

Abstract
Intracerebral‐ventricular (ICV) injection of streptozotocin (STZ) induces an insulin‐resistant brain state that may underlie the neural pathogenesis of sporadic Alzheimer disease (AD). Our previous work showed that prior ICV treatment of glucagon‐like peptide‐1 (GLP‐1) could prevent STZ‐induced learning memory impairment and tau hyperphosphorylation in the rat brain. The Chinese herbal medicine geniposide is known to relieve symptoms of type 2 diabetes. Because geniposide is thought to act as a GLP‐1 receptor agonist, we investigated the potential therapeutic effect of geniposide on STZ‐induced AD model in rats. Our result showed that a single injection of geniposide (50 μM, 10 μL) to the lateral ventricle prevented STZ‐induced spatial learning deficit by about 40% and reduced tau phosphorylation by about 30% with Morris water maze test and quantitative immunohistochemical analysis, respectively. It has been known that tau protein can be phosphorylated by glycogen synthase kinase‐3 (GSK3) and STZ can increase the activity of GSK3β. Our result with Western blot analysis showed that central administration of geniposide resulted in an elevated expression of GSK3β(pS‐9) but suppressed GSK3β(pY‐216) indicating that geniposide reduced STZ‐induced GSK3β hyperactivity. In addition, ultrastructure analysis showed that geniposide averted STZ‐induced neural pathology, including paired helical filament (PHF)‐like structures, accumulation of vesicles in synaptic terminal, abnormalities of endoplasmic reticulum (ER) and early stage of apoptosis. In summary, our study suggests that the water soluble and orally active monomer of Chinese herbal medicine geniposide may serve as a novel therapeutic agent for the treatment of sporadic AD.
Keywords: Alzheimer's disease, geniposide, glucagon‐like peptide‐1 receptor, glycogen synthase kinase‐3β, tau hyperphosphorylation, type 2 diabetes
Introduction
Alzheimer's disease (AD) is a debilitating neurodegenerative illness without a cure. There are five medications currently approved by the Food and Drug Administration (FDA) for the treatment of AD: tacrine, rivastigmine, galantamine, donepezil, and memantine, but none provided any indication for halting or delaying the progression of the disease 3. Thus, it is of great importance to seek novel therapy.
Three mutated genes including app (amyloid precursor protein), presenilin (PS)‐1 and PS‐2 are tightly associated with early onset (<60 years) or familial AD which accounts for less than 1% of AD cases 4. However, there are many other factors associated with the rest of 99% late onset or sporadic AD cases. Recent studies suggest that AD and type 2 diabetes may share some common molecular mechanisms 2, 14. The presence of type 2 diabetes mellitus (T2DM) is associated with an approximately twofold increased risk of AD (risk ratios vary between 1.5 and 4.0) 10, 29, 30. Although the exact mechanism underlying the neuropathogenesis of sporadic AD remains unknown, the insulin‐resistant brain state hypothesis has received strong experimental support since it was proposed in the late 1990s 11. Cumulative evidence suggests that central administration of low doses of streptozotocin (STZ), can induce an insulin‐resistant‐like brain state and is regarded as a credible method to trigger sporadic AD, especially in the early stages of the disease process 36, 37, 43. Central application of STZ produced both behavioral, neurochemical and histological features that resembled those found in human AD 7, 38.
Our previous work demonstrated that medications that are used for the treatment of diabetes could benefit in AD animal models such as glucagon‐like peptide‐1 (GLP‐1) and that resensitizes insulin signaling could ameliorate AD symptoms in animal models 14, 22. Geniposide is an iridoid glycoside present in the fruit of Gardenia jasmindides Ellis 15. Recent studies show that geniposide may act as a GLP‐1 receptor agonist 24 that can be used in diabetes therapy 26. Thus, we investigated whether geniposide can serve as a therapeutic agent for the treatment of AD in animal models.
The precise mechanism that underlies the therapeutic effect of geniposide is unknown. However, in vitro studies suggest that the antioxidative effect of geniposide may involve multiple signal transduction pathways through a cascade of interactions among phosphoinositol‐3 kinase (PI3K), protein kinase‐B (also known as Akt), Akt308, Akt473, Glycogen synthase kinase‐3β (GSK‐3β) and pyruvate dehydrogenase kinase (PDK1) under conditions of oxidative stress 25.
GSK3 may be involved in the pathogenesis of both T2DM and AD 21. GSK3 was originally discovered in 1984 as a key regulator in glycogen synthesis 46. The enzymatic activity of GSK3 is tightly regulated; phosphorylation at tyrosine‐216 in GSK3β or tyrosine‐279 in GSK3α enhances the enzymatic activity, while phosphorlyation of serine‐9 in GSK3β or serine‐21 in GSK3α decreases the activity 20, 45. It has been suggested that GSK3 may play a role in both Aβ accumulation and tau phosphorylation, which are the main pathological hallmarks of AD 13. As previous studies indicate that GSK3 may play an important role in AD pathogenesis and geniposide may interact with GSK3 pathway, we hypothesize that geniposide may serve as a GSK3 inhibitor to exert its neuroprotective effect in the AD animal model.
Materials and Methods
Animals and drug treatment
Adult male Sprague Dawley (SD) rats (220–250 g) were obtained from the animal center of Shanxi Medical University. All animal procedures were performed in accordance to National Institutes of Health (NIH) guideline (National Institutes of Health Publications, No. 80–23, revised 1978). Rats were housed in a quiet and humidity‐controlled room (22 ± 3°C and 62 ± 7% relative humidity) with 12:12 light dark cycle, and were fed ad libitum with standard rodent diet and water 18. Rats were randomly divided into five groups, (i) control group: artificial cerebral spinal fluid (aCSF) intracerebral‐ventricular (ICV) injection; (ii) geniposide group: single geniposide ICV injection; (iii) STZ group: STZ ICV injection; (iv) geniposide + STZ group: STZ + geniposide ICV injected; (v) Wortmannin (WT) group: WT + Geniposide + STZ ICV injection (n = 9–11 each group). After behavioral test, rats were sacrificed with overdose anesthesia [10% Chloral hydrate intraperitoneal (IP), 0.4 mL/100 g] and followed with intracardiac perfusion with phosphate buffered saline (PBS). One hemisphere of the cortex was quickly dissected and stored in −80°C for Western blot assay (n = 5–6 each group). The other hemisphere was fixed with 4% paraformaldehyde for immunohistochemistry (n = 5–6 each group). For transmission electron microscopy (TEM), the hippocampus was fixed with 2.5% gluteraldehyde (see details in the TEM section).
