

Abstract
Huntington's disease (HD) is an autosomal dominantly inherited, and currently untreatable, neuropsychiatric disorder. This progressive and ultimately fatal disease is named after the American physician George Huntington and according to the underlying molecular biological mechanisms is assigned to the human polyglutamine or CAG‐repeat diseases. In the present article we give an overview of the currently known neurodegenerative hallmarks of the brains of HD patients. Subsequent to recent pathoanatomical studies the prevailing reductionistic concept of HD as a human neurodegenerative disease, which is primarily and more or less exclusively confined to the striatum (ie, caudate nucleus and putamen) has been abandoned. Many recent studies have improved our neuropathological knowledge of HD; many of the early groundbreaking findings of neuropathological HD research have been rediscovered and confirmed. The results of this investigation have led to the stepwise revision of the simplified pathoanatomical and pathophysiological HD concept and culminated in the implementation of the current concept of HD as a multisystem degenerative disease of the human brain. The multisystem character of the neuropathology of HD is emphasized by a brain distribution pattern of neurodegeneration (i) which apart from the striatum includes the cerebral neo‐and allocortex, thalamus, pallidum, brainstem and cerebellum, and which (ii) therefore, shares more similarities with polyglutamine spinocerebellar ataxias than previously thought.
Keywords: brainstem, cerebral cortex, cerebellum, pathoanatomy, polyglutamine diseases, thalamus
HUNTINGTON'S DISASES (HD): MOLECULAR BIOLOGICAL FEATURES
Huntington's disease (HD) represents a slowly progressive and ultimately fatal neuropsychiatric disorder, which is inherited in an autosomal dominant manner and associated with a large spectrum of clinical symptoms. Identified only in 1993 and located on chromosome 4p16.3, the human HD gene (also called IT15) contains meiotically unstable sequences of CAG‐repeat sequences in its exon 1. These unstable CAG‐repeats in exon 1 of IT15 encode the large 350 kDa protein huntingtin with its 3100 amino acids. On account of its recently identified molecular biological features, HD has been assigned to the class of human polyglutamine or CAG‐repeat diseases. As with other members of the growing group of human polyglutamine diseases (eg, specific forms of spinocerebellar ataxias), the estimated prevalence of HD is comparatively low (5–8 individuals per 100 000). Furthermore, as with families affected by other polyglutamine diseases, in HD patients and preclinical HD gene carriers the unstable CAG‐repeats typically are prone to undergoing pathological elongations during meiosis. These pathologically elongated CAG‐repeat sequences are translated into abnormally long polyglutamine stretches in the mutated huntingtin protein. The pathological elongation of polyglutamine stretches in mutant huntingtin, in turn, are currently held to be responsible for the tendency of this multifunctional protein to lose its normal solubility in nerve cells and to aggregate into intra‐neuronal inclusions in different compartments of vulnerable brain regions (Figures 1 and 2) 2, 17, 20, 25, 36, 48, 51, 58, 59, 60, 64, 67, 72, 73, 78, 79, 81, 82.
Figure 1.
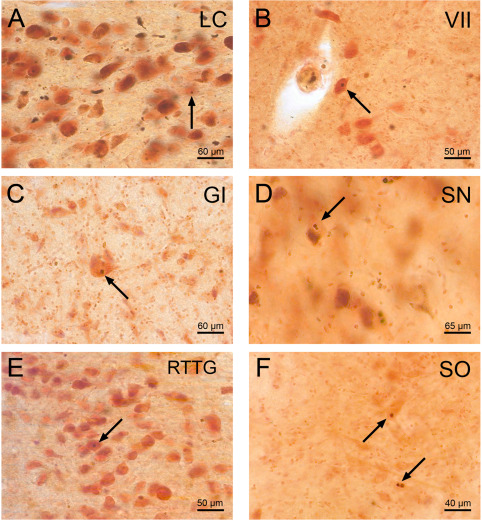
p62 immunoreactive neuronal intranuclear inclusions in well‐preserved and degenerated brainstem nuclei of Huntington's disease (HD) patients. P62 immumopositive neuronal intranuclear inclusions (NI) in spared brainstem nuclei in HD: A. locus coeruleus (LC), B. facial nucleus (VII), C. gigantocellular reticular nucleus (GI). NI in degenerated brainstem nuclei in HD: D. dopaminergic substantia nigra (SN), E. reticulotegmental nucleus of the pons (RTTG), F. superior olive (SO). Arrows point to NI. (A, D, E: HD patient with 49 CAG‐repeats in the mutated HD allele; age at HD onset: 33 years; duration of HD: 18 years; Vonsattel grade 4 of striatal atrophy—B, C: HD patient with 49 CAG‐repeats in the mutated HD allele; age at HD onset: 33 years; duration of HD: 18 years; Vonsattel grade 4 of striatal atrophy—F: HD patient with 45 CAG‐repeats in the mutated HD allele; age at HD onset: 53 years; duration of HD: 12 years; Vonsattel grade 4 of striatal atrophy) (A–F: p62 immunohistochemistry, counterstaining with aldehyde‐fuchsin Darrow red; 100 μm PEG sections).
Figure 2.
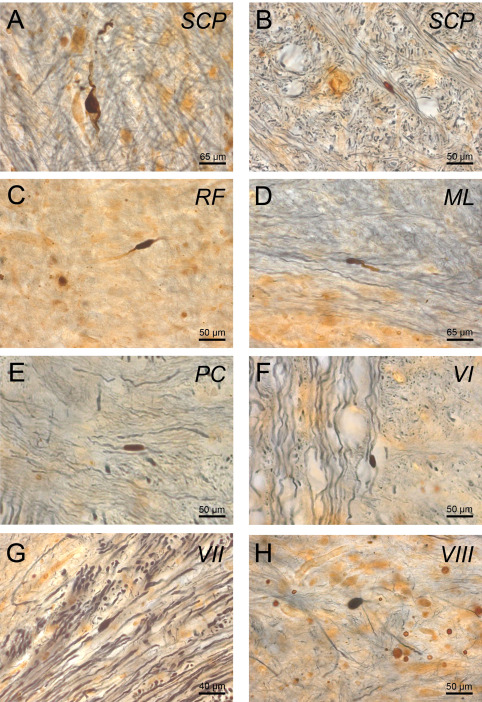
p62 immunoreactive axonal neuronal inclusions in brainstem fiber tracts in Huntington's disease (HD). P62 immumopositive axonal neuronal inclusions in HD: (A, B) superior cerebellar peduncles (SCP), (C) reticular formation (RF), (D) medial lemniscus (ML), (E) pontocerebellar fibers (PC), (F) abducens nerve (VI), (G) facial nerve (VII), (H) vestibulocochlear nerve (VIII), (I) (A–H: p62/AT270 double immunohistochemistry; 100 μm PEG sections). (A, B: HD patient with 44 CAG‐repeats in the mutated HD allele; age at HD onset: 50 years; duration of HD: 20 years; Vonsattel grade 2 of striatal atrophy—C, E, G: HD patient with 49 CAG‐repeats in the mutated HD allele; age at HD onset: 33 years; duration of HD: 18 years; Vonsattel grade 4 of striatal atrophy—D: HD patient with 45 CAG‐repeats in the mutated HD allele; age at HD onset: 35 years; duration of HD: 26 years; Vonsattel grade 3 of striatal atrophy—F, H: HD patient with 45 CAG‐repeats in the mutated HD allele; age at HD onset: 53 years; duration of HD: 12 years; Vonsattel grade 4 of striatal atrophy).
In healthy individuals the length of the CAG‐repeat sequences commonly ranges from 6 to 35 CAG triplets. However, recent research has revealed that the apparently healthy sequences of 28–35 CAG‐repeats are already associated with unstable behavior during meiosis and therefore may be dangerous and pathologically relevant. Accordingly, on account of their newly detected tendency to further expand, these apparently healthy but unstable CAG‐repeats may cause clinically relevant symptomatic HD mutations. In contrast, sequences of 35 or more CAG‐repeats are definitely in the pathological range and in any event provoke HD disease symptoms. Thirty‐six to forty CAG‐repeat triplets are associated with an incomplete clinical penetrance of HD, and forty‐one or more CAG‐repeats to a fully developed clinical picture 2, 3, 5, 9, 17, 20, 36, 48, 49, 51, 55, 58, 59, 60, 64, 67, 68, 73, 78, 81, 82.
