamide‐bis gel. For RhoA affinity binding assays, 50 µL (20 µg) of GST‐RTK‐RBD prefixed on beads was loaded according to the same protocol. For analysis of total Cdc42 and RhoA expression, 20 µg of total protein lysates from each group was subjected to 12.5% SDS‐PAGE, followed by Western blot analysis. Pulldown assays were digitized and densitometrically assessed.
Rho GTPase G‐LISA™ activation assay
To study the activation of RhoA and Cdc42 in microvessels of individual animals, a G‐LISA™ Rho activation assay (BK124; Cytoskeleton Inc.) and G‐LISA™ Cdc42 activation assay (BK127; Cytoskeleton Inc.) were established. Briefly, isolated brain microvessels were lysated using the G‐LISA™ cell lysis buffer and protein concentrations were estimated using Precision Red™ advanced protein assay reagent (GL50; Cytoskeleton Inc.). Five micrograms of brain microvessel lysates was equilibrated to an equal volume of 50 µL using G‐LISA™ cell lysis buffer. Fifty microliters of blank control buffer, 50 µL of RhoA or Cdc42 positive controls, and 50 µL of equalized brain microvessel lysates were added into the 96‐well Rho‐GTP or Cdc42‐GTP binding microplate and incubated at 4°C for 30 minutes. Wells were washed, and 200 µL of antigen presenting buffer was added into each well for 2 minutes. After washing, 50 µL of anti‐RhoA antibody, diluted to 1:250 in antibody dilution buffer, or 50 µL of anti‐Cdc42 antibody, diluted to 1:20 in antibody dilution buffer, was added to each well and incubated at room temperature for 45 minutes (for RhoA) or 30 minutes (for Cdc42). After primary antibody incubation, wells were washed and 50 µL of secondary HRP labeled antibody, diluted to 1:62.5 in antibody dilution buffer, were added to each well and incubated at room temperature for 45 minutes (RhoA) or 30 minutes (Cdc42). Afterwards, wells were washed and 50 µL of freshly prepared HRP detection reagent (in case of RhoA) or 70 µL (in case of Cdc42) were added to each well and incubated for at 37°C for 15 minutes (RhoA) or 10 minutes (Cdc42), respectively. Finally, 50 microliters or 140 µL of HRP stop buffer (for RhoA and Cdc42, respectively) were added and absorbance was measured at 490 nm using the iMark microplate reader. RhoA and Cdc42 activation was expressed as absorbance at 490 nm units (A490nm).
Co‐precipitation assay with constitutively inactive Cdc42
To study the interaction between inactive Cdc42 with p120 catenin, 50 µL (50 µg) of recombinant dominant negative (i.e. inactive) GST‐Cdc42‐(T17N) prefixed on agarose beads was processed with samples of equal protein quantities (600 µg) from lysates obtained using MgCl2 lysis buffer, as described in the affinity binding assay protocol. Samples were subjected to SDS‐PAGE using 10% acrylamide‐bis gel, following Western blot analysis for p120 catenin.
Immunoprecipitation assay
Equal protein quantities (800 µg) of lysates obtained using an NP‐40 lysis buffer containing 150 mM NaCl, 1% NP‐40, 50 mM Tris base (pH 8.0) were supplemented with sodium orthovanadate (final concentration: 1 mM) and complemented with three equal volumes of NET buffer (100 mM Tris, 200 mM NaCl, 5 mM EDTA, 5% NP‐40, pH 7.4). Six micrograms of anti‐RhoA antibody, 10 µg of occludin or 6 µg of p‐120 catenin were added to each sample, and incubated overnight at 4°C under slight rotation. The next day, 20 µL of protein A/G plus‐agarose was added to the samples and incubated over 1 h at 4°C. Finally, samples were centrifuged for 30 s at 15 000 rpm at 4°C. Supernatants were dispersed and pellets were washed three times in ice‐cold NET buffer. Twenty microliters of 2× SDS loading buffer was added to each pellet and boiled for 5 minutes (Heater Plate), followed by a short centrifugation at 4000 rpm to precipitate beads. Supernatants were subjected to SDS‐PAGE using 10% acrylamide‐bis gel followed by Western blot analysis for p120 catenin, ZO‐1, phospho‐tyrosine, phospho‐threonine or Vav2. As control experiments for the immunoprecipitation, ischemic tissues from vehicle‐treated ischemic mice were used. A/G plus‐agarose beads were added in order to clarify if the beads used nonspecifically bind endogenous IgG or the proteins of interest.
Statistics
Data were analyzed by unpaired t‐tests (comparisons between two groups) or one‐way analysis of variance followed by least significant differences tests (comparisons between ≥3 groups) using SPSS for Windows (SPSS Inc., Chicago, IL, USA). Results are presented as means ± SD. P‐values less than 0.05 were considered significant.
RESULTS
Effect of LXR agonist T0901317 on ischemic injury and vascular permeability
To evaluate effects of LXR activation on brain edema, vascular integrity and ischemic injury, we first analyzed cresyl violet stainings and histochemistries against extravasated serum IgG of animals pretreated with T0901317. Reduced brain swelling (Figure 1A) and decreased serum IgG extravasation (Figure 1B) were noticed in ischemic animals receiving T0901317, indicating preservation of the BBB by the LXR agonist. Infarct area did not show any difference between groups (Figure 1C). LDF recordings also did not differ between groups (not shown), indicating the absence of hemodynamic differences between T0901317 treated and vehicle‐treated animals.
Figure 1.
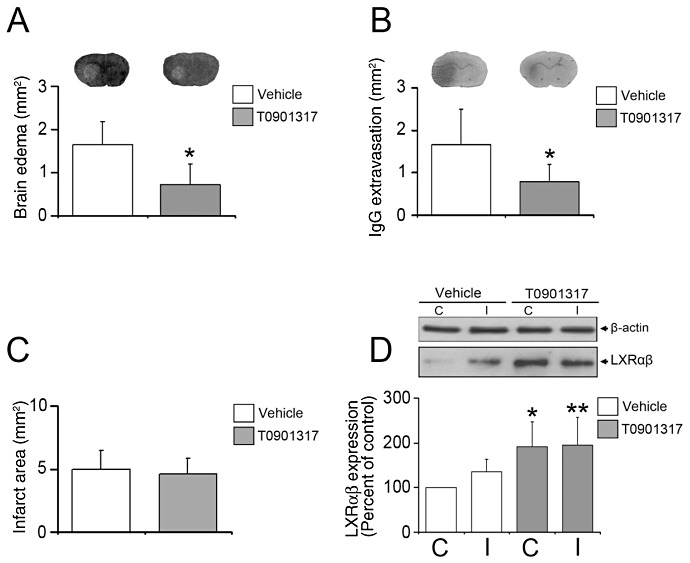
LXR agonist T0901317 decreases brain edema and leakage, once administered prior to stroke. Histochemical studies analyze brain swelling (A) and serum IgG extravasation (B), which are both reduced in animals receiving the LXR agonist T0901317, which in this study was delivered 1 week prior to MCA occlusion. Note that infarct area (C) is not influenced by LXR activation. Western blots using brain capillary extracts show that T0901317 robustly increases LXRαβ expression (D). In (A) and (B), representative cresyl violet and IgG extravasation sections are also shown. Data are means ± SD [n = 8 animals per group (A–C); n = 4 Western blots (D) using pools of tissue samples from animals belonging to the same group]. *P < 0.05/**P < 0.01 compared with nonischemic vehicle. C = contralateral nonischemic microvessels; I = ischemic microvessels; LXR = liver X receptor; MCA = middle cerebral artery.