For ICV injection, the rats were anesthetized with IP injection of 10% Chloral hydrate (0.3 mL/100 g), and placed on the stereotaxic frame with the following coordinates (0.8 mm posterior to bregma; 1.5 mm lateral to the sagittal suture; 3.6 mm ventral, unilateral injection) for the left side of lateral ventricle injection 1. All drug injection to the lateral ventricle was carried out with a Hamilton syringe in 10 μL volume with duration of 15 minutes. Both STZ and WT were purchased from Sigma (St. Louis, MO, USA) and geniposide was from the Marker Inc (Tianjin, China). All the drugs were dissolved in 10 μL aCSF (NaCl 140 mM; KCl 3.0 mM; CaCl2 2.5 mM; MgCl 1.2 mM; NaH2PO4 1.2 mM, pH 7.4). STZ (3 mg/kg), geniposide (50 μM), WT (100 μM) were prepared right before use.
Morris water maze (MWM) test
MWM task 32 was performed 14 days after the ICV injection. The apparatus (Zhenghua Bio Instrument Ltd, Shanghai, China) is a circular pool with a diameter of 150 cm and a wall height of 60 cm, which was made of stainless steel and painted black on the inner surface. The pool surrounded by a white curtain around was filled to a depth of 30 cm with warm water at a temperature of 25 ± 2°C to avoid hypothermia. A black platform (diameter, 14 cm) was positioned at the midpoint of the target quadrant and submerged approximately 1.0 cm below the surface of the water. A video camera connected to the computer was located above the pool to record the movement and the trail of the rats. The navigation task was performed with four trials per day for 5 consecutive days (detailed procedure was published previously) 22. Briefly, the rat was placed in the water facing the wall in one of the four quadrants and was allowed to swim freely to find the hidden platform. If the rat failed to find the platform within 120 s, it would be piloted onto the platform (only on the first test day). A 10 s session was recorded while the rats were on the platform before placing them in the holding cage for 20 s until the next trial. The starting quadrant was randomly chosen each day. The navigation latency (ie, the length of time to reach the platform, in seconds) was recorded and analyzed by a behavior software system (Ethovision 3.0, Noldus Information Technology, Wageningen, Netherlands). The probe trial was performed on the day after the navigation task (day 6). In this phase, hidden platform was removed and rats were given 120 s to swim in the pool for the spatial bias measurement. The percentage of swimming time spent in the target quadrant was recorded. To control the bias because of different animal's individual athletic ability, such as swimming speed and visual acuity, we recorded the swimming speed and conducted the visible platform test for each rat following the probe trials, which consisted of two sessions of each rat seeking the platform clearly elevated above the water surface (approximately 2 cm) (n = 9–11 each group).
Immunohistochemistry
The brains were removed and fixed in 4% paraformaldehyde for 2 h. The brains were then embedded with paraffin and 2 μm coronal sections were cut with the microtome. Then the sections were steamed in 0.001 mM ethylenediaminetetraacetic acid (EDTA) (pH = 9.0) for 2 minutes 16. Sections were incubated with rabbit monoclonal antibody against phosphorylated tau protein at Ser 396 (a common phosphorylation site in AD brain, 1:500, Abcam, Cambridge, MA, USA) overnight at 4°C and then incubated with goat‐anti‐rabbit IgG‐H&L (1:5000, Abcam) at 37°C for 40 minutes. The peroxidase was visualized with 3,3′,4,4′‐biphenyltetramine tetrahydrochloride (DAB) color reaction and counterstained with hematoxylin 6. The photomicrograph was captured with Axio Scope1 microscope (Carl Zeiss, Jena, Germany) and quantitatively analyzed with optical density.
Western blot assay
Fifty milligrams of tissue mainly from the temporal lobe per animal was homogenized (20 s homogenization and 10 s pause × three times) in cold lysis buffer [produced by Beyotime Inc, Shanghai, China, containing 1% Triton X‐100, 1% deoxycholate, 0.1% sodium dodecyl sulfate (SDS)] and phenylmethanesulfonyl fluoride (PMSF), and then centrifuged (12 000 × g for 5 minutes at 4°C), before taking the supernatant. Protein concentration was measured using the Bradford method 28. Samples were then added with loading buffer to the same concentration. Samples containing 6–10 μg were run on a 10% Tris‐Tricine gel and electrophoretically transferred to a polyvinylidene fluoride (PVDF) membrane. After blocking with 0.3% gelatin in TBST (TBS contains 0.05% Tween‐20) for 1 h. Membranes were probed overnight with primary antibodies [rabbit polyclonal GSK3‐β(pS‐9), 1:1000, Cell Signaling Technology Inc, Danvers, MA, USA; GSK3‐β(pY‐216, Abcam, 1:1000; GSK3‐β(total), Abcam, 1:1000; mouse monoclonal β‐Actin, Abcam, 0.5 μg/mL] under 4°C overnight, then labeled with secondary antibodies [goat‐anti‐rabbit IgG‐H&L (horseradish peroxidase, HRP), Abcam, 1:20 000; and goat‐anti‐mouse IgG‐H&L (HRP), Abcam, 1:20 000, respectively]. Immunoreactive bands were visualized with enhanced chemiluminescent (ECL, Pierce, Rockford, IL, USA) and analyzed with the image system of Quantity One 4.31 (Bio‐Rad, Hercules, CA, USA). Quantification of protein was performed by calculating the density of each individual band. Band densities were normalized for protein loading, using β‐Actin as loading control.
Preparation of TEM specimen
Specimens of hippocampus were prepared following our previous protocol 22. The brain specimens were fixed in 2.5% gluteraldehyde for 2 h at 4°C and were then washed twice in sodium cocodylate buffer (0.1 M; pH 7.4) for 10 minutes each. Samples were then post‐fixed in 1% osmium tetroxide for 2 h and then dehydrated in a series of graded acetone. The specimens were embedded in Epon 618 (ChemIndustry, Monrovia, CA, USA) and cut with LKB ultramicrotome set (Leica, Solms, Germany) at 500 Å ultrathin sections. The sections were stained with uranyl acetate‐lead citrate. Samples were observed under JEOL 1101 transmission electron microscope (Olympus, Tokyo, Japan).
