

Abstract
Radiotherapy is an essential component of glioma standard treatment. Glioblastomas (GBM), however, display an important radioresistance leading to tumor recurrence. To improve patient prognosis, there is a need to radiosensitize GBM cells and to circumvent the mechanisms of resistance caused by interactions between tumor cells and their microenvironment. STAT3 has been identified as a therapeutic target in glioma because of its involvement in mechanisms sustaining tumor escape to both standard treatment and immune control. Here, we studied the role of STAT3 activation on tyrosine 705 (Y705) and serine 727 (S727) in glioma radioresistance. This study explored STAT3 phosphorylation on Y705 (pSTAT3‐Y705) and S727 (pSTAT3‐S727) in glioma cell lines and in clinical samples. Radiosensitizing effect of STAT3 activation down‐modulation by Gö6976 was explored. In a panel of 15 human glioma cell lines, we found that the level of pSTAT3‐S727 was correlated to intrinsic radioresistance. Moreover, treating GBM cells with Gö6976 resulted in a highly significant radiosensitization associated to a concomitant pSTAT3‐S727 down‐modulation only in GBM cell lines that exhibited no or weak pSTAT3‐Y705. We report the constitutive activation of STAT3‐S727 in all GBM clinical samples. Targeting pSTAT3‐S727 mainly in pSTAT3‐Y705‐negative GBM could be a relevant approach to improve radiation therapy.
Keywords: glioma, Gö6976, radiotherapy, resistance, STAT3
Introduction
Gliomas are the most common primary tumors of the central nervous system. Among them, the most frequent and malignant type is glioblastoma multiforme (GBM). GBM displays an outstanding resistance to anticancer therapies. Despite the gold standard treatment that encompasses surgery and radiotherapy with temozolomide‐based concurrent and adjuvant chemotherapy, the median survival of selected GBM patients is almost 14.6 months 53. As the tumor recurrence usually occurs within the treated volume 13, 36, radioresistance of glioma cells is involved in treatment failure. Tumor resistance to radiation involves radioresistance of tumor cells themselves that is, intrinsic radioresistance, as well as interactions between tumor cells and their microenvironment 2, 4. Understanding the molecular mechanisms involved in GBM radioresistance might help improve GBM treatment effectiveness.
Concordant studies have reported Signal Transducer and Activator of Transcription 3 (STAT3) as a potential therapeutic target in glioma because of its functions at a focal point of multiple converging signaling pathways involved in cancer aggressiveness 6. There are several isoforms of STAT3 including alpha (α) and beta (β), which are generated by alternative mRNA splicing of a single gene transcript STAT3 11, 12, 48 or by proteolytic cleavage 12. Some of them are c‐terminal truncated proteins that act as dominant negatives of the corresponding full‐length protein. Early studies revealed that activation of STAT3 functions requires phosphorylation of its tyrosine (pSTAT3‐Y705) residue 47, 66. However, it has been shown that besides Y705 and independently of gp130/JAK pathway, STAT3 can be phosphorylated also on serine 727 (pSTAT3‐S727) residue 44. The canonical Y705 activation can be induced by the binding of the Interleukin 6‐family cytokine receptors to the gp130 receptor and the consecutive Janus kinase 2 (JAK2) activation 26. The pSTAT3‐Y705 then form homodimers or heterodimers with partner proteins such as STAT1 and NF‐κB (p65) 30. Those dimers translocate into the nucleus where they regulate expression of target‐genes 5. pSTAT3‐Y705 inhibits apoptosis through up‐regulation of Survivin 39, Mcl‐1 29, BCL‐XL 5, 9, 29 and promotes cell proliferation by up‐regulating c‐myc 5, 9. Through increased VEGF expression, pSTAT3‐Y705 stimulates angiogenesis 40. It also supports cell migration by augmenting MMP9 52 and ICAM‐1 30 expression. It has been also shown that pSTAT3‐Y705 activation in tumor cells participates in locally blocking immune cell maturation and activation by modifying tumor secretome 49. Moreover, pSTAT3‐Y705 expression in glioma tumors has been positively correlated to tumor grade as well as to a worse patient prognosis 1, 38. Those findings suggested that STAT3 activation is involved in glioma tumorigenesis and tumor aggressiveness. Furthermore, in vivo study of glioma molecular response to ionizing radiation showed a time‐dependent increase in pSTAT3‐Y705 expressing tumor cells in a spontaneous GBM model generated in mice 24. Irradiation also induced a dose‐dependent increase in pSTAT3‐Y705 nuclear accumulation in glioma xenografts 30. The above published data suggest that STAT3 activation might participate in influencing glioma sensitivity to radiotherapy. However, using JSI‐124, a JAK/STAT3 inhibitor or specific gp130 blocking antibodies, we showed that down‐regulating pSTAT3‐Y705 level in GBM cells did not impact cell intrinsic radioresistance 15.
In most studies, STAT3 pathway activation has been assessed through Y705 residue phosphorylation. In contrast to Y705 activation, upstream regulating events and downstream consequence of pSTAT3‐S727 are not clearly understood. In T98G and MO59K human glioma cell lines, pSTAT3‐S727 activation depends in part on PKCε 3. In GBM stem cells, pSTAT3‐S727 is influenced by the NOTCH pathway as inhibiting NOTCH by GSI‐18 impairs pSTAT3‐S727 accumulation 21. Some early studies reported that pSTAT3‐S727 might have a critical biological role in GBM. It has been shown that pSTAT3‐S727 was accumulated in GBM tumors 7 and was correlated with malignancy 50. Other studies reported that pSTAT3‐S727 and pSTAT3‐Y705 are independent bad prognosis markers in GBM and their co‐expression was associated to a worse patient clinical outcome 37, 38. These findings were consistent with others, which reported that pSTAT3‐S727 and pSTAT3‐Y705 were playing a critical role in GBM stem cells, as disrupting both STAT3 dimerization and binding to DNA resulted in apoptosis 56, decreased cell proliferation and reduced tumorigenic capacity 51, 56. Nevertheless, it remains a crucial need of a specific inhibitor of pSTAT3‐S727 to help decipher the role of STAT3 in malignancy. In addition to Y705 and S727 phosphorylation, it has been reported that lysine acetylation 43, 45, 64 and methylation 62 are involved in STAT3 functional regulation. Furthermore, STAT proteins, lacking phosphorylation on Y705 or mutated at this position, can still form dimers and induce transcription 8, 33, 34, 35, 61. To date, little is known about the relationship between the glioma response to radiation and the phosphorylation status of STAT3 on both S727 and Y705.
In this study, we evaluated, by Western blotting, STAT3 pathway activation in a panel of 15 glioma cell lines. We found a correlation between pSTAT3‐S727 accumulation and intrinsic radioresistance. Moreover, Gö6976, a chemical multiple kinase inhibitor, reduced pSTAT3‐S727 level in GBM cell lines that exhibited no or weak levels of pSTAT3‐Y705. Interestingly, this decrease in pSTAT3‐S727 was accompanied by a highly significant radiosensitization. Immunohistochemical analyses confirmed that pSTAT3‐S727 is a common feature in GBM clinical samples, strengthening that it might be a relevant therapeutic target.
Materials and Methods
Cell culture
SF763, SF767, U87MG and U251MG human glioma cell lines were kindly provided by Dr C. Delmas (Centre Claudius Regaud, Toulouse, France). SW1783, SNB19 and U373MG human glioma cell lines were given by N. Auger (Institut Curie, Paris, France). T98G, U118MG and CB193 cell lines were given by G. Pennarun (CEA, Grenoble, France). M059K, MO59J, CCF, LN229 and U138MG cell lines were given by N. Foray (Centre Léon Bérard, Lyon, France). All culture reagents were purchased from GIBCO (Invitrogen, Cergy‐Pontoise, France). Gö6976 (No 365250), a cell‐permeable, reversible and ATP‐competitive inhibitor of protein kinase C, was purchased from Merck Millipore (Nottingham, UK). Cells were grown in DMEM (with 4500 mg/L glucose and L‐glutamine) supplemented with sodium pyruvate 1%, non‐essential amino acids 1%, gentamicin 10 μg/mL and 10% fetal calf serum (20% for U138MG and CCF) in a humidified incubator containing 5% CO2 at 37°C.