HUNTINGTON'S DISASES (HD): CLINICAL FEATURES
The clinical course of HD begins insidiously in adulthood, typically in the mid‐40s. Initial disease symptoms of affected patients comprise progressive cognitive impairment and uncharacteristic motor symptoms often described as “clumsiness,” “tremor,” “balance trouble” and “jerkiness.” Involuntary choreatic movements are the pathognomonic and best known movement abnormalities of HD patients. They often evolve step‐by‐step during the early symptomatic phase of HD and at first occur only in circumscribed regions of the body or head. Their manifestation may be preceded by or accompany a large variety of personality changes, behavioral abnormalities, psychiatric symptoms (eg, depression, schizophrenia‐like symptoms, disinhibition, restlessness, irritability, anxiety) and/or neuropsychological symptoms. Subsequent to their early and subtle manifestation, choreatic movements become more and more severe. They then also tend to generalize and to run completely out of control and thus interfere considerably with voluntary movement and activity of everyday life. During the advanced clinical phases of HD choreatic movements may plateau, and may even disappear and be superseded by other motor symptoms (eg, rigidity). In addition to rigidity further somatomotor abnormalities (eg, bradykinesia, ataxia, dystonia, muscle hypotonia, postural instability, impaired fine motor skills, dysarthria and dysphagia), abnormalities of evoked potentials, a broad range of oculomotor symptoms, sensory dysfunction and severe weight loss may complement and complicate the clinical picture of HD. Impairments of eye movements are often among the early disease signs of HD. They belong to the most common and obvious disease symptoms of HD patients upon clinical examination and may include disturbances of saccades, smooth pursuits, vergence, vestibulo‐ocular reaction, optokinetic nystagmus and fixation. The severe weight loss which often occurs during the later clinical phases of HD is still unexplained, while it sooner or later leads to cachexia and is therefore life‐determining for many HD patients. The psychiatric and neuropsychological symptoms of HD patients commonly worsen during the clinical course of HD and ultimately discharge into severe and irreversible psychoorganic or dementing syndromes. Finally, when the cognitive decline and cachexia are at their zenith the care‐dependent and bed‐ridden patients most frequently die from complications of a dysphagia‐related aspiration pneumonia 4, 18, 20, 23, 45, 46, 47, 51, 58, 59, 60, 64, 67, 73, 78, 79, 81, 82.
HUNTINGTON'S DISASES (HD): CURRENT THERAPEUTICAL APPROACHES
Currently, there are no causative treatment options or effective disease‐modifying therapies available for HD patients. Treatment in HD is still solely based on symptomatic approaches to ameliorate some disease symptoms (eg, chorea, dystonia, seizures and psychiatric disease symptoms). Although the symptomatic treatment methods (eg, medication, surgical procedures, speech therapy, occupational therapy, exercise therapy, music therapy and cognitive interventions) have no positive effects on the natural course of HD they can help to improve the quality of life of affected patients and their caregivers 5, 36, 64, 81, 82.
HUNTINGTON'S DISEASE (HD): THE DISEASE PROTEIN
The wild‐type of the disease protein huntingtin is expressed in all neurons and glial cells of the human brain. In nerve cells this multifunctional protein is localized in the cytoplasm, axonal processes and synapses, and is also associated with many cell organelles (eg,. nucleus, endoplasmic reticulum, Golgi complex, mitochondria, endosomes), microtubules, synaptic and autophagic vesicles, caveolae and synaptosomes. Huntingtin can interact with over 200 other cellular proteins and participates in the regulation of transcription. Furthermore, huntingtin is an integral component of the endocytotic pathway and can unfold anti‐apoptotic activity. Acting as a scaffold in axonal transport processes, huntingtin is crucial for the anterograde and retrograde movement of mitochondria along neuronal axons and ensures the timely and local ATP demands in various parts of the nerve cells 3, 5, 13, 20, 36, 48, 49, 53, 59, 60, 64, 78, 81, 82.
The elongated polyglutamine stretch in mutant huntingtin is associated with conformational changes of this protein, triggers its misfolding and leads to reduced solubility, which confers a striking tendency on the huntingtin protein of forming insoluble aggregations in different compartments of affected nerve cells (eg, nucleus, cytoplasm, axon) (Figures 1 and 2). Despite progress, understanding of the molecular biological basis and exact pathophysiological mechanisms of the formation of the various forms of aggregations in diseased nerve cells, as well as their relevance to the demise of nerve cells that takes place in HD, remain only incompletely understood 3, 5, 9, 20, 36, 39, 48, 49, 64, 67, 68, 78, 81, 82.
HUNTINGTON'S DISEASE (HD): SOME MILESTONES OF EARLY RESEARCH
In his famous essay “On chorea” from 1872 the American physician George Huntington (1850–1916) provided a detailed description of the clinical picture of adult chorea, its hereditary transmission and the relentless clinical progress of this neurological disease. Although this widely known essay by George Huntington was preceded by earlier clinical descriptions of adult‐onset hereditary chorea (such as those of Waters, Lund and Lyon), hereditary chorea has been called Huntington's disease (HD) since the late 1880s 26, 35, 43, 44, 64, 71.
Many of the postmortem studies on hereditary chorea or HD from the early 1900s already pointed to a widespread brain neurodegeneration in patients suffering from hereditary chorea or HD. However, the long neglect of most of these pioneering postmortem findings and early assumptions resulted in a reductionist, pathoanatomical and pathophysiological notion that the pathological process of HD was more or less confined to the striatum (caudate nucleus and putamen) (Figures 3, 4 and 6−8). Although this unilateral concept, unfortunately, prevailed for more than a hundred years, it could offer no adequate explanations for the large variety of HD disease symptoms. Recent neuropathological HD research, however, led to the rediscovery and confirmation of many of the initial, groundbreaking postmortem findings. On account of the results of these recent re‐investigations, the oversimplified concept of HD as a neurodegenerative disease predominantly affecting the striatum was replaced by the current view of HD as a multisystem degenerative disease of the human brain (Figures 3, 4, 5) 32, 42, 59, 64, 71, 78. The early descriptions of macroscopical brain alterations (reduced brain weight and brain atrophy) and widespread visible microscopical degenerative changes in the striatum, thalamus, pallidum, brainstem and circumscribed regions of the cerebral cortex (Figures 3, 4, 5, 6, 7, 8, 9, 10, 11) in patients suffering from Huntington's chorea by both well‐known (Alois Alzheimer, Gerbrandus Jelgersma, Cécile and Oskar Vogt) and less prominent neuroscientists (Ewald Stier) were already compatible with the currently favored view of HD as a multisystemic neurodegenerative disease. Cécile and Oskar Vogt, in their classic comprehensive work on the human striatum and its diseases in 1920 confirmed the disease‐specific histopathological feature of striatal neurodegeneration in HD (differential vulnerability of small and large nerve cells of the striatum in the degenerative process) which was initially delineated by James Ramsey Hunt in 1917. However, Cécile and Oskar Vogt had already questioned the attribution of the occurrence of involuntary choreatic and incoordinated movements to striatal degeneration, as suggested by Gerbrandus Jelgersma in 1908 and by Alois Alzheimer in 1911 1, 26, 34, 35, 37, 42, 64, 70, 71, 76, 77.
Figure 3.
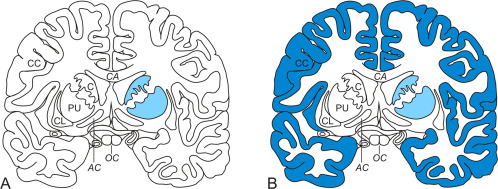
Previous ideas and current knowledge about the distribution and severity of neuronal loss in the basal forebrain of Huntington's disease (HD) patients. Schematized frontal sections through the basal forebrain at the level of the anterior commissure (AC). (A) Previous idea regarding the selective involvement of the striatum (caudate nucleus, C and putamen, PU). (B) Current pathoanatomical HD knowledge which includes the additional severe involvement of the cerebral cortex (brain regions undergoing severe neuronal loss are highlighted with blue shading). (Abbreviations: AC, anterior commissure; C, caudate nucleus; CL, claustrum; CC, cerebral cortex; CA, corpus callosum; HD, Huntington's disease; P, pallidum; PU, putamen; OC, optic chiasm).
Figure 4.
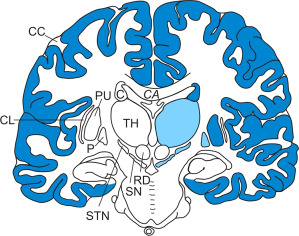
Distribution and severity of neuronal loss in select brain regions of Huntington's disease (HD) patients. Schematized frontal section through the cerebral hemispheres at the level of the red nucleus (RD) (brain regions undergoing severe neuronal loss are highlighted with blue shading and brain regions with marked loss of nerve cells with lighter blue shading). (Abbreviations: C, caudate nucleus; CC, cerebral cortex; CA, corpus callosum; CL, claustrum; HD, Huntington's disease; P, pallidum; PU, putamen; RD, red nucleus; SN, substantia nigra; STN, subthalamic nucleus; TH, thalamus).
Figure 6.