Effect of T0901317 on LXR expression
To check whether T0901317 indeed elevates LXRαβ levels, we used Western blots with capillary extracts obtained from tissue samples harvested from the MCA territories ipsilateral and contralateral to the stroke demonstrating that LXRαβ expression was indeed increased by T0901317, both in the ischemic and nonischemic brain tissue (Figure 1D).
Regulation of calpain‐1/2 and MMP‐2/9 activities by T0901317
To elucidate the role of calpain‐1/2 and MMP‐2/9 in BBB preservation induced by LXR activation, we evaluated their expression and activity in cerebral microvessels by Western blots, calpain‐1/2 caseinase microplate assay, gelatin zymography and MMP‐2/9 gelatinase microplate assay. Western blots revealed that the overall expression of calpain‐1/2 and MMP‐2/9 was neither influenced by ischemia nor by T0901317 (Figure 2A).
Figure 2.
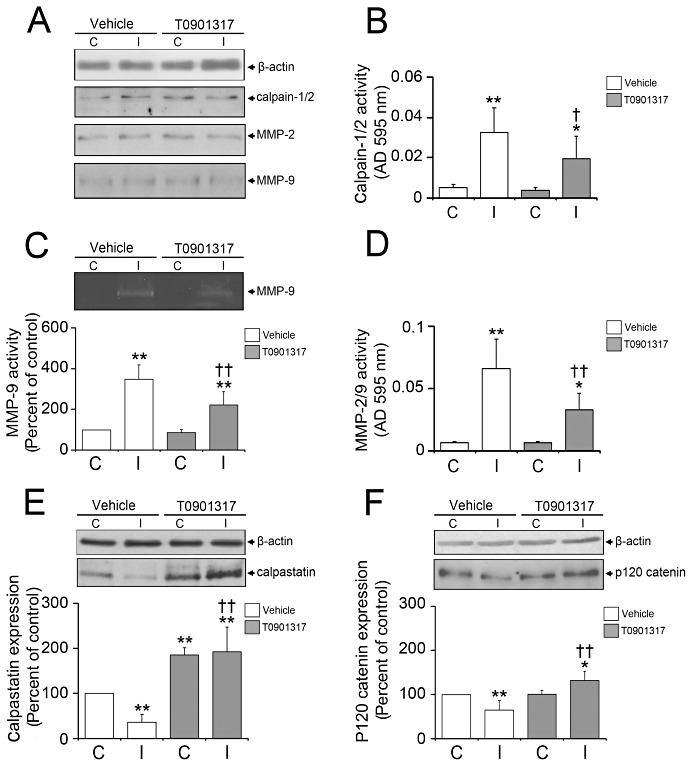
LXR agonist T0901317 induces calpastatin expression, inhibits calpain‐1/2 activity, stabilizing p120 catenin, and decreases MMP‐2/9 activity. Western blots (A, E, F), calpain‐1/2 caseinase assay (B), MMP‐9 gelatin zymography (C) and MMP‐2/9 gelatinase assay (D) showed that calpain‐1/2 and MMP‐2/9 activity (B–D), but not expression (A), is reduced in ischemic microvessels of T0901317‐treated mice, calpain‐1/2 being deactivated by its endogenous inhibitor calpastatin, which is overexpressed in the presence of the LXR agonist (E). In response to the reduced activity of calpain‐1/2, p120 catenin expression is preserved (F). In this study, T0901317 was again delivered prior to the stroke. Data are means ± SD (n = 4 Western blots, microplate assays or zymographies using pools of tissue samples). *P < 0.05/**P < 0.01 compared with nonischemic vehicle; † P < 0.05/ †† P < 0.01 compared with ischemic vehicle. C = contralateral nonischemic microvessels; I = ischemic microvessels; LXR = liver X receptor; MMP = matrix metalloproteinase.
The activities of calpain‐1/2 and MMP‐2/9, meanwhile, strongly increased after stroke (Figure 2B–D), which is in line with earlier studies from other groups 4, 39, 42. Importantly, calpain‐1/2 and MMP‐2/9 activities were attenuated by the LXR agonist (Figure 2B–D). In case of MMP‐2 and ‐9, the inhibition of protease activity was detected by means of gelatin zymography and gelatinase microplate assay (Figure 2C,D) that evaluate MMP‐2/9 activity in the absence or presence of its endogenous inhibitors, respectively. Based on the fact that both methods rendered similar results, endogenous inhibitors of MMP‐2/9 are probably not essential for the inhibitory effect of LXR activation on MMP‐2/9.
T0901317 increases the expression of LXR's target gene calpastatin
To identify factors responsible for calpain‐1/2 deactivation, we studied the expression of its specific inhibitor calpastatin, which is transcriptionally regulated by LXR (21), by means of Western blots. Reduced expression levels of calpastatin were observed in ischemic microvessels of vehicle‐treated mice (Figure 2E). T0901317 increased calpastatin expression above levels in nonischemic vessels (Figure 2E). Our data suggest that calpastatin may be responsible for the inhibition of calpain‐1/2 by the LXR agonist.
LXR activation by T0901317 stabilizes p120 catenin
To define downstream targets of calpain‐1/2 that might mediate BBB preservation induced by LXR activation, we assessed the expression of p120 catenin, a target protein degraded by calpain‐1/2 (31), using Western blots. In correlation with calpain‐1/2 activation, p120 catenin expression was reduced in ischemic microvessels of vehicle‐treated mice (Figure 2F). Conversely, T0901317, which inhibited calpain‐1/2, elevated p120 catenin expression in ischemic brain capillaries even above levels in nonischemic vessels (Figure 2F).