Antibodies
For Western blot, immunofluorescence and immunohistochemistry, anti‐pSTAT3‐Y705 monoclonal antibody (mAb, No 9145) was purchased from Ozyme (Saint‐Quentin‐en‐Yvelines, France) and anti‐pSTAT3‐S727 mAb (No 1121‐1) from Euromedex (Souffelweyersheim, France). For Western blot, rabbit anti‐STAT3 mAb (No 4904) was purchased from Ozyme and anti‐β‐actin polyclonal antibodies (pAbs, No A2066) from Sigma (Saint‐Quentin‐Fallavier, France). Anti‐phospho‐JAK2‐Y1007/1008 pAbs (No 3771) and anti‐JAK2 mAb (No 3230) were purchased from Ozyme. Anti‐EGFR pAbs (No RB‐1417‐P1) were purchased from Fisher Scientific (Illkirch, France) and anti‐phospho‐EGFR‐Y1068 mAb (No 1138‐S) from Euromedex. Anti‐PKCα pAbs (No 06‐870) and anti‐phospho‐PKCα‐S657/Y658 pAbs (No 07‐790) were purchased from Millipore (Molsheim, France). Anti‐rabbit conjugated to horseradish peroxidase was from P.A.R.I.S. (Compiègne, France). For immunofluorescence, Cy3‐conjugated goat anti‐rabbit pAbs (No 111‐165‐144) were purchased from Interchim (Montluçon, France).
Western blotting
Cells in exponential phase of growth were harvested and whole‐cell extract was prepared with Buffer C supplemented with NP‐40, phosphatase inhibitors (NaF, Na3VO4) and Complete Protease Inhibitor Cocktail (Roche Diagnostics, Meylan, France) and subjected to SDS‐PAGE as previously reported 15. Protein accumulation was normalized to β‐actin appropriate loading control. Phosphorylation of STAT3 was normalized to the corresponding total STAT3. Signal detection was performed using ECL Immobilon system (Millipore, Saint‐Quentin‐en‐Yvelines, France). Three independent experiments were used for analysis with Quantity One (BioRad, Marnes‐la‐Coquette, France).
Immunofluorescence
Glioblastoma cell lines were seeded in 9 cm2 slide‐flasks. 6 h after plating, they were treated with Gö6976. Fixation was done with methanol after 24 h or with PFA 4% after 72 h, respectively, for pSTAT3‐Y705 and pSTAT3‐S727 detection. Fixed cells were then permeabilized with PBS‐triton 0.5% and incubated with antibody in PBS‐BSA 0.5% before image acquisition and analysis. All images were acquired by a Leica SPE microscope (Leica Microsystemes SAS, Nanterre, France) with a 40×/1.15 oil immersion objective. Signal analysis was performed by ImageJ software (16). Nuclei were selected in DAPI channel as region of interest (ROI). Threshold of fluorescence intensity was applied in Cy3 channel and fluorescence intensity of all ROI was measured by the plug‐in “Measure.” Results were corrected by a factor which integrated the background and the area of each ROI according to the formula: Corrected total cell fluorescence (CTCF) = Integrated density—(Area of selected cell × Mean fluorescence of background). A mean of 100 cells in each condition was analyzed.
Cell growth assay
Global cell growth was assessed using the colorimetric MTT [3(4, 5‐dimethylthiazol‐2‐yl)‐diphenyltetrazolium bromide] assay (Sigma‐Aldrich, Saint‐Quentin‐Fallavier, France). Cells were seeded in 96‐well plates at a density adjusted to cell lines. Gö6976 was added 6 h after seeding and remained in culture medium until the experiment ended. At 24‐h incubation with Gö6976, irradiation was performed at room temperature as single exposure doses delivered by an Elekta SL18 linear accelerator with a nominal energy of 6 MV at a dose rate of about 400 cGy/min. Doses ranging from 2 to 10 Gy were calculated using a dedicated treatment planning system to a depth of water of 1.5 cm. Intensity was collected at 490 nm using a Bio‐Rad 11885 spectrophotometer. Cell growth was calculated as following: (ODtest sample − ODblank/ODcontrol − ODblank) × 100. Each experimental condition was analyzed at least in triplicate. Three independent experiments were used for analysis.
Clonogenic cell survival assay
Clonogenic cell survival assay was performed as previously reported 15. Briefly, cells were seeded at a density of 1000 to 6000 per 25‐cm2 flask. Gö6976 was added in growth medium 6 h after seeding, for 72‐h incubation before irradiation. Irradiation was performed as described in the preceding chapter. Gö6976 containing medium was replaced by growth medium 24 h after irradiation. The plating efficiency (PE) represents the percentage of seeded cells that grew into colonies. Colonies with more than 50 cells were counted by microscopic inspection, and PE as well as the radiation‐surviving fraction (PE of experimental group/PE of control group) was determined. Each experimental condition was performed in triplicate. Survival curves were obtained with data from three independent experiments in accordance with linear‐quadratic model (Kaleidagraph 4.0, Synergy Software, Reading, Pennsylvania, USA).
Glioblastoma clinical samples
GBM samples (Biobank N° DC‐2012‐1584) were taken from the material of surgical resection during the course of standard diagnosis procedure. Thirty tumors, classified as GBM (grade IV, WHO, 2007) according to histological diagnosis and grading criteria, were used in this study. Patients were treated by surgery followed by radiotherapy and concomitant temozolomide followed by at least six monthly cycles of adjuvant temozolomide 53.
Immunohistochemistry
Immunostaining of pSTAT3‐Y705 and pSTAT3‐S727 was performed with a Benchmark XT immunostainer (Ventana, Illkirch, France) on 5‐μm paraffin sections. After deparaffinization, antigen retrieval was carried out in CCI buffer. Antibodies were incubated at 37°C and the revelation was made with the Ultraview detection kit (Ventana). Sections were semiquantitatively scored by light microscopy by two pathologists. Cytoplasmic and nuclear staining, percentage of stained tumor cells and staining intensity (no, weak, moderate, strong) for each localization were specified.
Statistical analysis
Statistical analysis was performed using Stata 13 software (StataCorp LP, College Station, TX, USA). The tests were two‐sided, with a Type I error set at α = 0.05. Quantitative data were presented as mean ± standard error of mean. Comparisons between groups were realized using ANOVA or the Kruskal–Wallis test (assumption of normality assessed using the Shapiro–Wilk test and homoscedasticity verified by Bartlett test) followed, when appropriate, by Dunnett's test (multiple comparison procedure to compare each of a number of treatments with a single control). In case of non‐multiple comparison and because of sample size, Mann–Whitney's test was proposed. The study of relation between quantitative parameters was explored by correlation coefficients (Pearson or Spearman according to statistical distribution, noted r). Finally, correlated repeated data were analyzed using random‐effects models, which allow taking into account between and within subject variability and to study fixed effects: groups, dose and their interaction. The normality of residuals was verified for each considered model. For each of pSTAT‐Y705 and pSTAT3‐S727 staining, the percentage of stained tumor cells was multiplied by the intensity (1, 2 or 3) and added to get a score between 0 and 300. Survival rates were estimated by the Kaplan–Meier method. The study of pSTAT3‐Y705 and pSTAT3‐S727 (considered as quantitative parameters) staining impact on survival rate was considered, in first time, with Cox model. These parameters were then categorized according to the statistical distribution (rank indicators). Then, survival rates were compared by groups with log‐rank test.