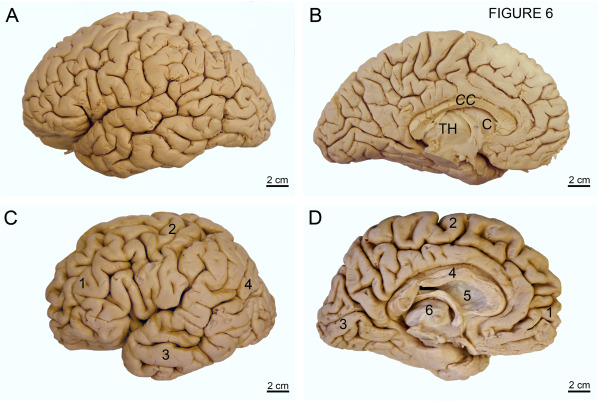
Atrophy of the cerebral lobes and subcortical regions in Huntington's disease (HD). (A) Dorsolateral aspect of the left cerebral hemisphere of a typical control individual without a medical history of neurological or psychiatric diseases. (B) Medial aspect of the left cerebral hemisphere of the same control individual. (C) Dorsolateral aspect of the left cerebral hemisphere of clinically diagnosed and genetically confirmed male Huntington's disease (HD) patient (Vonsattel grade 4 of neostriatal atrophy). Note the marked atrophy of the frontal 1, parietal 2, and temporal lobes 3 with widened sulci and narrowed gyri. Additionally, the occipital lobe of this patient is severely reduced 4. (D) Medial aspect of the left cerebral hemisphere of the same HD patient: atrophy of the frontal 1, parietal 2, and occipital lobes 3, and corpus callosum 4, head of the caudate nucleus 5 and the thalamus 6. (Abbreviations: C, caudate nucleus; CC, corpus callosum; TH, thalamus).
Figure 5.
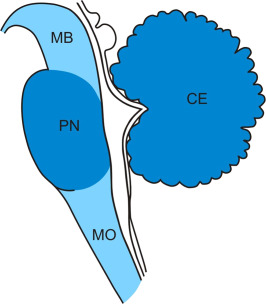
Distribution and severity of neuronal loss in the regions of the brainstem and cerebellum of Huntington's disease (HD) patients. Schematized sagittal section through the midbrain (MB), pons (PN), medulla oblongata (MO) and cerebellum (CE) (brainstem and cerebellar regions undergoing severe neuronal loss are highlighted with blue shading and brain regions with marked loss of nerve cells with lighter blue shading). For the exact localization of neurodegeneration in the brainstem and cerebellum please see text. (Abbreviations: CE, cerebellum; HD, Huntington's disease; MB, midbrain; MO, medulla oblongata; PN, pons).
Figure 7.
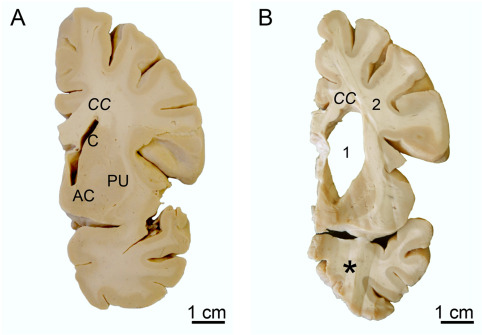
Atrophy of the striatum and white matter loss in Huntington's disease (HD). (A) Frontal section through the basal forebrain of a representative control individual at the level of the accumbens nucleus (AC) with the caudate nucleus (C) and putamen (PU). (B) Frontal section through the same basal forebrain level of a clinically diagnosed and genetically confirmed Huntington's disease (HD) patient (Vonsattel grade 3 of striatal atrophy). Along with the striatum, the corpus callosum (CC) and the temporal lobe are atrophic (asterisk). Note the widened lateral ventricle 1, reduction of the cerebral white matter 2, and flattened outline of the C. (Abbreviations: AC, accumbens nucleus; C, caudate nucleus; CC, corpus callosum; PU, putamen).
Figure 8.
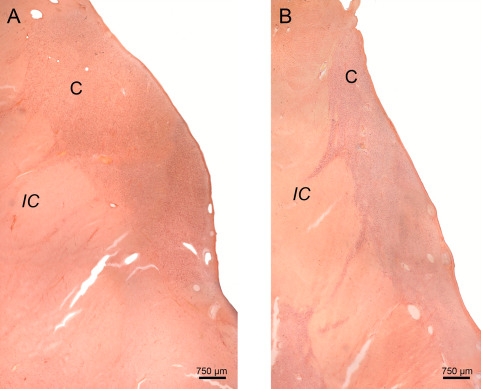
Neuronal loss in the caudate nucleus in Huntington's disease (HD). Frontal section through the head of the caudate nucleus (C) at the level of the rostral hypothalamus of a representative control individual. (B) Marked neuronal loss of the C of a representative Huntington's disease (HD) patient with Vonsattel grade 2 of neostriatal atrophy. Note the reduction of the IC and the fact that the convex outline of the head of the C is still discernable. (A, B: Aldehyde‐fuchsin Darrow red staining; 100 µm polyethylene glycol section). (Abbreviations: C, caudate nucleus; HD, Huntington's disease; IC, internal capsule).
Figure 9.
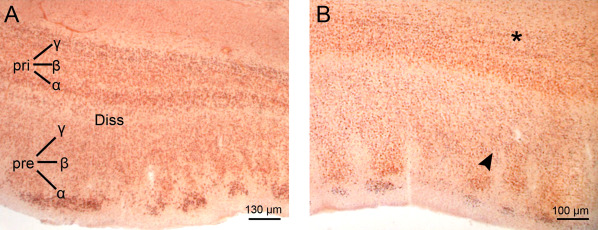
Neuronal loss in the entorhinal cortex in Huntington's disease (HD). Frontal section through the entorhinal cortex at the mid‐rostrocaudal level of the hypothalamus of a typical control individual. (B) Reduced neuronal density of the entorhinal cortex of a representative Huntington's disease (HD) patient with Vonsattel grade 2 of neostriatal atrophy. Note the predominant neuronal loss in the pre‐beta (arrowhead) and pri‐gamma layers (asterisk) of the entorhinal cortex (A, B: Aldehyde‐fuchsin Darrow red staining; 100 µm polyethylene glycol section). (Abbreviations: Diss, lamina dissecans; HD, Huntington's disease Pre, principal external layer; Pri, principal internal layer).
Figure 10.
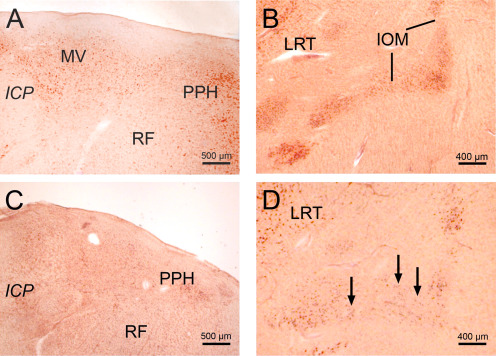
Neuronal loss in the brainstem in Huntington's disease (HD). (A) Horizontal section through the pontomedullary junction of a typical control individual showing the medial vestibular (MV) and prepositus hypoglossal nuclei (PPH). (B) Horizontal section through the caudal portion of the medulla oblongata of a representative control individual showing the medial subnucleus of the inferior olive (IOM) and the lateral reticular nucleus (LRT). (C) Considerable neuronal loss in the MV of an HD patient burdened by Vonsattel grade 3 of striatal atrophy. (D) Marked neuron loss of the IOM of a representative Huntington's disease (HD) patient with Vonsattel grade 3 atrophy. Arrows point to reaming extraneuronal lipofuscin granules. (A–D: Aldehyde‐fuchsin Darrow red staining; 100 µm polyethylene glycol section). (Abbreviations: HD, Huntington's disease; ICP, inferior cerebellar peduncle; LRT, lateral reticular nucleus; PPH, prepositus hypoglossal nucleus; RF, reticular formation).
Figure 11.
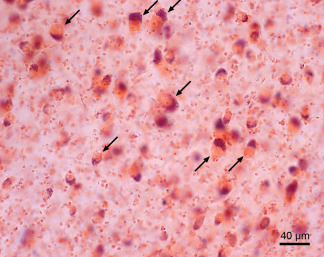
Surviving ballooned nerve cells in the affected ventrolateral thalamic nucleus of Huntington's disease (HD) patients. These remaining, morphologically altered ballooned neurons are swollen and display a massively enlarged cytoplasm with a homogenous central basophilic substance. Their Nissl substance, lipofuscin granules, and flattened nucleus are concentrated along the cytoplasmic membrane, thus mimicking central chromatolysis (arrows). (Aldehyde‐fuchsin Darrow red staining; 100 µm polyethylene glycol section). (HD patient with 45 CAG‐repeats in the mutated HD allele; age at HD onset: 35 years; duration of HD: 26 years; Vonsattel grade 3 of striatal atrophy).
HUNTINGTON'S DISEASE (HD): PROGRESSIVE DEGENERATION OF THE STRIATUM
Bilateral symmetrical neuronal loss in the neostriatum is a common prominent neuropathological feature of patients in the late clinical stages of HD. This dramatic neostriatal neuronal loss is mainly caused by the extensive demise of GABA‐ergic medium spiny stellate projections neurons (Figures 3, 4 and 6−8), which belong to the main targets of striatal input and provide the main efferent output of the striatum. These highly vulnerable nerve cells of the neostriatum coexpress substance P, encephalin and dynorphin, and they account for approximately 95% of all neurons occurring in the neostriatum. This tremendous demise of striatal nerve cells has been used in the past to explain nearly the entire spectrum of the disease symptoms of HD patients, including psychiatric, oculomotor and choreatic symptoms 2, 5, 7, 8, 9, 10, 12, 15, 18, 19, 20, 27, 29, 30, 31, 32, 36, 40, 42, 48, 51, 55, 56, 58, 59, 60, 63, 64, 69, 76, 77, 78, 79, 80, 81, 82.
