

Abstract
In Lewy body disease (LBD) such as dementia with LBs and Parkinson's disease, several lines of evidence show that disrupted proteolysis occurs. p62/SQSTM1 (p62) is highly involved with intracellular proteolysis and is a component of ubiquitin‐positive inclusions in various neurodegenerative disorders. However, it is not clear whether p62 deficiency affects inclusion formation and abnormal protein accumulation. To answer this question, we used a mouse model of LBD that lacks p62, and found that LB‐like inclusions were observed in transgenic mice that overexpressed α‐synuclein (Tg mice) with or without the p62 protein. p62 deficiency enhanced α‐synuclein pathology with regard to the number of inclusions and staining intensity compared with Tg mice that expressed p62. To further investigate the molecular mechanisms associated with the loss of p62 in Tg mice, we assessed the mRNA and protein levels of several molecules, and found that the neighbor of the brca1 gene (NBr1), which is functionally and structurally similar to p62, is increased in Tg mice without p62 compared with control Tg mice. These findings suggest that p62 and NBR1 affect the pathogenesis of neurodegenerative diseases through the cooperative modulation of α‐synuclein aggregation.
Keywords: α‐synuclein, Lewy body disease, p62/Sequestsome 1/SQSTM1, Parkinson's disease, proteolysis, stress
Introduction
Lewy body disease (LBD) including dementia with LBs and Parkinson's disease (PD) is pathologically characterized by the presence of intracellular inclusions called LBs. α‐Synuclein has been identified as a component of LBs 41, and the duplication and triplication of the α‐synuclein gene are found in both sporadic and early onset forms of PD 40. Mutations (A30P and A53T) in the α‐synuclein gene are linked to autosomal dominant forms of PD 20, 31. Originally, α‐synuclein is a proteinase K (PK)‐soluble protein that localizes at presynaptic terminals; however, α‐synuclein becomes resistant to PK and widely deposited throughout the brain of patients with LBD 19, 42. These findings suggest that α‐synuclein is significantly involved in the pathogenesis of both familial and sporadic cases of LBD.
α‐Synuclein is physiologically processed by two intracellular degradation systems, including the ubiquitin–proteasome and autophagy–lysosome systems. In case of α‐synuclein overload, the autophagy–lysosome system, including chaperone‐mediated autophagy, predominantly aids in the degradation of excess α‐synuclein 6, 22, 46. Thus, it is possible that dysfunction of intracellular degradation system results in the up‐regulation of α‐synuclein expression and contributes to abnormal protein accumulation. Indeed, several lysosomal‐related genes were identified as a causative mutation in familial PD, including leucine‐rich repeat kinase 2 and adenosine‐3‐phosphate 13A2. Furthermore, PD has been genetically linked to rare lysosomal storage diseases, including Gaucher's disease 25 and Sanfilippo syndrome 47.
p62/SQSTM1/sequestosome 1 (referred to as p62) is a multifunctional protein that is strongly associated with the intracellular degradation system. P62 knockout (KO) mice exhibit mature‐onset obesity, insulin and leptin resistance 37. Pathologically, loss of p62 results in the accumulation of hyperphosphorylated tau and insoluble K63‐linked polyubiquitin chains 33, 48. p62 contains a ubiquitin‐associated (UBA) domain at the C‐terminus that enables its interaction with ubiquitinated and misfolded proteins. Additionally, p62 possesses a Phox and Bem1p (PB1) domain at the N‐terminus and a LC3 interacting region, suggesting that p62 is able to interact with proteasome components and autophagosomal membranes 29, 38. Thus, it has been suggested that p62 can efficiently degrade ubiquitinated and misfolded proteins through the proteasome and autophagy–lysosome systems. It has been reported that p62 is an inducible protein that easily aggregates under several pathological conditions, such as oxidative stress and neurodegeneration 1, 11, 27. Accordingly, dysfunction of the intracellular degradation systems induces p62 aggregation in vivo 2, 16. Furthermore, loss of p62 suppressed ubiquitin‐positive inclusions in neurons of brain‐specific autophagy‐deficient mice 17. Additionally, ubiquitin‐ and p62‐positive protein aggregates were abrogated in Atg8 and p62 double‐mutant flies 26. These findings suggest that p62 may be responsible for the formation of cytoplasmic inclusions and abnormal protein accumulation.