Results
Serine 727 phosphorylated STAT3 correlates with intrinsic radioresistance
By Western blots, we assessed STAT3 phosphorylation status in 15 human glioma cell lines that were grown in basal conditions. Levels of protein activation were calculated by the phospho‐protein/total protein expression ratio. All cell lines harbored variable level of pSTAT3‐S727. By contrast, pSTAT‐Y705 is not found in all cell lines because it was hardly or not detectable in SF767, U87MG, U118MG and U138MG (Figure 1, Table 1). There was no correlation between amounts of accumulated pSTAT3‐S727, pSTAT3‐Y705 and total STAT3. Previously described intrinsic radioresistance determination by standard clonogenic cell survival assay 15 was then extended to the selected glioma cell lines. Statistical analysis revealed that pSTAT3‐S727 levels, but neither pSTAT3‐Y705 nor total STAT3, were correlated (Spearman, r = 0.57, P < 0.05) with the surviving fractions at 2 Gy (SF2). We observed that in human glioma, STAT3 pathway was differently activated in vitro and that pSTAT3‐S727 and intrinsic radioresistance positively correlated.
Figure 1.
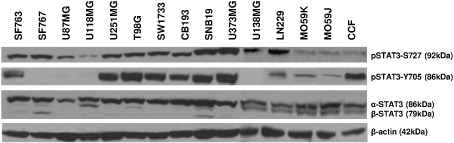
STAT3 serine 727 and tyrosine 705 phosphorylation in human malignant glioma cell lines. Cells were harvested during exponential growth phase. Thirty micrograms of total cell lysate were loaded per lane and electrophoresed by SDS‐PAGE. Transfer membranes were immunoblotted with anti‐STAT3, anti‐pSTAT3‐Y705 and anti‐pSTAT3‐S727 specific antibodies. To ensure equal protein loading, β‐actin was used as control. One representative immunoblot is shown. Three independent experiments were achieved for densitometric analyses.
Table 1.
STAT3 pathway activation and intrinsic radioresistance of human malignant glioma cell lines. Densitometric analyses of the blots are presented as relative ratio of phosphoprotein/total protein. Data are presented as mean values ± standard error of triplicate determinations (arbitrary units). Abbreviation: SF2 = surviving fraction at 2 Gy; ND = not determined
| Cell line | Origin | Western blot | SF2 | |
|---|---|---|---|---|
| pSTAT3‐Y705/STAT3 | pSTAT3‐S727/STAT3 | |||
| SF763 | Glioblastoma | 8.46 ± 4.83 | 1.55 ± 0.96 | 0.84* |
| SF767 | Glioblastoma | 0 | 1.18 ± 0.46 | 0.68* |
| U87MG | Glioblastoma | 0 | 1.08 ± 0.51 | 0.46 |
| U118MG | Glioblastoma | 0 | 0.46 ± 0.09 | 0.44 |
| U251MG | glioblastoma | 1.33 ± 0.96 | 0.6 ± 0.24 | 0.58* |
| T98G | Glioblastoma | 1.8 ± 0.86 | 0.79 ± 0.23 | 0.64* |
| SW1783 | Astrocytoma grade III | 2.22 ± 2.08 | 0.92 ± 0.33 | 0.46* |
| CB193 | Glioma grade III | 1.25 ± 1.13 | 1.68 ± 0.8 | 0.52* |
| SNB19 | Glioblastoma | 0.95 ± 0.64 | 1.05 ± 0.48 | 0.5* |
| U373MG | Glioblastoma | 1.65 ± 0.98 | 1.11 ± 0.67 | 0.58* |
| U138MG | Glioblastoma | 0.08 ± 0.08 | 0.26 ± 0.08 | 0.39 |
| LN229 | Glioblastoma | 5.31 ± 0.4 | 0.3 ± 0.04 | 0.52 |
| MO59K | Glioblastoma | 1.48 ± 0.2 | 0.17 ± 0.02 | ND |
| MO59J | Glioblastoma | 0.58 ± 0.14 | 0.27 ± 0.06 | 0.02 |
| CCF | Glioblastoma | 7.96 ± 0.84 | 0.27 ± 0.02 | 0.53 |
*Previously published 15.
Gö6976 reduces in vitro growth of human glioblastoma
As STAT3 is differently activated in glioma cell lines, we hypothesized that glioma might respond differently to STAT3 pathway inhibition. We tested Gö6976, a multiple kinase inhibitor that was previously reported to inhibit in diverse cell types upstream regulators of STAT3 activation such as PKCs and JAK 3, 18, 23. We selected 3 GBM cell lines that displayed both pSTAT3‐S727 and pSTAT3‐Y705 activation (SF763, U251MG and T98G) and 3 others that did not exhibit detectable pSTAT3‐Y705 (SF767, U118MG and U87MG). 90‐h incubation with Gö6976 resulted in a dose‐dependent growth inhibition, regardless of STAT3 activation status (Figure 2). However, the cell lines displayed different sensitivity to Gö6976 as shown by estimated relative IC50 ranging from 73 to 209 nM.
Figure 2.
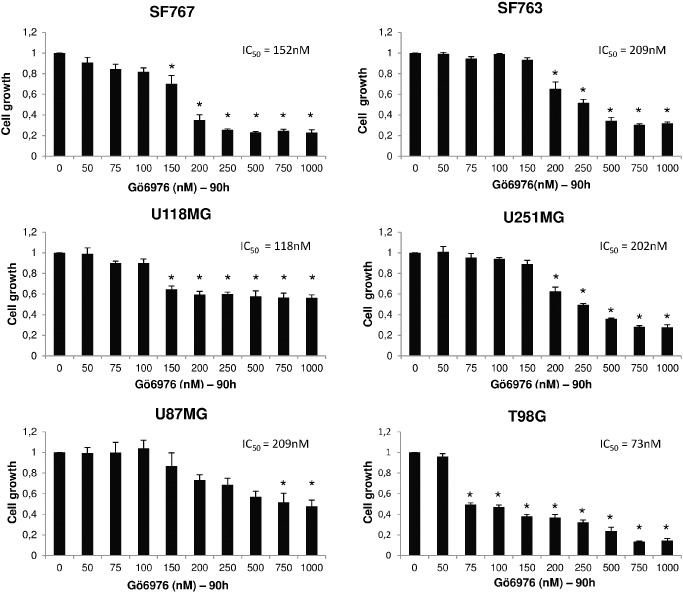
Gö6976 reduces in vitro growth of human glioblastoma. Cells were seeded at 7500 per well of 96‐well plates. Indicated quantity of Gö6976 has been added to growth medium 6 h after seeding. Cells were then grown 90 h before MTT assay. Results represent mean values ± SEM of relative cell growth normalized to untreated cells from three independent experiments done in triplicate; relative IC50 are specified; *significantly different from the control, Dunnett's test.
Gö6976 radiosensitizes pSTAT3‐Y705‐negative human glioblastoma cells
We further explored effect of Gö6976 on intrinsic radioresistance of GBM. MTT tests were done on cells treated with Gö6976, at 72 h after 4‐Gy irradiation. Data were normalized to controls (cells treated with Gö6976 without irradiation) to exclude the effect of Gö6976 alone on cell growth. Incubation with Gö6976 significantly enhanced growth‐inhibiting effect of irradiation only on the three pSTAT3‐Y705‐negative cell lines (Mann–Whitney test, P < 0.05, Figure 3A–F). Thus, Gö6976 radiosensitized at 4 Gy GBM cells depending on the activation status of STAT3.
Figure 3.