The progression of the slowly on‐going neuronal loss and accompanying reactive astrogliosis in the neostriatum does not take place in a random manner or by chance. On the contrary, demise of striatal nerve cells and concomitant astrogliosis occur according to rigorous rules and follow stereotypical chronological and spatial sequences. Since the destructive process in the striatum consistently moves simultaneously in caudo‐rostral, dorso‐ventral and medio‐lateral directions, this highly systematic damage to the neostriatum serves as the empirical basis for the universally acknowledged Vonsattel grading system of the HD‐associated striatal neuropathology 64, 78, 79, 80.
HUNTINGTON'S DISEASE (HD): THE VONSATTEL GRADING SYSTEM
The Vonsattel grading system for striatal degeneration is based on the macroscopic appearance of striatal atrophy and the visible microscopic extent of the striatal neuronal loss and degree of reactive astrogliosis. This widely applied grading system describes the chronological and topographical evolution and severity of HD‐related degeneration in the striatum, distinguishes among five grades (grades 0–4) of striatal involvement in HD, and is characterized by its simple practicability and high reliability 64, 78, 79, 80.
Vonsattel grade 0—Brains graded as Vonsattel 0 are commonly macroscopically unremarkable. Upon microscopic examination grade 0 brains show a 30%–40% loss of neurons in the head of the caudate nucleus in the absence of reactive astrocytes. Vonsattel grade 1—In brains burdened by grade 1 neuropathology the tail and body of the caudate nucleus are considerably atrophic. At this grade the head of the caudate nucleus and the putamen may still have a normal appearance upon macroscopic examination. Neuronal loss and reactive astrogliosis are present predominantly in the tail of the caudate nucleus. The body and head of the caudate nucleus and the adjacent putamen are less severely involved, while the accumbens nucleus at the junction of the rostral caudate nucleus and putamen still appears macroscopically and microscopically unremarkable. Vonsattel grade 2—Although the remarkable loss of cerebral white matter and striatal atrophy leads to an enlargement of the lateral ventricles, the convex outline of the medial ventricular surface of the head of the caudate nucleus is still retained. Unequivocal neuronal loss and reactive astrogliosis are found in the dorsal half (medial > lateral) of the caudate nucleus and adjacent putamen (Figure 8). The accumbens nucleus no longer appears microscopically normal. In brains with grade 2 striatal neuropathology, initial neurodegeneration is also present in the pallidum (external segment > internal segment). Vonsattel grade 3—Upon macroscopic inspection the head of the caudate nucleus appears atrophic. Its medial outline now forms a straight line, which parallels the internal capsule. Neuronal loss and fibrillary astrocytosis are commonly severe within the caudate nucleus and putamen (rostral > caudal; dorsal > ventral), and in the accumbens nucleus they are at best mild (Figure 7). Vonsattel grade 4—In patients with grade 4 neuropathology the striatum is severely atrophic. On account of their considerable volume loss, the medial contours of the internal capsule and head of the caudate nucleus are now concave. Microscopic investigation reveals severe striatal neuronal loss, which amounts to 95% of neostriatal nerve cells. The volume reduction of the pallidum by about 50% is mainly because of the loss of fiber connections in the neutrophil. In more than 50% of diseased HD patients the accumbens nucleus is among the targets of the neurodegenerative process (Figures 3, 4 and 6) 64, 78, 79, 80.
HUNTINGTON'S DISEASE (HD): ATROPHY OF THE CEREBRAL CORTEX AND ITS AREA, AND LAYER‐SPECIFIC NEURONAL LOSS
Some of the early neuropathological researchers (Alois Alzheimer, Gerbrandus Jelgersma, Cécile and Oskar Vogt, Ewald Stier) already described the degeneration and nerve cell loss of distinct regions of the cerebral cortex of patients with hereditary chorea or HD. Although these researchers were convinced that the pathological process of hereditary chorea or HD was not restricted to the striatum, the progressing striatal degeneration has for a long time been regarded by many neuroscientists as the neuropathological hallmark of HD (Figures 3, 4 and 6−8) 64.
Recent investigation in HD, however, has shown that all four cerebral lobes undergo thinning of their cortical mantles along with a severe atrophy, and it has demonstrated layer‐specific neuronal loss in various areas of the cerebral allocortex and neocortex (Figures 3, 4, 6 and 9). These findings have sounded the death knell of the traditional, reductionistic pathoanatomical and pathophysiological concepts of the polyglutamine disease HD as a selective disease of the striatum and they have resulted in the definitive appreciation of the cerebral neocortex and allocortex as the main targets of the HD disease process (Figures 3−6 and 9) 1, 3, 5, 6, 7, 10, 12, 18, 26, 27, 28, 29, 30, 31, 32, 34, 36, 37, 40, 42, 48, 50, 51, 56, 59, 60, 63, 64, 67, 68, 69, 70, 71, 76, 77, 78, 79, 80, 81, 82. The layer‐specific neuronal loss in the neocortical and allocortical targets of the destructive process of HD is now regarded as the most important morphological correlate of the psychiatric symptoms, neuropsychological deficits, cognitive decline and neurophysiological abnormalities (ie, alterations of the somatosensory and visual evoked potentials) of affected HD patients and some at‐risk gene carriers. Although the occurrence of involuntary choreatic movements has been widely interpreted as an immediate consequence of striatal degeneration, some renowned HD researchers have attributed the occurrence of involuntary choreatic movement to cortical neurodegeneration. Accordingly, the manifestation of chorea in HD patients is currently believed to result from functional disorder of corticostriatal projection neurons and/or impairment of their afferent input 18, 64, 66, 77, 81, 82.
The first systematic volumetric studies of serial tissue sections from the brains of HD patients remain among the most important postmortem HD investigations. The pioneering and groundbreaking findings of these studies performed nearly 30 years ago pointed to a widespread area‐specific vulnerability of the cerebral cortex in HD. The findings of these studies included atrophy and volume loss in the following areas or regions of the cerebral cortex: peristriate and parastriate visual areas in the occipital lobe (Brodmann areas 18 and 19); primary visual striate area in the occipital lobe (Brodmann area 17); primary somatosensory cortex (Brodmann areas 1, 2 and 3); anterior cingulate cortex (Brodmann area 24); posterior cingulate cortex (Brodmann area 23); primary auditory cortex (Brodmann areas 41 and 42); and insula, temporal neocortex, parahippocampal gyrus including the transentorhinal and entorhinal regions. These findings in the cerebral cortex contributed substantially to the subsequent paradigm shift in neuropathological HD research and laid the groundwork for the current concept of HD as a multisystem neurodegenerative disease (Figures 3 and 4) 8, 32, 40, 41, 42, 59, 63, 64, 78.
Further sophisticated studies supported the re‐emergent opinion that degeneration of the cerebral neocortex and allocortex is among the neuropathological hallmarks of HD and offered appropriate explanations for the psychiatric and neuropsychological HD symptoms, cognitive decline, basic and complex visual dysfunction and the abnormalities of somatosensory (SEP) and visual evoked potentials (VEP). Their findings demonstrated an overall volume and neuronal loss in the cerebral cortex of HD patients and also provided additional evidence of the area‐ and layer‐specific vulnerability of the cerebral cortex 12, 27, 41, 42, 50, 63, 64, 69.
Quantitative estimations revealed an overall neuronal loss in the cerebral cortex of HD patients of 33%, which is most pronounced in the isocortical neuronal layers III, V and VI of the affected areas. In addition, an overall neuronal loss of 32% has been discovered in the primary visual striate area of the occipital lobe (Brodmann area 17) of HD patients, which is mainly because of the predominant neurodegeneration of its outer pyramidal layer III, inner granular layers IVa and IVc and multiform layer VI 8, 63, 64.
In the primary motor cortex (Brodmann area 4) of HD patients a significant reduction in the total number of pyramidal cells of 24% and of the subpopulation of SMI‐32 immunoreactive pyramidal cells of 27% was found, while in the posterior cingulate gyrus (Brodmann area 24) an overall loss of pyramidal cells of 36% and of SMI‐32 immunopositive pyramidal cells of 34% was seen 8, 64, 74.
According to recent studies the following neocortical regions of HD patients are affected by layer‐specific neuronal loss: primary somatosensory cortex (Brodmann areas 3, 1, 2): outer pyramidal layer III, inner granular layer IV, multiform layer VI; frontal, temporal and parietal association cortices: outer pyramidal layer III, inner pyramidal layer V; primary visual area 17 (Brodmann area 17): outer pyramidal layer III, inner granular layers IVa and IVc, multiform layer VI; primary auditory cortex (Brodmann area 41): outer pyramidal layer III, inner granular layer IV, multiform layer VI 6, 7, 8, 10, 18, 27, 28, 29, 30, 31, 32, 48, 52, 63, 64, 69, 78, 79, 81, 82.