LXR activation by T0901317 differentially regulates RhoA and Cdc42 activity
To understand whether and how LXR activation influences the activity of Rho GTPases involved in the regulation of BBB integrity, we next performed pull‐down assays for RhoA and Cdc42. These studies revealed that both RhoA and Cdc42 are activated upon ischemia in cerebral microvessels (Figure 3A,B). Interestingly, T0901317 inhibited RhoA (Figure 3A), at the same time further elevating Cdc42 activity in ischemic, but not non‐ischemic vessels (Figure 3B).
Figure 3.
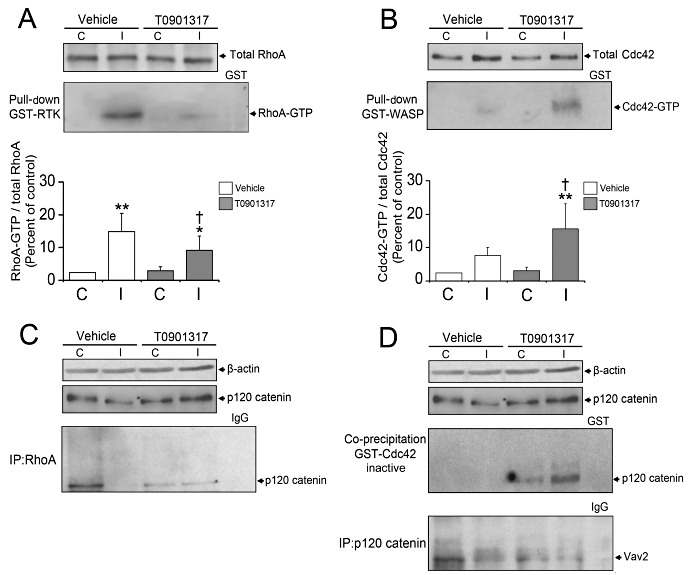
p120 catenin controls RhoA and Cdc42 activation. Pulldown assays detecting GTP‐bound (ie, activated) RhoA and Cdc42 reveal that LXR induction reduces RhoA activation (A) and increases Cdc42 activation (B) in ischemic microvessels. Western blots of total RhoA and Cdc42 expression reveal no differences among groups (A, B). Immunoprecipitation experiments demonstrate that RhoA binding to p120 catenin is abolished upon ischemia, but restored upon T0901317 treatment (C). Co‐precipitation studies show that inactive Cdc42 does not bind p120 catenin neither under normal nor ischemic microvessels of vehicle‐treated mice (D). Interestingly, LXR activation induces binding of inactive Cdc42 to p120 catenin, which also binds Vav2, a Cdc42 GEF (D). For these experiments, T0901317 was administered prior to the stroke. Data are means ± SD (n = 4 pull‐down assays using pools of tissue samples). p120 catenin Western blot from Figure 2F is used for illustration purposes. *P < 0.05/ **P < 0.01 compared with nonischemic vehicle; † P < 0.05 compared with ischemic vehicle. C = contralateral nonischemic microvessels; I = ischemic microvessels; LXR = liver X receptor.
p120 catenin controls RhoA and Cdc42 activity in ischemic microvessels
As p120 catenin acts as GDI for RhoA 1, 40 and also binds GEFs that specifically activate Cdc42 (29), we investigated the role of p120 catenin in the differential regulation of RhoA and Cdc42 by T0901317. In immunoprecipitation studies, we showed that RhoA binds physically to p120 catenin in vehicle‐treated, nonischemic cerebral microvessels, whereas this RhoA interaction with p120 catenin was absent in ischemic brain capillaries (Figure 3C). T0901317 restored the RhoA binding to p120 catenin in ischemic microvessels (Figure 3C), suggesting that RhoA interaction with this GDI is responsible for the RhoA inhibition by the LXR agonist.
However, we found that inactive GST‐Cdc42 binds p120 catenin in T0901317, but not in vehicle‐treated nonischemic and ischemic microvessels (Figure 3D), which may have been facilitated by the restoration of p120 catenin expression in ischemic capillary cells. Active GST‐Cdc42 did not reveal interaction with p120 catenin (not shown). Moreover, we observed that Vav2 interacts with p120 catenin (Figure 3D), suggesting that p120 catenin promotes Cdc42 activation by enhancing the access of Cdc42 to its GEF.
Regulation of occludin and ZO‐1 by LXR agonist T0901317
To further elucidate how LXR activation influences BBB integrity, we studied the expression, phosphorylation and interaction of the tight junction proteins occludin and ZO‐1 by Western blots and immunoprecipitation studies. In accordance to previous studies 6, 38, the overall expression and threonine phosphorylation of occludin decreased, the tyrosine phosphorylation of occludin increased and the association of occludin with ZO‐1 decreased upon ischemia in vehicle‐treated microvessels (Figure 4A–D), reflecting the breakdown of BBB integrity following stroke. Interestingly, T0901317 selectively prevented the downregulation of occludin and ZO‐1 on ischemic microvessels (Figure 4A,B), at the same time reversing occludin threonine phosphorylation and abolishing occludin tyrosine phosphorylation, thus enhancing the assembly of occludin with ZO‐1 (Figure 4C,D).
Figure 4.
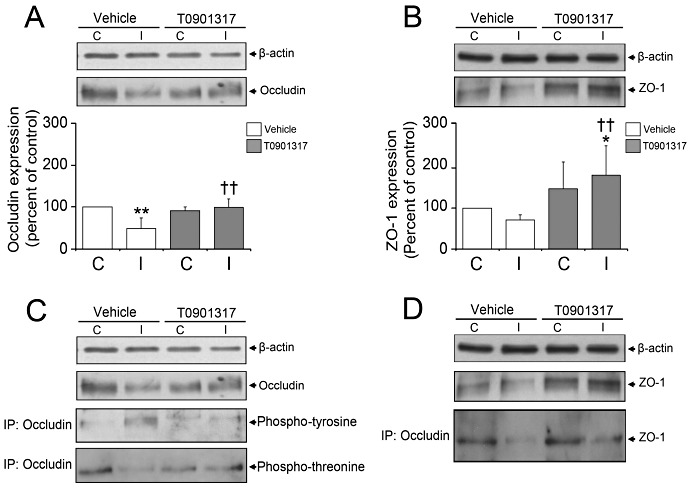
LXR activation by T0901317 modulates the expression and phosphorylation of occludin, and its assembly with ZO‐1. Western blot analysis (A, B) and immunoprecipitation experiments (C, D) demonstrate that T0901317 restores occludin and ZO‐1 expression in ischemic microvessels that are otherwise reduced in vehicle‐treated mice (A, B), decreasing occludin tyrosine phosphorylation (C), increasing occludin threonine phosphorylation (C) and enhancing occludin interaction with ZO‐1 (D). T0901317 was delivered prior to the stroke. Data are means ± SD (n = 4 Western blots using pools of tissue samples). C, contralateral non‐ischemic microvessels; I, ischemic microvessels. *P < 0.05/**P < 0.01 compared with nonischemic vehicle; †† P < 0.01 compared with ischemic vehicle. C = contralateral nonischemic microvessels; I = ischemic microvessels; LXR = liver X receptor; ZO = zona occludens.