Statistics analysis
All values were presented as mean ± standard error of the mean and displayed with the graphic software Prism 5 (GraphPad, GraphPad Software Inc, La Jolla, CA, USA). The statistical software SPSS 16.0 (Graphpad) was used for statistical analysis. Two‐way analysis of variance (ANOVA) was used to analyze the result of acquisition task in MWM, one‐way ANOVA was used for the rest of the analyses, with statistical significance level set at P < 0.05.
Results
Geniposide partially prevented STZ‐induced learning and memory impairment through modulation of PI3K/GSK3 signaling pathway
We used MWM test to investigate the effect of geniposide to STZ‐induced learning and memory deficit in rats. Ten rats were treated with vehicle alone as controls with 10 μL of aCSF slowly injected to one side of the lateral ventricle over 15 minutes; 11 rats received geniposide injection alone (50 μM, 10 μL); nine rats received STZ alone (3 mg/kg, 10 μL); 10 rats received STZ (3 mg/kg, 5 μL) and then followed with a second injection 5 minutes later with geniposide (50 μM, 5 μL) on the left side of the lateral ventricle (the syringe was switched between drugs without removing the needle inside the brain); and the final group of 10 rats were treated with STZ (3 mg/kg, 5 μL), geniposide (50 μM, 3 μL) and wortmannin (WT, 100 μM, 2 μL) on the same side of the lateral ventricle with 5 minutes interval between different drugs. For all the ICV treatment groups, the syringe needle remained inside the ventricle for additional 10 minutes after the injection was completed to ensure effective drug delivery to the brain. After the ICV procedure, rats were put in a clean and warm (40°C) cage with cotton on the bottom to recover from the surgery.
The MWM test was performed 14 days after the ICV injection, starting off with the navigation task to study spatial learning of the rats. The pool was filled with warm water with markings on the inside wall. On the computer screen, the maze was divided into four quadrants (hidden platform was located in the center of a certain quadrant under the water). Rats were gently placed into the water facing the opposite side of the pool and were given a maximum time of 120 s to find the hidden platform. Four trials were conducted for each rat, starting from a different quadrant each time, and the rats were allowed to rest for 30 s between trials. The task continued for 5 consecutive days. The navigation latency on each day was recorded to calculate a mean latency for each quadrant. Consistent with previous findings, the navigation latency decreases with training days until there is virtually no difference on the fourth and fifth day (Figure 1A). Based the two‐way ANOVA analysis, we found that there was a significant difference between groups (F = 378, P < 0.001), and over time (F = 8.56, P < 0.001). There was no interaction between the two variables, so the drug effect was not changing over time (F = 1.69, P = 0.506).
Figure 1.
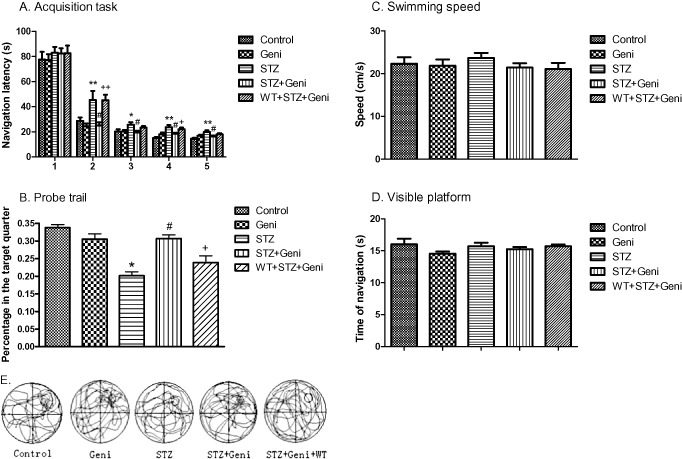
Geniposide (Geni) prevented STZ (Streptozotocin)‐induced spatial learning and memory impairment. Navigation latency was significantly increased in the STZ‐treated group compared with controls on the second, third, fourth and fifth day (*, compared with control, P < 0.05; **, P < 0.01), but geniposide treatment prevented STZ‐induced learning deficit (#, compared with STZ group, P < 0.01). However, PI3K inhibitor wortmannin (WT) blocked the protective effect of geniposide (+, compared with STZ + Geniposide group, P < 0.05; ++, P < 0.01). A. STZ treatment significantly decreased the percent of time each rat spent in the target quadrant (P < 0.05), and geniposide treatment prevented STZ‐induced memory deficit. B. The probe trial did not affected by the variation of swim speed or visual acuity, respectively, of each individual rat (C,D). Video recording of navigation pattern of the five different drug treatment groups (E).
Consistent with previous findings, on the first day of the navigation task, we did not find any significant difference in navigation latency among any of the five groups. However, major differences in navigation latency between different treatment groups started to emerge on the second day. Rats injected with geniposide alone (77.217 ± 4.569 s, 24.475 ± 2.188 s, 20.242 ± 1.204 s, 18.142 ± 1.293 s, 16.983 ± 0.758 s, for each of the 5 days respectively) did not differ from vehicle controls (77.475 ± 4.8 s, 28.500 ± 2.8 s, 19.858 ± 1.6 s, 15.550 ± 0.7 s, 14.642 ± 0.6 s) (Figure 1A). Consistent with previous findings, rats that received STZ alone (89.926 ± 4.675 s, 45.380 ± 7.256 s, 25.583 ± 2.153 s, 23.713 ± 1.644 s, 19.889 ± 1.294 s) exhibited major impairment in spatial learning as the navigation latency was much longer than the control rats (P < 0.01 on the second day and P < 0.05 on all the other days) (Figure 1A). Much to our delight, rats that received both geniposide and STZ (82.417 ± 4.259 s, 25.037 ± 2.483 s, 19.463 ± 1.350 s, 18.643 ± 0.712 s, 16.389 ± 0.617 s) exhibited robust improvement in navigation latency compared with rats that received STZ alone (P < 0.01 on the second day and P < 0.05 on all the other days). To better understand the potential mechanism underlying the neural protective effect of geniposide, we examined the role of PI3K, using a potent and selective PI3K inhibitor WT 17. Rats treated with WT + STZ + geniposide showed longer navigation latency (82.574 ± 6.151 s, 45.111 ± 4.467 s, 23.398 ± 1.207 s, 22.028 ± 1.324 s, 18.037 ± 0.941 s respectively) compared with the STZ + geniposide rats (P < 0.01 on the second day and P < 0.05 on the fourth day, although no significant difference was found on the third and fifth day) (Figure 1A).