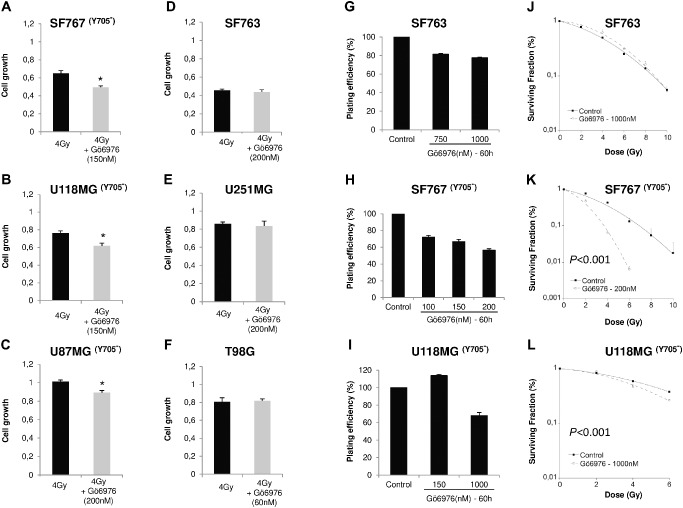
Gö6976 radiosensitizes pSTAT3‐Y705‐negative human glioblastoma cells. A–F. Cells were seeded at a density of 5000 per well of 96‐well plates. 6 h later, Gö6976 was added to growth medium. Single 4‐Gy dose irradiation was performed at 24‐h time course incubation with Gö6976. After 90 h of cell growth in Gö6976, cell number was estimated by MTT assay. Results represent mean values ± SEM of relative cell normalized to untreated cells (n = 5, *P < 0.05, Mann–Whitney test). G–I. SF763, SF767 or U118MG cells were incubated 60 h in different concentrations of Gö6976 and PE was determined 10 days after seeding. J–L. For clonogenic survival assay, SF763 and U118MG cells were treated with 1000 nM of Gö6976 and 200 nM for SF767. After 72 h of incubation, cells were irradiated and the surviving fraction was compared with that of control. Data are fitted to linear‐quadratic model. They were represented by their mean ± SEM of values of three independent experiments, each condition performed in triplicate (random‐effects model). Y705‐ : pSTAT3‐Y705 negative.
To confirm these results, we performed standard clonogenic cell survival assay on SF763, SF767 and U118MG cell lines. In order to reveal radiosensitizing effect of Gö6976, we used doses reducing only slightly cell PE. We first tested Gö6976 effect on PE and observed less than 25% decrease of SF763 PE with 1000 Nm, whereas 200 nM induced 43% decrease on SF767 (Figure 3G,H). Because 150 nM of Gö6976 did not reduce the PE of U118MG cells, we increased the drug concentration to 1000 nM that reduced 32% of cell PE (Figure 3I). Treating SF767 cells with Gö6976 before irradiation at doses ranging from 2 to 10 Gy resulted in a highly significant radiosensitization even at lowest doses, in comparison to untreated irradiated controls (random‐effects model, P < 0.001, Figure 3K). For U118MG cell line, we had to increase cell density at plating in order to obtain enough colonies after treatment with Gö6976 combined to radiation. As previously reported 10, 14, heightening cell density at plating increased resistance to irradiation alone (SF of 0.8 vs. 0.4, Figure 3I and Table 1). When U118MG cells were irradiated at doses higher than 6 Gy, no colony was observed. Irradiation of Gö6976‐pretreated U118MG cells resulted in a significant radiosensitization (random‐effects model, P < 0.001, Figure 3L). These results confirmed that Gö6976 radiosensitized SF767 and U118MG cell lines that are pSTAT3‐Y705 negative. However, SF763 cells that were pSTAT3‐Y705 positive were not radiosensitized by Gö6976 treatment (Figure 3J).
Gö6976 down‐modulates pSTAT3‐S727 only in pSTAT3‐Y705‐negative cells
To investigate the mechanisms involved in the effects of Gö6976, we explored STAT3 pathway activation in cells treated with Gö6976. As shown in Figure 4, treatment with Gö6976 resulted in a decrease of pSTAT3‐Y705 in SF763, T98G and U251MG after 72‐h incubation and earlier (24 h, data not shown). Both pSTAT3‐S727 and total STAT3 levels were unaffected in those cells. By contrast, pSTAT3‐Y705‐negative cells showed decreased levels of pSTAT3‐S727 with no change in total STAT3. To confirm Western blot results and examine the subcellular distribution of pSTAT3‐S727 and pSTAT3‐Y705 following Gö6976 treatment, we performed immunofluorescence assay. Analysis confirmed that, in untreated cells, pSTAT3‐S727 and pSTAT3‐Y705 were distributed in cytoplasm and nuclei with a strong predominance in nuclei (Figure 5). The pSTAT3‐S727 signal was observed in all cell lines but pSTAT3‐Y705 was not detectable in SF767, U118MG and U87MG (data not shown). 72‐h incubation of cells with Gö6976 resulted in a decreased pSTAT3‐S727 specific signal in the pSTAT3‐Y705‐negative cell lines (Figure 5A). Treatment with Gö6976 also reduced early (at 24 h) STAT3‐Y705 phosphorylation in SF763, T98G and U251MG (Figure 5B). Moreover, Gö6976 did not induce any change in subcellular location of neither pSTAT3‐S727 nor pSTAT3‐Y705. Nuclear pSTAT3‐S727 and pSTAT3‐Y705 signal quantification confirmed these observations (Figure 5, histograms). These results confirmed that Gö6976 down‐modulated pSTAT3‐Y705 in GBM cells and also reduced pSTAT3‐S727 only in the pSTAT3‐Y705‐negative cell lines.
Figure 4.
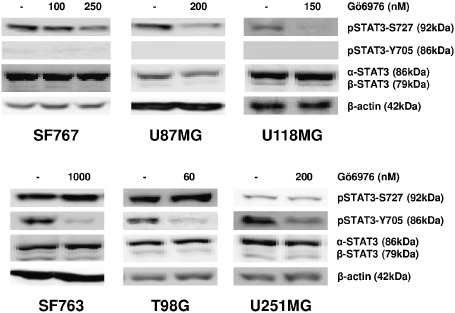
Gö6976 down‐modulates pSTAT3‐S727 only in pSTAT3‐Y705‐negative human glioblastoma cells. Cells were grown 72 h in indicated concentration of Gö6976 or DMSO (control) before harvest. Thirty micrograms of total proteins were loaded per lane and electrophoresed by SDS‐PAGE. Transfer membranes were immunoblotted with anti‐STAT3, anti‐pSTAT3‐Y705 and anti‐pSTAT3‐S727 specific antibodies. To ensure equal protein loading, β‐actin was used as control.
Figure 5.
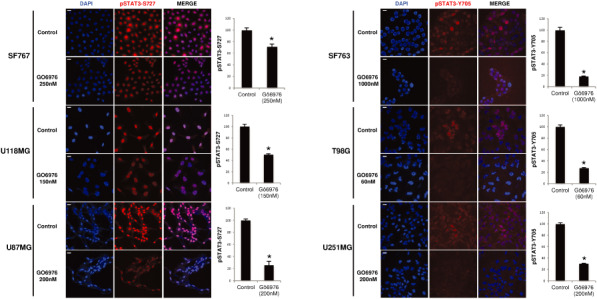
Subcellular distribution of phosphorylated STAT3 in human glioblastoma cell lines treated by Gö6976. Cells were treated with Gö6976 or DMSO (control) and fixed at 24‐ and 72‐h incubation with Gö6976, respectively, for pSTAT3‐Y705 and pSTAT3‐S727 detection. Anti‐pSTAT3‐Y705 and anti‐pSTAT3‐S727 antibodies were used. DAPI and Cy3‐conjugated secondary goat anti‐rabbit antibody were used. Histograms represent the mean observed intensity of fluorescence in each condition (scale bars are equal to 20 μm, 40×/1.15).
Gö6976‐induced pSTAT3‐S727 down‐modulation remains 24 h after irradiation
Gö6976 effects on GBM cells have been investigated also in irradiated conditions (Figure 6). Cells were incubated in Gö6976 for 72 h, then irradiated at 4 Gy and harvested 24 h later. After irradiation, a decrease of total STAT3 is described in Gö6976‐pretreated (compared with only irradiated), in SF767 and T98G cells but not in the other cell lines. We also observed a radio‐induction of pSTAT3‐Y705 in U118MG and U87MG cells without Gö6976 treatment but not in SF767 cells. We still detected pSTAT3‐Y705 in U251, SF763 and T98G cells following irradiation. The Gö6976‐induced pSTAT3‐Y705 inhibition (in non‐irradiated cells, Figure 4) still remained in SF763 after irradiation (Figure 6). Moreover, Gö6976 impeded the radio‐induced pSTAT3‐Y705 phosphorylation in Gö6976‐pretreated and irradiated U118MG and U87MG cells. However, Gö6976 failed to maintain pSTAT3‐Y705 inhibition after irradiation in Gö6976‐pretreated U251MG and T98G cells. For all cell lines, pSTAT3‐S727 was still detected in cells untreated with Gö6976 and irradiated. But after irradiation, the Gö6976‐induced down‐modulation of pSTAT3‐S727 remained 24 h following 4‐Gy irradiation. We concluded that 4‐Gy irradiation did not affect the Gö6976‐induced pSTAT3‐S727 down‐modulation in pSTAT3‐Y705‐negative GBM cells.
