

Abstract
Neuropilin-1 (NRP-1) is a multifunctional transmembrane receptor for ligands that affect developmental axonal growth and angiogenesis. In addition to a role in cancer, NRP-1 is a reported entry point for several viruses, including severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causal agent of coronavirus disease 2019 (COVID-19). The furin cleavage product of SARS-CoV-2 Spike protein takes advantage of the vascular endothelial growth factor A (VEGF-A) binding site on NRP-1 which accommodates a polybasic stretch ending in a C-terminal arginine. This site has long been a focus of drug discovery efforts for cancer therapeutics. We recently showed that interruption of the VEGF-A/NRP-1 signaling pathway ameliorates neuropathic pain and hypothesize that interference of this pathway by SARS-CoV-2 spike protein interferes with pain signaling. Here, we report confirmed hits from a small molecule and natural product screen of nearly 0.5 million compounds targeting the VEGF-A binding site on NRP-1. We identified nine chemical series with lead- or drug-like physico-chemical properties. Using ELISA, we demonstrate that six compounds disrupt VEGF-A-NRP-1 binding more effectively than EG00229, a known NRP-1 inhibitor. Secondary validation in cells revealed that all tested compounds inhibited VEGF-A triggered VEGFR2 phosphorylation. Further, two compounds displayed robust inhibition of a recombinant vesicular stomatitis virus protein that utilizes the SARS-CoV-2 Spike for entry and fusion. These compounds represent a first step in a renewed effort to develop small molecule inhibitors of the VEGF-A/NRP-1 signaling for the treatment of neuropathic pain and cancer with the added potential of inhibiting SARS-CoV-2 virus entry.
Keywords: Neuropilin-1, VEGF-A, SARS-CoV-2, natural compounds, ELISA, VSV, small molecules, pain, cancer
Graphical Abstract
Introduction
As of February, 2021 COVID-19 has infected more than 110 million people and caused over 2 million deaths worldwide1. This disease is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) which primarily gains entry to cells via binding of SARS-CoV-2 Spike glycoprotein to angiotensin converting enzyme 2 (ACE-2) and subsequent endocytosis2–4. Recent reports have identified additional entry points, including neuropilin 1 (NRP-1)5–7.
Neuropilins are cell surface receptors for secreted glycoproteins with roles in neural outgrowth, cardiovascular development, immune response, as well as tumor growth and vascularization8, 9. Two neuropilin isoforms, NRP-1 and NRP-2, share ~44% sequence identity in humans and function in different pathways8, 9. Both share a modular architecture with three extracellular domains, a single transmembrane helix, and a short cytoplasmic tail (Figure 1A)10. The a1a2 modules, homologous to CUB (for complement C1r/C1s, Uegf, Bmp1) domains, interact with semaphorin 3A (SEMA3A) to mediate stimulation of growth cone collapse in developing neurons8. The b1b2 modules are homologous to the C-terminal domains of blood coagulation Factors V and VIII8. The c domain, homologous to meprin, A5, and mu-phosphatase (MAM), was initially thought to be involved in dimerization, but is more likely to contribute to complex assembly by positioning the preceding domains away from the membrane11. The single transmembrane helix contributes to homo and heterodimerization12 and the C-terminal cytoplasmic tail is thought to contribute to signaling through synectin, stimulating receptor-mediated endocytosis9.
Figure 1. Schematics of NRP-1 domains, VEGF-A165 binding, and CendR interaction network.
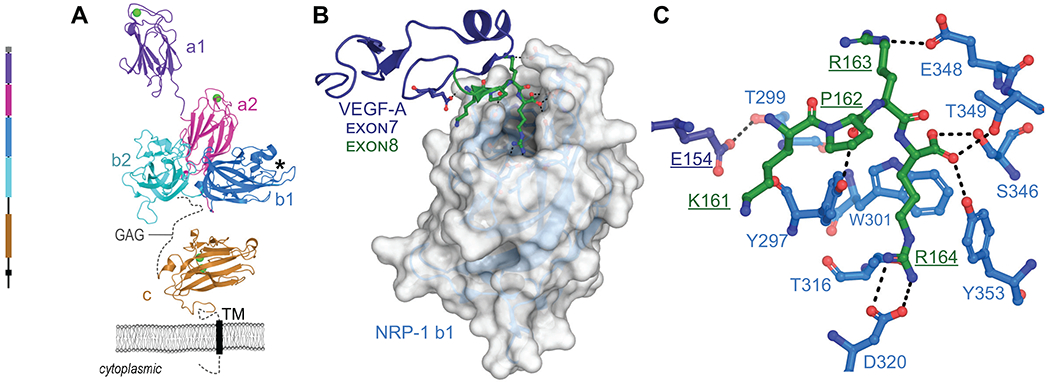
A. Domain architecture of NRP-1 with domain a1 from mouse (PDB ID 4GZ9)94, domains a2, b1, b2 (PDB ID 2QQM)10 and MAM (PDB ID 5L73)11 from human. Bound Ca2+ shown as green spheres, missing loops as dashes, transmembrane domain as a rectangle. GAG indicates region of glycosaminoglycan modification. *Indicates VEGF-A interaction pocket. B. Structure of NRP-1 b1 domain, shown as white surface, in complex with the heparin binding domain (exon 7/8) of VEGF-A164, shown as cartoon with exon 7 in dark blue, exon 8 in green (PDB ID 4DEQ)15 C. Details of VEGF-A Glu154 and KPRR164 interactions with NRP-1 (close up of view B). Dashes indicate polar or salt-bridge contacts within 3.0 Å.
It is through the b1 module that SARS-CoV-2 may gain entry, by taking advantage of the interaction site for vascular endothelial growth factor A (VEGF-A). VEGF-A isoforms and other growth factors that terminate in a polybasic stretch ending with an obligatory arginine residue, termed the C-terminal end arginine (CendR) rule13, interact with an acidic pocket formed by loops extending from the beta-barrel of b1 (Figure 1B)14, 15. The structure of the heparin binding domain of the 164 residue isoform of VEGF-A (VEGF-A164) confirmed that the C-terminal Arg (Arg164) engages the NRP-1 b1 domain pocket with the guanidine forming a bidentate salt-bridge with conserved Asp320 and the carboxylate forming hydrogen bonds to conserved Ser346, Thr349, and Tyr353 (Figure 1C)15. It is notable that VEGF-A binds to both NRP-1 and NRP-2, but has higher affinity for NRP-1 due to amino acid substitutions within the first loop region of NRP-1 that provide additional contacts between NRP-1 Thr299 and VEGF-A Glu154 (Figure 1B, C)15. Furthermore, although b1 and b2 are homologous domains, critical residues within the loops of the binding domain are not conserved with the result that the CendR interaction site is not present on b2.
VEGF-A has long been known for its essential role in blood vessel growth and function, acting as a selective endothelial cell mitogen that promotes angiogenesis, primarily via interaction with the VEGF receptor VEGFR2, also known as kinase insert domain-containing receptor (KDR)16. In addition, VEGF-A has a complicated role in nociception with both pro- and anti-algesic findings reported in the literature17, 18. These disparate effects are thought to be due to variation in tissue specific expression levels of two key VEGF-A splice variants and their differential effects on sensory neurons17, 19. Alternative splicing produces isoforms of varying length, indicated as VEGF-Axxx, where xxx refers to the length of the mature protein19. The most well-studied pain-related isoforms result from alternative splicing of exon 8 with VEGF-A165a generally found to be pro-nociceptive and VEGF-A165b found to be anti-nociceptive17, 18, with one exception20. This alternative splicing is dependent on serine-arginine rich protein kinase 1 (SRPK1) which mediates the phosphorylation of serine-arginine rich splice factor (SRSF1)17, 21, 22. Current thought is that the balance of the a and b isoforms is what determines the effect on sensory neurons17, 19. The pro-nociceptive action of the VEGF-A165a isoform17 occurs through sensitization of transient receptor potential (TRP) channels23 and ATP-gated purinergic P2X2/3 receptors24 on dorsal root ganglion (DRG) neurons. VEGF binding to VEGFR2, a co-receptor for NRP-1, is associated with receptor dimerization and activation that triggers downstream signaling pathways including phosphatidylinositol 3-kinase (PI3-K)/Akt and phospholipase C gamma/extracellular signal-regulated kinase (PLCg/ERK)16. Clinical findings that VEGF-A contributes to pain are supported by observations that in osteoarthritis increased VEGF expression in synovial fluids has been associated with higher pain scores25. VEGF-A has been reported to enhance pain behaviors in normal, nerve-injured and diabetic animals17, 26. In this present study, we seek to target the VEGF-A/NRP1 pathway, which we recently identified as a novel pain pathway27. All exon 8 splice variants differ in the composition of the six C-terminal amino acids: isoforms a end with CDKPRR and isoforms b end with the sequence SLTRKD19. This difference is key, as VEGF isoforms without the CendR motif do not bind to NRP119, 28. Thus, the compounds we present here are expected to affect the VEGF-A165a/NRP1 pathway through NRP1 rather than broadly inhibiting VEGF-A interactions with its receptors.
In addition to a role in pain, NRP-1 has an emerging role in the immune system29, 30. NRP-1 is expressed in several types of immune cells including macrophages, thymocytes, dendritic cells (DC) and T cells where it participates in different functions both in physiological and pathological conditions. It is crucial for regulating the immune response since it is involved in several important functions such as the formation of immune synapses at antigen presentation29. In mice, NRP-1 is essential to the first steps of the primary immune response since it acts as an additional mediator of the long contact interaction between antigen-presenting cells such as immature dendritic cells and regulatory T cells in immune synapses31. Likewise, in humans, blocking this interaction with a specific NRP-1 antibody resulted in a reduced T cell proliferation induced by dendritic cell stimulation32. On the other hand, a subpopulation of plasmacytoid DC that express NRP-1 is associated with fighting viral infections through the secretion of IFN-α. NRP-1 has an immunoregulatory role in these cells since studies show that incubation with an anti-NRP-1 antibody suppresses INF-α production33. As to VEGF, it is known as an immunosuppressive cytokine that inhibits DC maturation in an NRP-1 dependent manner inducing immune tolerance34.
It is known that neuropilins are entry points for several viruses, including human T-lymphotropic virus-1 (HTLV-1)35 and Epstein-Barr virus (EBV)36. In both cases, furin processing of viral glycoproteins results in polybasic CendR motifs that directly interact with the VEGF-A site on the NRP-1 b1 domain37. Compared to SARS-CoV-1, the causative agent of SARS, the SARS-CoV-2 Spike protein contains a furin consensus site which was shown to be essential for infection of lung cells38. Furin cleavage at this site by the host cell results in production of a surface-exposed CendR motif (682RRAR685)38 which was shown to bind to the neuropilin b1 VEGF-A site, suggesting NRP-1 as a possible route of viral entry5, 6. The importance of NRP-1 is supported by recent evidence of upregulated NRP-1 in lung samples from COVID-19 patients5.
These connections raise an interesting question: does interference with the VEGF-A/NRP-1 signaling pathway by SARS-CoV-2 result in dampened pain? This question has been examined by our laboratory; we recently showed that SARS-CoV-2 Spike protein binding to NRP-1 prevents VEGF-A signaling and reduces neuropathic pain in an animal model27. Thus, NRP-1 represents a novel target for treating neuropathic pain27. Furthermore, targeting NRP-1 also presents a unique approach to inhibiting viral entry and/or re-entry into cells to reduce viral load. Due to its role in cancer, NRP-1 has been a target for drug design for over 20 years. During this time, discovery efforts have focused on development of NRP-1 antibody therapies10, 39–44, including a recent dual-specificity antibody to VEGFA and NRP-143, 44, peptides that target transmembrane domain interactions12, 45–48 or the CendR interaction site13, 42, 49–64, as well as small-molecules that target the CendR site 65–72. With one exception54, the CendR site targeting peptides contain a CendR motif, including those with cyclical50, 56, 57, 60 or branched62, 64 backbones. Two of the most well-known small molecule NRP-1 inhibitors, EG00229 and EG01377, contain a terminal Arg-like moiety and carboxyl group known to be key to interaction with NRP-165, 72 as does the newly developed fluorescent compound based on EG0137773. The remaining known small molecule compounds consist of an arginine-derivative series71 and several diverse chemotypes such as acylthioureas67, benzamidosulfonamides70, bis-guanidines66, or aryl benzylethers69, discussed further below.