After the navigation task, the rats were followed with the probe trial to test their spatial memory. The platform was removed from the pool, and we monitored the time percentage of rats swim in the original platform zone. The rats were allowed to swim spontaneously for 120 s, and we recorded the percentage of time they spent in the target quadrant. The results show that geniposide injection alone (30.6% ± 1.5%) did not differ from vehicle controls (33.8% ± 0.9%) (Figure 1B), but rats treated with STZ alone resulted in a 25% decrease in spatial memory (19% ± 0.3%) compare with controls (P < 0.01), which was consistent with our previous findings. Interestingly, geniposide + STZ prevented STZ‐induced memory deficit (30.7% ± 1.04%) (P < 0.01). The probe trial of the WT + STZ + geniposide group (23.9% ± 1.9%) performed better than the STZ‐alone group (P < 0.05) but not as good as the STZ + geniposide group (P < 0.01) (Figure 1B).
It is important to note that the probe trial was not affected by the variation of swimming speed or visual acuity of each individual rat (Figure 1C,D).
Geniposide attenuated STZ‐induced hyperphosphorylation of tau protein
Because tau protein hyperphosphorylation is an important feature of AD pathology as well as other neurodegenerative diseases associated with cognitive deficits 33, we sought to investigate the effect of geniposide on tau protein phosphorylation.
Rats were sacrificed with overdose anesthesia and then received intracardiac perfusion with PBS after the completion of the MWM test (21 days after surgery). Following perfusion, one hemisphere of the rat brain, chosen by random order, was fixed with 4% paraformaldehyde for 2 h and then embedded with low melting point paraffin (the other hemisphere was store in −80°C for Westernblot assay). Coronal sections (2 μm thick) of the rat brain were cut with a sliding microtome for immunohistochemical study. Briefly, the tissue sections were incubated with primary antibody against phosphorylated tau protein (Abcam, anti‐tau‐Sp396, 1:500) overnight at 4°C and then incubated with goat‐anti‐rabbit IgG‐H&L secondary antibody (1:5000, Abcam) at 37°C for 40 minutes. The peroxidase was visualized with the DAB color reaction and counterstained with hematoxylin.
The immunoreactivity of phosphorylated tau protein in the brain was presented as a prominent brown staining with DAB around the blue stained nucleus with hematoxylin. The phospho‐tau immunoreactivity was quantitatively analyzed with average optical density (Figure 2).
Figure 2.
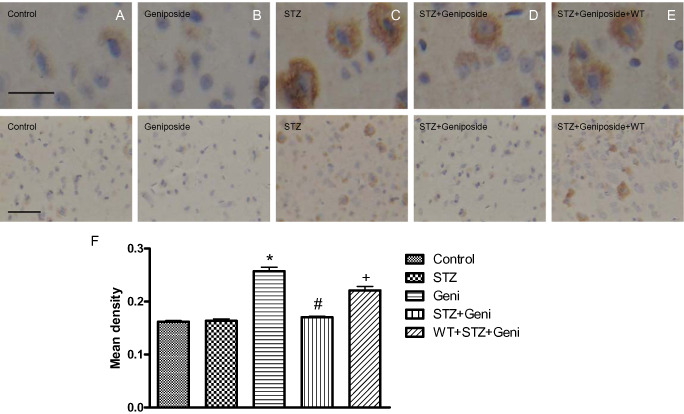
Photomicrographs and quantitative analysis of immunohistochemistry of phospho‐tau in the rat cerebral cortex. Immunoreactivity of phospho‐tau was presented as brown staining detected under higher and lower magnification, respectively (A–E, above bar = 50 μm; below bar = 150 μm). STZ treatment significantly increased phospho‐tau compared with controls (A,C,F; *, compared with control, P < 0.01), but geniposide (Geni) treatment prevented STZ‐induced increase in phospho‐tau (C,D,F; #, compared with STZ group, P < 0.01); even injection of geniposide alone did not induced the remarkable change of phosphor‐tau (A,B). PI3K inhibitor wortmannin (WT) partially prevented the protective effect of geniposide (E,F; +, compared with STZ + Geniposide group, P < 0.01).
Interestingly, the group average of phospho‐tau immunoreactivity largely correlated with the severity of cognitive impairment. More phospho‐tau was found in the STZ‐treated group (0.257 ± 0.008) than the vehicle control group (0.162 ± 0.002) (P < 0.01). (Figure 2A,B). Injection with geniposide alone (0.163 ± 0.004) did not change the level of phospho‐tau in the brain (Figure 2B,F). However, the geniposide + STZ group showed significant attenuation of phospho‐tau (0.170 ± 0.002) compared with the STZ‐alone group (P < 0.01) (Figure 2C,F). In contrast, PI3K inhibitor WT averted the protective effect of geniposide, the phospho‐tau was higher in the WT + STZ + geniposide group (0.221 ± 0.007) than the STZ + geniposide group (P < 0.01) (Figure 2E,F).
Geniposide may exert neural protective effect through inhibition of GSK3 activity
GSK3 is thought to play an important role in tau protein phosphorylation and dysregulation of GSK3 is associated with multiple pathological changes in AD and diabetes 12. We used Western blot assay to investigate the effect of geniposide to GSK3β regulation, including the total protein expression level and its functional modification. Protein samples were prepared from the temporal lobe of one hemisphere of the rat brain (the other hemisphere was used for histology).
To investigate the effect of geniposide on total GSK3 expression, we used primary antibody against total GSK3β (Abcam, 1:1000). Given the activity of GSK3β is regulated by phosphorylation of the inhibitory site of Ser9 and the active site of Tyr216, we used specific antibodies against GSK3β (pY216) (Abcam, 1:1000) as well as antibodies against GSK3β (pS9) (Abcam, 1:1000) to study the dynamic changes of these two phosphorylation sites in response to geniposide. β‐Actin (Abcam, 0.5 μg/mL) was used as loading controls (Figure 3A).