In this study, we used transgenic (Tg) mice overexpressing α‐synuclein with a A53T mutation as a model for LBD. We crossed the Tg mice with p62 KO mice to examine the involvement of p62 in abnormal α‐synuclein pathology. Immunohistochemical analyses showed that p62 deficiency enhanced α‐synuclein pathology, as shown by an increase in inclusion number and staining intensity. We assessed several genes and proteins related to stress response and proteolysis. These data revealed that the expression of neighbor of brca1 gene (NBR1), which is a functional homologue to p62, was increased in p62‐deficient mice.
Materials and Methods
Animals and experimental design
α‐Synuclein Tg mice have been widely used as an animal model for LBD 7, 14, 21, 23, 24, 34, 36, 45. To create this LBD model in a p62‐deficient background, we used mice overexpressing human α‐synuclein with the A53T mutation under the prion promoter (Jackson Laboratories, Bar Harbor, ME, USA) 7 and p62 KO mice with exon 1–4 deleted as previously described 17. The p62 KO mice lacked abnormal tau pathology. α‐Synuclein Tg and p62 KO mice were backcrossed with C57BL/6J mice for at least 10 generations. First, heterozygous α‐synuclein Tg mice were bred with p62 KO mice to generate α‐synuclein+/−/p62+/− mice. Second, α‐synuclein+/−/p62+/− mice were inbred to generate wild type, p62 KO, α‐synuclein+/−/p62+/+ and α‐synuclein+/−/p62−/− mice (Figure 1A). Hereafter, wild‐type, p62 KO, α‐synuclein+/−/p62+/+ and α‐synuclein+/−/p62−/− mice are simply referred to as WT, KO, Tg and Tg/KO mice, respectively. All comparisons were made among littermates to minimize confounding effects by different genetic backgrounds. Mice were housed with a light/dark cycle of 12 h and were given food and water ad libitum. The experimental protocol was approved by the Institutional Animal Care and Use Committee at the Hirosaki University Graduate School of Medicine in Japan. Tg mice were genotyped using real‐time polymerase chain reaction (PCR) analysis (forward primer, 5′‐TGT AGG CTC CAA AAC CAA GG‐3′; reverse primer, 5′‐TAT GCC TGT GGA TCC TGA CA‐3′), and verified by backcrossing. Conventional PCR was used for p62 genotyping (primer pair for wild type, forward, 5′‐CTT ACG GGT CCT TTT CCC AAC‐3′; reverse, 5′‐TCC TCC TTG CCC AGA AGA TAG‐3′; primer for p62 KO, forward; 5′‐CTG CAT GTC TTC TCC CAT GAC‐3′; reverse, 5′‐TAG ATA CCT AGG TGA GCT CTG‐3′). Mice were transcardially perfused with phosphate‐buffered saline. The brain was removed, and the right hemisphere was fixed with 4% paraformaldehyde for 48 h. After dehydrating through a graded ethanol series, the right hemisphere was embedded in paraffin and cut into 4‐μm thick sections. The left hemisphere was frozen at −80°C for subsequent biochemical analyses.
Figure 1.

Characterization of p 62 protein deficiency in an animal model of L ewy body disease. A. Breeding strategies to generate p62 deficiency in α‐synuclein transgenic (Tg) mice. Initially, heterozygous α‐synuclein Tg and homozygous p62‐knockout (KO) mice were crossed. Next, littermates and heterozygous p62‐deficient mice were mated to generate Tg mice without p62 (Tg/KO), of which four groups were used in this study (black circles). B. Immunoblot analysis confirmed that α‐synuclein was overexpressed in Tg and Tg/KO mice and that p62 signals were diminished in KO and Tg/KO mice (9 weeks of age, n = 6 per group). The molecular mass is indicated on the left side of the panel. β‐Actin was used as a loading control. C. A quantitative analysis shows that human α‐synuclein is expressed in Tg and Tg/KO mice and that p62 is absent in KO and Tg/KO mice. The values of Tg mice are defined as 100%. D. The weight changes of Tg (black circle) and Tg/KO mice (grey circle) are shown (mean ± standard deviation, n = 6–8 per group).