In order to identify unique compounds that could be used to interrogate the role of these signaling complexes in pain, we conducted a virtual screen of nearly 0.5 million compounds against the NRP-1 CendR site, resulting in about 1,000 hits. Here, we present 9 chemical series of synthetic and natural compounds with lead- or drug-like physico-chemical properties and identify a pharmacophore model which will guide future design of compounds. We also show that six of the compounds interfere with VEGF-A binding more effectively than EG00229, a known NRP-1 inhibitor. Furthermore, our compounds show inhibition of VEGF-A triggered VEGFR2 phosphorylation in a cell-based assay. Finally, two of the compounds inhibited SARS-CoV-2 mediated viral entry into cells.
Results and Discussion
We conducted virtual screens against the VEGF-A binding site on the NRP-1 b1 domain using three libraries: a ~211 K synthetic compound library (DIV) from ChemBridge; a ~257 K natural compound library (NC1) obtained from the COlleCtion of Open NatUral producTs (COCONUT) resource74; and a ~20 K (NC2) natural compound library from the ZINC15 database75. The screens were run once without ligand-receptor interaction constraints and repeated with the constraint that compounds form a hydrogen bond to Asp 320, a key residue for coordinating the terminal arginine in the CendR motif15. This constraint was used in attempt to select for compounds that interact in a similar way as observed for VEGF-A15, known inhibitors53, 65, 71, 72 and modeled SARS-CoV-219 CendR terminal arginine6. However, application of this constraint led to reduced overall scores and strained conformations for most compounds in the DIV and NC1 libraries. Therefore, we report only the unconstrained screen results for these libraries and both constrained and unconstrained results for the NC2 screen.
Selection of top compounds
The combined output of the four screens produced a total of 1,147 hits. Compounds from each screen were sorted by Glide XP GScore (kcal/mol) and visually inspected for substructure match of core scaffolds and patterns of chemically reactive moieties while considering the diversity of chemotypes. Compound representatives with scaffold decoration that suggested initial structure activity relationships (SAR) were extracted and grouped into series resulting in a set of nine diverse chemotypes (series). These series include both small synthetic molecules and natural products (Table 1). While calculated property ranges vary for the chemotype and drug indication, we intend to keep compounds in our series such that molecular weight (Mw) < 400 Da, calculated octanol-water partition coefficient (clogP) < 3.5, and calculated solubility (clogS) at pH 7.4 > −5M. Consequently, we expect the initial absorption, distribution, metabolism, and excretion (ADME) profile of the series to be acceptable (e.g., good hepatocyte clearance and bioavailability). To assess the probability that hit series compounds are orally available, we determined components and overall compliance with Lipinski rule of 5 (Ro5)76, 77. Since the Ro5 guidelines were derived for orally available small molecules, we did not use this binary parameter for the classification of natural products. Also, since solubility and CNS penetration rules originated from small molecule sets, these calculations are not applicable to natural products and were also omitted. Furthermore, detailed inspection of data of Table 1 revealed that natural product compounds 11, 12, and 15 are in fact small molecule chemotypes. Correspondingly, all molecular descriptors were calculated for these compounds and they were considered part of the small molecule set.
Table 1. Docking scores and physico-chemical properties of hit and reference compounds.
Compound ranking (Glide Gscore (kcal/mol)) and calculated properties of the top twenty hits (1-20) and reference compounds (A-F).
| Cmpd | Glide Gscore | Set | Source Idx | Series | Bnd Mode | BBB score | logS (7.4) | Mw | cLogP | HBD | HBA | Lipinski | NHOH | RotB | TPSA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | −12.1 | DIV | 29267 | 1 | I | 4.2 | −5.2 | 293.33 | 3.2 | 2 | 4 | Y | 2 | 4 | 67 |
| 2 | −11.5 | DIV | 2147 | 2 | I | 4.1 | −3.3 | 279.34 | 0.7 | 2 | 4 | Y | 2 | 5 | 84 |
| 3 | −10.8 | DIV | 37503 | 2 | I | 3.9 | −4.2 | 342.4 | 0.7 | 1 | 6 | Y | 1 | 4 | 95 |
| 4 | −10.4 | DIV | 41088 | 1 | I | 2.7 | −5.0 | 364.41 | 2.1 | 2 | 7 | Y | 2 | 4 | 96 |
| 5 | −10.3 | DIV | 45423 | 1 | I | 2.7 | −5.2 | 378.44 | 2.9 | 2 | 7 | Y | 2 | 5 | 96 |
| 6 | −10.3 | DIV | 42130 | 2 | I | 4.3 | −2.5 | 329.4 | 1.5 | 1 | 5 | Y | 1 | 6 | 92 |
| 7 | −9.8 | DIV | 8757 | 3 | I | 3.2 | −1.7 | 218.22 | 0.9 | 2 | 5 | Y | 2 | 3 | 80 |
| 8 | −9.7 | DIV | 8892 | 3 | I | 3.0 | −1.5 | 204.19 | 0.6 | 3 | 5 | Y | 3 | 2 | 91 |
| 9 | −9.2 | DIV | 9167 | 4 | II | 3.4 | −2.3 | 250.26 | 0.0 | 2 | 5 | Y | 2 | 4 | 95 |
| 10 | −11.3 | NC1 | 15860 | 6 | I | 1.9 | 0.0 | 209.17 | −0.9 | 4 | 5 | Y | 4 | 3 | 124 |
| 11 | −11.0 | NC1 | 34346 | 1 | I | 3.4 | 0.6 | 231.21 | 0.5 | 2 | 4 | Y | 2 | 3 | 96 |
| 12 | −10.9 | NC1 | 21399 | 5 | I | 3.7 | −2.3 | 347.37 | −0.9 | 2 | 5 | Y | 2 | 4 | 99 |
| 13 | −10.7 | NC1 | 36469 | 9 | I | na | na | 358.35 | 2.4 | 5 | 6 | Y | 5 | 7 | 135 |
| 14 | −9.8 | NC2 | 13787 | 7 | II | na | na | 312.27 | −0.1 | 5 | 7 | Y | 5 | 6 | 145 |
| 15 | −9.6 | NC2 | 7454 | 5 | I | 4.6 | 0.6 | 222.29 | 0.4 | 2 | 3 | Y | 2 | 2 | 56 |
| 16 | −9.6 | NC1 | 16482 | 9 | II | na | na | 453.46 | 0.2 | 5 | 5 | Y | 6 | 8 | 171 |
| 17 | −9.4 | NC2 | 13777 | 7 | II c | na | na | 312.23 | −0.4 | 5 | 7 | Y | 5 | 6 | 162 |
| 18 | −9.1 | NC2 | 13788 | 7 | II c | na | na | 312.27 | −0.1 | 5 | 7 | Y | 5 | 6 | 145 |
| 19 | −8.6 | NC2 | 15222 | 8 | II c | na | na | 396.44 | 0.6 | 4 | 5 | Y | 6 | 13 | 176 |
| 20 | −8.4 | NC2 | 15223 | 8 | II | na | na | 396.44 | 0.6 | 4 | 5 | Y | 6 | 13 | 176 |
| A | −6.4 | Ref 63 | EG00229 | 10 | II c | na | na | 497.58 | 0.8 | 5 | 9 | Y | 7 | 10 | 203 |
| B | −5.8 | Ref 70 | EG01377 | 10 | II | na | na | 586.7 | 1.8 | 6 | 8 | N | 9 | 12 | 212 |
| C | −5.2 | Ref 65 | 1 | 11 | II c | 3.2 | −7.9 | 444.52 | 4.4 | 3 | 5 | Y | 3 | 3 | 88 |
| D | −4.3 | Ref 64 | 32 | 12 | II | na | na | 500.6 | 0.0 | 6 | 6 | Y | 9 | 10 | 190 |
| E | −4.1 | Ref 67 | CB-7739526 | 13 | na | 5.0 | −3.5 | 390.69 | 4.3 | 2 | 4 | Y | 2 | 8 | 51 |
| F | −3.5 | Ref 68 | NRPa-308 | 14 | II | 4.1 | −6.2 | 424.52 | 4.8 | 2 | 4 | Y | 2 | 7 | 85 |
Set: screening library; Source Idx: reference to internal ID in the screening set; Series: chemotype assignment to hit series 1-9 and known compound series 10-14; Bnd Mode: binding Mode I or II (c denotes use of constrained docking); BBB score: score for compound probability of having CNS exposure; LogS(7.4) predicted solubility (M) at pH 7.4; Mw molecular weight (Da); cLogP predicted lipophilicity coefficient in octanol/water; HBD number of hydrogen-bond donors; HBA number of hydrogen bond acceptors; Lipinski - a binary (Y/N) assignment of complying with Lipinski rule-of-5; NHOH number of polar NH and OH hydrogens; RotB number of rotatable bonds; TPSA total polar surface area (Å2); na denotes not applicable.
Properties were calculated using RDKit and ChemAxon Aqueous solubility module.
Since one objective of inhibiting the VEGF-A/NRP-1 interaction is disruption of pain signals, the full therapeutic effect will require drug exposure in the central nervous system (CNS). Accordingly, the optimization strategy for these compounds will include modifications beneficial for crossing the blood-brain barrier (BBB), for example, decreasing the number of hydrogen bond donors and polarity/charge. To supplement traditional medicinal chemistry approaches to increasing CNS exposure, we implemented the BBB score algorithm of Weaver et al.78 as one of the optimization parameters. Values of the BBB score in the range of 4 to 6 correctly predict 90% of CNS drugs78. While the brain/plasma ratio will be experimentally assessed for series representatives, optimization will be additionally guided by surrogate estimates of passive diffusion (PAMPA) and assessment of efflux and transporter proteins using assays in MDCK cell lines. Overall, predicted physico-chemical properties of all small molecule hit series (series 1-6) fall within ranges of lead-like and/or drug-like molecules (Table 1). Moreover, several representatives of series 1 and 2 are expected to have acceptable CNS exposure.
Series analysis and docked binding modes
After initial ranking and selection of the top 20 hits, we proceeded to a more detailed analysis of structural and chemical features of the compounds. Chemical structures of all twenty hits and one virtual SAR analog are shown in Figure 2. Hits are grouped by common core motifs and molecular fragments predicted to engage in productive hydrogen bond (HB) and alkyl/aryl π-contacts within the binding pocket. Inspection of aligned 2-D structures makes it apparent that the 2(1H)-pyridone core of structure 15 (highlighted in Figure 2) is the minimal motif of all small molecule hits (discussed further below). This core binds near the top of a central hydrophobic box formed by residues Tyr297, Tyr253, Trp301, and the methyl group of Thr316 (Figure 3). As expected, all of the hits bind within this box with the aryl or alkyl (e.g., isobutyl) groups engaged in hydrophobic interactions with these residues. Indeed, it is these hydrophobic interactions that are drivers of the overall binding affinity as judged by the Glide XP Gscore.
Figure 2. Chemical structures of the top 20 hits.