Figure 3.
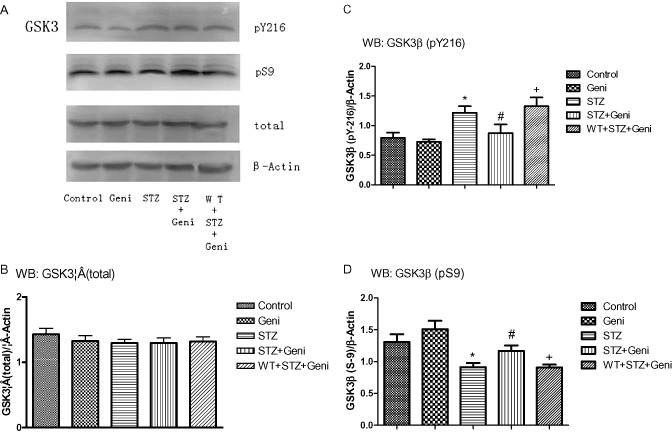
Western blot assay of GSK3β at different phosphorylation sites in response to STZ, geniposide and WT treatment was shown in A. Total GSK3β protein was shown in the histograms of brain cortex (B). Quantitative analysis shows that no significant difference on the total expression of GSK3β among the five treatment groups (B). Phospho‐GSK3β (inactive) and (active) was presented as the ratio over loading control (β‐Actin). GSK3β activity did not differ between rats treated with aCSF and Geniposide alone (C,D). STZ alone significantly elevated GSK3β activity compared with controls as reflected with increased pGSK3βY216 (C, *, compared with control, P < 0.01) and decreased pGSK3βS9 (D, *, compared with control, P < 0.01). STZ + Geniposide prevented STZ‐induced increase in GSK3β activity, reflected as lower ratio of pGSK3βY216 (C, #, compared to STZ‐alone group, P < 0.01) and higher ratio of pGSK3βS9 (D, # compared to STZ‐alone group, P < 0.01). PI3K inhibitor WT blocked the protective effect of geniposide, reflected as higher ratio of pGSK3βY216 (C, + compared to STZ + Geni, P < 0.01) and lower ratio of pGSK3βS9 (D, + compared to STZ + Geni, P < 0.01).
Total protein of GSK3β did not differ significantly among the five different treatment groups. The data were presented as the ratio of total GSK3β/β‐Actin, 1.429 ± 0.092 for the vehicle control group, 1.325 ± 0.082 for the geniposide alone group, 1.294 ± 0.059 for the STZ alone group, 1.294 ± 0.080 for the STZ + geniposide group and 1.318 ± 0.073 for the WT + STZ + geniposide group (Figure 3A,B).
To assess the functional state of GSK3β, we probed phospho‐GSK3β at different phosphorylation sites, specifically S9 and Y216, with specific antibodies. Our results show that geniposide injection alone did not alter the phosphorylation on either sites (0.685 ± 0.049 for pY‐216/β‐Actin, and 1.463 ± 0.081for pS‐9/β‐Actin) compared with vehicle controls (0.791 ± 0.044 for pY‐216/β‐Actin, and 1.324 ± 0.073 for pS‐9/β‐Actin).
As expected, STZ treatment elevated the active form GSK3β (pY216) (pY‐216/β‐Actin: 1.293 ± 0.091) but suppressed the inactive form GSK3β (pS9) (pS‐9/β‐Actin: 0.838 ± 0.019) (P < 0.01) (Figure 3C,D). However, geniposdie attenuated the STZ‐induced activation of GSK3β. Rats treated with STZ + geniposide decreased the level of GSK3β (pY216) by about 25% (pY‐216/β‐Actin: 0.871 ± 0.087), but increased the level of GSK3β (pS9) by about 20% (pS‐9/β‐Actin: 1.179 ± 0.056) compared with the STZ‐alone group (P < 0.01) (Figure 3C,D). Based on previous studies, the activity of GSK3β was increased when PI3K was inhibited by WT. Compared with the STZ + geniposide group, WT + STZ + geniposide decreased Ser9 phosphorylation by about 30% (pS‐9/β‐Actin: 0.869 ± 0.027, P < 0.01) and increased Tyr216 phosphorylation by about 20% (pY‐216/β‐Actin: 1.315 ± 0.074, P < 0.01) (Figure 3C,D).
Geniposide prevented STZ‐induced ultrastructure pathology
Because it is hard to get the statistic value, we made three repeats (n = 3) for the observation, results shown in the figure are all those that emerged frequently during the test. Alternation of ultrastructure in nerve cells is another important feature in the early stage of AD 5, 8, 27, 41. We prepared ultrathin sections for TEM from the rat hippocampus (mainly the CA1 and DG region) after the completion of the MWM test. Geniposide injection alone exhibited normal ultrastructure virtually identical to controls (Figure 4A,B). However, in the STZ‐alone injected group, we observed several ultrastructural changes that are thought to be early signs of apoptosis, including dim cells, nuclear granules, high electronic density of nucleus and nucleolus (Figure 4C,D). We also detected the nuclear and cytoplasmic condensation and clumping of chromatin, as well as altered subcellular organelles, such as mitochondria vacuolization (white arrow) and abnormal vacuolization of dendrites (black arrow) in the hippocampal neurons of STZ‐treated rats (Figure 4C,D). Interestingly, geniposide treatment abolished the occurrence of these ultrastructure alternations (Figure 4E,F).
Figure 4.