LXR agonist T0901317 deactivates JNK1/2 and caspase‐3 pathways
The activation of JNK‐1/2 via phosphorylation represents a cellular stress signal that promotes endothelial apoptosis (35). To analyze how LXR activation influences JNK1/2 phosphorylation state, we performed Western blots using antibodies detecting either phosphorylated or total (ie, phosphorylated and unphosphorylated) JNK1/2. In vehicle‐treated mice, JNK phosphorylation was decreased in ischemic microvessels (Figure 5A). T0901317 further reduced JNK1/2 phosphorylation levels in ischemic microvessels (Figure 5A).
Figure 5.
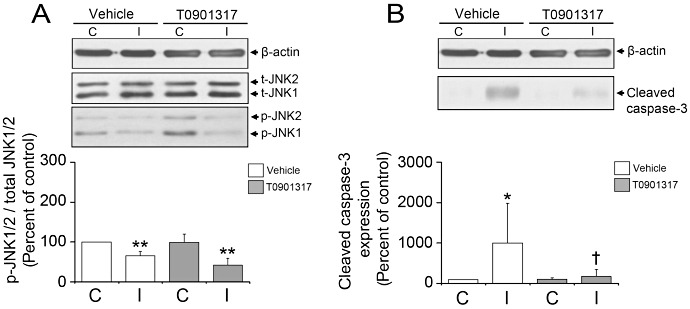
LXR agonist T0901317 deactivates JNK1/2 and caspase‐3. Western blot analysis shows that LXR activation decreases JNK phosphorylation (A) and caspase‐3 cleavage (B) in ischemic microvessels. T0901317 was administered prior to the stroke. Data are means ± SD (n = 4 Western blots using pools of tissue samples). *P < 0.05/**P < 0.01 compared with non‐ischemic vehicle; † P < 0.05 compared with ischemic vehicle. C = contralateral nonischemic microvessels; I = ischemic microvessels; JNK = Jun N‐terminal kinase; LXR = liver X receptor.
It has been shown that calpain‐1/2 facilitates the activation of caspase‐3, thus promoting neuronal apoptotic injury (2). To investigate how T0901317, which deactivates calpain‐1/2, influences caspase‐3 activity, we examined the expression of cleaved, that is, activated caspase‐3 by Western blots. Our results showed that caspase‐3, which is strongly activated upon ischemia, is deactivated by the LXR agonist (Figure 5B). Our data reveal a survival‐promoting effect of T0901317 on cerebral microvessels.
T0901317 upregulates ABC transporters at the BBB
It has previously been shown that the cholesterol transporters ABCA1 and ABCG1 are upregulated on noncerebral microvessels upon LXR activation (28). By means of Western blots, we now observed that the cholesterol transporters ABCA1 and ABCG1 are also upregulated on cerebral microvessels upon T0901317 treatment (Figure 6A,B). Strikingly, LXR activation also elevated ABCB1 expression (Figure 6C) and restored the reduced ABCC1 expression that was observed in ischemic vehicle‐treated mice (Figure 6D). As such, T0901317 increased the overall abundance of ABCB1, ABCC1, ABCA1 and ABCG1 transporters without influencing luminal (ABCB1, ABCA1, ABCG1) and abluminal (ABCC1) transporters in a differential way.
Figure 6.
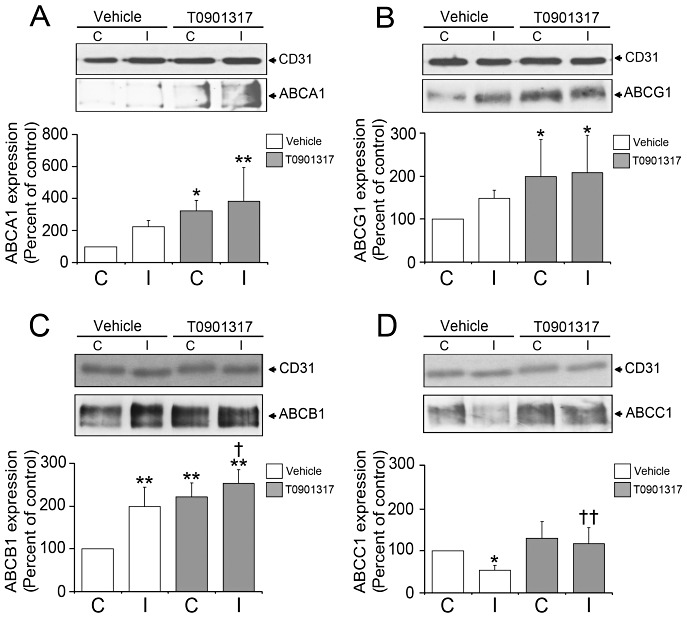
T0901317 increases the abundance of ABC transporters ABCA1, ABCG1, ABCB1 and ABCC1 on cerebral endothelial cells. Western blot analysis reveals that the LXR agonist increases the abundance of the cholesterol transporters ABCA1 (A) and ABCG1 (B) on brain microvessels, at the same time elevating the abundance of the drug transporters ABCB1 (C) and ABCC1 (D) on ischemic capillaries. T0901317 was delivered prior to the stroke. Data are means ± SD (n = 4 Western blots using pools of tissue samples). *P < 0.05/**P < 0.01 compared with non‐ischemic vehicle; † P < 0.05/†† P < 0.01 compared with ischemic vehicle. ABC = ATP‐binding cassette; C = contralateral nonischemic microvessels; I = ischemic microvessels; JNK = Jun N‐terminal kinase; LXR = liver X receptor.
T0901317 decreases brain edema also when delivered after the stroke, and its effect is mimicked by a calpain‐1/2 inhibitor
To test whether postischemic LXR activation preserves BBB integrity similar to pretreatment, we evaluated brain edema, infarct area, calpain‐1/2 activity, MMP‐2/9 activity and RhoGTPase activity in animals receiving T0901317 1 h after MCA occlusion by means of cresyl violet stainings, calpain‐1/2 caseinase and MMP‐2/9 gelatinase microplate assays, and RhoGTPase G‐LISA. Similar to LXR agonist pretreatment, brain edema (Figure 7A) was reduced by T0901317 in animals receiving the agonist during reperfusion. In addition, a reduction of infarct area was seen (Figure 7B). Furthermore, calpain‐1/2 activity was decreased in ischemic microvessels (Figure 7C) as was MMP‐2/9 activity, which was nonsignificantly reduced (Figure 7D). The deactivation of calpain‐1/2 went along with the deactivation of the Rho GTPase RhoA (Figure 7E) and overactivation of Cdc42 (Figure 7F). Importantly, calpain‐1/2 inhibition using the pharmacological inhibitor MDL28170 mimicked the effects of T0901317 on brain swelling (Figure 7A), calpain‐1/2 (Figure 7C), MMP‐2/9 (Figure 7D), RhoA (Figure 7E) and Cdc42 (Figure 7F) activities, indicating that calpain‐1/2 inhibition may causally be involved in T0901317's effects at the blood–brain interface. Conversely, calpain‐1/2 inhibition incompletely mimicked the effect of the LXR agonist on infarct area (Figure 7B), indicating that the neuroprotective effect of the LXR agonist involves mechanisms other than calpain‐1/2.