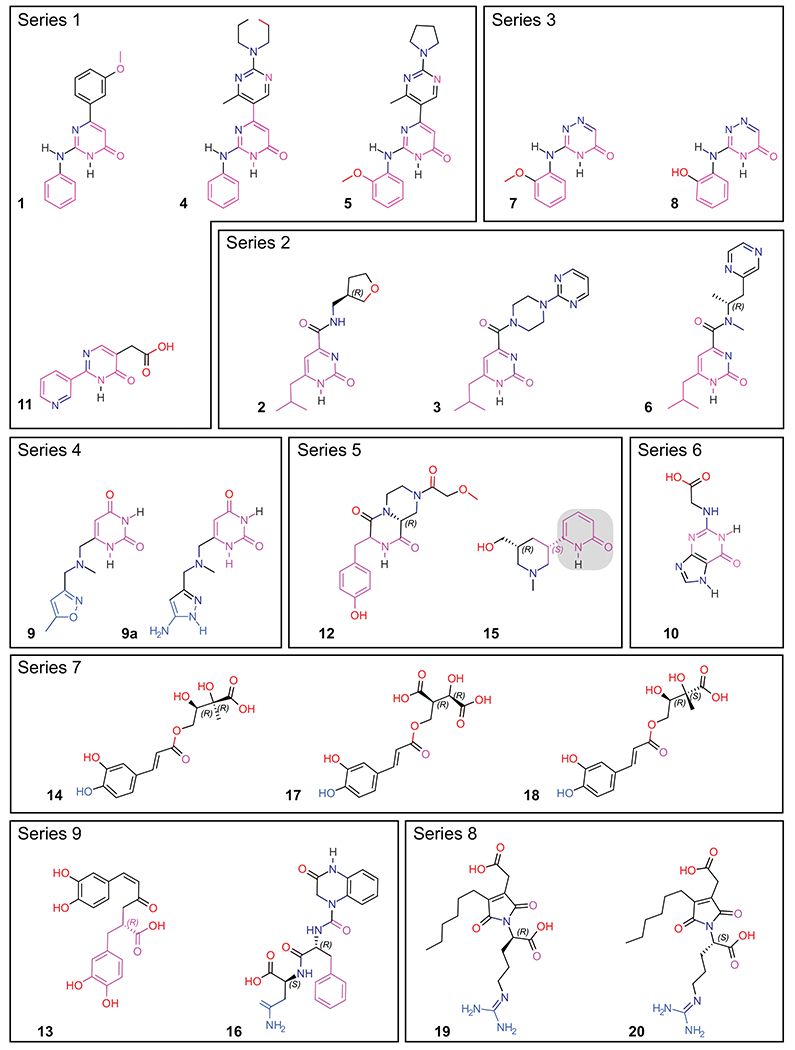
Compounds are grouped by common core motif, shown in magenta. Molecules that adopt binding Mode II (9, 9a, 14, 17, 18, 16, 19, 20) have the atoms that form potential hydrogen bonds with Asp320, E319, or G318 colored in blue. The common 2(1H)-pyridone core is highlighted with a gray box in structure 15.
Figure 3. NRP-1 hydrophobic box.
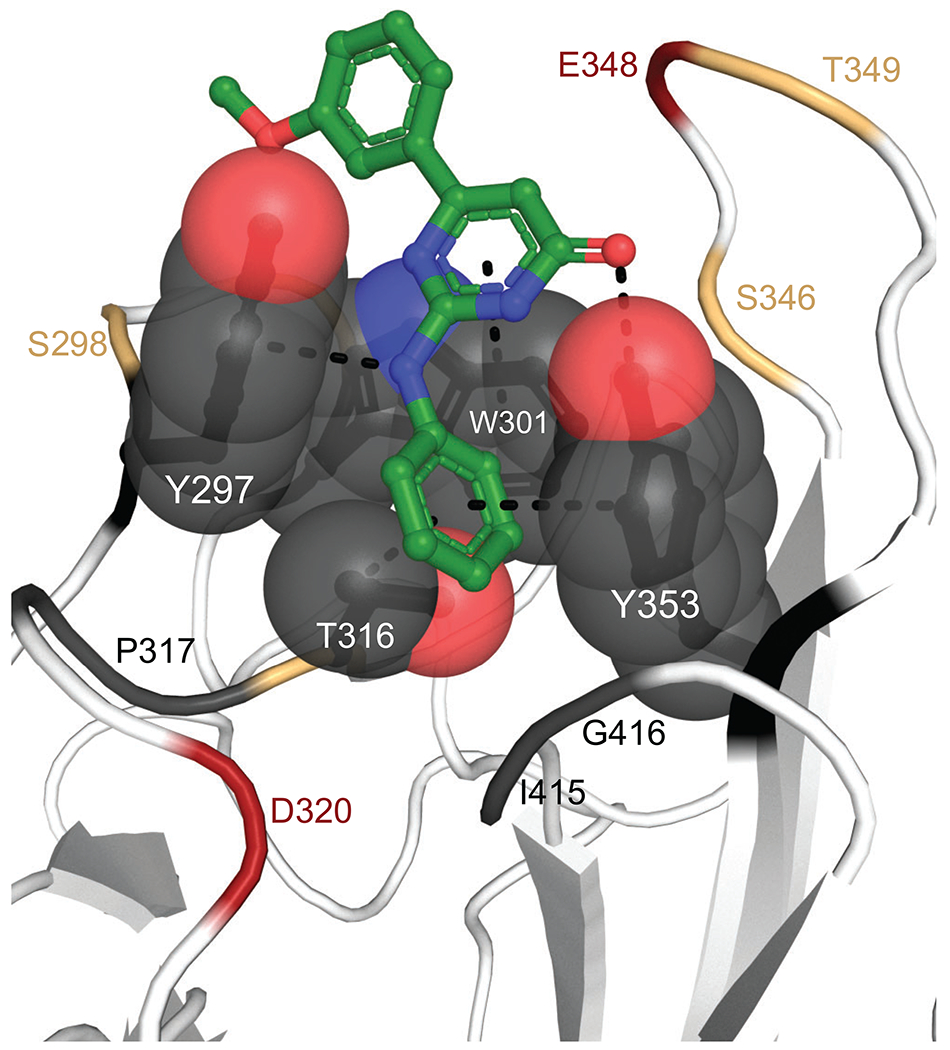
The hydrophobic groove formed by residues Tyr297, Tyr253, Trp301, and Thr316 are shown as gray spheres. Compound 1 is shown as green sticks and contacts with hydrophobic box residues within 5 Å shown as dashes. Remaining binding site residues colored red if acidic, yellow if polar, and gray if hydrophobic.
From this central position, the hits extend out of the aryl box in two general binding modes, which we refer to as Mode I (Figure 4A) and Mode II (Figure 4B). Interestingly, it is the carbonyl group of the core lactam in compounds 1-12, 15, 19-20 that makes potential hydrogen bonds with the hydroxyl groups of Thr349 and Tyr353 (Figure 4C, D) whereas in 14, 16-18 the carbonyl in the carboxylic group makes these contacts, and in 13, 19, 20 the carboxyl makes contacts with S346, T349, and Y353 (Figure 4E). Thus, all of compounds are able to partially mimic the terminal carboxyl contacts that are considered to be critical for anchoring the terminal Arg of CendR peptides13, 42, 49–53, 55–64 (Figure 1C) or known small molecule mimetics and inhibitors that contain a terminal guanidyl from Arg moiety and carboxylic group65, 72, 73.
Figure 4. Hit compound poses in the NRP-1 b1-domain pocket.
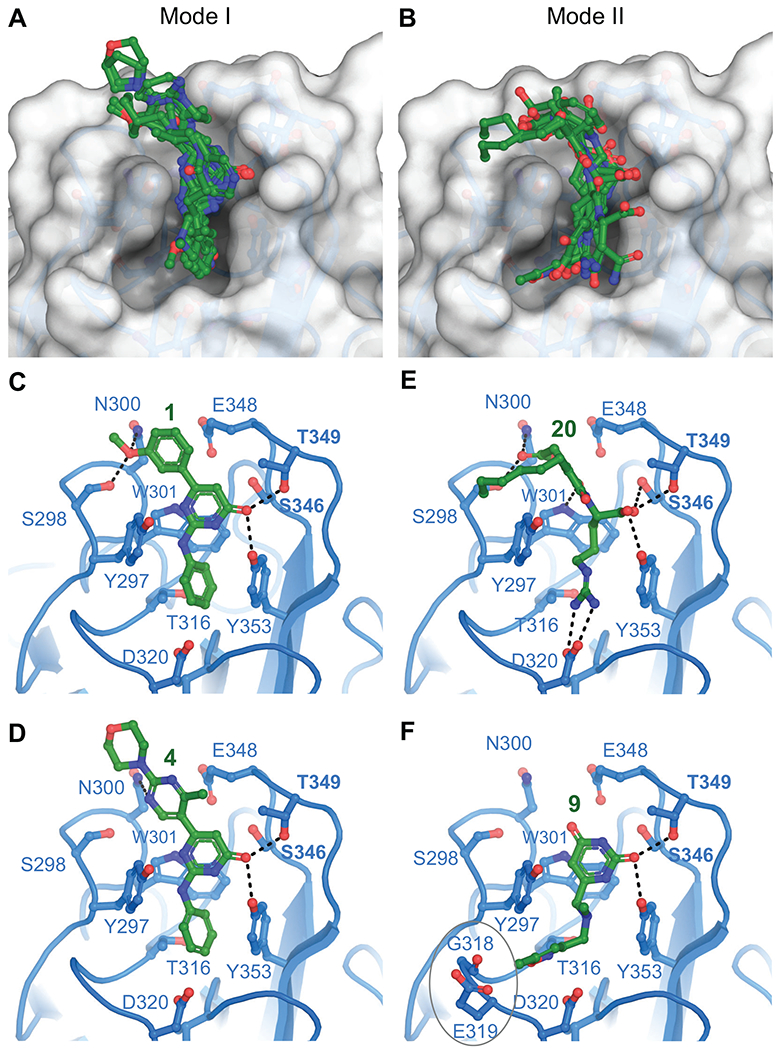
A. Overlay of hits that adopt binding Mode I. B. Overlay of hits that adopt binding Mode II. Polar interactions for representative compounds are shown as dashes for Mode I in C. (compound 1) and D. (compound 4) and for Mode II in E. (compound 20) and F. (compound 9).
All of the hit molecules occupying Mode I are synthetic compound chemotypes, including 11, 12, and 15 from the NC1 and NC2 libraries, as noted above. Functional groups in 1, 2, 4, 5, 10, 12, 13 are involved in one or more potential hydrogen bond contacts with polar side chains Ser298 and Asn300 (Figure 4C, D). No other heteroatoms nor N-H hydrogen in pyrimidone (1, 4, 5, 10, 11), 2-pyrimidone (2, 3, 6), or 4H-1,2,4-triazin-5-one (7, 8) scaffolds are seemingly engaged in productive binding. Such “silent” polar sites provide the opportunity for replacement and optimization of ADME properties (e.g., oral absorption, systemic/CNS distribution) of these compounds. Interestingly, upon detailed analysis of hydrogen-bond patterns, we noticed that compounds in series 1-6 feature H-donor/H-acceptor topology, a canonical feature of kinase inhibitors, including the presence of hydrophobic residues found in ATP mimics. Potential consequences are discussed further below. Nevertheless, such features warrant the inclusion of kinase selectivity panels in the optimization stage. While kinase activity might be a feature to optimize out, our validation experiments do support binding of compounds from our series to the CendR site on NRP-1 (discussed below).
Mode II, with the exception of compound 9, features skeletons of natural compounds. The functional groups in molecules 14 and 16-20 possess extensions toward the base of the pocket that form ionic or hydrogen bond contacts to residue Asp320 (Figure 4E). Furthermore, compound 9 extends toward the open region of the binding pocket bordered by Gly318 and Glu319 (Figure 4F). In preliminary SAR for compound 9, we found that augmenting these interactions by the replacement of 5-methylisoxazole with 5-aminopyrazole (9a, Figure 2) led to an improvement in the Glide XP Gscore of 1.4 kcal/mol. Notably, compound 17, in the series of esters of 3,4-dihydroxycinnamic acid , showed a robust disruption of the VEGF-A/NRP-1 interaction (IC50 = 2.1 nM). Unfortunately, the limited commercial availability of analogs did not permit direct comparison within Series 7. On the other hand, two-component synthesis of dihydroxycinnamic esters is well established and will allow rational expansion of series SAR. Exploration of the interaction patterns observed in both binding modes is expected to improve binding affinity and compound selectivity.
We note that molecules 13 and 16, while having the key pharmacophores present, had their geometry altered during the ligand preparation, likely a result of missing or incorrect chiral information in the COCONUT library, a known potential issue74. The hydroxycinnamyl group in 13 is present in the less stable Z-conformation and the chiral center at phenylalanine in 16 has inverted to (R)-configuration. Such alterations made both analogs unavailable from commercial sources of natural products. However, due to the availability of both precursors, derivatives 13 and 16 can be synthesized. Further, we were able to obtain the S-Phe diastereomer of 16, denoted 16a and this compound was included during experimental validation. Finally, the last two compounds (19, 20) are ionic, moderately reactive compounds which are not considered to be drug-like. Nevertheless, since both match the key features of CendR peptides (N-acylarginine), they provide valuable points for SAR.