A typical predictable pattern of layer‐specific neuronal loss has been repeatedly demonstrated in the allocortical transentorhinal and entorhinal regions of HD patients (Figure 9). In our experience, the dramatic neuronal loss consistently present in the pre‐ß and pri‐γ layers of these allocortical regions represents an outstanding and reliable pathognomonic degenerative feature of HD, which on account of its layer‐specificity is suggestive and pathbreaking for the neuropathological diagnosis of HD 6, 7, 28, 64.
HUNTINGTON'S DISEASE (HD): DEGENERATION OF SELECT NUCLEI OF THE THALAMUS
Degeneration of the thalamus of HD patients (eg, global atrophy and volume loss of the thalamus; thalamic astrogliosis; diminished density of microneurones in the precerebellar ventrolateral nucleus; reactive astrogliosis in the thalamic centromedian and parafascicular nuclei; shrinkage of the centromedian nucleus and its nerve cells) was described in some early neuropathological HD reports. In the period that follow the involvement of this large diencephalic nuclear complex in HD patients, however, it experienced only minor attention (Figures 4 and 6) 5, 15, 40, 42, 52, 64, 71, 78, 79, 80, 81, 82. Recent re‐investigations applying modern stereological methods, however, have demonstrated that thalamic neurodegeneration is not confined to the cerebellar territory of the thalamus (ie, motor ventrolateral nucleus), but also occurs in the thalamic centromedian‐parafascicular complex and mediodorsal nucleus 15, 30, 31, 64. These recent quantitative thalamic studies have demonstrated a volume reduction, significant neuronal loss and concomitant astrogliosis in both nuclear complexes of the thalamus of HD patients 30, 31, 64.
The intralaminar centromedian–parafascicular complex of the human thalamus is anatomically associated with the striatum 31, 33, 38, 54, 64. Its centromedian nucleus represents a nodal point in the reciprocal neostriatal–pallido‐centromedian–neostriatal loop of the basal ganglia. It is mainly linked with the sensorimotor territory of the striatum and helps to integrate sensorimotor functions and to process motor information 31, 38, 64. The parafascicular nucleus of this intralaminar complex is closely connected with the associative and limbic territories of the striatum. On account of its additional connections with the frontal and supplementary eye fields of the cerebral cortex, the parafascicular nucleus is crucial for cognitive, emotional and oculomotor functions 31, 33, 38, 54, 64. Following this, the somatomotor, cognitive, psychiatric, emotional and oculomotor symptoms of HD patients are caused in part by neurodegeneration in the centromedian–parafasciular complex 31, 64.
The voluminous mediodorsal nucleus of the thalamus is intimately related with the prefrontal cortex and integrated into the associative, limbic and oculomotor circuits of the human brain. On account of this integration, the thalamic mediodorsal nucleus plays an essential role in the performance of cognitive, learning and memory processes, as well as executive and oculomotor functions 30, 33, 38, 54, 64. Accordingly, damage to this central thalamic nucleus most likely plays a role in the pathogenesis of the cognitive, amnestic, psychiatric and emotional disease symptoms of HD patients and may also be conducive to the oculomotor impairments that develop in HD patients and their at‐risk relatives 30, 31, 64.
HUNTINGTON'S DISEASE (HD): WIDESPREAD DEGENERATION OF THE CEREBELLUM
The disease symptoms in HD patients may also include dysarthria, impaired rapid alternating movements and fine motor skills, postural and gait instability, ataxia and falls. Although the occurrence of these disease symptoms clearly points to involvement of the cerebellum during the course of HD neurodegeneration, the cerebellum of HD patients has also only been fragmentarily investigated in the past. Despite the occurrence of these disease symptoms unequivocally pointing to its involvement in HD patients, degeneration of the cerebellum in HD has been controversial for several decades 10, 23, 51, 59, 64, 78, 79, 80, 81, 82.
However, recent systematic investigation of the cerebella of clinically diagnosed and genetically confirmed HD patients (Vonsattel grades 2–4 of striatal atrophy) has revealed its consistent and widespread involvement (Figure 5). Upon macroscopic investigation the following pathological abnormalities of the cerebellum were present: a reduction of the cerebellar volume; a reduction of the surface area of the cerebellar arbor vitae; atrophy of the anterior and posterior cerebellar lobes; widening of the primary fissure. Light‐microscopy disclosed widespread neuronal loss in the cerebellar cortex and in the four deep cerebellar nuclei. While the molecular and granular cell layers of the cerebellar cortex typically appear unremarkable, its Purkinje cell layer is affected at circumscribed predilection sites. In addition, the four deep cerebellar nuclei (ie, fastigial, globose, emboliform and dentate nuclei) consistently undergo conspicuous neurodegeneration in HD, with the fastigial nucleus being the most affected (Figure 5) 59, 64.
This damage to the cerebellum of HD patients (i) also underscores the multisystem nature of the polyglutamine disease HD, (ii) is highly reminiscent of that present in the cerebellum of patients suffering from autosomal dominantly inherited polyglutamine ataxias (spinocerebellar ataxias types 1, 2 and 3) and (iii) indicates that brain neurodegeneration in HD is more closely related to these congeneric polyglutamine ataxic disorders than believed so far 3, 5, 6, 7, 10, 12, 15, 16, 18, 27, 28, 29, 30, 31, 32, 36, 40, 41, 51, 52, 56, 58, 59, 60, 61, 62, 63, 64, 65, 68, 69, 77, 78, 79, 80, 81, 82.
Considering the well‐known functional neuroanatomy of the human cerebellum, its involvement during HD together with neuronal loss in the precerebellar motor ventrolateral thalamic nucleus (Figure 11) 15 for the first time offers credible explanations for the following HD symptoms: disturbances of rapid alternating movements and fine motor skills of the upper limbs, dysarthria, postural instability and ataxia, impaired gait balance, broad‐based gait and stance, falls, disturbed smooth pursuits, muscle hypotonia and abnormalities of the Hoffmann reflex (H‐reflex) 23, 45, 47, 51, 59, 64, 81, 82.
HUNTINGTON'S DISEASE (HD): DEGENERATION OF SELECT BRAINSTEM NUCLEI
HD patients also commonly suffer from disease symptoms (eg, various oculomotor dysfunctions) which cannot be convincingly explained by striatal neurodegeneration but rather point to involvement of select brainstem nuclei. Despite these pathbreaking disease symptoms of HD patients, brainstem neurodegeneration and its possible relevance for the clinical picture of HD has been controversially discussed for many decades. In addition, early studies suggesting degeneration of distinct brainstem nuclei during HD (dopaminergic and GABA‐ergic substantia nigra, auditory superior olive, lateral vestibular nucleus, precerebellar inferior olive) have been widely disregarded by contemporary researchers. Therefore, brainstem neurodegeneration was for a long time not included among the established neuropathological hallmarks of HD 10, 40, 42, 45, 46, 58, 60, 61, 64, 78, 79, 80, 81, 82.
Despite its unremarkable macroscopic appearance the brainstem of HD patients (Vonsattel grades 1–4 of striatal atrophy) consistently shows a widespread neuronal loss upon light‐microscopic examination (Figure 5). Commonly affected in a marked way by neuronal loss are the dopaminergic compact and GABA‐ergic reticulate parts of the substantia nigra in the midbrain, the precerebellar pontine nuclei and inferior olive in the medulla oblongata and the oculomotor reticulotegmental nucleus of the pons (nucleus Bechterew). In addition, a severe demise of nerve cells can be observed in the premotor oculomotor area of the excitatory burst neurons for horizontal saccades and raphe interpositus nucleus, in the auditory superior olive, as well as in the lateral and medial vestibular nuclei (Figure 10) 60, 64.
Degeneration of these brainstem nuclei does not merely offer appropriate explanations for a variety of hitherto unexplained disease symptoms of HD patients. Additionally, their involvement lends credence to the new view of HD as a multisystem neurodegenerative disease and shows that the distribution pattern of brain neurodegeneration in HD patients has been largely underestimated for a long time. It has more in common with the pattern in the molecular biologically related polyglutamine ataxias (spinocerebellar ataxias types 1, 2 and 3) than believed so far 3, 5, 6, 7, 10, 12, 18, 27, 28, 29, 30, 31, 32, 36, 40, 42, 48, 51, 56, 58, 59, 60, 61, 62, 63, 64, 65, 68, 69, 78, 79, 80, 81, 82.
Demise of dopaminergic nerve cells of the compact part of the substantia nigra is a neuropathological feature shared by neurological diseases known to be associated with the occurrence of motor parkinsonian features 60, 64. Although the pars compacta of their substantia nigra regularly undergoes neuronal loss, adult HD patients commonly do not suffer from Parkinsonism. This indicates that damage to the dopaminergic substantia nigra in HD patients does not reach the critical threshold of neuronal loss beyond which clinically relevant parkinsonian motor features requiring treatment usually emerge 40, 42, 60, 64, 80, 81, 82.