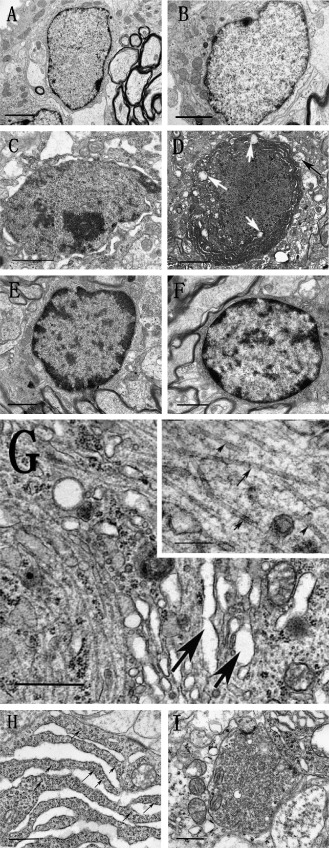
Photomicrograph reveals ultrastructure alterations in the rat hippocampus under TEM in response to different drug treatment. Normal structure of hippocampal neurons that received aCSF and geniposide alone (A, B) (bar = 1 μ). Dim cells, indicative of early stages of apoptosis, that were characterized with whole cell condensation (C), dark colored mitochondria vacuolation (white arrow), and dendritic vacuolation (black arrow) (D) (bar = 1 μ) were seen in animals treated with STZ, but these signs of early apoptosis were absent in animal that were co‐administered with STZ and geniposide (E, F) (bar = 1 μ). STZ also induced the PHFs‐like structures (G, bar = 1 μ; bar = 200 nm in the higher magnification insert), which are indicative of early signs of intracellular NFTs formation. Furthermore, STZ also induced increased cleft of ER and with agglutinated ribosome (arrow) (H, bar = 1 μ), as well as elevated accumulation of neurotransmitter vesicles (I, bar = 1 μ).
Another hallmark of AD pathology is the presence of intracellular neurofibrillary tangles (NFTs), which are formed by aberrantly hyperphosphorylated tau proteins which could self‐aggregate as paired helical filaments (PHFs) 42. Under TEM magnification, the STZ‐treated rats exhibited PHF‐like structures that were arranged in parallel, forming small clumps or strands in neurons and nerve fibers (Figure 4G), similar to previous report by van den Bosch et al 44. In addition to the alteration of cytoskeletons, abnormally accumulated synaptic vesicles were found in the terminals in the STZ‐treated group (Figure 4H), which may indicate impairment in vesicular regulation in the early stages of AD. Furthermore, STZ treatment also increased cleft of ER and agglutinated ribosome, which may indicate oxidative impairment (Figure 4I) 23. It is important to note that none of the above ultrastructure changes were found in the geniposide treated group, which further support our hypothesis that geniposide may exert neural protective effect in the early stages of the disease process.
Discussion
AD is a debilitating illness currently affecting over 30 million people worldwide. With extended life expectancy and increasingly aging society, the global patient population may reach 100 million in 2050 39. Although there is no cure for AD at present, successful management of this disease depends on early diagnosis and early intervention before the onset of significant cognitive impairment.
The STZ‐induced insulin‐resistant brain state provides a credible animal model that produced pathological changes may help identify some previously unknown aspect of early stage AD. In addition to behavioral and biochemical changes that have been documented before, we discovered that central administration of STZ induced ultrastructure changes that may occur in the early stage of AD before irreversible neuronal loss with apoptosis. Besides early stages of apoptosis, we also discovered early signs of tangle formation (Figure 4G). NFTs is the insoluble form of aggregated tau protein which could be observed under microscope with certain chemical stain and it is one of the important hallmarks in AD and other neurodegenerative stresses. What we showed in Figure 4G is the PHF structure which is considered as the early stage of tau self‐aggregation which could currently only detected under TEM scope 42. Much to our delight, injection of geniposide prevented the development of all three types of ultrastructure abnormalities that were induced by STZ.
According to the molecular structure of geniposide and STZ, it is quite hard for the two to reach chemical reaction in the regular environment (there is a possibility that geniposide and STZ could reach a Williamson reaction under approximately 140°C). Therefore, the only way geniposide could prevent neuronal ultrastructure changes that may underlie the early stages of AD neuropathogenesis in animal models is through activation the GLP‐1 receptor in the brain. Our research strongly suggests that early use of geniposide could halt the progression of the disease process before the onset of permanent cell death and synaptic loss that ultimately lead to irreversible cognitive impairment.
Our ultrastructure findings are consistent with the biochemical data with tau protein phosphorylation, as the NFTs are formed by hyperphosphorylation of a microtubule‐associated protein known as tau, causing it to an aggregation form 9. These aggregations of hyperphosphorylated tau protein are referred to as PHF under electron microscopy. Although the precise mechanism of tangle formation is not completely understood, our data convincingly show that geniposide could greatly attenuate STZ‐induced tau protein hyperphosphorylation, and render the PHFs below detectable level under TEM. Our findings suggest that geniposide may prevent the formation of PHFs in the early stages of the pathogenesis before they accumulate to a certain point to become insoluble tangles. There are several cellular signaling pathways that regulate tau phosphorylation. One which is related with tau phosphorylation and insulin resistance is the PI3K/GSK3 pathway 12.
GLP‐1 receptor is one kind of the G‐protein coupled receptor (GPCR). Activation of GLP‐1 receptor by geniposide could induce multiple cellular signaling pathways. Studies showed that some kinds of GLP‐1 analogs could prevent animal models from AD pathologies, including improving the learning and memory deficit induced by Aβ or STZ 22, 31. And the chronic treatment of liraglutide (one of the analogs of GLP‐1) could promote the cell proliferation and differentiation into neuron in AD mouse model 34. In our study, we proved that application of geniposide could improve the spatial cognitive ability of STZ‐induced rats model. However, most of the neuroprotective mechanism remains unknown. By western assay, our data showed that geniposide attenuated STZ‐induced activation of GSK3β. Geniposide treatment decreased the active form of GSK3β (pY216), but increased the inactive form of GSK3β (pS9), resulting in a reduction of GSK3β activity. The decrease activation of GSK3β could promote the ameliorating of the tau hyperphosphorylation, which is shown in our results. GSK3 plays an important role in learning and memory. Its upstream protein, PI3K, is phosphorylated upon N‐methyl‐D‐aspartate (NMDA) receptor‐dependent CaMKII activity 19. GSK3β, on the other hand, its activity is enhanced during long‐term depression (LTD) via activation of PP1; conversely, following the induction of LTP, there is inhibition of GSK3β activity 35. In behavioral study, by WT interruption, we proved that PI3K/GSK3 pathway also plays an important role in the neuroprotective effects of geniposide.