Figure 6.
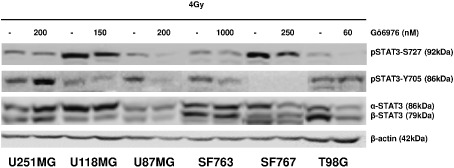
Gö6976 effects after irradiation. Cells were grown 72 h in growth medium with indicated concentration of Gö6976 or DMSO (control) prior to 4‐Gy irradiation. Cells were then harvested 24 h later. Thirty micrograms of total proteins were loaded per lane and electrophoresed by SDS‐PAGE. Transfer membranes were immunoblotted with anti‐STAT3, anti‐pSTAT3‐Y705 and anti‐pSTAT3‐S727 specific antibodies. To ensure equal protein loading, β‐actin was used as control.
STAT3 pathway activation in glioblastoma clinical samples
To confirm clinical relevance of targeting the STAT3 pathway in glioma patients, we assessed the activation level of pSTAT3‐Y705 and pSTAT3‐S727 in human GBM paraffin‐embedded sections (Figure 7). The calculated staining score for pSTAT3‐Y705 ranged from 0 to 150. In the positively stained GBM cells, pSTAT3‐Y705 was located mainly in the nuclei (Figure 7B–D), whereas cytoplasm was not or weakly stained. The pSTAT3‐Y705 positive staining was found in 24/30 patients (Table 2). In these positive specimens, pSTAT3‐Y705 was accumulated in 38% of total GBM cells. Staining intensity was variable; the majority (45%) of GBM cells showed a moderate staining. Interestingly, in pSTAT3‐Y705‐positive samples, we observed pSTAT3‐Y705 staining of some blood vessel endothelial cells. Like in tumor cells, staining intensity was variable and mainly located in the nuclei (Figure 7C,D). We did not find any pSTAT3‐Y705 stained stromal cell in the tumor sections that did not display pSTAT3‐Y705 stained GBM cells (Figure 7A). In the vicinity of vessels, we found few lymphocytes and histiocytes that were negative for pSTAT3‐Y705 staining. The pSTAT3‐S727 staining was found in GBM cells of all specimens (Table 2) with a score ranging from 180 to 300. The majority of GBM cells displayed a moderate or a high intensity of positive pSTAT3‐S727 staining (Table 2). The pSTAT3‐S727 staining was located in cytoplasm and nuclei, but nuclei were more intensely stained than cytoplasm (Figure 7F–H). Moreover, all GBM cells of all tumor specimens were positively stained for pSTAT3‐S727. However, Figure 7E shows one transitional zone with invasive stained malignant cells among non‐stained non‐neoplastic brain cells. Thus, unlike pSTAT3‐Y705, pSTAT3‐S727 is accumulated by all GBM cells in all GBM specimens of our cohort. Interestingly, blood vessel endothelial cells were highly positive for pSTAT3‐S727 staining (Figure 7G). In case of capillary proliferation, endothelial cells also displayed intense pSTAT3‐S727 staining (Figure 7H). Like for pSTAT3‐Y705, the rare lymphocytes and histiocytes were negative for pSTAT3‐S727 staining. We further explored the relationship between pSTAT3‐S727 and pSTAT3‐Y705 expression and overall survival. Neither pSTAT3‐Y705 (P = 0.34) nor pSTAT3‐S727 staining (P = 0.42) were associated with patient overall survival.
Figure 7.
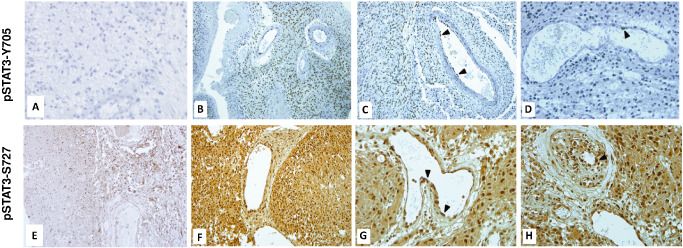
STAT3 activation in glioblastoma clinical samples. Immunostaining of pSTAT3 was performed on paraffin‐embedded sections. A. Negative pSTAT3‐Y705 stained section (×40). B,C. Positive pSTAT3‐Y705 staining of GBM and vascular endothelial cells (×20). D. Heterogeneous pSTAT3‐Y705 staining of endothelial cells (×40). E. pSTAT3‐S727 staining of a transitional zone in a tumor periphery, displaying positively stained GBM cells and unstained non‐neoplastic glial cells (×20). F. pSTAT3‐S727 staining of GBM cells (×20). G. pSTAT3‐S727 staining of vascular endothelial cells (×40). H. pSTAT3‐S727 staining in case of capillary endothelial cell proliferation (×40).
Table 2.
STAT3 activation in glioblastoma clinical samples. Percentage of stained tumor cells and staining intensity is presented for pSTAT3‐Y705 and pSTAT3‐S727. Negative = no staining; positive + = weak staining; positive ++ = moderate staining; positive +++ = strong staining
| Positive specimens | Cell staining intensity in positive specimens | ||
|---|---|---|---|
| pSTAT3‐Y705 | 24/30 | Positive +++ | 11% |
| Positive ++ | 17% | ||
| Positive + | 10% | ||
| Negative | 62% | ||
| pSTAT3‐S727 | 30/30 | Positive +++ | 55% |
| Positive ++ | 41% | ||
| Positive + | 4% | ||
| Negative | 0% | ||
Discussion
Radiotherapy is a main part of the gold standard treatment of GBM patients despite a demonstrated tumor radioresistance 53. GBM cells release, in vitro and in vivo, cytokines such as IL‐6, which modulate their environment to support their growth. IL‐6 and STAT3 are involved together in a same vicious regulatory loop in glioma as IL‐6 initiates STAT3 activation via the complex gp130/IL‐6Rα 26 and activated STAT3 in turn induces IL‐6 secretion 59. The role of IL‐6 in mediating immune system inactivation through activation of STAT3 in GBM and glioma stromal cells is established 49, 60, 65 but it does not explain all the bad clinical outcome of neither IL‐6 nor STAT3 activation in GBM cells. Inhibition of gp130/pSTAT3‐Y705 signaling axis failed to radiosensitize glioma despite evidence that STAT3 pathway activation supports several hallmarks of glioma, including resistance to irradiation and to cell death induction 5, 24, 30, 32. As STAT3 functions might be at least in part driven by pSTAT3‐S727 as reported in other malignancies including prostate cancer 44, colorectal cancer 17, chronic lymphocytic leukemia 25 and mouse hepatocarcinoma 42, we postulated that pSTAT3‐S727 might be involved in glioma intrinsic radioresistance.
In this study, we found that STAT3 pathway was differently activated in cell lines, as, on one hand, some clearly exhibited variable levels of pSTAT3‐Y705 while the others did not (Figure 1 and Table 1). On the other hand, pSTAT3‐S727 was expressed in a large‐amplitude scale in all 15 glioma cell lines. Interestingly, we reported for the first time that pSTAT3‐S727 correlated with intrinsic radioresistance. Those analyses have been achieved before the American Type Culture Collection (ATCC) revealed that U251MG, U373MG and SNB19 originated from a same cell line. However, they are grown in different laboratories worldwide and they express phenotypic variations such as IL‐6 secretion (data not shown). We further hypothesized that targeting pSTAT3‐S727 would affect glioma radiosensitivity.