Comparison to known small molecules
To enable comparison with small molecules reported by others we docked and calculated physico-chemical properties of six compounds that also target the NRP-1 CendR site65–67, 69, 70, 72. Based on chemotypes, we assigned these six molecules into unique series 10-14 (Figure 5). These molecules all exhibited lower docking scores than our hits (Table 1). Moreover, several of compounds A-F feature functional groups known for contributing to suboptimal physico-chemical and ADME properties, such as low solubility of arylsulfonamides and benzamides (C, F), low intestinal absorption due to the ionic character of zwitterions, ammonium and/or guanidinium cations (A, B, D), metal chelation by acylthioureas (C), and the propensity of benzylamines, benzylethers or thiophenes to lower stability in liver microsomes or hepatocytes (E, A, B) (Table 1). Docking poses for all compounds except E adopted Mode II binding.
Figure 5. Structures of selected NRP-1 targeting compounds.
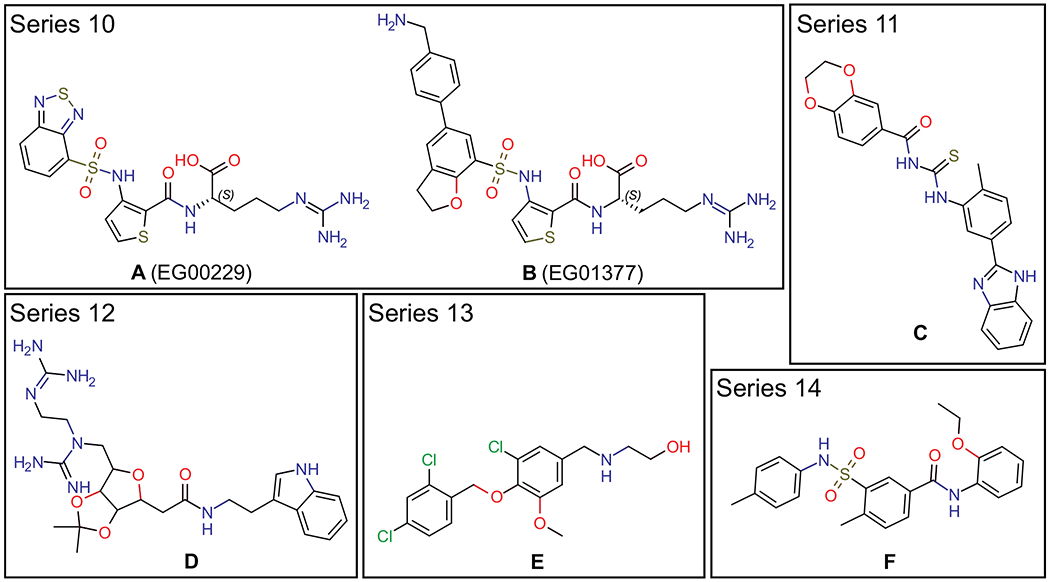
Compound A (EG00229) from Ref65, compound B (EG01377) from Ref72, compound C is “compound 1” from Ref67, compound D is “bis-guanidinylated compound 32” from Ref66, compound E is ChemBridge ID: 7739526 from Ref69, and compound F is “NRPa-308” from Ref70.
Validation of selected hits
We evaluated the ability of hit compounds to interfere with the NRP-1/VEGF-A interaction using an enzyme linked immunosorbent assay (ELISA). We coated plates with the extracellular domain of human NRP-1 (containing the a1a2 and the b1b2 regions) and added a selection of our compounds (based on SAR and commercial availability) to disrupt the NRP-1/VEGF-A interaction. Examples of concentration-response curves of inhibition of the NRP-1/VEGF-A interaction for two compounds (4, 5) are shown in Figure 6A. Compounds 2, 4, 5, 8, 9 and 17 exhibited IC50 <12 nM (Figure 6B). In comparison, EG00229 inhibited the NRP-1/VEGF-A interaction with an IC50 value of 930 nM in our cell-free assay (Figure 6B). Thus, five of the synthetic and one of the natural compounds significantly inhibited the interaction between VEGF-A and NRP-1, confirming that they compete for binding to the CendR site (Table 1).
Figure 6. Identification of nanomolar inhibitors of the NRP-1/VEGF-A interaction.
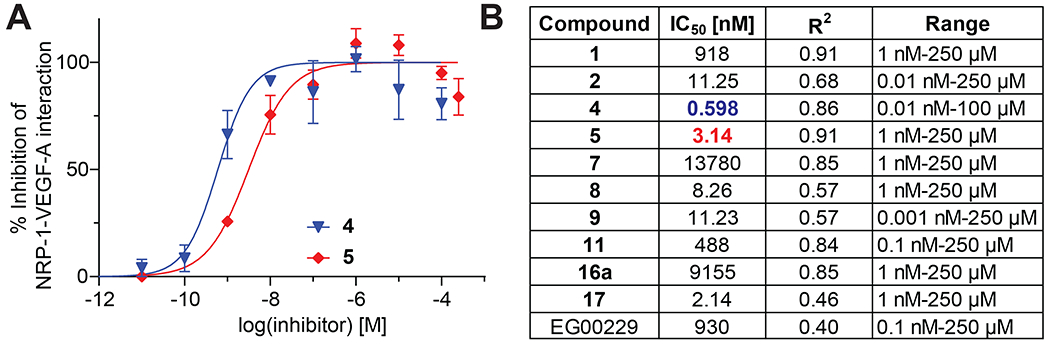
A. Representative concentration curves for inhibition of the NRP-1/VEGF-A interaction assessed by ELISA (see Methods for details). B. Table listing the calculated IC50 values of inhibition in the presence of the indicated compounds. The good of fit (r2) values are also indicated. Compounds were tested in the concentration ranges shown (n=3 replicates per concentration).
Compounds for biochemical evaluation were selected such that each structural feature was complementary to the overall SAR. Unfortunately, compounds 12-16 and 18-20 were not commercially available, though, as noted above, we were able to obtain 16a, the S-Phe diastereoisomer of 16. Since many of those compounds can be synthesized in two to five steps, we intend to make essential representatives during future SAR optimization.
Next, we set out to test the compounds for their capacity to inhibit the activation of the VEGF-A pathway. VEGF-A binding to the dimeric complex of its receptor VEGFR2 and co-receptor NRP-1 triggers phosphorylation of the VEGFR2 cytoplasmic domain at Y1175 (Figure 7A). Using an in-cell western assay, we tested the compounds for their ability to inhibit increased phosphorylation of VEGFR2 by VEGF-A. In this assay, VEGF-A doubled the level of VEGFR2 phosphorylation at Y1175 (Figure 7B, C) which could be blocked by SARS-CoV-2 Spike protein as well as by the reference compound EG00229 (Figure 7C). All 10 of our tested compounds significantly blocked the VEGF-A stimulated increased phosphorylation of VEGFR2 (Figure 7C). In the absence of stimulation by VEGF-A, only one of the 10 compounds (4) showed significant inhibition of basal VEGFR2 phosphorylation.
Figure 7. Screening of NRP-1/VEGF-A165 inhibitors by in cell western.
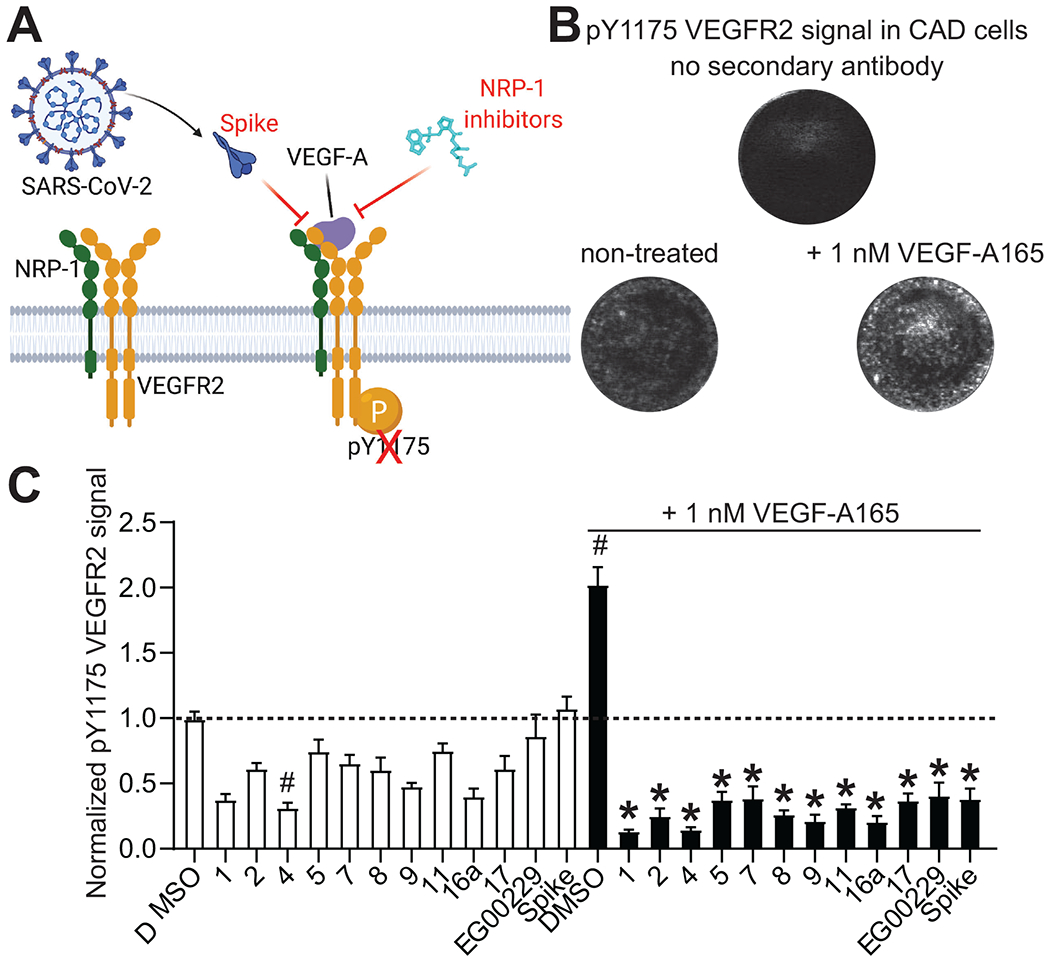
A. Schematic of VEGF-A triggered phosphorylation of VEGF-R2. B. Representative micrographs of Cathecholamine A differentiated (CAD) cells showing the lack of signal in controls with omission of the secondary antibody. Phosphorylated VEGFR2 was increased by the addition of 1 nM VEGF-A165 on the cells. Cells were stained for pY1175 VEGFR2 as a marker of the activation of the pathway by VEGF-A165 (see Methods). C. Bar graph showing the levels of pY1175 VEGFR2 normalized to the quantity of cells in each well. Cells were treated with the NRP-1/VEGF-A165 inhibitors at 12.5 μM or SARS-CoV-2 Spike (100 nM) in combination with 1 nM VEGF-A165 as indicated. # p<0.05 compared to 0.1% DMSO (vehicle) treated cells without VEGF-A165. * p<0.05 compared to 0.1% DMSO + 1 nM VEGF-A165 treated cells, Kruskal-Wallis test with Dunn’s multiple test correction (n=7 replicates per condition). Data was analyzed by a repeated measures one-way analysis of variance (post hoc: Dunnett’s), *p<0.05.
As mentioned above, the donor/acceptor pattern featured in our compounds does exhibit features consistent with hinge-binding groups of kinase inhibitors. It is thus vital that kinase selectivity be addressed during the optimization of these hits. To address possible interaction in kinase active site, we ran a substructure search against known ATP site binders using the KLIFS database79. We were encouraged to find only one structural match (out of 3,447 ligands) of 2-aminopyrimidones (Series 1) having a similar hinge-binding motif in IRAK4 kinase (4ztm.pdb). The absence of substitution at position 5 of compounds 1, 4, 5 (Figure 2) would lead to a lack of important hydrophobic contacts in the gate area should the chemotypes bind in a similar fashion.