Antibodies and immunohistochemistry
Rabbit antibodies against Keap1 (ProteinTech Group, Inc., Chicago, IL, USA), p62 (MBL, Nagoya, Japan), NBR1 (Sigma, St. Louis, MO, USA and Santa Cruz Biotechnology, Santa Cruz, CA, USA), NAD(P)H quinone oxidoreductase 1 (NQO1) (Sigma), LC3 (Sigma and MBL), ubiquitin (DAKO, Glostrup, Denmark), UBQLN1 (Lifespan Biosciences, Seattle, WA, USA), phosphorylated α‐synuclein (Abcam, Cambridge, UK) and β‐actin (Sigma) were used in this study. Mouse antibodies against p62 (BD Biosciences, Franklin Lakes, NJ, USA), SNAP25 (Chemicon, Temecula, CA, USA), synaptophysin (DAKO), human α‐synuclein (LB509; Zymed, South San Francisco, CA, USA), human and mouse α‐synucleins (4D6; GeneTex, Irvine, CA, USA) and phosphorylated α‐synuclein (pSyn#64; Wako, Osaka, Japan) were also used.
The sections were dehydrated and pretreated with heat retrieval using an autoclave for 10 minutes in 10 mM citrate buffer (pH 6.0) for rabbit anti‐Keap1 and anti‐NBR1 antibodies. The sections were then subjected to immunohistochemical processing using the avidin‐biotin‐peroxidase complex method with diaminobenzidine (Sigma). In addition, the sections were counterstained with hematoxylin. For the staining of presynaptic PK‐resistant α‐synuclein, sections were pretreated with PK (Gibco BRL, Gaithersburg, MD, USA; 50 μg/mL) in a PK buffer containing 10 mM Tris‐HCl, pH 7.8, 100 mM NaCl, 0.1% Nonidet‐P40 at 37°C for 5 minutes. The total number of inclusions immunostained with anti‐phosphorylated α‐synuclein was quantified in contiguous sections. Immunohistochemical studies were performed at 9 weeks of age (n = 6 per group).
Quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR)
Total RNA was extracted from the right hemisphere of the brain using the RNeasy lipid tissue mini kit (Qiagen, Hilden, Germany) at 9 weeks of age (n = 3 per group). cDNA was synthesized from 1 μg of total RNA using the PrimeScript® II first‐strand cDNA synthesis kit (Takara Bio Inc., Otsu, Japan). An aliquot of cDNA was used for gene expression analysis with the SYBR® Premix Ex Taq™ II (Perfect Real Time) (Takara Bio Inc.) and CFX Real‐Time PCR Detection System (Bio‐Rad, Hercules, CA, USA) using the following primer sets: heme oxygenase‐1 (Ho‐1) (5′‐CCA GCA ACA AAG TGC AAG ATT C‐3′; 5′‐TCA CAT GGC ATA AAG CCC TAC AG‐3′), Nqo1 (5′‐GTC ATT CTC TGG CCA ATT CAG AGT‐3′; 5′‐TTC CAG GAT TTG AAT TCG GG‐3′), glutamate‐cysteine ligase catalytic subunit (Gclc) (5′‐AAA ATG CGG AGG CAT CAA‐3′; 5′‐ATA TGC TGC AGG CTT GGA AT‐3′), p62 (5′‐AGC TGC CTT GTA CCC ACA TC‐3′; 5′‐CAG AGA AGC CCA TGG ACA G‐3′), Cyclophilin A (5‐ATG CTG GAC CCA ACA CAA AT‐3′; 5‐TCT TTC ACT TTG CCA AAC ACC‐3′), Keap1 (5′‐CAC AGC AGC GTG GAG AGA‐3′; 5′‐CAA CAT TGG CGC GAC TAG A‐3′), Lamp1 (5′‐CCT ACG AGA CTG CGA ATG GT‐3′; 5′‐CCA CAA GAA CTG CCA TTT TTC‐3′), Cathepsin D (5′‐CCC TCC ATT CAT TGC AAG ATA C‐3′; 5′‐ TGC TGG ACT TGT CAC TGT TGT‐3′), transcription factor EB (TfEB) (5′‐GAG CTG GGA ATG CTG ATC C‐3′; 5′‐GGG ACT TCT GCA GGT CCT T‐3′); Rab7l1 (5′‐GCT GCA GCT CTG GGA TAT TG‐3′; 5′‐TAG TAG AGT CGT GTC ATG GAT GTG‐3′) and Nbr1 (5′‐TCA ACA GGA CTC GCA AAC AG‐3′; 5′‐ATG CTG CTC CCA TTG TGG‐3′). Cyclophilin A was used for normalization.