The pontine nuclei and the inferior olive are intimately connected with the cerebellum. They are therefore assigned to the so‐called precerebellar brainstem nuclei, which acts as a gateway for all afferent input destined for the cerebellum. Considering their physiological functions, damage to the cerebellum, brainstem and thalamic precerebellar nuclei (ie, motor ventrolateral nucleus) most likely underlies the following disease symptoms of HD patients: disturbances of rapid alternating movements and fine motor skills, dysarthria, muscle hypotonia, instability of posture, ataxia and impaired balance of gait, falls and broad‐based gait and stance) (Figure 10) 15, 23, 51, 59, 60, 64, 78, 79.
Since the human reticulotegmental nucleus of the pons (nucleus Bechterew) is involved in the generation of smooth pursuit eye movements, neuronal loss in this precerebellar premotor oculomotor brainstem nucleus together with damage to the interconnected cerebellar fastigial nucleus may account for the slowing and corrective saccades during smooth pursuits of HD patients 58, 59, 60, 61, 64.
The area of the excitatory burst neurons for horizontal saccades in the pontomedullary junction is crucial for the initiation of and provides the immediate premotor signals for horizontal saccades. The adjacent raphe interpositus nucleus harbors the omnipause neurons related to saccades in all directions. Accordingly, damage to the area of the excitatory burst neurons for horizontal saccades may account for the initiation deficits and slowing of horizontal saccades of HD patients, while loss of the premotor omnipause neurons in the raphe interpositus nucleus contributes to their slowed horizontal and vertical saccades 45, 46, 47, 58, 60, 61, 64.
The superior olive is a crucial component of the auditory network of the human brainstem and is involved in the performance of a variety of auditory functions (eg, perception of the frequency, intensity and temporal patterns of sounds; analysis of the location, distance and movement of sound sources; discriminative signal/noise hearing; speech discrimination) and in the generation of brainstem auditory evoked potentials (BAEP). Degeneration of the superior olive, therefore, not only may contribute to the pathogenesis of altered BAEP of HD patients, but also suggests that impairment of these auditory functions—although to date not reported in the HD literature—may belong to the clinical picture of HD patients 60, 64.
The lateral vestibular nucleus subserves the optokinetic nystagmus and the medial vestibular nucleus the vestibulo‐ocular reflex. Therefore, the neuronal loss present in these vestibular nuclei of HD patients most likely plays a substantial role in the pathogenesis of impairments of these kinds of eye movements that may be observed in HD patients (Figure 10) 45, 46, 47, 58, 60, 61, 64.
PATHOLOGICAL NERVE CELL ALTERATIONS IN HUNTINGTON'S DISEASE (HD)
Along with atrophic neurons in the striatum (ie, so‐called neostriatal dark neurons) no other alterations of vulnerable brain nerve cells have been reported during more than hundred years of neuropathological HD research. To the best of our knowledge, to date no degenerating nerve cells displaying the typical morphological features of classical apoptosis (ie, condensation of nuclear chromatin; fragmentation of the nucleus; presence of apoptotic bodies) have been described in the neuropathological HD literature 22, 59, 64, 78, 79, 80. The fact that up to now no apoptotic nerve cells have been described in the brains of HD patients indicates (i) that the programmed cell death associated with classical apoptosis has no significant pathophysiological relevance for the neurodegenerative process of HD, and (ii) that other destructive mechanisms may lead to the demise of post‐mitotic nerve cells in HD 20, 22, 59, 64.
In recent studies we described an abundance of surviving ballooned neurons (ie, chromatolytic nerve cells) in affected brain regions of HD patients (Figure 11). Ballooned nerve cells are among the pathological features of distinct human tauopathies (ie, Alzheimer's disease; argyrophilic grain disease; corticobasal degeneration; progressive supranuclear palsy). Their brain distribution pattern in these tauopathies, however, is different from that observed in the brains of HD patients. Ballooned neurons are characterized by their striking morphological alterations and, together with extremely shrunken nerve cells, constitute the vast majority of surviving nerve cells in diseased brain regions of HD patients 14, 24, 59, 64. The cytoplasm of ballooned nerve cells is considerably enlarged and is filled with a homogeneous basophilic substance. The nucleus of ballooned nerve cells is flattened, and together with their Nissl substance and lipofuscin granules is moved to the cytoplasmic membrane (Figure 11). Since the massive enlargement of the perikarya of ballooned neurons may result from the structural damage to their axons and/or deterioration of their intra‐axonal transport mechanisms, the manifestation of ballooned nerve cells may provide a new approach to understanding the unknown pathogenetic mechanisms of nerve cell death in HD 59, 64.
The pathophysiological explanation for the manifestation of ballooned nerve cells from the neuropathological literature undoubtedly moves the intra‐axonal protein aggregations that occur in HD into the center of possible pathophysiological considerations of the cascades that lead to nerve cell swelling, shrinkage and, eventually, demise (Figure 2) 3, 5, 20, 36, 48, 49, 59, 64, 82. The axonal inclusions that develop in the fiber tracts of the brains of HD patients most likely physically hamper the essential transport mechanisms in affected axons. These physical impairments of axonal transport mechanisms among others may be detrimental to a continuous mitochondrial trafficking along axons and may interfere with an exact positioning of mitochondria at the axonal sites of local energy demands. Since damage to mitochondrial transport along axons is therefore detrimental to timely ATP production and energy supply in nerve cells, such damage may contribute to the well‐known deterioration of the energy situation and may have a significant pathophysiological role in neuronal dysfunction and death in HD. Moreover, impairment of intra‐axonal transport mechanisms may also lead to retention and conglomeration of newly synthesized mitochondria in neuronal perikarya, which could explain perikaryal enlargement and the formation of ballooned neurons in HD 5, 13, 20, 36, 48, 49, 53, 59, 60, 64.
As in other human neurodegenerative diseases and their experimental models, molecular chaperones (ie, heat shock proteins) may be up‐regulated in the cytoplasm of chromatolytic nerve cells of HD patients. Since heat shock proteins thus can serve as an efficient suppressor of neurodegeneration, their neuroprotective up‐regulation in ballooned or chromatolytic nerve cells could also represent a promising therapeutic interventional strategy in HD 3, 5, 9, 21, 36, 39, 59, 64.
CONCLUSIONS AND FUTURE DIRECTIONS
Initial, pioneering postmortem studies on hereditary chorea or HD yielded many valuable conjectures and neuropathological findings, which have been neglected for a long time and not integrated into appropriate pathophysiological concepts of HD. In recent decades, however, many of these early conjectures and findings have been revisited and may be confirmed by postmortem re‐investigations. These re‐investigations enabled detailed clinico‐pathological correlations and provided a number of neuroanatomically based explanations for HD disease symptoms, previously unilaterally associated with the functional consequences of the striking striatal degeneration (psychiatric, oculomotor, cognitive and neuropsychological symptoms and neurophysiological abnormalities) 64.
According to these re‐investigations, brain neurodegeneration in HD is more widespread than previously assumed. It goes far beyond the striatal predilection site and also targets the cerebral cortex, pallidum, thalamus, brainstem and cerebellum. The destructive process of HD ultimately leads to a disease‐specific distribution pattern of brain neurodegeneration, which shares many neuropathological characteristics with the congeneric polyglutamine spinocerebellar ataxias types 1 (SCA1), 2 (SCA2) and 3 (SCA3). Taken together, the findings of these recent investigations have facilitated the breakthrough to the currently favored concept of the polyglutamine disease HD as a multisystem degenerative disease of the human brain 32, 42, 59, 60, 62, 64, 65, 78.
Despite progress in pathoanatomical research, further re‐investigation, however, is required, in order to (i) analyze the additional involvement of further subcortical regions (eg, amygdala, hypothalamus, subthalamic nucleus, claustrum), anatomically linked with the established targets of the disease process 19, 40, 42, 64, 75, 78, 79, 80, and (ii) elucidate the morphological basis of some of the more poorly understood disease symptoms of HD patients (eg, severe unintended weight loss during the advanced disease stages of HD, and dysfunction of vertical saccades, vergence and steady fixation) 4, 5, 20, 45, 46, 47, 58, 60, 81, 82.
Currently, there is no convincing explanation for the enigmatic phenomenon that the destructive process of HD targets only selective cortical and subcortical brain regions and ultimately leads to a disease‐specific brain distribution of neurodegeneration. Since, however, the cortical and subcortical targets of the pathological process are anatomically interconnected via fiber tracts, it is believed by some that the pattern of brain neurodegeneration in HD and its evolution follow the interconnecting brain fiber tracts. Accordingly, recent explanations for the high vulnerability of select cortical and subcortical brain sites have focused on the possible pathophysiological role of intact anatomical interconnections. According to this pathoanatomically based idea, the anatomical interconnections of a given brain structure represent a crucial factor for the spread of the pathological process of HD throughout the brain and account for its susceptibility or resistance to the neurodegenerative process of HD. Following from this, the anatomical interconnections of the diseased brains may be the rails along which the pathological process of HD expands in a predictable trans‐neuronal spread throughout the brain, leaving behind a characteristic brain distribution pattern of neuronal loss 9, 11, 21, 39, 53, 57, 60, 64.