Finally, we screened the compounds for antiviral activity using a GFP-expressing vesicular stomatitis virus (VSV) recombinant protein, encoding the SARS-CoV-2 spike protein rather than the native envelope glycoprotein80. This VSV-eGFP-SARS-CoV-2 mimics SARS-CoV-2 entry in a convenient BSL2 platform to assess SARS-CoV-2 Spike-dependency. Vero-E6-TMPRSS2 cells, which overexpress the transmembrane serine protease 2 (TMPRSS2)80, were infected in the presence of individual compounds or DMSO vehicle. GFP fluorescence was measured 36 hours post infection by automated microscopy (Figure 8). Two compounds (1 and 5) displayed >50% inhibition of VSV-eGFP-SARS-CoV-2 entry into cells while another compound (17) demonstrated ~15% inhibition. Spike inhibited viral activity by ~35% while the known NRP-1 inhibitor EG00229 was ineffective in this assay.
Figure 8. Screening for VSV-eGFP-SARS-CoV-2 inhibition.
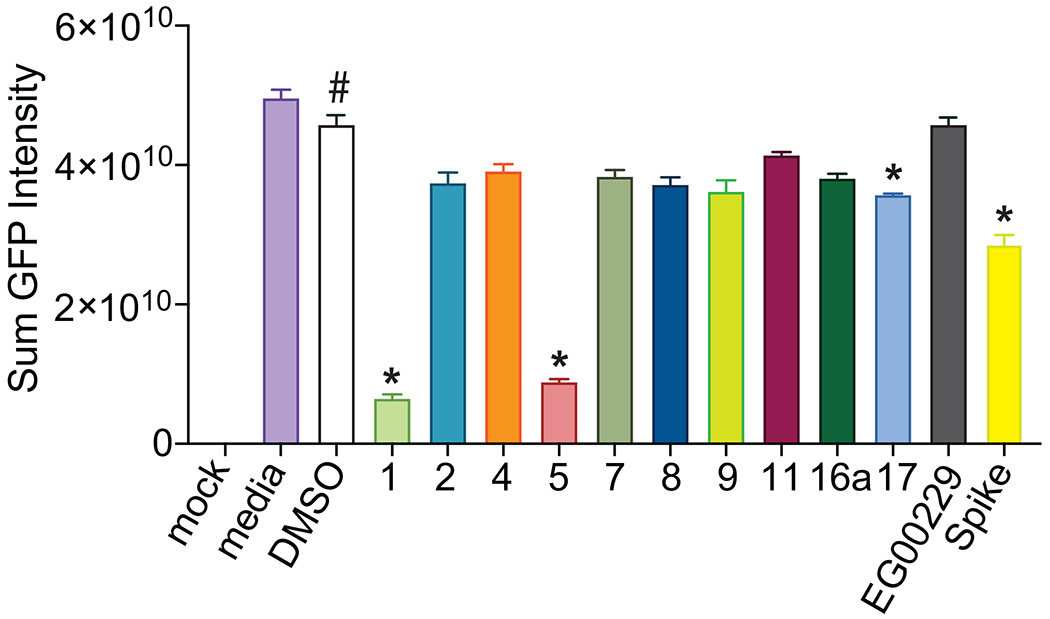
Compounds were screened at 25 μM for inhibition of VSV-eGFP-SARS-CoV-2 infection of Vero-E6-TMPRSS2 cells. Recombinant Spike S1 domain was included at 68 nM. Cells were infected for 36 h prior to live cell automated microscopy and quantification of sum GFP fluorescence intensity, normalized to cell count by HCS CellMask Blue, was measured and for each well and plotted with Prism 6. Results are presented as mean intensity ± SEM, # P <0.05 vs. mock; *P < 0.05 versus DMSO (n=3 replicates). Data was analyzed by a one-way analysis of variance (post hoc: Dunnett’s), *p<0.05.
Initial inspection of docking scores (Table 1) reveals no clear correlation between structural subtype and extent of the inhibition of NRP-1/VEGF-A interaction. However, the ELISA IC50 data suggest that the 2-aminopyrimidones, 6-alkyl-2-pyrimidones, and 3-aminotriazin-5-ones of Series 1, 2, 3, and 4 show the most robust inhibitory response. Most importantly, when appropriately decorated, compounds 4, 5, 8, 2, and 9 were found to disrupt the VEGF-A/NRP-1 interaction more effectively than EG00229. As could be expected based on the model, additional functional groups at positions 6 in Series 1 (4, 5) or 4 in Series 2 (2) (Figure 2) were essential for increasing affinity. On the other hand, the carbonyl group at position 4 in Series 4 (9) was sufficient to maintain the inhibitory activity of this pyrimidine-2,4-dione. Such a trend is important for designing new analogs that will expand on underexplored scaffolds (Series 3 or compound 11). In addition, the natural product compound 17, an ester of 3,4-dihydroxycinnamic acid which docked in binding in Mode II, was also more effective in disrupting the VEGF-A/NRP-1 interaction than EG00229. As discussed below, we intend to improve binding and inhibition by borrowing structural features from Modes I and II (e.g., as shown in compound 9a).
Pharmacophore models
A pharmacophore model was derived from the identified hits, considering both steric and electronic requirements (Figure 9). The most critical features are the aromatic rings A1, A2, and the hydrogen bond acceptor HBA. This HBA is typically a carbonyl oxygen engaged in contacts with the hydroxyl groups of Tyr353 and Thr349. The aryl group in A1 directs the carbonyl oxygen of the HBA toward those residues. Alternatively, A1 can be presented in an edge-to-face contact with Tyr297. There are two additional acceptor sites on the opposite side of the A1 ring, relative to acceptor HBA. These can form hydrogen bonds with the side chain of the Asn300 amide or the Ser298 hydroxyl. The area between the HBA and these two additional acceptors (where the label A1 is located) is expected to accommodate a structural molecule of water10, 14, 71, 72. This water has been proposed to be important in ligand binding as it may bridge interactions to Trp30171. However, our screens were conducted in the absence of water molecules. Nevertheless, we observe that polar groups in several analogs occupy the position of this structural water and get involved in the corresponding hydrogen bond network, suggesting they could displace it. Aromatic ring A2 is sandwiched in the hydrophobic box formed by residues Y353, W301, Y297, and the methyl group of T316. Binding mode II includes A1/HBA, A2, and features an expansion toward polar residues (E319, D320) and additional stabilizing contacts in the lower part of the pocket. It is this area (donor, donor, acceptor) which accepts the guanidino group of the CendR Arg and contains the open lower left pocket seen in Mode I (Figure 4A). Interestingly, we found that the validated hits are consistent with both modes of binding (Table 1), lending support to this model. By connecting all important pharmacophore features, hybrid synthetic molecules can be envisioned that will merge binding modes I and II and extend into the lower left to fully occupy the available binding pocket.
Figure 9. Schematic of pharmacophore models.
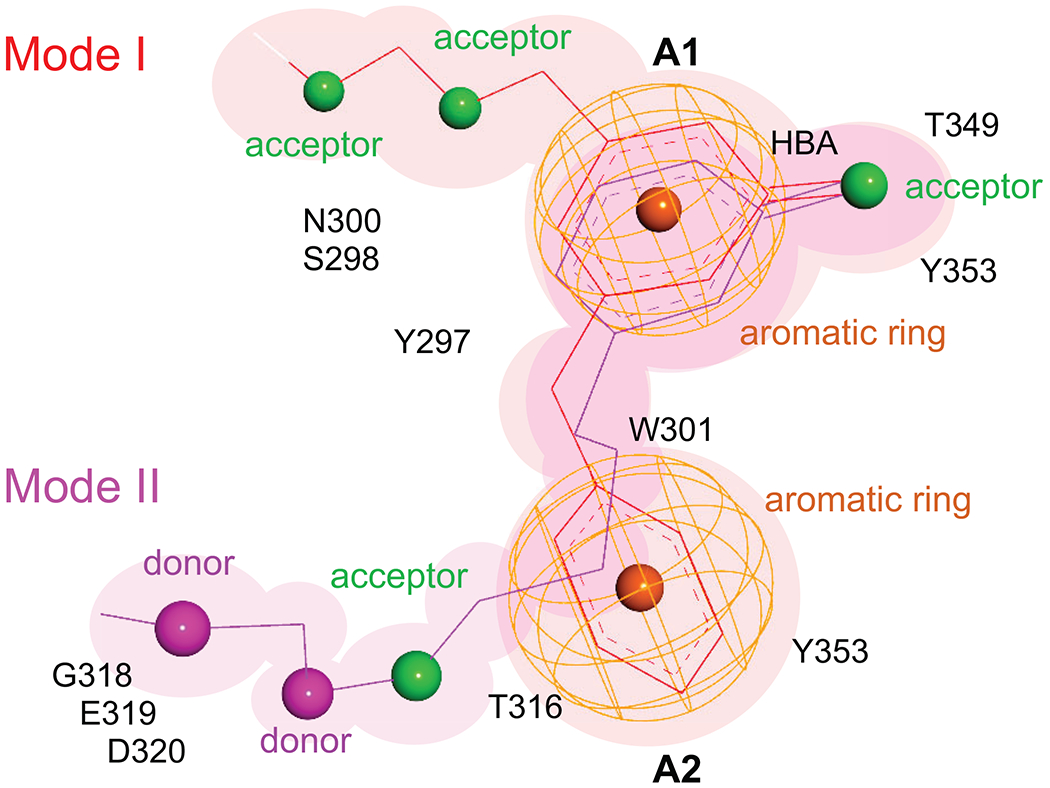
Thin lines indicate bond distances between pharmacophores while the color indicates the mode of binding (Mode I in red, Mode II in magenta). Pharmacophore features are shown in color (acceptors in green, donors in magenta, and aromatic rings as orange mesh). The neighboring or contact protein residues are listed.
Conclusions
From a virtual screen of nearly 0.5 M compounds, we identified nine chemical series comprising small molecules and natural products that target the CendR binding site on NRP-1. All compounds identified in our series fall within ranges of lead-like and/or drug-like molecules which enhances their potential for efficacy in in vitro and in vivo assays. The in silico results predict two modes of binding within the CendR pocket. To guide future drug discovery efforts, we propose a hybrid pharmacophore model that will enable design of small molecules that will maximize the pocket occupancy. ELISA validation experiments confirmed that a subset (about 25%) of our top hits compete with binding of VEGF-A, supporting direct binding to the CendR site on the b1 domain. A second validation experiment revealed that all of the tested hits interfered with VEGF-A induced phosphorylation of VEGFR2. While such interference might suggest a general inhibition of the VEGFR2 kinase domain, the probability of all different chemotypes to effectively block the ATP site is marginal. Also, as discussed above, the substructure similarity of our series to known kinase inhibitors found only one partial match. Moreover, visual inspection of all known VEGFR2 (KDR) inhibitors showed no similarity to any chemotypes of Series 1-8. In a third experiment, two of the CendR-blocking compounds inhibited Spike-dependent infection of cells by VSV-eGFP-SARS-CoV-2 and may have potential for further development, although additional studies are needed to understand their antiviral mechanisms and involvement of NRP-1 and ACE-2 receptors. Since the VEGF-A/NRP-1 signaling pathway participates in multiple pathologies, including neuropathic pain and cancer, our series of lead compounds represent a first step in a renewed effort to develop small molecule inhibitors for the treatment of these diseases.
Finally, we mention one additional interesting aspect of this system that is still being explored. Heparin, the widely used anticoagulant drug is routinely used for hospitalized SARS-CoV-2 patients to lower the probability of blood clothing and embolism81. It is also known that heparin prevents infection by a range of viruses82 and has been reported to inhibit invasion by SARS-CoV-2 in cell-based assays83, 84. Further, it has recently been shown that binding of heparan sulfate to the SARS-CoV-2 Spike protein stabilizes the open conformation of the receptor binding domain and acts as a co-receptor for interaction with ACE284. Heparin is known to be a required co-receptor for VEGF-A signaling85, 86 and NRP-1 also binds heparin, mainly through the b1b2 domain, through sites distal to the CendR pocket85, 87. This raises the possibility that the interaction of SARS-CoV-2 Spike with NRP-1 is also facilitated by heparan sulfate and invites speculation of a potential synergistic effect of heparin and NRP-1 inhibitors as an efficacious drug combination to prevent viral entry.
Methods
Preparation of receptor protein and grid for virtual screening
Preparation and virtual screening steps were conducted using Schrödinger Release 2019-3 (Schrödinger, LLC, New York, NY, 2020). The highest resolution structure of the NRP-1 b1 domain was selected for docking (PDB ID: 6fmc)72. This structure was prepared using the Protein Preparation Wizard88 to remove all water molecules and alternate conformations, add and refine hydrogen atoms, and conduct restrained minimization (OPLS3e force field, convergence to 0.30 Å). There were no residues with alternate conformations within the binding pocket. A 20x20x20 Å grid box was centered on the co-crystallized inhibitor EG01377 to target the VEGF-A165 site. An optional, symmetric constraint was generated that required hit compounds to form a hydrogen bond to the side-chain of Asp 320.