To our knowledge, to date, there is no reported specific inhibitor of pSTAT3‐S727 capable of discriminating pSTAT3‐Y705 and pSTAT3‐S727 41, 54. Because pSTAT3‐S727 regulation involved several pathways depending on cell types, broad‐spectrum kinase inhibitors are useful in exploring STAT3 pathway activation. Western blots and immunofluorescence analyses showed that Gö6976 significantly decreased pSTAT3‐Y705 level in cells. Interestingly, Gö6976 reduced pSTAT3‐S727 level only in pSTAT3‐Y705‐negative cell lines. The subcellular location of both phosphorylated STAT3 was unaffected by Gö6976. The discrimination in pSTAT3‐S727 inhibition depending on STAT3 status strengthened the relevance of testing Gö6976 on glioma cells. STAT3 pathway activation by Y705 and/or S727 phosphorylation has been reported to play a critical role on cell growth by stimulating proliferation, blocking cell death induction and supporting cell survival 5, 9, 28, 32, 44. We tested Gö6976 on GBM cell growth, choosing three cell lines that displayed pSTAT3‐Y705 and three others that did not. Unlike a previous study that used lower doses of Gö6976 on U251MG 20, we report here that treating cells with Gö6976 resulted in a dose‐dependent decrease of cell growth regardless of STAT3 activation status. Confirming growth inhibition, Gö6976 reduced SF763 and SF767 cell PE. Impact of Gö6976 on cell growth should not be attributed to inhibition of the one rather than the other of pSTAT3‐S727 and pSTAT3‐Y705 because each of them has been reported to sustain cell growth 21, 29, 56. As Gö6976 is an inhibitor of multiple kinases including Chk1 and the T790M mutant EGFR 18, 22, 55, Gö6976‐induced cell growth decrease might also be due to another property. It remains unclear how Gö6976 did act in down‐modulating pSTAT3‐S727. Inhibition of pSTAT3‐Y705 was clear as earlier as 6‐h incubation (data not shown), whereas the decrease in pSTAT3‐S727 was not obvious before 72 h. This latency regarding pSTAT3‐S727 is compatible with an effect on any long‐lasting activated pathway such as PKCs 18, 31. However, diverse mechanisms have been attributed to Gö6976 in different cancers. Gö6976 abrogated Chk1 activity and abolished DNA damage‐induced intra‐S and G2/M cell cycle checkpoints in nasopharyngeal carcinoma and breast cancer cells 22, 31. Checkpoint abolition by Gö6976 resulted in sensitizing in vitro and in vivo nasopharyngeal carcinoma to irradiation or cisplatin treatment 22. Gö6976 also inhibited the T790M mutant EGFR activation when reducing growth of xenografted non‐small cell lung cancers 55. Thus, Gö6976 might interfere with several signaling pathways in radiosensitizing human glioma. Further investigation should help understand the mechanism of STAT3 pathway modulation in the GBM cell response to Gö6976.
Consistent with our hypothesis, a single dose of Gö6976 radiosensitized at a 4‐Gy irradiation the three pSTAT3‐Y705‐negative cell lines concomitantly with pSTAT3‐S727 down‐regulation. This is strengthened by the persistence of Gö6976‐induced pSTAT3‐S727 down‐modulation in the 24 h following irradiation of Gö6976‐pretreated pSTAT3‐Y705‐negative cells. Radiosensitization was confirmed on SF767 and U118MG by clonogenic cell survival assay. Radiosensitizing effect of Gö6976 was irradiation dose‐dependent. Gö6976 was a potent radiosensitizer even at the lowest irradiation dose. More interestingly, Gö6976 failed to radiosensitize pSTAT3‐Y705‐positive cell lines. This observation suggests that radiosensitization was a result of pSTAT3‐S727 inhibition. Moreover, these results might be influenced by genetic background of GBM tumors. It has been shown that STAT3 may play in some tumors, a pro‐ or an anti‐oncogenic role, depending on the mutational status of PTEN 19. We showed here that STAT3 status in turn can influence tumor responsiveness to a chemical treatment (Gö6976) and to irradiation. STAT3 activation status is more likely independent on the mutational status of PTEN, TP53, p16, p14ARF or EGFR according to genetic data compiled from different laboratories 27, 57, 63. Moreover, we did not find any variation in PKCα, JAK2 and EGFR signaling, confirming that the down‐modulation of both pSTAT3‐S727 and cell radioresistance by Gö6976 may not be mediated through those pathways (Supporting Information Figure S1).
To confirm the relevance of targeting the STAT3 pathway in GBM, we assessed pSTAT3‐Y705 and pSTAT3‐S727 accumulation in GBM clinical specimens collected in our tumor bank. All specimens exhibited variable level of pSTAT3‐S727 in all neoplastic cells as well as in some stromal cells, while some samples were pSTAT3‐Y705 negative. Our description of pSTAT3‐S727 and pSTAT3‐Y705 in GBM is consistent with other previous reports 37. Some reported that stem‐like cells in tumors can differentiate into endothelial cells, thereby generating the tumor vasculature 46, 58. Here, we found that endothelial cells in tumor specimens displayed the similar staining in pSTAT3‐S727 and pSTAT3‐Y705 as GBM cells. However, we did not find any correlation between pSTAT3‐Y705 and pSTAT3‐S727. We did not also find any correlation linking neither pSTAT3‐S727 nor pSTAT3‐Y705 with patient overall survival as it has been recently reported 37, 38. The size of the cohort, the constitutive activation of S727 in all samples, the sensitivity of immunostaining and clinical data heterogeneity might explain such discrepancies. As far as pSTAT3‐S727 is constitutively accumulated in all GBM cell lines and clinical samples, it might drive oncogenesis 42 and furthermore support constant radioresistance. However, targeting pSTAT3‐Y705 in glioma also remains a pertinent strategy at least in part because of its critical role, in relation with IL‐6, in modulating tumor microenvironment 49, 60, 65.
In conclusion, our description of STAT3 pathway activation in vitro was consistent with our findings in clinical samples. We identified that pSTAT3‐S727 is involved in intrinsic radioresistance and unphosphorylated STAT3‐Y705 is a predicting marker of glioma response to Gö6976 as a radiosensitizer. Altogether, our results suggest that unphosphorylated STAT3‐Y705 might be also in vivo a predictive biomarker of radiosensitization by Gö6976 or its related compounds. They also strengthen the need of a specific inhibitor to neutralize pSTAT3‐S727 that could be a relevant target to overcome glioma resistance.
Conflict of Interest
None.
Supporting information
Figure S1. Effect of Gö6976 on PCKα, JAK2 and EGFR pathways in human glioblastoma cell lines. Cells were grown 72 h in indicated concentration of Gö6976 or DMSO (control) before harvest. Thirty micrograms of total proteins were loaded per lane and electrophoresed by SDS‐PAGE. Transfer membranes were immunoblotted with anti‐PKCα, anti‐pPKCα, anti‐JAK2, anti‐pJAK2, anti‐EGFR or anti‐pEGFR specific antibodies. To ensure equal protein loading, β‐actin was used as control.
Acknowledgments
This work was supported by the Ligue Nationale Contre le Cancer (Comité du Puy‐De‐Dôme), by the Institut National du Cancer and by the Region Auvergne. Z.G.O. was the recipient of a fellowship from the Ministère des Enseignements Secondaire et Supérieur, Burkina Faso. We thank the imaging facility Imagerie Confocale de Clermont‐Ferrand (ICCF) for provision of Leica SPE microscope. We are grateful to Dr Nicolas Foray for fruitful discussions, Angélique DeHaze, Annette Quinsat and Jean‐Paul Saru for technical assistance.