Figure 7.
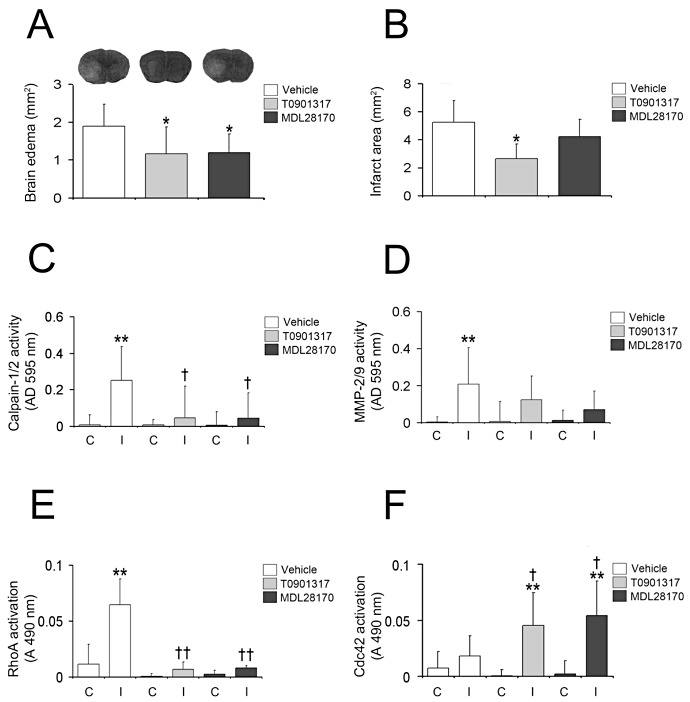
T0901317 decreases brain edema, when delivered after MCA occlusion, and its effect is mimicked by a calpain‐1/2 inhibitor. Histochemical studies examining brain swelling (A) and infarct area (B), microplate assays evaluating calpain‐1/2 (C) and MMP‐2/9 (D) activity, and G‐LISA analyzing RhoA (E) and Cdc42 (F) activity in animals receiving the LXR agonist T0901317 or the calpain‐1/2 inhibitor MDL28170 that were administered 1 h after MCA occlusion. Note that unlike preischemic delivery, postischemic T0901317 both reduces brain swelling (A) and infarct size (B), furthermore deactivating calpain‐1/2 (C), MMP‐2/9 (D) and RhoA (E), and activating Cdc42 (F) in ischemic tissues. Note that the calpain‐1/2 inhibitor MDL28170 mimics the effect of T0901317 on brain swelling (A), calpain‐1/2 (C), MMP‐2/9 (D), RhoA (E) and Cdc42 (F), but not on infarct size (B). In (A), representative cresyl violet sections are also shown. Data are means ± SD [n = 5 animals per group (A, B); n = 5 Western blots, microplate assays or G‐LISA (C–F) using tissue samples from individual animals]. *P < 0.05/**P < 0.01 compared with nonischemic vehicle; † P < 0.05/†† P < 0.01 compared with ischemic vehicle. C = contralateral nonischemic microvessels; I = ischemic microvessels; LXR = liver X receptor; MCA = middle cerebral artery; MMP = matrix metalloproteinase.
LXR agonist GW3965 also decreases brain edema and increases calpastatin and p120 catenin expression
To verify that the changes observed are indeed induced by LXR activation, we also administered a second LXR agonist, GW3965, 1 h after MCA occlusion, and examined brain edema, infarct volume and the expression of LXRα, LXRβ, calpastatin and p120 catenin by means of cresyl violet stainings and Western blots. Similar to T0901317, GW3965 reduced edema volume (Figure 8A) and infarct volume (Figure 8B). LXR activation induced LXRα expression (Figure 8C) on cerebral endothelial cells, without affecting LXRβ expression (Figure 8D). Calpastatin expression was increased on ischemic brain capillaries by GW3965 (Figure 8E) as was p120 catenin (Figure 8F).
Figure 8.
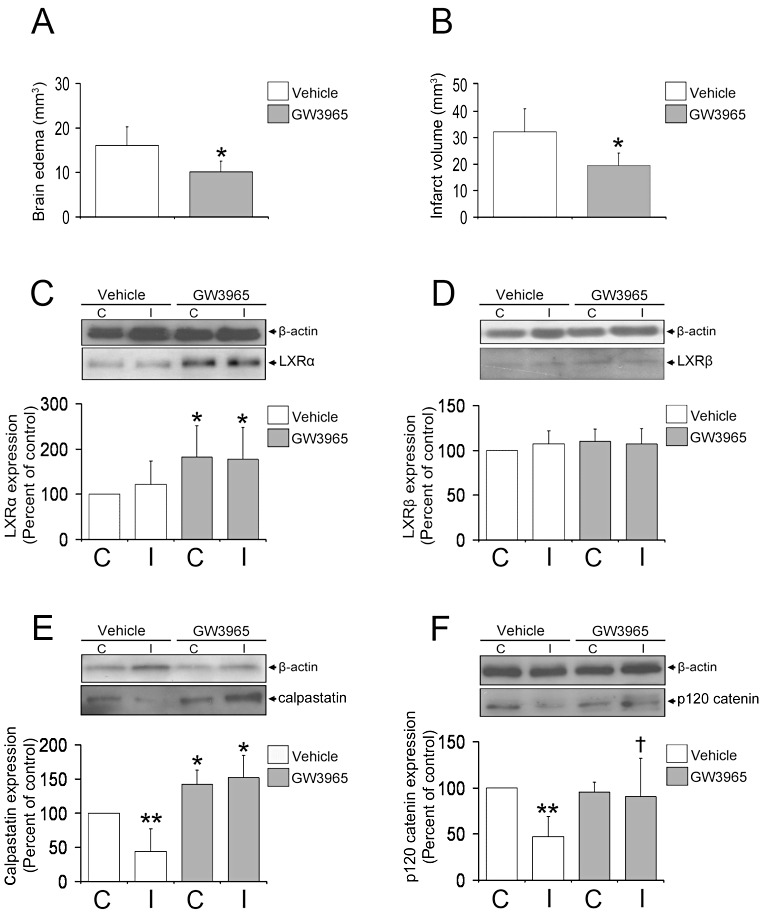
LXR agonist GW3965 also reduces brain edema, increasing LXRα, calpastatin and p120 catenin. Histochemical studies examine brain edema (A) and infarct volume (B), as well as Western blots evaluating the expression of LXRα (C), LXRβ (D) calpastatin (E) and p120 catenin (F) in animals receiving the LXR agonist GW3965 that was administered 1 h after MCA occlusion. Similar to postischemic T0901317, GW3965 delivery reduces brain swelling (A) and infarct size (B). Besides, GW3965 increases LXRα expression (C) without affecting LXRβ (D), and potently induces the expression of calpastatin (E), thereby stabilizing p120 catenin (F) in ischemic microvessels. Data are means ± SD [n = 5 animals per group (A, B); n = 4 Western blots]. *P < 0.05/**P < 0.01 compared with nonischemic vehicle; † P < 0.05 compared with ischemic vehicle. C = contralateral nonischemic microvessels; I = ischemic microvessels; LXR = liver X receptor; MCA = middle cerebral artery.