Immunoblot analysis
Western blot analysis was performed as previously described 43. For total cell lysate, we used a lysis buffer with 4% sodium dodecyl sulfate (SDS; 75 mM Tris‐HCl, pH 6.8, 4% SDS, 25% glycerol, 5% β‐mercaptoethanol) and passed sample through 21 gauge needle attached on a 1 mL syringe. For an experiment using insoluble sample of detergent, samples were weighted and lysed with 10‐fold volume of Tris‐based buffer (pH 7.4) containing 0.1% Triton X‐100 on ice. After homogenization with a pestle 20 times, they were passed 10 times through 21 gauge needle attached on a 1 mL syringe. Lysates were incubated for 5 minutes on ice, and centrifuged at 12 000 × g for 10 minutes. Supernatant was used as a soluble fraction. The pellets were resuspended with 8 M urea and sonicated (insoluble fraction). Signal detection was performed according to the protocol provided with the ECL or ECL prime detection systems (Amersham Pharmacia Biotech, Piscataway, NJ, USA). We performed each immunoblot analysis a minimum of three times, and all data were quantified and collected.
Animal behavioral testing
The Morris water maze
Spatial learning was assessed in a round tank of water (0.95 m in diameter) at 30°C. An escape platform (10 cm in diameter) was placed 1 cm below the water surface. A camera (Primetech Engineering Corp., Tokyo, Japan) was mounted above the maze and attached to a computer running the Smart software (Primetech Engineering Corp.). The training paradigm for the hidden platform version of the Morris water maze consisted of two trials per day for five consecutive days. The time taken to reach the platform (latency to escape) was recorded for each trial. The time limit was 120 s, and the intertrial interval was 1 h. If the animal could not find the platform, it was placed on the platform for 20 s. After removing the platform, the probe trial was carried out 2 h after the completion of training on the fifth day. The latency to reach the former location of the platform and the percentage of total time spent in each quadrant were recorded.
Forced swim test
Immobility time was analyzed using a forced swim test. Animals were individually placed in a transparent acrylic cylindrical beaker (height: 25 cm, diameter: 18 cm) containing 4600 mL of clear water at 25 ± 1°C for 6 minutes. A mouse was judged to be immobile when it remained passively floating in the water for more than 2 s. Immobility time was quantified using a Forced Swim Scan software (Clever Sys Inc., Reston, VA, USA).
Quantitative analysis and statistical analysis
A semi‐quantitative analysis of protein levels was performed using the ImageJ software provided by the NIH. All data were represented as the mean + standard deviation. The statistical significance was evaluated using one‐way analysis of variance (ANOVA) with Bonferroni's post hoc test to analyze four genotypes and Student's t‐test to analyze two genotypes. A probability value of less than 0.05 (P < 0.05) was considered to be significant.
Results
Characterization of α‐synuclein Tg mice with or without p62
To test the possibility that p62 is responsible for the formation of cytoplasmic inclusions and abnormal protein accumulation, we generated mice that overexpressed human α‐synuclein (Tg) on a p62‐deficient background (Figure 1A). First, we crossed Tg mice with p62 KO mice. Next, littermates with or without p62 and/or human α‐synuclein were selected by genotyping and crossed to generate Tg mice lacking p62. Consequently, littermates with or without endogenous p62 and/or human α‐synuclein expression were born at the expected Mendelian ratio. For our studies, we used WT, KO, Tg and Tg/KO mice.
We confirmed that α‐synuclein was robustly expressed in the brains of Tg mice and Tg/KO mice (Figure 1B). We used a human α‐synuclein‐specific antibody, LB509, to confirm that human α‐synuclein expression was present only in Tg and Tg/KO mice. There were no differences in the endogenous and human α‐synuclein levels between the Tg and Tg/KO mice. We also confirmed that p62 protein levels were diminished in the brains of KO and Tg/KO mice. Interestingly, the amount of p62 was slightly higher in Tg mice than it was in WT mice (Figure 1C). An increase of p62 was also supported by immunohistochemical studies that showed an increase in p62 immunoreactivity in Tg mice compared with WT mice (Figure 2A). α‐Synuclein expression was mainly observed in the presynapses in the brains of WT and p62 KO mice; however, additional staining of α‐synuclein was observed in the cytoplasm and presynapses in the brains of Tg and Tg/KO mice (Figure 2B). Consistent with previous papers 28, 37, KO mice exhibited mature‐onset obesity. As they aged, Tg/KO mice had a heavier average body weight than did Tg mice (Figure 1D). The majority of Tg mice remained healthy until at least 70 weeks of age. Tg and Tg/KO mice were behaviorally indistinguishable and displayed lower food intake and activity at the end stage of the disease.