On the aspect of diabetic treatment, the remarkable advance of GLP‐1 is that it could enhance insulin secretion and inhibits glucagon in a glucose‐dependent manner 40. In the single geniposide treatment group, the activity of GSK3β did not show any significant change compared with control group (Figure 4C,D). The result suggested an interesting phenomenon that GSK3β regulated by geniposide hinges on the impairment of STZ, which could be an indication about the safety advance of this drug. And the underlying mechanism of this phenomena might have some sophisticated relationship with the glucose‐dependent manner of GLP‐1, which is worth exploring. Geniposide is water soluble and orally active that can readily cross the blood–brain barrier. In addition to reliving the symptoms of diabetes, geniposide has been used to ameliorate many other types of illness in traditional Chinese medicine, such as improving mood and sleep that are both common problems in AD. In summary, the Chinese herbal medicine geniposide may serve as a novel therapeutic agent for the treatment of sporadic AD in the early stage of the disease process to delay or prevent the onset of irreversible cognitive impairment.
Acknowledgments
This study is supported by the Shanxi Science and Technology Department (2010081062) and Shanxi Scholarship Council of China (2007‐46).
References
- 1. Agrawal R, Tyagi E, Shukla R, Nath C (2011) Insulin receptor signaling in rat hippocampus: a study in STZ (ICV) induced memory deficit model. Eur Neuropsychopharmacol 21:261–273. [DOI] [PubMed] [Google Scholar]
- 2. Akter K, Lanza EA, Martin SA, Myronyuk N, Rua M, Raffa RB (2011) Diabetes mellitus and Alzheimer's disease: shared pathology and treatment? Br J Clin Pharmacol 71:365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Atri A (2011) Effective pharmacological management of Alzheimer's disease. Am J Manag Care 17:S346–355. [PubMed] [Google Scholar]
- 4. Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M et al (1999) Early‐onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet 65:664–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carvalho C, Cardoso S, Correia SC, Santos RX, Santos MS, Baldeiras I et al (2012) Metabolic alterations induced by sucrose intake and Alzheimer's disease promote similar brain mitochondrial abnormalities. Diabetes 61:1234–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cornish TC, Halushka MK (2009) Color deconvolution for the analysis of tissue microarrays. Anal Quant Cytol Histol 31:304–312. [PubMed] [Google Scholar]
- 7. Dhull DK, Jindal A, Dhull RK, Aggarwal S, Bhateja D, Padi SS (2012) Neuroprotective effect of cyclooxygenase inhibitors in ICV‐STZ induced sporadic alzheimer's disease in rats. J Mol Neurosci 46:223–235. [DOI] [PubMed] [Google Scholar]
- 8. Dikranian K, Kim J, Stewart FR, Levy MA, Holtzman DM (2012) Ultrastructural studies in APP/PS1 mice expressing human ApoE isoforms: implications for Alzheimer's disease. Int J Clin Exp Pathol 5:482–495. [PMC free article] [PubMed] [Google Scholar]
- 9. Duan Y, Dong S, Gu F, Hu Y, Zhao Z (2012) Advances in the pathogenesis of Alzheimer's disease: focusing on tau‐mediated neurodegeneration. Transl Neurodegener 1:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP et al (2003) Insulin‐degrading enzyme regulates the levels of insulin, amyloid beta‐protein, and the beta‐amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci USA 100:4162–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Frolich L, Blum‐Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S et al (1998) Brain insulin and insulin receptors in aging and sporadic Alzheimer's disease. J Neural Transm 105:423–438. [DOI] [PubMed] [Google Scholar]
- 12. Gao C, Holscher C, Liu Y, Li L (2012) GSK3: a key target for the development of novel treatments for type 2 diabetes mellitus and Alzheimer disease. Rev Neurosci 23:1–11. [DOI] [PubMed] [Google Scholar]
- 13. Hernandez F, Nido JD, Avila J, Villanueva N (2009) GSK3 inhibitors and disease. Mini Rev Med Chem 9:1024–1029. [DOI] [PubMed] [Google Scholar]
- 14. Holscher C (2011) Diabetes as a risk factor for Alzheimer's disease: insulin signalling impairment in the brain as an alternative model of Alzheimer's disease. Biochem Soc Trans 39:891–897. [DOI] [PubMed] [Google Scholar]
- 15. Hou YC, Tsai SY, Lai PY, Chen YS, Chao PD (2008) Metabolism and pharmacokinetics of genipin and geniposide in rats. Food Chem Toxicol 46:2764–2769. [DOI] [PubMed] [Google Scholar]
- 16. Hu S, Begum AN, Jones MR, Oh MS, Beech WK, Beech BH et al (2009) GSK3 inhibitors show benefits in an Alzheimer's disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol Dis 33:193–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang CY, Hsiao JK, Lu YZ, Lee TC, Yu LC (2011) Anti‐apoptotic PI3K/Akt signaling by sodium/glucose transporter 1 reduces epithelial barrier damage and bacterial translocation in intestinal ischemia. Lab Invest 91:294–309. [DOI] [PubMed] [Google Scholar]
- 18. Isik AT, Celik T, Ulusoy G, Ongoru O, Elibol B, Doruk H et al (2009) Curcumin ameliorates impaired insulin/IGF signalling and memory deficit in a streptozotocin‐treated rat model. Age (Dordr) 31:39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Joyal JL, Burks DJ, Pons S, Matter WF, Vlahos CJ, White MF, Sacks DB (1997) Calmodulin activates phosphatidylinositol 3‐kinase. J Biol Chem 272:28183–28186. [DOI] [PubMed] [Google Scholar]
- 20. Lee J, Kim MS (2007) The role of GSK3 in glucose homeostasis and the development of insulin resistance. Diabetes Res Clin Pract 77:S49–57. [DOI] [PubMed] [Google Scholar]
- 21. Li L, Holscher C (2007) Common pathological processes in Alzheimer disease and type 2 diabetes: a review. Brain Res Rev 56:384–402. [DOI] [PubMed] [Google Scholar]