Screening libraries
The synthetic compound library (DIV) was obtained by combining ChemBridge Diversity Core and Express sets of drug-like compounds. These were prepared for screening in LigPrep using the OPLS3e force field, neutral ionization, desalting, and tautomer generation. If specified, chirality centers were maintained, otherwise up to three chiral variations were generated per atom and ligand. This library contained a total of 210,677 compounds (293,251 conformers). The COlleCtion of Open Natural producTs (COCONUT) set of open-access natural compounds74 was downloaded from https://zenodo.org/record/3778405#.Xs1D6mhKiUk (on 5/26/20) and prefiltered by excluding compounds with molecular weight ≥ 500 Da and alogP ≥ 5. LigPrep settings were the same as for the DIV set and the resulting library (NC1) consisted of 257,166 natural compounds (50,686 conformers). The smaller natural compound library (NC2) library was a curated set of 20,088 natural compounds (23,846 conformers) originally obtained from ZINC1575. The NC2 library had some overlap with the NC1 library, but nevertheless produced useful results.
Virtual screening and scoring
Virtual screens were run for each library against the VEGF-A165 binding site of NRP-1 using the Glide virtual screening workflow (Schrödinger, LLC, New York, NY, 2020)89. For the DIV and NC1 libraries, the default docking settings were accepted, with 10% of compounds at each stage (high-throughput virtual screen, standard precision docking, extra precision docking) resulting in 293 hits for the DIV library and 550 hits for the NC1 library. Because the NC2 library was smaller, it was set to retain 25%, 20%, 15% of the hits at each stage, resulting in 152 hits. The virtual screens were first run without and then with the use of the Asp 320 constraint, but only the constrained hits from NC2 were retained due to strained conformations and lower docking scores for the DIV and NC1 screens. Thus, a total number of 1,147 virtual hit compounds were obtained from 4 screens.
Docking of known NRP-1 targeting compounds
Representatives of known compound series65–67, 69, 70, 72 were prepared for screening in LigPrep using the OPLS3e force field, neutral ionization, desalting, and tautomer generation. Docking was run against the VEGF-A165 binding site of NRP-1 using Glide XP (Schrödinger, LLC, New York, NY, 2020)89.
Compound property calculations
The following physico-chemical properties were calculated using RDKit90: molecular weight in Daltons (Mw, Da), partition coefficient (clogP), number of hydrogen-bond donors and acceptors (HB-D, HB-A), total number of N-H and OH groups (NHOH), number of rotatable bonds (RotB), and total polar surface area (TPSA, Å2). To estimate compound solubility, calculated logS (M) at pH=7.4 was obtained using ChemAxon Aqueous solubility module91.
ELISA-based NRP1-VEGF-A165 protein binding assay
The assay was performed as described previously92. Plates (96-well, Nunc Maxisorp; Thermo Fisher Scientific, Waltham, MA, USA) were coated with human Neuropilin-1-Fc (200 ng per well, Cat# 50-101-8343, Fisher, Hampton, NH) and incubated at 4°C overnight. The following day, the plates were washed and blocked with 3% BSA in PBS to minimize non-specific adsorptive binding to the plates. EG00229 (Cat#6986, Tocris) or the indicated compounds were added at different concentrations and incubated for 30 min at room temperature prior to adding biotinylated human VEGF-A165 (Cat#BT293, R&D systems) at 20 nM with 4 μg/ml of heparin. As a negative control, some wells received PBS containing 3% BSA and as a positive control only hVEGF-A165 was used. The plates were incubated at room temperature with shaking for 2 h. Next, the plates were washed with PBS 0.05% Tween to eliminate unbound protein. Bound biotinylated VEGF was detected by streptavidin-HRP (Cat#016-030-084, Jackson immunoResearch). Tetramethylbenzidine (Cat#DY999, R&D Systems, St. Louis, MO) was used as the colorimetric substrate. The optical density of each well was determined immediately, using a microplate reader (Multiskan Ascent; Thermo Fisher Scientific) set to 450 nm with a correction wavelength of 570 nm. Percent inhibition was calculated by the following formula:
where S is the optical density measured in the wells with the compounds, N is the optical density measured in the negative control wells, and P is the optical density in the positive control wells. The half-maximal inhibitory concentration (IC50) value was calculated using a non-linear regression function with GraphPad Prism Version 9. The data are presented as log (inhibitor) [M] versus normalized response-variable slope. Data are the means of three independent experiments.
In cell western for detecting inhibition of VEGFR2 activation by VEGF-A165
Mouse neuron derived Cathecholamine A differentiated CAD (ECACC, Cat# 08100805)93 were grown in standard cell culture conditions, 37 °C in 5% CO2. All media was supplemented with 10% fetal bovine serum (Hyclone) and 1% penicillin/streptomycin sulfate from 10,000 μg/ml stock. CAD cells were maintained in DMEM/F12 media. Cells were plated in a 96 well plate and left overnight. The next day, indicated compounds (at 12.5μM), or SARS-CoV-2 Spike (100 nM, S1 domain) were added in CAD cell complete media supplemented with 1 nM of mouse VEGF-A165 (Cat# RP8672, Invitrogen) and left at 37°C for 1 hour. Media was removed and the cells rinsed three times with PBS before fixation using ice cold methanol (5 min). Methanol was removed and cells were left to dry completely at room temperature. Anti-VEGFR2 pY1175 was used to detect the activation of the pathway triggered by VEGF-A165 in the cells. The antibody was added in PBS containing 3% BSA and left overnight at room temperature. The cells were washed three times with PBS and then incubated with Alexa Fluor® 790 AffiniPure Goat Anti-Rabbit IgG (Cat# 111-655-144, Jackson immunoResearch) in PBS, 3% BSA for 1 hour at room temperature. Cells were washed three times with PBS and stained with DAPI. Plates were imaged on an Azure Sapphire apparatus. Wells that did not receive the primary antibody were used a negative control. The signal was normalized to the cell load in each well (using DAPI) and to control wells not treated with VEGF-A165.
Cell and viral culture
Vero-E6-TMPRSS2 cells were cultured in high glucose DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were supplemented with 20 μg/mL blasticidin (Invivogen, ant-bl-1) to maintain stable expression of TMPRSS2 during routine culture. Cells were maintained at 37°C with 5% CO2 and passaged every 2-3 days. These African green monkey kidney cells express NRP-1 which is 99.3% similar within its b1 domain to the human NRP-1. Homology models (not shown) also reveal no differences in NRP-1 passage between these species. Infectious VSV-eGFP-SARS-CoV-2 stock was a generous gift from Sean P.J. Whelan (Washington University, St. Louis, MO, USA). VSV-eGFP-SARS-CoV-2 was passaged once by infecting Vero-E6-TMPRSS2 cells at MOI = 0.01 in DMEM + 2% FBS and 1% penicillin/streptomycin for 72 hr at 34°C. Cell-free supernatant was collected and concentrated 10-fold through Amicon-Ultra 100 kDa MWCO spin filter units (Millipore UFC905008) prior to aliquoting and storage at −80°C. Titer was determined to be approximately 1x107 PFU/mL as determined by median tissue culture infectious dose (TCID50) assay on Vero-E6-TMPRSS2 cells.
Screening compounds for VSV-eGFP-SARS-CoV-2 inhibition
African green monkey kidney (Vero)-E6-TMPRSS2 cells were plated at 15,000 cells per well in black/clear bottom 96 well tissue culture plates (Thermo Fisher Scientific 165305). The next day cells were infected ± 0.001% DMSO, 25 μM compounds, or 68 nM recombinant Spike protein, at an MOI of 0.05 in 100 μL DMEM + 10% FBS and 1% penicillin/streptomycin for 36 hr at 37°C prior to live cell fluorescent microscopy on a Nikon Eclipse Ti2 automated microscopy system with 4x objective and 488/532 nm filters. Sum GFP fluorescence intensity, normalized to cell count by HCS CellMask Blue (Thermo #H32720), was measured and for each well and plotted with Prism 6 (GraphPad Software). Significant differences were determined by a repeated measures one-way ANOVA followed by multiple comparisons test. An alpha of 0.05 was used to determine the statistical significance of the null-hypothesis.
Supplementary Material
Acknowledgments
We thank Sean P.J. Whelan and Paul W. Rothlauf for generously sharing the VSV-eGFP-SARS-CoV-2 clone and the Vero-E6-TMPRSS2 cells.
Funding:
Supported by NINDS (NS098772 (R.K.)), NIDA (DA042852, R.K.), NIGMS (GM136853, S.K.C.), a Tech Launch Arizona asset development award (UA21-038, R.K., A.M., S.P.M.) and a UA COVID-19 seed grant (002196, S.K.C.).
Abbreviations
- ACE-2
angiotensin converting enzyme-2
- ADME
absorption, distribution, metabolism, and excretion
- BBB
blood brain barrier
- CNS
central nervous system
- COCONUT
COlleCtion of Open NatUral producTs
- COVID-19
coronavirus disease 2019
- ELISA
enzyme linked immunosorbent assay
- NC
natural compound
- NRP-1
Neuropilin 1
- PDB
protein data bank
- Ro5
Lipinski rule of 5
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- VEGF-A
vascular endothelial growth factor-A
- VSV
vesicular stomatitis virus
Footnotes
Competing interests
R. Khanna is the co-founder of Regulonix LLC – a wholly owned subsidiary of Regulonix Holding Inc. – a company developing non-opioids drugs for chronic pain. In addition, R. Khanna has patents US10287334 and US10441586 issued to Regulonix LLC. R. Khanna, A. Moutal, and S. Perez-Miller have filed a provisional application on compositions and methods for blocking neuropilin receptor 1 for the treatment of pain and prevention of viral entry (USPTO Application No. 63/117,336). The other authors declare no competing financial interests.
Supporting Information
List of the top 100 hits from the virtual screen sorted by Glide docking score in csv format.
Data and materials availability
All data is available in the main text, figures, and supplementary materials.