Figure 2.
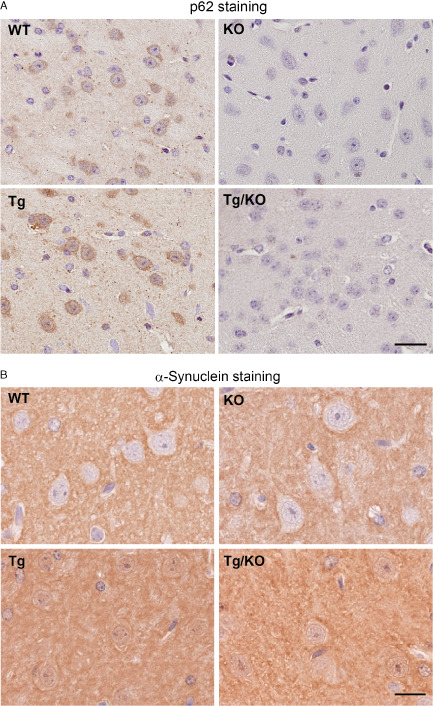
p 62 and α‐synuclein staining in wild‐type ( WT ), knockout ( KO ), T g and T g/ KO mice. A. Immunohistochemical analysis shows that p62 immunoreactivity is observed in WT and Tg mice but not in KO mice (9 weeks of age, n = 6 per group). Bar = 20 μm. B. Human and mouse α‐synuclein is strongly expressed in the presynapse and cytoplasm of cortical neurons in Tg and Tg/KO mice. Bar = 10 μm.
Tg/KO mice exhibit an increase in phosphorylated α‐synuclein staining and inclusion number compared with Tg mice
Similar to the human pathological conditions, there are two types of abnormal α‐synuclein in the brains of Tg mice 7, 42, including phosphorylated α‐synuclein (P‐syn) and PK‐resistant α‐synuclein (PK‐syn). Immunohistochemical analyses showed that P‐syn is observed in both Tg/KO and Tg mice (Figure 3A). We compared the number of P‐syn‐positive inclusions in the thalamus of Tg and Tg/KO mice. Quantitative data indicated that the number of inclusions was higher in Tg/KO mice compared with Tg mice (Figure 3B). Furthermore, the intensity of P‐syn staining was increased in the hippocampus and cerebral cortex of Tg/KO mice compared with Tg mice (Figure 3C). Unlike human pathological conditions, p62 was not localized in the cytoplasmic inclusions in the brains of Tg mice. Immunohistochemical studies demonstrated that PK treatment abolished normal α‐synuclein immunoreactivity, and PK‐syn was found in the presynapses of the brain of both Tg and Tg/KO mice (Figure 3D). Western blot analysis verified that P‐syn signal intensity was higher in Tg/KO than Tg mice using two kinds of antibodies against P‐syn (Figure 3E). Furthermore, we fractionated samples of Tg and Tg/KO mice by buffer with 0.1% Triton X‐100 detergent, and found that insoluble P‐syn level was increased in Tg/KO compared with Tg mice (Figure 3F). Thus, p62 deficiency modulates α‐synuclein pathology with regard to P‐syn staining intensity, the number of P‐syn inclusions and solubility.
Figure 3.