References
- 1. Abou‐Ghazal M, Yang DS, Qiao W, Reina‐Ortiz C, Wei J, Kong L‐Y et al (2008) The incidence, correlation with tumor‐infiltrating inflammation, and prognosis of phosphorylated STAT3 expression in human gliomas. Clin Cancer Res 14:8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amberger‐Murphy V (2009) Hypoxia helps glioma to fight therapy. Curr Cancer Drug Targets 9:381–390. [DOI] [PubMed] [Google Scholar]
- 3. Aziz MH, Hafeez BB, Sand JM, Pierce DB, Aziz SW, Dreckschmidt NE, Verma AK (2010) Protein kinase Cε mediates Stat3Ser727 phosphorylation, Stat3‐regulated gene expression and cell invasion in various human cancer cell lines via integration with MAPK cascade (RAF‐1, MEK1/2, and ERK1/2). Oncogene 29:3100–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barcellos‐Hoff MH, Newcomb EW, Zagzag D, Narayana A (2009) Therapeutic targets in malignant glioblastoma microenvironment. Semin Radiat Oncol 19:163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bowman T, Garcia R, Turkson J, Jove R (2000) STATs in oncogenesis. Oncogene 19:2474–2488. [DOI] [PubMed] [Google Scholar]
- 6. Brantley EC, Benveniste EN (2008) STAT‐3: a molecular hub for signaling pathways in gliomas. Mol Cancer Res 6:675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brantley EC, Nabors LB, Gillespie GY, Choi Y‐H, Palmer CA, Harrison K et al (2008) Loss of PIAS3 expression in glioblastoma multiforme tumors: implications for STAT‐3 activation and gene expression. Clin Cancer Res 14:4694–4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Braunstein J, Brutsaert S, Olson R, Schindler C (2003) STATs dimerize in the absence of phosphorylation. J Biol Chem 278:34133–34140. [DOI] [PubMed] [Google Scholar]
- 9. Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE Jr (1999) Stat3 as an oncogene. Cell 98:295–303. [DOI] [PubMed] [Google Scholar]
- 10. Buronfosse A, Thomas CP, Ginestet C, Doré JF (1994) Radiosensitivity in vitro of clonogenic and non‐clonogenic glioblastoma cells obtained from a human brain tumour. C R Acad Sci III 317:1031–1041. [PubMed] [Google Scholar]
- 11. Caldenhoven E, van Dijk TB, Solari R, Armstrong J, Raaijmakers JAM, Lammers J‐WJ et al (1996) STAT3β, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. J Biol Chem 271:13221–13227. [DOI] [PubMed] [Google Scholar]
- 12. Chakraborty A, White SM, Schaefer TS, Ball ED, Dyer KF, Tweardy DJ (1996) Granulocyte colony‐stimulating factor activation of Stat3 alpha and Stat3 beta in immature normal and leukemic human myeloid cells. Blood 88:2442–2449. [PubMed] [Google Scholar]
- 13. Chan JL, Lee SW, Fraass BA, Normolle DP, Greenberg HS, Junck LR et al (2002) Survival and failure patterns of high‐grade gliomas after three‐dimensional conformal radiotherapy. J Clin Oncol 20:1635–1642. [DOI] [PubMed] [Google Scholar]
- 14. Chandna S, Dwarakanath BS, Khaitan D, Mathew TL, Jain V (2002) Low‐dose radiation hypersensitivity in human tumor cell lines: effects of cell‐cell contact and nutritional deprivation. Radiat Res 157:516–525. [DOI] [PubMed] [Google Scholar]
- 15. Chautard E, Loubeau G, Tchirkov A, Chassagne J, Vermot‐Desroches C, Morel L, Verrelle P (2010) Akt signaling pathway: a target for radiosensitizing human malignant glioma. Neuro‐Oncol 12:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rasband WS, ImageJ US (1997–2014) National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/ [Google Scholar]
- 17. Courapied S, Sellier H, de Carne Trecesson S, Vigneron A, Bernard A‐C, Gamelin E et al (2010) The cdk5 kinase regulates the STAT3 transcription factor to prevent DNA damage upon topoisomerase I inhibition. J Biol Chem 285:26765–26778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Davies SP, Reddy H, Caivano M, Cohen P (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351:95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. de la Iglesia N, Konopka G, Lim KL, Nutt CL, Bromberg JF, Frank DA et al (2008) Deregulation of a STAT3‐IL8 signaling pathway promotes human glioblastoma cell proliferation and invasiveness. J Neurosci 28:5870–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Donson AM, Banerjee A, Gamboni‐Robertson F, Fleitz JM, Foreman NK (2000) Protein kinase C zeta isoform is critical for proliferation in human glioblastoma cell lines. J Neurooncol 47:109–115. [DOI] [PubMed] [Google Scholar]
- 21. Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N et al (2010) NOTCH pathway blockade depletes CD133‐positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 28:5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Feng Z, Xu S, Liu M, Zeng Y‐X, Kang T (2010) Chk1 inhibitor Gö6976 enhances the sensitivity of nasopharyngeal carcinoma cells to radiotherapy and chemotherapy in vitro and in vivo . Cancer Lett 297:190–197. [DOI] [PubMed] [Google Scholar]
- 23. Grandage VL, Everington T, Linch DC, Khwaja A (2006) Gö6976 is a potent inhibitor of the JAK 2 and FLT3 tyrosine kinases with significant activity in primary acute myeloid leukaemia cells. Br J Haematol 135:303–316. [DOI] [PubMed] [Google Scholar]
- 24. Halliday J, Helmy K, Pattwell SS, Pitter KL, LaPlant Q, Ozawa T, Holland EC (2014) In vivo radiation response of proneural glioma characterized by protective p53 transcriptional program and proneural‐mesenchymal shift. PNAS 111:5248–5253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hazan‐Halevy I, Harris D, Liu Z, Liu J, Li P, Chen X et al (2010) STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood 115:2852–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Heinrich PC, Behrmann I, Muller‐Newen G, Schaper F, Graeve L (1998) Interleukin‐6‐type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J 334:297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ishii N, Maier D, Merlo A, Tada M, Sawamura Y, Diserens A‐C, Van Meir EG (1999) Frequent co‐alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol 9:469–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Isomoto H, Kobayashi S, Werneburg NW, Bronk SF, Guicciardi ME, Frank DA, Gores GJ (2005) Interleukin 6 upregulates myeloid cell leukemia‐1 expression through a STAT3 pathway in cholangiocarcinoma cells. Hepatology 42:1329–1338. [DOI] [PubMed] [Google Scholar]
- 29. Iwamaru A, Szymanski S, Iwado E, Aoki H, Yokoyama T, Fokt I et al (2007) A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo . Oncogene 26:2435–2444. [DOI] [PubMed] [Google Scholar]
- 30. Kesanakurti D, Chetty C, Maddirela DR, Gujrati M, Rao JS (2013) Essential role of cooperative NF‐κB and Stat3 recruitment to ICAM‐1 intronic consensus elements in the regulation of radiation‐induced invasion and migration in glioma. Oncogene 32:5144–5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kohn EA, Yoo CJ, Eastman A (2003) The protein kinase C inhibitor Gö6976 is a potent inhibitor of DNA damage‐induced S and G2 cell cycle checkpoints. Cancer Res 63:31–35. [PubMed] [Google Scholar]
- 32. Konnikova L, Kotecki M, Kruger MM, Cochran BH (2003) Knockdown of STAT3 expression by RNAi induces apoptosis in astrocytoma cells. BMC Cancer 3:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Koo MY, Park J, Lim JM, Joo SY, Shin S‐P, Shim HB et al (2014) Selective inhibition of the function of tyrosine‐phosphorylated STAT3 with a phosphorylation site‐specific intrabody. Proc Natl Acad Sci U S A 111:6269–6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kretzschmar AK, Dinger MC, Henze C, Brocke‐Heidrich K, Horn F (2004) Analysis of Stat3 (signal transducer and activator of transcription 3) dimerization by fluorescence resonance energy transfer in living cells. Biochem J 377:289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kumar A, Commane M, Flickinger TW, Horvath CM, Stark GR (1997) Defective TNF‐α‐induced apoptosis in STAT1‐null cells due to low constitutive levels of caspases. Science 278:1630–1632. [DOI] [PubMed] [Google Scholar]