The exact principles that become important in interconnected nerve cells and enable the targeted trans‐neuronal brain spread of the pathological process of HD along anatomical pathways, however, is still a matter for investigation. Nevertheless, some HD researchers believe that the essential pathophysiological mechanisms may include (i) multifactorially conditioned impairment of axonal transport mechanisms and (ii) self‐sustained auto‐catalytic interneuronal spreading according to prion‐like mechanisms. Although the concept that some human neurodegenerative disorders represent prion‐like diseases is currently popular and well‐received, further studies are necessary to demonstrate that the polyglutamine disease HD is indeed a chronically progredient prion‐like protein misfolding disease 9, 11, 21, 39, 53, 57, 60, 64.
In addition, future studies should be undertaken to identify the primary target of the disease process in the brains of HD patients. Currently, we have two alternative scenarios with respect to the possible origin of the pathological process of HD: (i) The striatum is the primary target from which the destructive process subsequently spreads along anatomical pathways to other vulnerable subcortical and cortical brain sites, or (ii) the primary damage to the cerebral cortex triggers the additional involvement of subcortical regions via transneuronal, anterograde or retrograde mechanisms 30, 32, 36, 42, 59, 60, 64, 81.
ACKNOWLEDGMENTS
This study was supported by grants from Dr. Senckenbergische Stiftung (Frankfurt/Main, Germany), the Deutsche Huntington Hilfe e.V., and the Huntington‐Selbsthilfe Nordrhein‐Westfalen e.V. We thank M. Bouzrou (tissue preparation and staining) and I. Szasz (graphics) for their excellent work and assistance.
REFERENCES
- 1. Alzheimer A (1911) Über die anatomische Grundlage der Huntington'schen Chorea und der choreatischen Bewegung überhaupt. Neurol Centralblatt 30:891–892. [Google Scholar]
- 2. Andrew SE, Goldberg YP, Kremer B, Telenius H, Theilmann J, Adam S et al (1993) The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nat Genet 4:398–403. [DOI] [PubMed] [Google Scholar]
- 3. Atkin G, Paulson H (2014) Ubiquitin pathways in neurodegenerative disease. Front Mol Neurosci 7:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aziz NA, van der Marck MA, Pijl H, Olde Rikkert MG, Bloem BR, Roos RA (2008) Weight loss in neurodegenerative disorders. J Neurol 255:1872–1880. [DOI] [PubMed] [Google Scholar]
- 5. Borrell‐Pagès M, Zala D, Humbert S, Saudou F (2006) Huntington's disease: from huntingtin function and dysfunction to therapeutic strategies. Cell Mol Life Sci 63:2642–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Braak H, Braak E (1992) The human entorhinal cortex: normal morphology and lamina‐specific pathology in various diseases. Neurosci Res 15:6–31. [DOI] [PubMed] [Google Scholar]
- 7. Braak H, Braak E (1992) Allocortical involvement in Huntington's disease. Neuropathol Appl Neurobiol 18:539–547. [DOI] [PubMed] [Google Scholar]
- 8. Brodmann K (1909) Vergleichende Lokalisationslehre der Großhirnrinde. Johann Ambrosius Barth: Leipzig. [Google Scholar]
- 9. Brundin P, Melki R, Kopito R (2010) Prion‐like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol 11:301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bruyn GW, Bots GTAM, Dom R (1979) Huntington's chorea: current neuropathological status. Adv Neurol 23:83–93. [Google Scholar]
- 11. Costanzo M, Zurzolo C (2013) The cell biology of prion‐like spread of protein aggregates: mechanisms and implication in neurodegeneration. Biochem J 452:1–17. [DOI] [PubMed] [Google Scholar]
- 12. De la Monte SM, Vonsattel JP, Richardson EP Jr (1988) Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington's disease. J Neuropathol Exp Neurol 47:516–525. [DOI] [PubMed] [Google Scholar]
- 13. De Vos KJ, Grierson AJ, Ackerley S, Miller CCJ (2008) Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci 31:1151–1173. [DOI] [PubMed] [Google Scholar]
- 14. Dickson DW (1999) Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J Neurol 246:6–15. [DOI] [PubMed] [Google Scholar]
- 15. Dom R, Malfroid M, Baro F (1976) Neuropathology of Huntington's chorea. Studies of the ventrobasal complex of the thalamus. Neurology 26:64–68. [DOI] [PubMed] [Google Scholar]
- 16. Dunlap CB (1927) Pathologic changes in Huntington's chorea: with special reference to the corpus striatum. Arch Neurol Psychiatry 18:867–943. [Google Scholar]
- 17. Duyao M, Ambrose C, Myers R, Novelletto A, Persichetti F, Frontali M et al (1993) Trinucleotide repeat length instability and age of onset in Huntington's disease. Nat Genet 4:387–392. [DOI] [PubMed] [Google Scholar]
- 18. Estrada‐Sanchez AM, Rebec GV (2013) Role of cerebral cortex in the neuropathology of Huntington's disease. Front Neural Circuits 7:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gabery S, Murphy K, Schultz K, Loy CT, McCusker E, Kirik D et al (2010) Changes in key hypothalamic neuropeptide populations in Huntington disease revealed by neuropathological analyses. Acta Neuropathol 120:777–788. [DOI] [PubMed] [Google Scholar]
- 20. Gil JM, Rego AC (2008) Mechanisms of neurodegeneration in Huntington's disease. Eur J Neurosci 27:2803–2820. [DOI] [PubMed] [Google Scholar]
- 21. Goedert M, Clavaguera F, Tolnay M (2010) The propagation of prion‐like protein inclusions in neurodegenerative diseases. Trend Neurosci 33:317–325. [DOI] [PubMed] [Google Scholar]
- 22. Graeber MB, Moran LB (2002) Mechanisms of cell death in neurodegenerative diseases: fashion, fiction, and facts. Brain Pathol 12:385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grimbergen YA, Knol MJ, Bloem BR, Kremer BP, Roos RA, Munneke M (2008) Falls and gait disturbances in Huntington's disease. Mov Disord 23:970–976. [DOI] [PubMed] [Google Scholar]
- 24. Grinberg LT, Heinsen H (2009) Argyrophilic grain disease. An update on a frequent cause of dementia. Dement Neuropsych 3:2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harper PS (1992) The epidemiology of Huntington's disease. Hum Genet 89:365–376. [DOI] [PubMed] [Google Scholar]
- 26. Heathfield KW (1973) Huntington's chorea: a centenary review. Postgrad Med J 49:32–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hedreen JC, Peyser CE, Folstein SE, Ross CA (1991) Neuronal loss in layers V and VI of cerebral cortex in Huntington's disease. Neurosci Lett 133:257–261. [DOI] [PubMed] [Google Scholar]
- 28. Heinsen H, Bauer M, Ulmar G, Gangnus D, Jungkunz G (1992) The entorhinal region in Huntington's disease: a cytoarchitectonic and quantitative investigation in five cases. Clin Neuropathol 11:226–226. [Google Scholar]
- 29. Heinsen H, Rüb U (1997) Quantitative investigations of neural circuits in Huntington's disease. J Neural Transm 104:1139–1140. [Google Scholar]
- 30. Heinsen H, Rüb U, Bauer M, Ulmar G, Bethke B, Schüler M et al (1999) Nerve cell loss in the thalamic mediodorsal nucleus in Huntington's disease. Acta Neuropathol 97:613–622. [DOI] [PubMed] [Google Scholar]
- 31. Heinsen H, Rüb U, Gangnus D, Jungkunz G, Bauer M, Ulmar G et al (1996) Nerve cell loss in the thalamic centromedian‐parafascicular complex in patients with Huntington's disease. Acta Neuropathol 91:161–168. [DOI] [PubMed] [Google Scholar]
- 32. Heinsen H, Strik M, Bauer M, Luther K, Ulmar G, Gangnus D et al (1994) Cortical and striatal neurone number in Huntington's disease. Acta Neuropathol 88:320–333. [DOI] [PubMed] [Google Scholar]
- 33. Hirai T, Jones EG (1989) A new parcellation of the human thalamus on the basis of histochemical staining. Brain Res Rev 14:1–34. [DOI] [PubMed] [Google Scholar]
- 34. Hunt JR (1917) Progressive atrophy of the globus pallidus. Brain 40:58–148. [Google Scholar]
- 35. Huntington G (1872) On chorea. Med Surg Reporter 26:317–321. [Google Scholar]