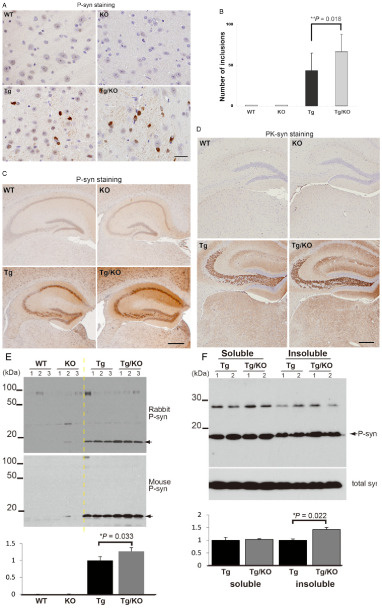
The effect of p 62 deficiency on abnormal α‐synuclein expression. A. Cytoplasmic inclusions are positive for phosphorylated α‐synuclein (P‐syn) in the thalamus of Tg and Tg/knockout (KO) mice. Bar = 20 μm. B. A quantitative analysis shows that the number of cytoplasmic inclusions is significantly increased in Tg/KO mice when compared with Tg mice (9 weeks of age, n = 6 per group). The groups differed significantly [analysis of variance (ANOVA), F(3, 11) = 160.81, P < 0.01]. C. P‐syn staining is observed in the neurons of the cerebral cortex and hippocampus in Tg and Tg/KO mice. An increased staining intensity is observed in Tg/KO mice compared with Tg mice. Bar = 500 μm. D. No obvious differences in proteinase K‐resistant α‐synuclein (PK‐syn) are found between Tg and Tg/KO mice. Bar = 250 μm. E. P‐syn level is significantly increased in Tg/KO mice compared with Tg mice. Ratio of P‐syn to β‐actin was calculated, and the values of Tg mice are defined as 1.0. The groups differed significantly [ANOVA, F(3, 11) = 147.1, P < 0.01]. F. Triton X‐100 soluble and insoluble samples were prepared from Tg and Tg/KO mice (9 weeks of age, n = 2 per Tg and Tg/KO groups). Insoluble P‐syn level is increased in Tg/KO mice compared with Tg mice. P‐syn levels were normalized by total synuclein, and the values of Tg mice were defined as 1.0 in a soluble or insoluble sample.
Behavioral tests revealed a longer immobility time for p62‐deficient mice
Given the presynaptic aggregation of PK‐syn in the hippocampus of Tg and Tg/KO mice, we sought to determine whether memory function was also affected in these mice. We performed the Morris water maze test using mice at a younger age (9 weeks old) to exclude differences in body weight. The average weight was comparable between Tg and Tg/KO mice (21.1 g in Tg, 22.0 g in Tg/KO) at 9 weeks of age. During the training phase of the Morris water maze test, WT and Tg mice showed a gradual decrease in escape latency over time; however, KO and Tg/KO mice exhibited longer escape latencies (Supporting Information Video Clip S1 and S2). When the platform was removed, 80% of WT mice and 70% of Tg mice found the platform location. In contrast, less than 50% of KO and Tg/KO mice found the platform location. KO and Tg/KO mice took a longer time to reach to the platform location (Figure 4A) and spent less time in the target quadrant (Figure 4B) than did WT mice. The lower rate of platform crossing in the KO mice was due to their immobility (Figure 4C,D), which is consistent with previous results showing that KO mice exhibited immobility during training and probe trials 33. During only the first minute of a forced swim test, KO mice showed a significantly increased immobility that lasted longer than 2 s when compared with WT mice (Figure 4E,F). Thereafter, the time course for floating behavior (the percentage of immobility) was similar between groups. There was no significant difference between Tg/KO and WT mice. These results suggest that p62 plays a role in maintaining neurological functions, such as stress responses and motivation to escape.
Figure 4.

p 62‐deficient mice exhibit longer escape latencies due to lower activity. A. The probe trial was completed after 5 days of hidden platform training in the Morris water maze. Wild type (WT, n = 10), p62 knockout (KO, n = 11), α‐synuclein Tg (Tg, n = 9) and α‐synuclein mice lacking p62 (Tg/KO, n = 9) were tested at 9 weeks of age. KO mice take longer to reach the platform location. The groups differed significantly [analysis of variance (ANOVA), F(3, 39) = 4.53, P < 0.01]. B. The percentage of time spent in the target quadrant (black) during a 60 s probe trial of the Morris water maze test. KO mice spend less time in the target quadrant. C. The immobility time of the Morris water maze. Longer immobility times are evident in KO mice. D. Representative path tracings are shown. Light pink indicates the position of the platform. E. A forced swim test was performed at 9–10 weeks of age (n = 9–11 per group) and shows a significant difference in immobility latency, with KO mice lasting longer than 2 s and WT mice remaining mobile for the first 1 minute. F. KO mice exhibit higher immobility times for the first 6 minutes. The groups differed significantly [ANOVA, F(3, 39) = 2.14, P < 0.05]. *P < 0.05.