DISCUSSION
Using in vivo experiments of mice submitted to focal cerebral ischemia, which we combined with protein expression and interaction studies, we demonstrate that LXR activation promotes BBB integrity by mechanisms involving upregulation of LXR's target gene calpastatin, deactivation of calpain‐1/2, a known target of calpastastin, and stabilization of p120 catenin, calpain‐1/2's downstream target. p120 catenin specifically interacts with RhoA and Cdc42, the former of which is inhibited and the latter overactivated, thus restoring the postischemic expression and phosphorylation of tight junction proteins and promoting their assembly. Our data provide a detailed analysis of signaling pathways involved in BBB regulation after stroke. In parallel to its effects on tight junction integrity, LXR activation decreases the activation of the stress kinase JNK1/2 and caspase‐3 and increases the abundance both of the luminal endothelial drug transporter ABCB1 and the abluminal endothelial drug transporter ABCC1 on ischemic brain capillaries. Our results show that LXR activation promotes the integrity of cerebral endothelial cells in several ways.
It has previously been shown that the cysteine protease calpain‐1/2 increases BBB permeability and exacerbates brain edema 4, 36. The signal pathways via which this protease leads to BBB breakdown remained unknown. We now provide evidence that ischemia induces calpain‐1/2 activation in brain microvessels by downregulating its endogenous inhibitor calpastatin 13, 36, which reduces p120 catenin levels in ischemic microvessels, thus resulting in the loss of RhoA binding to its GDI p120 catenin 1, 29 and inducing RhoA overactivation. Indeed, the delivery of LXR agonists restored calpastastin, a known LXR target based on transcriptional profiling studies (21), deactivated calpain‐1/2 and stabilized p120 catenin, reconstituting p120 catenin interaction with RhoA and Cdc42, which resulted in RhoA deactivation and, as p120 catenin binds GEFs specifically activating Cdc42, such as Vav2, in Cdc42 overactivation. The effects of LXR activation on brain swelling, RhoA and Cdc42 activity were mimicked by MDL28170, a calpain‐1/2 inhibitor, indicating that calpain‐1/2 is causally involved in the stabilization of the BBB. That LXR activation deactivates calpain‐1/2 is new, and modulation of Rho GTPases by LXR agonists to the best of our knowledge, has not yet been shown.
In migrating cells, RhoA activation is followed by Cdc42 activation (25). That both Rho GTPases differentially respond to LXR activation is noteworthy and may offer a clue about why the LXR agonist treatment promotes BBB integrity. RhoA and Cdc42 have opposite effects on BBB integrity. RhoA activation destabilizes tight junction complexes via stress fiber formation, thereby inducing BBB leakage 7, 18, 33, whereas Cdc42 activation promotes the assembly of tight junction proteins, thus enabling BBB tightness 5, 12. In our studies, the shift between RhoA and Cdc42 balance was accompanied by profound changes in the expression, phosphorylation and interaction of tight junction proteins occludin and ZO‐1. We presume that the differential response of RhoA and Cdc42 to LXR activation is a consequence of specific interactions with p120 catenin, which has differential effects on the activation of both small GTPases 1, 30.
Besides modulating the activation of Rho GTPases, LXR activation also deactivated MMP‐2/9, JNK1/2 and caspase‐3 in cerebral microvessels, which may at least partly represent downstream signals of the reduced calpain‐1/2 activity. Previous studies already suggested a role of calpains in MMP‐9 activation in the stroke brain (39). In microvascular endothelial cells, calpain‐1 was shown to translocate into mitochondria upon activation, inducing oxidative bursts activating MMP‐2/9 (27). Indeed, calpain‐1 inhibitors blocked the formation of reactive oxygen species and prevented the MMP‐9 activation (38). Regarding caspase‐3, the proteolytic capacity of calpain‐2 to facilitate the activation of this executer caspase is well known. As such, calpain‐2 degrades the caspase‐3 full‐length proform into a 29 kDa fragment that may subsequently be cleaved more easily to active forms (2). This degradation has been shown to contribute to neonatal hypoxic–ischemic neuronal injury (2).
In our study, the LXR agonist T0901317 protected the brain from ischemic injury when the compound was delivered after but not prior to the stroke. Interestingly, calpain‐1/2 deactivation by MDL28170 only partly mimicked the effect of LXR activation on infarct area, indicating that mechanisms other than inhibition of calpain‐1/2 are involved in their neuroprotective effects. Neuroprotection induced by LXR activation has previously been described in models of focal cerebral ischemia (26). In that earlier study, T0901317‐induced neuroprotection involved anti‐inflammatory effects, namely reduction of nuclear factor κB transcriptional activity. That in our study neuroprotection was noticed only following postischemic, but not preischemic, T0901317 delivery might suggest that inflammatory responses are influenced by single vs. repeated LXR activation in a different way. Our data argue in favor of a specific effect of LXR agonists at the ischemic BBB, which is independent of neuronal survival.
We did not use LXR‐deficient mice in our present study to assess the effect of LXR activation on BBB integrity. As LXR activation by T0901317 agonist has been shown to activate other nuclear receptors to some extent, namely farnesoid X receptor (FXR) (19), we administered a second LXR agonist, GW3965, which was shown to be a selective activator of LXR based on reporter gene assays (8). We found that GW3965 mimicked the effects of T0901317 on brain edema, ischemic injury, and endothelial calpastatin and p120 catenin expression. The combined evidence of the two agonists strongly suggests that the BBB changes we observed are indeed specific for LXR.