References
- 1.Dong E, Du H, and Gardner L (2020) An interactive web-based dashboard to track COVID-19 in real time, Lancet Infect Dis 20, 533–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, et al. (2020) SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor, Cell 181, 271–280 e278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Q, Zhang Y, Wu L, Niu S, Song C, Zhang Z, Lu G, Qiao C, Hu Y, Yuen KY, et al. (2020) Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2, Cell 181, 894–904 e899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yan R, Zhang Y, Li Y, Xia L, Guo Y, and Zhou Q (2020) Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2, Science 367, 1444–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cantuti-Castelvetri L, Ojha R, Pedro LD, Djannatian M, Franz J, Kuivanen S, van der Meer F, Kallio K, Kaya T, Anastasina M, et al. (2020) Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity, Science 370, 856–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daly JL, Simonetti B, Klein K, Chen KE, Williamson MK, Antón-Plgáro C, Shoemark DK, Simón-Gracia L, Bauer M, Hollandi R, et al. (2020) Neuropilin-1 is a host factor for SARS-CoV-2 infection, Science 370, 861–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies J, Randeva HS, Chatha K, Hall M, Spandidos DA, Karteris E, and Kyrou I (2020) Neuropilin1 as a new potential SARSCoV2 infection mediator implicated in the neurologic features and central nervous system involvement of COVID19, Mol Med Rep 22, 4221–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pellet-Many C, Frankel P, Jia H, and Zachary I (2008) Neuropilins: structure, function and role in disease, Biochem J 411, 211–226. [DOI] [PubMed] [Google Scholar]
- 9.Plein A, Fantin A, and Ruhrberg C (2014) Neuropilin regulation of angiogenesis, arteriogenesis, and vascular permeability, Microcirculation 21, 315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Appleton BA, Wu P, Maloney J, Yin J, Liang WC, Stawicki S, Mortara K, Bowman KK, Elliott JM, Desmarais W, et al. (2007) Structural studies of neuropilin/antibody complexes provide insights into semaphorin and VEGF binding, The EMBO journal 26, 4902–4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yelland T, and Djordjevic S (2016) Crystal Structure of the Neuropilin-1 MAM Domain: Completing the Neuropilin-1 Ectodomain Picture, Structure 24, 2008–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacob L, Sawma P, Garnier N, Meyer LA, Fritz J, Hussenet T, Spenle C, Goetz J, Vermot J, Fernandez A, et al. (2016) Inhibition of PlexA1-mediated brain tumor growth and tumor-associated angiogenesis using a transmembrane domain targeting peptide, Oncotarget 7, 57851–57865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teesalu T, Sugahara KN, Kotamraju VR, and Ruoslahti E (2009) C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration, Proceedings of the National Academy of Sciences of the United States of America 106, 16157–16162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee CC, Kreusch A, McMullan D, Ng K, and Spraggon G (2003) Crystal structure of the human neuropilin-1 b1 domain, Structure 11, 99–108. [DOI] [PubMed] [Google Scholar]
- 15.Parker MW, Xu P, Li X, and Vander Kooi CW (2012) Structural basis for selective vascular endothelial growth factor-A (VEGF-A) binding to neuropilin-1, The Journal of biological chemistry 287, 11082–11089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Djordjevic S, and Driscoll PC (2013) Targeting VEGF signalling via the neuropilin co-receptor, Drug discovery today 18, 447–455. [DOI] [PubMed] [Google Scholar]
- 17.Hulse RP. (2017) Role of VEGF-A in chronic pain, Oncotarget 8, 10775–10776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Llorian-Salvador M, and Gonzalez-Rodriguez S (2018) Painful Understanding of VEGF, Frontiers in pharmacology 9, 1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peach CJ, Mignone VW, Arruda MA, Alcobia DC, Hill SJ, Kilpatrick LE, and Woolard J (2018) Molecular Pharmacology of VEGF-A Isoforms: Binding and Signalling at VEGFR2, Int J Mol Sci 19, 1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Cesare Mannelli L, Tenci B, Micheli L, Vona A, Corti F, Zanardelli M, Lapucci A, Clemente AM, Failli P, and Ghelardini C (2018) Adipose-derived stem cells decrease pain in a rat model of oxaliplatin-induced neuropathy: Role of VEGF-A modulation, Neuropharmacology 131, 166–175. [DOI] [PubMed] [Google Scholar]
- 21.Hulse RP, Drake RA, Bates DO, and Donaldson LF (2016) The control of alternative splicing by SRSF1 in myelinated afferents contributes to the development of neuropathic pain, Neurobiology of disease 96, 186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oltean S, Gammons M, Hulse R, Hamdollah-Zadeh M, Mavrou A, Donaldson L, Salmon AH, Harper SJ, Ladomery MR, and Bates DO (2012) SRPK1 inhibition in vivo: modulation of VEGF splicing and potential treatment for multiple diseases, Biochemical Society transactions 40, 831–835. [DOI] [PubMed] [Google Scholar]
- 23.Hulse RP, Beazley-Long N, Hua J, Kennedy H, Prager J, Bevan H, Qiu Y, Fernandes ES, Gammons MV, Ballmer-Hofer K, et al. (2014) Regulation of alternative VEGF-A mRNA splicing is a therapeutic target for analgesia, Neurobiology of disease 71, 245–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joseph EK, Green PG, Bogen O, Alvarez P, and Levine JD (2013) Vascular endothelial cells mediate mechanical stimulation-induced enhancement of endothelin hyperalgesia via activation of P2X2/3 receptors on nociceptors, The Journal of neuroscience : the official journal of the Society for Neuroscience 33, 2849–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takano S, Uchida K, Inoue G, Matsumoto T, Aikawa J, Iwase D, Mukai M, Miyagi M, and Takaso M (2018) Vascular endothelial growth factor expression and their action in the synovial membranes of patients with painful knee osteoarthritis, BMC Musculoskelet Disord 19, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verheyen A, Peeraer E, Nuydens R, Dhondt J, Poesen K, Pintelon I, Daniels A, Timmermans JP, Meert T, Carmeliet P, et al. (2012) Systemic anti-vascular endothelial growth factor therapies induce a painful sensory neuropathy, Brain : a journal of neurology 135, 2629–2641. [DOI] [PubMed] [Google Scholar]
- 27.Moutal A, Martin LF, Boinon L, Gomez K, Ran D, Zhou Y, Stratton HJ, Cai S, Luo S, Gonzalez KB, et al. (2021) SARS-CoV-2 spike protein co-opts VEGF-A/neuropilin-1 receptor signaling to induce analgesia, Pain 162, 243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cebe Suarez S, Pieren M, Cariolato L, Arn S, Hoffmann U, Bogucki A, Manlius C, Wood J, and Ballmer-Hofer K (2006) A VEGF-A splice variant defective for heparan sulfate and neuropilin-1 binding shows attenuated signaling through VEGFR-2, Cell Mol Life Sci 63, 2067–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roy S, Bag AK, Singh RK, Talmadge JE, Batra SK, and Datta K (2017) Multifaceted Role of Neuropilins in the Immune System: Potential Targets for Immunotherapy, Front Immunol 8, 1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chuckran CA, Liu C, Bruno TC, Workman CJ, and Vignali DA (2020) Neuropilin-1: a checkpoint target with unique implications for cancer immunology and immunotherapy, Journal for ImmunoTherapy of Cancer 8, e000967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarris M, Andersen KG, Randow F, Mayr L, and Betz AG (2008) Neuropilin-1 expression on regulatory T cells enhances their interactions with dendritic cells during antigen recognition, Immunity 28, 402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tordjman R, Lepelletier Y, Lemarchandel V, Cambot M, Gaulard P, Hermine O, and Roméo PH (2002) A neuronal receptor, neuropilin-1, is essential for the initiation of the primary immune response, Nat Immunol 3, 477–482. [DOI] [PubMed] [Google Scholar]
- 33.Grage-Griebenow E, Löseke S, Kauth M, Gehlhar K, Zawatzky R, and Bufe A (2007) Anti-BDCA-4 (neuropilin-1) antibody can suppress virus-induced IFN-alpha production of plasmacytoid dendritic cells, Immunol Cell Biol 85, 383–390. [DOI] [PubMed] [Google Scholar]
- 34.Oussa NA, Dahmani A, Gomis M, Richaud M, Andreev E, Navab-Daneshmand AR, Taillefer J, Carli C, Boulet S, Sabbagh L, et al. (2016) VEGF Requires the Receptor NRP-1 To Inhibit Lipopolysaccharide-Dependent Dendritic Cell Maturation, J Immunol 197, 3927–3935. [DOI] [PubMed] [Google Scholar]
- 35.Lambert S, Bouttier M, Vassy R, Seigneuret M, Petrow-Sadowski C, Janvier S, Heveker N, Ruscetti FW, Perret G, Jones KS, et al. (2009) HTLV-1 uses HSPG and neuropilin-1 for entry by molecular mimicry of VEGF165, Blood 113, 5176–5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang HB, Zhang H, Zhang JP, Li Y, Zhao B, Feng GK, Du Y, Xiong D, Zhong Q, Liu WL, et al. (2015) Neuropilin 1 is an entry factor that promotes EBV infection of nasopharyngeal epithelial cells, Nat Commun 6, 6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo HF, and Vander Kooi CW (2015) Neuropilin Functions as an Essential Cell Surface Receptor, The Journal of biological chemistry 290, 29120–29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoffmann M, Kleine-Weber H, and Pohlmann S (2020) A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells, Mol Cell 78, 779–784 e775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang WC, Dennis MS, Stawicki S, Chanthery Y, Pan Q, Chen Y, Eigenbrot C, Yin J, Koch AW, Wu X, et al. (2007) Function blocking antibodies to neuropilin-1 generated from a designed human synthetic antibody phage library, J Mol Biol 366, 815–829. [DOI] [PubMed] [Google Scholar]
- 40.Pan Q, Chanthery Y, Liang WC, Stawicki S, Mak J, Rathore N, Tong RK, Kowalski J, Yee SF, Pacheco G, et al. (2007) Blocking neuropilin-1 function has an additive effect with anti-VEGF to inhibit tumor growth, Cancer Cell 11, 53–67. [DOI] [PubMed] [Google Scholar]
- 41.Weekes CD, Beeram M, Tolcher AW, Papadopoulos KP, Gore L, Hegde P, Xin Y, Yu R, Shih LM, Xiang H, et al. (2014) A phase I study of the human monoclonal anti-NRP1 antibody MNRP1685A in patients with advanced solid tumors, Invest New Drugs 32, 653–660. [DOI] [PubMed] [Google Scholar]
- 42.Kim YJ, Bae J, Shin TH, Kang SH, Jeong M, Han Y, Park JH, Kim SK, and Kim YS (2015) Immunoglobulin Fc-fused, neuropilin-1-specific peptide shows efficient tumor tissue penetration and inhibits tumor growth via anti-angiogenesis, J Control Release 216, 56–68. [DOI] [PubMed] [Google Scholar]
- 43.Kim YJ, Baek DS, Lee S, Park D, Kang HN, Cho BC, and Kim YS (2019) Dual-targeting of EGFR and Neuropilin-1 attenuates resistance to EGFR-targeted antibody therapy in KRAS-mutant non-small cell lung cancer, Cancer Lett 466, 23–34. [DOI] [PubMed] [Google Scholar]
- 44.Ko JH, Kwon HS, Kim B, Min G, Shin C, Yang SW, Lee SW, Lee Y, Hong D, and Kim YS (2020) Preclinical Efficacy and Safety of an Anti-Human VEGFA and Anti-Human NRP1 Dual-Targeting Bispecific Antibody (IDB0076), Biomolecules 10, 919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roth L, Nasarre C, Dirrig-Grosch S, Aunis D, Cremel G, Hubert P, and Bagnard D (2008) Transmembrane domain interactions control biological functions of neuropilin-1, Mol Biol Cell 19, 646–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nasarre C, Roth M, Jacob L, Roth L, Koncina E, Thien A, Labourdette G, Poulet P, Hubert P, Cremel G, et al. (2010) Peptide-based interference of the transmembrane domain of neuropilin-1 inhibits glioma growth in vivo, Oncogene 29, 2381–2392. [DOI] [PubMed] [Google Scholar]
- 47.Arpel A, Gamper C, Spenle C, Fernandez A, Jacob L, Baumlin N, Laquerriere P, Orend G, Cremel G, and Bagnard D (2016) Inhibition of primary breast tumor growth and metastasis using a neuropilin-1 transmembrane domain interfering peptide, Oncotarget 7, 54723–54732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Albrecht C, Appert-Collin A, Bagnard D, Blaise S, Romier-Crouzet B, Efremov RG, Sartelet H, Duca L, Maurice P, and Bennasroune A (2020) Transmembrane Peptides as Inhibitors of Protein-Protein Interactions: An Efficient Strategy to Target Cancer Cells?, Front Oncol 10, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Binetruy-Tournaire R, Demangel C, Malavaud B, Vassy R, Rouyre S, Kraemer M, Plouet J, Derbin C, Perret G, and Mazie JC (2000) Identification of a peptide blocking vascular endothelial growth factor (VEGF)-mediated angiogenesis, The EMBO journal 19, 1525–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jia H, Bagherzadeh A, Hartzoulakis B, Jarvis A, Lohr M, Shaikh S, Aqil R, Cheng L, Tickner M, Esposito D, et al. (2006) Characterization of a bicyclic peptide neuropilin-1 (NP-1) antagonist (EG3287) reveals importance of vascular endothelial growth factor exon 8 for NP-1 binding and role of NP-1 in KDR signaling, J Biol Chem 281, 13493–13502. [DOI] [PubMed] [Google Scholar]