- 22. Li L, Zhang ZF, Holscher C, Gao C, Jiang YH, Liu YZ (2012) (Val(8)) glucagon‐like peptide‐1 prevents tau hyperphosphorylation, impairment of spatial learning and ultra‐structural cellular damage induced by streptozotocin in rat brains. Eur J Pharmacol 674:280–286. [DOI] [PubMed] [Google Scholar]
- 23. Ling ZQ, Tian Q, Wang L, Fu ZQ, Wang XC, Wang Q, Wang JZ (2009) Constant illumination induces Alzheimer‐like damages with endoplasmic reticulum involvement and the protection of melatonin. J Alzheimers Dis 16:287–300. [DOI] [PubMed] [Google Scholar]
- 24. Liu J, Zheng X, Yin F, Hu Y, Guo L, Deng X et al (2006) Neurotrophic property of geniposide for inducing the neuronal differentiation of PC12 cells. Int J Dev Neurosci 24:419–424. [DOI] [PubMed] [Google Scholar]
- 25. Liu JH, Yin F, Guo LX, Deng XH, Hu YH (2009) Neuroprotection of geniposide against hydrogen peroxide induced PC12 cells injury: involvement of PI3 kinase signal pathway. Acta Pharmacol Sin 30:159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu J, Yin F, Xiao H, Guo L, Gao X. (2012) Glucagon‐like peptide 1 receptor plays an essential role in geniposide attenuating lipotoxicity‐induced β‐cell apoptosis. Toxicol In Vitro 26:1093–1097 [DOI] [PubMed] [Google Scholar]
- 27. Long J, He P, Shen Y, Li R (2012) New evidence of mitochondria dysfunction in the female Alzheimer's disease brain: deficiency of estrogen receptor‐beta. J Alzheimers Dis 30:545–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu TS, Yiao SY, Lim K, Jensen RV, Hsiao LL (2010) Interpretation of biological and mechanical variations between the Lowry versus Bradford method for protein quantification. N Am J Med Sci 2:325–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luchsinger JA, Tang MX, Shea S, Mayeux R (2004) Hyperinsulinemia and risk of Alzheimer disease. Neurology 63:1187–1192. [DOI] [PubMed] [Google Scholar]
- 30. Luchsinger JA, Tang MX, Stern Y, Shea S, Mayeux R (2001) Diabetes mellitus and risk of Alzheimer's disease and dementia with stroke in a multiethnic cohort. Am J Epidemiol 154:635–641. [DOI] [PubMed] [Google Scholar]
- 31. McClean PL, Parthsarathy V, Faivre E, Holscher C (2011) The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer's disease. J Neurosci 31:6587–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Morris R (1984) Developments of a water‐maze procedure for studying spatial learning in the rat. J Neurosci Methods 11:47–60. [DOI] [PubMed] [Google Scholar]
- 33. Obulesu M, Venu R, Somashekhar R (2011) Tau mediated neurodegeneration: an insight into Alzheimer's disease pathology. Neurochem Res 36:1329–1335. [DOI] [PubMed] [Google Scholar]
- 34. Parthsarathy V, Holscher C (2013) Chronic treatment with the GLP1 analogue liraglutide increases cell proliferation and differentiation into neurons in an AD mouse model. PLoS One 8:e58784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J et al (2007) LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 53:703–717. [DOI] [PubMed] [Google Scholar]
- 36. Plaschke K, Kopitz J, Siegelin M, Schliebs R, Salkovic‐Petrisic M, Riederer P, Hoyer S (2010) Insulin‐resistant brain state after intracerebroventricular streptozotocin injection exacerbates Alzheimer‐like changes in Tg2576 AbetaPP‐overexpressing mice. J Alzheimers Dis 19:691–704. [DOI] [PubMed] [Google Scholar]
- 37. Plaschke K, Muller D, Hoyer S (2010) Insulin‐resistant brain state (IRBS) changes membrane composition of fatty acids in temporal and entorhinal brain cortices of rats: relevance to sporadic Alzheimer's disease? J Neural Transm 117:1419–1422. [DOI] [PubMed] [Google Scholar]
- 38. Ponce‐Lopez T, Liy‐Salmeron G, Hong E, Meneses A (2011) Lithium, phenserine, memantine and pioglitazone reverse memory deficit and restore phospho‐GSK3beta decreased in hippocampus in intracerebroventricular streptozotocin induced memory deficit model. Brain Res 1426:73–85. [DOI] [PubMed] [Google Scholar]
- 39. Pouryamout L, Dams J, Wasem J, Dodel R, Neumann A (2012) Economic evaluation of treatment options in patients with Alzheimer's disease: a systematic review of cost‐effectiveness analyses. Drugs 72:789–802. [DOI] [PubMed] [Google Scholar]
- 40. Repas T (2011) Next‐generation GLP‐1 therapy: an introduction to liraglutide. Postgrad Med 123:239–247. [DOI] [PubMed] [Google Scholar]
- 41. Ruben GC, Wang JZ, Iqbal K, Grundke‐Iqbal I (2005) Paired helical filaments (PHFs) are a family of single filament structures with a common helical turn period: negatively stained PHF imaged by TEM and measured before and after sonication, deglycosylation, and dephosphorylation. Microsc Res Tech 67:175–195. [DOI] [PubMed] [Google Scholar]
- 42. Rudrabhatla P, Jaffe H, Pant HC (2011) Direct evidence of phosphorylated neuronal intermediate filament proteins in neurofibrillary tangles (NFTs): phosphoproteomics of Alzheimer's NFTs. FASEB J 25:3896–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Salkovic‐Petrisic M, Hoyer S (2007) Central insulin resistance as a trigger for sporadic Alzheimer‐like pathology: an experimental approach. J Neural Transm Suppl 1:217–233. [DOI] [PubMed] [Google Scholar]
- 44. van den Bosch de Aguilar P, Goemaere‐Vanneste J (1984) Paired helical filaments in spinal ganglion neurons of elderly rats. Virchows Arch B Cell Pathol Incl Mol Pathol 47:217–222. [DOI] [PubMed] [Google Scholar]
- 45. Woodgett JR (1991) cDNA cloning and properties of glycogen synthase kinase‐3. Methods Enzymol 200:564–577. [DOI] [PubMed] [Google Scholar]
- 46. Woodgett JR, Cohen P (1984) Multisite phosphorylation of glycogen synthase. Molecular basis for the substrate specificity of glycogen synthase kinase‐3 and casein kinase‐II (glycogen synthase kinase‐5). Biochim Biophys Acta 788:339–347. [DOI] [PubMed] [Google Scholar]