Besides influencing paracellular BBB permeability, the LXR agonist T0901317 increased expression levels of ABC transporters on ischemic cerebral endothelial cells. As such, not only the cholesterol transporters ABCA1 and ABCG1, which are known targets of LXR based on transcriptional profiling studies 21, 28 and which have been shown to be induced by T0901317 on noncerebral vessels 3, 9, but also the drug transporters ABCB1 and ABCC1, were elevated on cerebral microvessels. It is noteworthy that both ABCB1, which is expressed predominantly on the luminal membrane of brain capillary cells (37), and ABCC1, which is found mainly on the abluminal endothelial membrane (22), were upregulated in response to T0901317. Both transporters are regulated in opposite ways after focal cerebral ischemia, ABCB1 being upregulated and ABCC1 being downregulated in response to stroke 22, 37.
We did not perform chromatin immunoprecipitation in this study. As such, we are unable to answer whether the abcb1 and abcc1 genes are direct or indirect targets of LXR. Transcriptional regulation of the abcb1 and abcc1 genes by LXR has so far not been shown. It has previously been reported in human primary blood monocytes that abcb1 mRNA is regulated by T0901317 (23). Whether the abcb1 gene was a direct target of LXR or whether it was controlled by downstream transcription factors was also not assessed in that earlier study. We did not perform ABC transporter functionality studies in this report. As such, we cannot draw any conclusions whether the changes of ABC transporter expression induced by LXR activation are relevant for drug biodistribution. As LXR activation stabilizes cerebral endothelial cells in several ways, increasing paracellular BBB tightness, reducing cellular stress responses and caspase‐3 activation, and increasing ABC transporter abundance, this receptor may represent an interesting target for therapeutic interventions aiming at the stabilization of blood vessels under conditions of pronounced BBB breakdown, for example, under conditions of malignant brain edema.
DISCLOSURE STATEMENT
There are no actual or potential conflicts of interests.
ACKNOWLEDGMENTS
We thank B. Karow for technical assistance. This study is supported by a German Research Foundation grant (HE3173/2‐1; to D.M.H.), a grant of the Dr‐Werner‐Jackstädt‐Foundation (to A.EA.) and an endowment of the Heinz‐Nixdorf‐Foundation (to D.M.H.).
REFERENCES
- 1. Anastasiadis PZ, Moon SY, Thoreson MA, Mariner DJ, Crawford HC, Zheng Y, Reynolds AB (2000) Inhibition of RhoA by p120 catenin. Nat Cell Biol 2:637–644. [DOI] [PubMed] [Google Scholar]
- 2. Blomgren K, Zhu C, Wang X, Karlsson JO, Leverin AL, Bahr BA et al (2001) Synergistic activation of caspase‐3 by m‐calpain after neonatal hypoxia‐ischemia: a mechanism of “pathological apoptosis”? J Biol Chem 276:10191–10198. [DOI] [PubMed] [Google Scholar]
- 3. Bradley MN, Hong C, Chen M, Joseph SB, Wilpitz DC, Wang X et al (2007) Ligand activation of LXR beta reverses atherosclerosis and cellular cholesterol overload in mice lacking LXR alpha and apoE. J Clin Invest 117:2337–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin XM et al (2007) Critical role of calpain I in mitochondrial release of apoptosis‐inducing factor in ischemic neuronal injury. J Neurosci 27:9278–9293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cau J, Hall A (2005) Cdc42 controls the polarity of the actin and microtubule cytoskeletons through two distinct signal transduction pathways. J Cell Sci 118:2579–2587. [DOI] [PubMed] [Google Scholar]
- 6. Chen Y, Lu Q, Schneeberger EE, Goodenough DA (2000) Restoration of tight junction structure and barrier function by downregulation of the mitogen‐activated protein kinase pathway in ras‐transformed Madin‐Darby canine kidney cells. Mol Biol Cell 11:849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chrissobolis S, Sobey CG (2006) Recent evidence for an involvement of rho‐kinase in cerebral vascular disease. Stroke 37:2174–2180. [DOI] [PubMed] [Google Scholar]
- 8. Collins JL, Fivush AM, Watson MA, Galardi CM, Lewis MC, Moore LB et al (2002) Identification of a nonsteroidal liver X receptor agonist through parallel array synthesis of tertiary amines. J Med Chem 45:1963–1966. [DOI] [PubMed] [Google Scholar]
- 9. Efanov AM, Sewing S, Bokvist K, Gromada J (2004) Liver X receptor activation stimulates insulin secretion via modulation of glucose and lipid metabolism in pancreatic beta‐cells. Diabetes 53:S75–S78. [DOI] [PubMed] [Google Scholar]
- 10. ElAli A, Hermann DM (2010) Apolipoprotein‐E controls ATP‐binding cassette transporters in the ischemic brain. Sci Signal 3:ra72. [DOI] [PubMed] [Google Scholar]
- 11. Etienne‐Manneville S, Hall A (2002) Rho GTPases in cell biology. Nature 420:629–635. [DOI] [PubMed] [Google Scholar]
- 12. Fukuhara A, Shimizu K, Kawakatsu T, Fukuhara T, Takai Y (2003) Involvement of nectin‐activated Cdc42 small G protein in organization of adherens and tight junctions in Madin‐Darby canine kidney cells. J Biol Chem 278:51885–51893. [DOI] [PubMed] [Google Scholar]
- 13. Goll DE, Thompson VF, Li H, Wei W, Cong J (2003) The calpain system. Physiol Rev 83:731–801. [DOI] [PubMed] [Google Scholar]
- 14. Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A et al (2002) S‐nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science 297:1186–1190. [DOI] [PubMed] [Google Scholar]
- 15. Hawkins BT, Davis TP (2005) The blood‐brain barrier/neurovascular unit in health and disease. Pharmacol Rev 57:173–185. [DOI] [PubMed] [Google Scholar]
- 16. Hermann DM, Bassetti CL (2007) Implications of ATP‐binding cassette transporters for brain pharmacotherapies. Trends Pharmacol Sci 28:128–134. [DOI] [PubMed] [Google Scholar]
- 17. Hermann DM, Kilic E, Spudich A, Krämer SD, Wunderli‐Allenspach H, Bassetti CL (2006) Role of drug efflux carriers in the healthy and diseased brain. Ann Neurol 60:489–498. [DOI] [PubMed] [Google Scholar]