- 36. Imarisio S, Carmichael J, Korolchuk V, Chen CW, Saiki S, Rose C et al (2008) Huntington's disease: from pathology and genetics to potential therapies. Biochem J 412:191–209. [DOI] [PubMed] [Google Scholar]
- 37. Jelgersma G (1908) Über anatomische Befunde bei Paralyis agitans und bei chronischer Chorea. Neurol Centralblatt 27:995–996. [Google Scholar]
- 38. Jones EG (1985) The Thalamus. Plenum Press: New York, London. [Google Scholar]
- 39. Jucker M, Walker LC (2011) Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol 70:532–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lange H, Thorner G, Hopf A, Schröder KF (1976) Morphometric studies of the neuropathological changes in choreatic diseases. J Neurol Sci 28:401–425. [DOI] [PubMed] [Google Scholar]
- 41. Lange HW (1981) Quantitative changes of telencephalon, diencephalon, and mesencephalon in Huntington's chorea, postencephalitic, and idiopathic Parkinsonism. Verh Anat Ges 73:923–925. [Google Scholar]
- 42. Lange HW, Aulich A (1986) Die Hirnatrophie bei der Huntingtonschen Krankheit. In: Die Huntingtonsche Krankheit. Oepen H (ed), pp. 25–41. Hippokrates: Stuttgart. [Google Scholar]
- 43. Lanska DJ (2000) George Huntington (1850‐1916) and hereditary chorea. J Hist Neurosci 9:76–89. [DOI] [PubMed] [Google Scholar]
- 44. Lanska DJ (2010) The history of movement disorders. In: Handbook of Clinical Neurology, Vol. 95. Finger S, Boller F, Tyler KL (eds), pp. 501–546. Elsevier: Amsterdam. [DOI] [PubMed] [Google Scholar]
- 45. Lasker AG, Zee DS (1997) Ocular motor abnormalities in Huntington's disease. Vis Res 37:3639–3645. [DOI] [PubMed] [Google Scholar]
- 46. Leigh RJ, Newman SA, Folstein SE, Lasker AG, Jensen BA (1983) Abnormal ocular motor control in Huntington's disease. Neurology 33:1268–1275. [DOI] [PubMed] [Google Scholar]
- 47. Leigh RJ, Zee DS (2006) The Neurology of Eye Movements, 4th edn. Oxford University Press: Oxford. [Google Scholar]
- 48. Li JY, Conforti L (2013) Axonopathy in Huntington's disease. Exp Neurol 246:62–71. [DOI] [PubMed] [Google Scholar]
- 49. Li XJ, Li S (2011) Proteasomal dysfunction in aging and Huntington disease. Neurobiol Dis 43:4–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mann DM, Oliver R, Snowden JS (1993) The topographic distribution of brain atrophy in Huntington's disease and progressive supranuclear palsy. Acta Neuropathol 85:553–559. [DOI] [PubMed] [Google Scholar]
- 51. Margolis RL, Ross CA (2003) Diagnosis of Huntington disease. Clin Chem 49:1726–1732. [DOI] [PubMed] [Google Scholar]
- 52. McCaughey WTE (1961) The pathologic spectrum of Huntington's chorea. J Nerv Ment Dis 133:91–103. [Google Scholar]
- 53. Millecamps S, Julien JP (2013) Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci 14:161–176. [DOI] [PubMed] [Google Scholar]
- 54. Morel A, Magnin M, Jeanmonod D (1997) Multiarchitectonic and stereotactic atlas of the human thalamus. J Comp Neurol 387:588–630. [DOI] [PubMed] [Google Scholar]
- 55. Myers RH (2004) Huntington's disease genetics. NeuroRx 1:255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Myers RH, Vonsattel JP, Stevens TJ, Cupples LA, Richardson EP, Martin JB, Bird ED (1988) Clinical and neuropathologic assessment of severity in Huntington's disease. Neurology 38:341–347. [DOI] [PubMed] [Google Scholar]
- 57. Norrby E (2011) Prions and protein‐folding diseases. J Intern Med 270:1–14. [DOI] [PubMed] [Google Scholar]
- 58. Rüb U, Heinsen H, Brunt ER, Landwehrmeyer B, den Dunnen WF, Gierga K, Deller T (2009) The human premotor oculomotor brainstem system ‐ can it help to understand oculomotor symptoms in Huntington's disease? Neuropathol Appl Neurobiol 35:4–15. [DOI] [PubMed] [Google Scholar]
- 59. Rüb U, Hoche F, Brunt ER, Heinsen H, Seidel K, Del Turco D et al (2013) Degeneration of the cerebellum in Huntington's disease (HD): possible relevance for the clinical picture and potential gateway to pathological mechanisms of the disease process. Brain Pathol 23:165–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rüb U, Hentschel M, Stratmann K, Brunt E, Heinsen H, Seidel K et al (2014) Huntington's disease (HD): degeneration of select nuclei, widespread occurrence of neuronal nuclear and axonal inclusions in the brainstem. Brain Pathol 24:247–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rüb U, Jen JC, Braak H, Deller T (2008) Functional neuroanatomy of the human premotor oculomotor brainstem nuclei: insights from postmortem and advanced in vivo imaging studies. Exp Brain Res 187:167–180. [DOI] [PubMed] [Google Scholar]
- 62. Rüb U, Schöls L, Paulson H, Auburger G, Kermer P, Jen JC et al (2013) Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog Neurobiol 104:38–66. [DOI] [PubMed] [Google Scholar]
- 63. Rüb U, Seidel K, Vonsattel JP, Lange HW, Eisenmenger W, Götz M et al (2014) Huntington's disease (HD): neurodegeneration of Brodmann's primary visual area 17 (BA17). Brain Pathol 25:701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rüb U, Vonsattel JPG, Heinsen H, Korf HW (2015) The neuropathology of Huntington's disease: classical findings, recent developments and correlation to functional neuroanatomy. Adv Anat Embryol Cell Biol 217:1–146. [PubMed] [Google Scholar]
- 65. Seidel K, Siswanto S, Brunt ER, den Dunnen W, Korf HW, Rüb U (2012) Brain pathology of spinocerebellar ataxias. Acta Neuropathol 124:1–21. [DOI] [PubMed] [Google Scholar]
- 66. Sheperd GM (2013) Corticostriatal connectivity and its role in disease. Nat Rev Neurosci 14:278–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shoulson I, Young AB (2011) Milestones in Huntington disease. Mov Disord 26:1127–1133. [DOI] [PubMed] [Google Scholar]
- 68. Sieradzan KA, Mann DM (2001) The selective vulnerability of nerve cells in Huntington's disease. Neuropathol Appl Neurobiol 27:1–21. [DOI] [PubMed] [Google Scholar]
- 69. Sotrel A, Paskevich PA, Kiely DK, Bird ED, Williams RS, Myers RH (1991) Morphometric analysis of the prefrontal cortex in Huntington's disease. Neurology 41:1117–1121. [DOI] [PubMed] [Google Scholar]
- 70. Stier E (1903) Zur pathologischen Anatomie der Huntingtonschen Chorea. Arch F Psychol 37:62–86. [Google Scholar]
- 71. Stone TT, Falstein EI (1938) Pathology of Huntington's chorea. J Nerv Ment Dis 88:602–628. [Google Scholar]
- 72. Tanner CM, Goldman SM (1994) Epidemiology of movement disorders. Curr Opin Neurol 7:340–345. [DOI] [PubMed] [Google Scholar]
- 73. The Huntington's disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72:971–983. [DOI] [PubMed] [Google Scholar]
- 74. Thu DC, Oorschot DE, Tippett LJ, Nana AL, Hogg VM, Synek BJ, Luthi‐Carter R, Waldvogel HJ, Faull RL (2010) Cell loss in the motor and cingulate cortex correlates with symptomatology in Huntington's disease. Brain 133:1094–1110 [DOI] [PubMed] [Google Scholar]
- 75. van Wamelen DJ, Aziz NA, Roos RA, Swaab DF (2014) Hypothalamic alterations in Huntington's disease patients: comparison with genetic rodent models. J Neuroendocrinol 26:761–775. [DOI] [PubMed] [Google Scholar]
- 76. Vogt C, Vogt O (1920) Zur Lehre der Erkrankungen des striären Systems. J Psychol Neurol 25:631–846. [Google Scholar]
- 77. Vogt C, Vogt O (1942) Morphologische Gestaltungen unter normalen und pathogenen Bedingungen. Ein hirnanatomischer Beitrag zu ihrer Kenntnis. J Psychol Neurol 50:161–524. [Google Scholar]
- 78. Vonsattel JP (2008) Huntington disease models and human neuropathology: similarities and differences. Acta Neuropathol 115:55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Vonsattel JP, DiFiglia M (1998) Huntington disease. J Neuropathol Exp Neurol 57:369–384. [DOI] [PubMed] [Google Scholar]
- 80. Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr. (1985) Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 44:559–577. [DOI] [PubMed] [Google Scholar]
- 81. Walker FO (2007) Huntington's disease. Semin Neurol 27:143–150. [DOI] [PubMed] [Google Scholar]
- 82. Walker FO (2007) Huntington's disease. Lancet 369:218–228. [DOI] [PubMed] [Google Scholar]