- 36. Lee SW, Fraass BA, Marsh LH, Herbort K, Gebarski SS, Martel MK et al (1999) Patterns of failure following high‐dose 3‐D conformal radiotherapy for high‐grade astrocytomas: a quantitative dosimetric study. Int J Radiat Oncol Biol Phys 43:79–88. [DOI] [PubMed] [Google Scholar]
- 37. Lin G‐S, Chen Y‐P, Lin Z‐X, Wang X‐F, Zheng Z‐Q, Chen L (2014) STAT3 serine 727 phosphorylation influences clinical outcome in glioblastoma. Int J Clin Exp Pathol 7:3141–3149. [PMC free article] [PubMed] [Google Scholar]
- 38. Lin G‐S, Yang L‐J, Wang X‐F, Chen Y‐P, Tang W‐L, Chen L, Lin Z‐X (2014) STAT3 Tyr705 phosphorylation affects clinical outcome in patients with newly diagnosed supratentorial glioblastoma. Med Oncol 31:1–7. [DOI] [PubMed] [Google Scholar]
- 39. Lin L, Hutzen B, Li PK, Ball S, Zuo M, DeAngelis S et al (2010) A novel small molecule, LLL12, inhibits STAT3 phosphorylation and activities and exhibits potent growth‐suppressive activity in human cancer cells. Neoplasia 12:39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Loeffler S, Fayard B, Weis J, Weissenberger J (2005) Interleukin‐6 induces transcriptional activation of vascular endothelial growth factor (VEGF) in astrocytes in vivo and regulates VEGF promoter activity in glioblastoma cells via direct interaction between STAT3 and Sp1. Int J Cancer 115:202–213. [DOI] [PubMed] [Google Scholar]
- 41. Miklossy G, Hilliard TS, Turkson J (2013) Therapeutic modulators of STAT signalling for human diseases. Nat Rev Drug Discov 12:611–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Miyakoshi M, Yamamoto M, Tanaka H, Ogawa K (2014) Serine 727 phosphorylation of STAT3: an early change in mouse hepatocarcinogenesis induced by neonatal treatment with diethylnitrosamine. Mol Carcinog 53:67–76. [DOI] [PubMed] [Google Scholar]
- 43. O'Shea JJ, Kanno Y, Chen X, Levy DE (2005) Stat acetylation—a key facet of cytokine signaling? Science 307:217–218. [DOI] [PubMed] [Google Scholar]
- 44. Qin HR, Kim H‐J, Kim J‐Y, Hurt EM, Klarmann GJ, Kawasaki BT et al (2008) Activation of Stat3 through a phosphomimetic serine 727 promotes prostate tumorigenesis independent of tyrosine705 phosphorylation. Cancer Res 68:7736–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ray S, Boldogh I, Brasier AR (2005) STAT3 NH2‐terminal acetylation is activated by the hepatic acute‐phase response and required for IL‐6 induction of angiotensinogen. Gastroenterology 129:1616–1632. [DOI] [PubMed] [Google Scholar]
- 46. Ricci‐Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T et al (2010) Tumour vascularization via endothelial differentiation of glioblastoma stem‐like cells. Nature 468:824–828. [DOI] [PubMed] [Google Scholar]
- 47. Sasse J, Hemmann U, Schwartz C, Schniertshauer U, Heesel B, Landgraf C et al (1997) Mutational analysis of acute‐phase response factor/Stat3 activation and dimerization. Mol Cell Biol 17:4677–4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schaefer TS, Sanders LK, Nathans D (1995) Cooperative transcriptional activity of Jun and Stat3 beta, a short form of Stat3. Proc Natl Acad Sci U S A 92:9097–9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. See AP, Han JE, Phallen J, Binder Z, Gallia G, Pan F et al (2012) The role of STAT3 activation in modulating the immune microenvironment of GBM. J Neurooncol 110:359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shen J, Li R, Li G (2009) Inhibitory effects of decoy‐ODN targeting activated STAT3 on human glioma growth in vivo . In Vivo 23:237–243. [PubMed] [Google Scholar]
- 51. Sherry MM, Reeves A, Wu JK, Cochran BH (2009) STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 27:2383–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Song Y, Qian L, Song S, Chen L, Zhang Y, Yuan G et al (2008) Fra‐1 and Stat3 synergistically regulate activation of human MMP‐9 gene. Mol Immunol 45:137–143. [DOI] [PubMed] [Google Scholar]
- 53. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996. [DOI] [PubMed] [Google Scholar]
- 54. Szeląg M, Czerwoniec A, Wesoly J, Bluyssen HAR (2014) Comparative screening and validation as a novel tool to identify STAT‐specific inhibitors. Eur J Pharmacol 740:417–420. [DOI] [PubMed] [Google Scholar]
- 55. Taube E, Jokinen E, Koivunen P, Koivunen JP (2012) A novel treatment strategy for EGFR mutant NSCLC with T790M‐mediated acquired resistance. Int J Cancer 131:970–979. [DOI] [PubMed] [Google Scholar]
- 56. Villalva C, Martin‐Lannerée S, Cortes U, Dkhissi F, Wager M, Le Corf A et al (2011) STAT3 is essential for the maintenance of neurosphere‐initiating tumor cells in patients with glioblastomas: a potential for targeted therapy? Int J Cancer 128:826–838. [DOI] [PubMed] [Google Scholar]
- 57. Wang C‐C, Liao Y‐P, Mischel PS, Iwamoto KS, Cacalano NA, McBride WH (2006) HDJ‐2 as a target for radiosensitization of glioblastoma multiforme cells by the farnesyltransferase inhibitor R115777 and the role of the p53/p21 pathway. Cancer Res 66:6756–6762. [DOI] [PubMed] [Google Scholar]
- 58. Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A et al (2010) Glioblastoma stem‐like cells give rise to tumour endothelium. Nature 468:829–833. [DOI] [PubMed] [Google Scholar]
- 59. Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S et al (2004) Regulation of the innate and adaptive immune responses by Stat‐3 signaling in tumor cells. Nat Med 10:48–54. [DOI] [PubMed] [Google Scholar]
- 60. Wei J, Barr J, Kong L‐Y, Wang Y, Wu A, Sharma AK et al (2010) Glioblastoma cancer‐initiating cells inhibit T cell proliferation and effector responses by the STAT3 pathway. Mol Cancer Ther 9:67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yang J, Chatterjee‐Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, Stark GR (2005) Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res 65:939–947. [PubMed] [Google Scholar]
- 62. Yang J, Huang J, Dasgupta M, Sears N, Miyagi M, Wang B et al (2010) Reversible methylation of promoter‐bound STAT3 by histone‐modifying enzymes. Proc Natl Acad Sci U S A 107:21499–21504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang L, Clarke MJ, Carlson BL, Mladek AC, Schroeder MA, Decker P et al (2008) PTEN loss does not predict for response to RAD001 (Everolimus) in a glioblastoma orthotopic xenograft test panel. Clin Cancer Res 14:3993–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yuan Z, Guan Y, Chatterjee D, Chin YE (2005) Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 307:269–273. [DOI] [PubMed] [Google Scholar]
- 65. Zhang J, Sarkar S, Cua R, Zhou Y, Hader W, Yong VW (2012) A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin‐6 axis. Carcinogenesis 33:312–319. [DOI] [PubMed] [Google Scholar]
- 66. Zhong Z, Wen Z, Darnell JE (1994) Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin‐6. Science (New York, NY) 264:95–98. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effect of Gö6976 on PCKα, JAK2 and EGFR pathways in human glioblastoma cell lines. Cells were grown 72 h in indicated concentration of Gö6976 or DMSO (control) before harvest. Thirty micrograms of total proteins were loaded per lane and electrophoresed by SDS‐PAGE. Transfer membranes were immunoblotted with anti‐PKCα, anti‐pPKCα, anti‐JAK2, anti‐pJAK2, anti‐EGFR or anti‐pEGFR specific antibodies. To ensure equal protein loading, β‐actin was used as control.