- 51.von Wronski MA, Raju N, Pillai R, Bogdan NJ, Marinelli ER, Nanjappan P, Ramalingam K, Arunachalam T, Eaton S, Linder KE, et al. (2006) Tuftsin binds neuropilin-1 through a sequence similar to that encoded by exon 8 of vascular endothelial growth factor, J Biol Chem 281, 5702–5710. [DOI] [PubMed] [Google Scholar]
- 52.Starzec A, Ladam P, Vassy R, Badache S, Bouchemal N, Navaza A, du Penhoat CH, and Perret GY (2007) Structure-function analysis of the antiangiogenic ATWLPPR peptide inhibiting VEGF(165) binding to neuropilin-1 and molecular dynamics simulations of the ATWLPPR/neuropilin-1 complex, Peptides 28, 2397–2402. [DOI] [PubMed] [Google Scholar]
- 53.Vander Kooi CW, Jusino MA, Perman B, Neau DB, Bellamy HD, and Leahy DJ (2007) Structural basis for ligand and heparin binding to neuropilin B domains, Proc Natl AcadSci U S A 104, 6152–6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Getz JA, Cheneval O, Craik DJ, and Daugherty PS (2013) Design of a cyclotide antagonist of neuropilin-1 and -2 that potently inhibits endothelial cell migration, ACS Chem Biol 8, 1147–1154. [DOI] [PubMed] [Google Scholar]
- 55.Nissen JC, Selwood DL, and Tsirka SE (2013) Tuftsin signals through its receptor neuropilin-1 via the transforming growth factor beta pathway, J Neurochem 127, 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jia H, Aqil R, Cheng L, Chapman C, Shaikh S, Jarvis A, Chan AW, Hartzoulakis B, Evans IM, Frolov A, et al. (2014) N-terminal modification of VEGF-A C terminus-derived peptides delineates structural features involved in neuropilin-1 binding and functional activity, Chembiochem 15, 1161–1170. [DOI] [PubMed] [Google Scholar]
- 57.Grabowska K, Puszko AK, Lipinski PFJ, Laskowska AK, Wilenska B, Witkowska E, and Misicka A (2016) Design, synthesis and in vitro biological evaluation of a small cyclic peptide as inhibitor of vascular endothelial growth factor binding to neuropilin-1, Bioorg Med Chem Lett 26, 2843–2846. [DOI] [PubMed] [Google Scholar]
- 58.Richard M, Chateau A, Jelsch C, Didierjean C, Manival X, Charron C, Maigret B, Barberi-Heyob M, Chapleur Y, Boura C, et al. (2016) Carbohydrate-based peptidomimetics targeting neuropilin-1: Synthesis, molecular docking study and in vitro biological activities, Bioorg Med Chem 24, 5315–5325. [DOI] [PubMed] [Google Scholar]
- 59.Fedorczyk B, Lipinski PFJ, Tymecka D, Puszko AK, Wilenska B, Perret GY, and Misicka A (2017) Conformational latitude - activity relationship of KPPR tetrapeptide analogues toward their ability to inhibit binding of vascular endothelial growth factor 165 to neuropilin-1, J Pept Sci 23, 445–454. [DOI] [PubMed] [Google Scholar]
- 60.Grabowska K, Puszko AK, Lipinski PF, Laskowska AK, Wilenska B, Witkowska E, Perret GY, and Misicka A (2017) Structure-activity relationship study of a small cyclic peptide H-c[Lys-Pro-Glu]-Arg-OH: a potent inhibitor of Vascular Endothelial Growth Factor interaction with Neuropilin-1, Bioorg Med Chem 25, 597–602. [DOI] [PubMed] [Google Scholar]
- 61.Tymecka D, Lipinski PFJ, Fedorczyk B, Puszko A, Wilenska B, Perret GY, and Misicka A (2017) Structure-activity relationship study of tetrapeptide inhibitors of the Vascular Endothelial Growth Factor A binding to Neuropilin-1, Peptides 94, 25–32. [DOI] [PubMed] [Google Scholar]
- 62.Tymecka D, Puszko AK, Lipinski PFJ, Fedorczyk B, Wilenska B, Sura K, Perret GY, and Misicka A (2018) Branched pentapeptides as potent inhibitors of the vascular endothelial growth factor 165 binding to Neuropilin-1: Design, synthesis and biological activity, Eur J Med Chem 158, 453–462. [DOI] [PubMed] [Google Scholar]
- 63.Fedorczyk B, Lipinski PFJ, Puszko AK, Tymecka D, Wilenska B, Dudka W, Perret GY, Wieczorek R, and Misicka A (2019) Triazolopeptides Inhibiting the Interaction between Neuropilin-1 and Vascular Endothelial Growth Factor-165, Molecules 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Puszko AK, Sosnowski P, Tymecka D, Raynaud F, Hermine O, Lepelletier Y, and Misicka A (2019) Neuropilin-1 peptide-like ligands with proline mimetics, tested using the improved chemiluminescence affinity detection method, Medchemcomm 10, 332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jarvis A, Allerston CK, Jia H, Herzog B, Garza-Garcia A, Winfield N, Ellard K, Aqil R, Lynch R, Chapman C, et al. (2010) Small molecule inhibitors of the neuropilin-1 vascular endothelial growth factor A (VEGF-A) interaction, Journal of medicinal chemistry 53, 2215–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Novoa A, Pellegrini-Moise N, Bechet D, Barberi-Heyob M, and Chapleur Y (2010) Sugar-based peptidomimetics as potential inhibitors of the vascular endothelium growth factor binding to neuropilin-1, Bioorg Med Chem 18, 3285–3298. [DOI] [PubMed] [Google Scholar]
- 67.Borriello L, Montes M, Lepelletier Y, Leforban B, Liu WQ, Demange L, Delhomme B, Pavoni S, Jarray R, Boucher JL, et al. (2014) Structure-based discovery of a small non-peptidic Neuropilins antagonist exerting in vitro and in vivo anti-tumor activity on breast cancer model, Cancer Lett 349, 120–127. [DOI] [PubMed] [Google Scholar]
- 68.Liu WQ, Megale V, Borriello L, Leforban B, Montes M, Goldwaser E, Gresh N, Piquemal JP, Hadj-Slimane R, Hermine O, et al. (2014) Synthesis and structure-activity relationship of non-peptidic antagonists of neuropilin-1 receptor, Bioorg Med Chem Lett 24, 4254–4259. [DOI] [PubMed] [Google Scholar]
- 69.Starzec A, Miteva MA, Ladam P, Villoutreix BO, and Perret GY (2014) Discovery of novel inhibitors of vascular endothelial growth factor-A-Neuropilin-1 interaction by structure-based virtual screening, Bioorg Med Chem 22, 4042–4048. [DOI] [PubMed] [Google Scholar]
- 70.Liu WQ, Lepelletier Y, Montes M, Borriello L, Jarray R, Grepin R, Leforban B, Loukaci A, Benhida R, Hermine O, et al. (2018) NRPa-308, a new neuropilin-1 antagonist, exerts in vitro anti-angiogenic and anti-proliferative effects and in vivo anti-cancer effects in a mouse xenograft model, Cancer Lett 414, 88–98. [DOI] [PubMed] [Google Scholar]
- 71.Mota F, Fotinou C, Rana RR, Chan AWE, Yelland T, Arooz MT, O’Leary AP, Hutton J, Frankel P, Zachary I, et al. (2018) Architecture and hydration of the arginine-binding site of neuropilin-1, The FEBS journal 285, 1290–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Powell J, Mota F, Steadman D, Soudy C, Miyauchi JT, Crosby S, Jarvis A, Reisinger T, Winfield N, Evans G, et al. (2018) Small Molecule Neuropilin-1 Antagonists Combine Antiangiogenic and Antitumor Activity with Immune Modulation through Reduction of Transforming Growth Factor Beta (TGFbeta) Production in Regulatory T-Cells, Journal of medicinal chemistry 61, 4135–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Conole D, Chou YT, Patsiarika A, Nwabo V, Dimitriou E, Soudy C, Mota F, Djordjevic S, and Selwood DL (2020) Discovery of a novel fluorescent chemical probe suitable for evaluation of neuropilin-1 binding of small molecules, Drug Dev Res 81, 491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sorokina M, and Steinbeck C (2020) Review on natural products databases: where to find data in 2020, Journal of Cheminformatics 12, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sterling T, and Irwin JJ (2015) ZINC 15--Ligand Discovery for Everyone, J Chem Inf Model 55, 2324–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lipinski CA (2004) Lead- and drug-like compounds: the rule-of-five revolution, Drug Discov Today Technol 1, 337–341. [DOI] [PubMed] [Google Scholar]
- 77.Shultz MD (2019) Two Decades under the Influence of the Rule of Five and the Changing Properties of Approved Oral Drugs, J Med Chem 62, 1701–1714. [DOI] [PubMed] [Google Scholar]
- 78.Gupta M, Lee HJ, Barden CJ, and Weaver DF (2019) The Blood-Brain Barrier (BBB) Score, Journal of medicinal chemistry 62, 9824–9836. [DOI] [PubMed] [Google Scholar]
- 79.Kanev GK, de Graaf C, Westerman BA, de Esch IJP, and Kooistra AJ (2020) KLIFS: an overhaul after the first 5 years of supporting kinase research, Nucleic acids research 49, D562–D569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Case JB, Rothlauf PW, Chen RE, Liu Z, Zhao H, Kim AS, Bloyet LM, Zeng Q, Tahan S, Droit L, et al. (2020) Neutralizing Antibody and Soluble ACE2 Inhibition of a Replication-Competent VSV-SARS-CoV-2 and a Clinical Isolate of SARS-CoV-2, Cell Host Microbe 28, 475–485 e475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Testa S, Paoletti O, Giorgi-Pierfranceschi M, and Pan A (2020) Switch from oral anticoagulants to parenteral heparin in SARS-CoV-2 hospitalized patients, Intern Emerg Med 15, 751–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cagno V, Tseligka ED, Jones ST, and Tapparel C (2019) Heparan Sulfate Proteoglycans and Viral Attachment: True Receptors or Adaptation Bias?, Viruses 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mycroft-West CJ, Su D, Pagani I, Rudd TR, Elli S, Gandhi NS, Guimond SE, Miller GJ, Meneghetti MCZ, Nader HB, et al. (2020) Heparin Inhibits Cellular Invasion by SARS-CoV-2: Structural Dependence of the Interaction of the Spike S1 Receptor-Binding Domain with Heparin, Thromb Haemost 120, 1700–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Clausen TM, Sandoval DR, Spliid CB, Pihl J, Perrett HR, Painter CD, Narayanan A, Majowicz SA, Kwong EM, McVicar RN, et al. (2020) SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2, Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Uniewicz KA, Ori A, Ahmed YA, Yates EA, and Fernig DG (2014) Characterisation of the interaction of neuropilin-1 with heparin and a heparan sulfate mimetic library of heparin-derived sugars, PeerJ 2, e461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Koehler L, Ruiz-Gomez G, Balamurugan K, Rother S, Freyse J, Moller S, Schnabelrauch M, Kohling S, Djordjevic S, Scharnweber D, et al. (2019) Dual Action of Sulfated Hyaluronan on Angiogenic Processes in Relation to Vascular Endothelial Growth Factor-A, Sci Rep 9, 18143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mamluk R, Gechtman Z, Kutcher ME, Gasiunas N, Gallagher J, and Klagsbrun M (2002) Neuropilin-1 binds vascular endothelial growth factor 165, placenta growth factor-2, and heparin via its b1b2 domain, J Biol Chem 277, 24818–24825. [DOI] [PubMed] [Google Scholar]
- 88.Sastry GM, Adzhigirey M, Day T, Annabhimoju R, and Sherman W (2013) Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments, J Comput Aided Mol Des 27, 221–234. [DOI] [PubMed] [Google Scholar]
- 89.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, et al. (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy, Journal of medicinal chemistry 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- 90.RDKit: Open-source cheminformatics, 2018.09.03 ed.
- 91.ChemAxon.
- 92.Puszko AK, Sosnowski P, Rignault-Bricard R, Hermine O, Hopfgartner G, Pulka-Ziach K, Lepelletier Y, and Misicka A (2020) Urea-Peptide Hybrids as VEGF-A165/NRP-1 Complex Inhibitors with Improved Receptor Affinity and Biological Properties, Int J Mol Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang Y, Brittain JM, Jarecki BW, Park KD, Wilson SM, Wang B, Hale R, Meroueh SO, Cummins TR, and Khanna R (2010) In silico docking and electrophysiological characterization of lacosamide binding sites on collapsin response mediator protein-2 identifies a pocket important in modulating sodium channel slow inactivation, The Journal of biological chemistry 285, 25296–25307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Janssen BJ, Malinauskas T, Weir GA, Cader MZ, Siebold C, and Jones EY (2012) Neuropilins lock secreted semaphorins onto plexins in a ternary signaling complex, Nat Struct Mol Biol 19, 1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is available in the main text, figures, and supplementary materials.