- 18. Hirase T, Kawashima S, Wong EY, Ueyama T, Rikitake Y, Tsukita S et al (2001) Regulation of tight junction permeability and occludin phosphorylation by RhoA‐p160 ROCK‐dependent and ‐independent mechanisms. J Biol Chem 276:10423–10431. [DOI] [PubMed] [Google Scholar]
- 19. Houck KA, Borchert KM, Hepler CD, Thomas JS, Bramlett KS, Michael LF, Burris TP (2004) T0901317 is a dual LXR/FXR agonist. Mol Genet Metab 83:184–187. [DOI] [PubMed] [Google Scholar]
- 20. Huber JD, Egleton RD, Davis TP (2001) Molecular physiology and pathophysiology of tight junctions in the blood‐brain barrier. Trends Neurosci 24:719–725. [DOI] [PubMed] [Google Scholar]
- 21. Hummasti S, Laffitte BA, Watson MA, Galardi C, Chao LC, Ramamurthy L et al (2004) Liver X receptors are regulators of adipocyte gene expression but not differentiation: identification of apoD as a direct target. J Lipid Res 45:616–625. [DOI] [PubMed] [Google Scholar]
- 22. Kilic E, Spudich A, Kilic U, Rentsch KM, Vig R, Matter CM et al (2008) ABCC1: a gateway for pharmacological compounds to the ischaemic brain. Brain 131:2679–2689. [DOI] [PubMed] [Google Scholar]
- 23. Langmann T, Mauerer R, Schmitz G (2006) Human ATP‐binding cassette transporter TaqMan low‐density array: analysis of macrophage differentiation and foam cell formation. Clin Chem 52:310–313. [DOI] [PubMed] [Google Scholar]
- 24. Löscher W, Potschka H (2005) Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci 6:591–602. [DOI] [PubMed] [Google Scholar]
- 25. Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P et al (2009) Coordination of Rho GTPase activities during cell protrusion. Nature 461:99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morales JR, Ballesteros I, Deniz JM, Hurtado O, Vivancos J, Nombela F et al (2008) Activation of liver X receptors promotes neuroprotection and reduces brain inflammation in experimental stroke. Circulation 118:1450–1459. [DOI] [PubMed] [Google Scholar]
- 27. Moshal KS, Singh M, Sen U, Rosenberger DS, Henderson B, Tyagi N et al (2006) Homocysteine‐mediated activation and mitochondrial translocation of calpain regulates MMP‐9 in MVEC. Am J Physiol Heart Circ Physiol 291:H2825–H2835. [DOI] [PubMed] [Google Scholar]
- 28. Naik SU, Wang X, Da Silva JS, Jaye M, Macphee CH, Reilly MP et al (2006) Pharmacological activation of liver X receptors promotes reverse cholesterol transport in vivo . Circulation 113:90–97. [DOI] [PubMed] [Google Scholar]
- 29. Neuwelt E, Abbott NJ, Abrey L, Banks WA, Blakley B, Davis T et al (2008) Strategies to advance translational research into brain barriers. Lancet Neurol 7:84–96. [DOI] [PubMed] [Google Scholar]
- 30. Noren NK, Liu BP, Burridge K, Kreft B (2000) p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J Cell Biol 150:567–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ohno H, Uemura K, Shintani‐Ishida K, Nakamura M, Inomata M, Yoshida K (2007) Ischemia promotes calpain‐mediated degradation of p120‐catenin in SH‐SY5Y cells. Biochem Biophys Res Commun 353:547–552. [DOI] [PubMed] [Google Scholar]
- 32. Park KP, Rosell A, Foerch C, Xing C, Kim WJ, Lee S et al (2009) Plasma and brain matrix metalloproteinase‐9 after acute focal cerebral ischemia in rats. Stroke 40:2836–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schreibelt G, Kooij G, Reijerkerk A, van Doorn R, Gringhuis SI, van der Pol S et al (2007) Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J 21:3666–3676. [DOI] [PubMed] [Google Scholar]
- 34. Schroepfer GJ Jr (2000) Oxysterols: modulators of cholesterol metabolism and other processes. Physiol Rev 80:361–554. [DOI] [PubMed] [Google Scholar]
- 35. Schulz E, Dopheide J, Schuhmacher S, Thomas SR, Chen K, Daiber A et al (2008) Suppression of the JNK pathway by induction of a metabolic stress response prevents vascular injury and dysfunction. Circulation 118:1347–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sedarous M, Keramaris E, O'Hare M, Melloni E, Slack RS, Elce JS et al (2003) Calpains mediate p53 activation and neuronal death evoked by DNA damage. J Biol Chem 278:26031–26038. [DOI] [PubMed] [Google Scholar]
- 37. Spudich A, Kilic E, Xing H, Kilic U, Rentsch KM, Wunderli‐Allenspach H et al (2006) Inhibition of multidrug resistance transporter‐1 facilitates neuroprotective therapies after focal cerebral ischemia. Nat Neurosci 9:487–488. [DOI] [PubMed] [Google Scholar]
- 38. Takenaga Y, Takagi N, Murotomi K, Tanonaka K, Takeo S (2009) Inhibition of Src activity decreases tyrosine phosphorylation of occludin in brain capillaries and attenuates increase in permeability of the blood‐brain barrier after transient focal cerebral ischemia. J Cereb Blood Flow Metab 29:1099–1108. [DOI] [PubMed] [Google Scholar]
- 39. Tsubokawa T, Solaroglu I, Yatsushige H, Cahill J, Yata K, Zhang JH (2006) Cathepsin and calpain inhibitor E64d attenuates matrix metalloproteinase‐9 activity after focal cerebral ischemia in rats. Stroke 37:1888–1894. [DOI] [PubMed] [Google Scholar]
- 40. Wildenberg GA, Dohn MR, Carnahan RH, Davis MA, Lobdell NA, Settleman J, Reynolds AB (2006) p120‐catenin and p190RhoGAP regulate cell‐cell adhesion by coordinating antagonism between Rac and Rho. Cell 127:1027–1039. [DOI] [PubMed] [Google Scholar]
- 41. Zechariah A, ElAli A, Hermann DM (2010) Combination of tissue‐plasminogen activator with erythropoietin induces blood‐brain barrier permeability, extracellular matrix disaggregation and DNA fragmentation after focal cerebral ischemia in mice. Stroke 41:1008–1012. [DOI] [PubMed] [Google Scholar]
- 42. Zhao BQ, Ikeda Y, Ihara H, Urano T, Fan W, Mikawa S et al (2004) Essential role of endogenous tissue plasminogen activator through matrix metalloproteinase 9 induction and expression on heparin‐produced cerebral hemorrhage after cerebral ischemia in mice. Blood 103:2610–2616. [DOI] [PubMed] [Google Scholar]
- 43. Zhao BQ, Wang S, Kim HY, Storrie H, Rosen BR, Mooney DJ et al (2006) Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med 12:441–445. [DOI] [PubMed] [Google Scholar]