Increased levels of the functional homologue, NBR1, in p62 KO and Tg/KO mice
To analyze the molecular mechanisms associated with the loss of p62 on Tg mice, we performed quantitative RT‐PCR analysis using primers for genes related to the stress response and proteolysis (Figure 5). Consistent with the genotype results, the p62 mRNA level was diminished in KO and Tg/KO mice. Keap1 is a binding partner of p62 and functions as a sensor for noxious stimuli such as oxidants and electrophiles. The mRNA level of Keap1 appeared to be different between the four groups; however, the data were not statistically significant (P = 0.069). Previous papers have reported that autophagy‐deficient mice display a higher expression of detoxifying enzymes, such as Ho‐1, Nqo1 and Gclc 18. There were no differences in the mRNA levels of these enzymes among the four groups. Recent evidence indicates that α‐synuclein overexpression causes dynamic changes in the autophagy–lysosomal system. Therefore, we assessed levels of TfEB, a major transcriptional regulator for this system 39, lysosomal enzymes (Lamp1 and cathepsin D), molecules responsible for membrane trafficking (Rab7l1) and selective autophagy markers (Nbr1). Among these genes, only the Nbr1 mRNA levels were significantly different (P < 0.01) between the four groups. Consistent with this result, the NBR1 protein levels were significantly increased in mice lacking p62 compared with mice with p62 (P < 0.05) (Figure 6A,B). Additionally, Keap1 protein levels were also significantly different among the four groups at the protein level. There were no alterations in NQO1, synaptic proteins and proteolysis‐related molecules, such as ubiquitin and LC3, which are essential to autophagosomal formation 13. Based on the increased NBR1 levels in mice lacking p62, we compared the distribution patterns of p62 and NBR1 in the mouse brain. Interestingly, immunoblotting showed that p62 and NBR1 are similarly distributed in distinct regions of the mouse brain (Figure 7A,B). NBR1 was mainly localized in the cytoplasm of neurons, and its intensity was higher in Tg/KO than in Tg mice (Figure 7C). These data are consistent with the qRT‐PCR and immunoblotting analyses.
Figure 5.

The effect of p62 deficiency on several kinds of genes. The mRNA levels of genes related to proteolysis and oxidative stress in the brains of WT, KO, Tg and TG/KO mice were determined at 9 weeks of age (n = 3 per group). mRNA was measured by quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR) using the right hemisphere of the brain. Data are normalized by the C yclophilin A mRNA level in each sample, and the average and standard deviation was calculated. qRT‐PCR reveals that there is no significant difference in the K eap1, glutamate‐cysteine ligase catalytic subunit (Gclc), NAD(P)H quinone oxidoreductase 1 (Nqo 1), heme oxygenase‐1 (Ho ‐1), Lamp 1, C athepsin D, TfEB and Rab7l1 levels among the four groups. In contrast, the mRNA level of p 62 (P < 0.01) and Nbr 1 (P < 0.05) are significantly different. The groups differed significantly [analysis of variance, F(3, 11) = 226.86, P < 0.01 in p 62 mRNA, F(3, 11) = 14.15, P < 0.01 in Nbr 1 mRNA]. The WT values are defined as 100%. *P < 0.05, **P < 0.01.
Figure 6.

The effect of p 62 deficiency on molecules related to proteolysis, oxidative stress and the synapse. A. Expression of Keap1 and NBR1 is significantly increased in Tg/KO mice compared with Tg mice. NAD(P)H quinone oxidoreductase 1 (NQO1), LC3, ubiquitin, synaptophysin and SNAP25 levels are not significantly different between the four groups (9 weeks of age, n = 6 per group). B. A quantitative analysis indicates that the Keap1 and NBR1 levels are significantly increased in p62‐deficient mice compared with mice with p62. The Tg values are defined as 100%. *P < 0.05, **P < 0.01. The groups differed significantly [analysis of variance, F(3, 11) = 7.44, P = 0.011 in Keap1, F(3, 11) = 4.27, P = 0.045 in NBR1].
Figure 7.

The spatial patterns of p 62 and NBR 1 in the mouse brain. A. Equal amounts of homogenates from the indicated regions were analyzed by immunoblotting (12 weeks of age, n = 2 in wild‐type mice). Antibodies against NBR1 (upper) or p62 (bottom) were used to detect endogenous proteins. NBR1 is mainly expressed in the olfactory bulb, temporal and occipital cortices, striatum, thalamus and hypothalamus. The highest expression of p62 is observed in the olfactory bulb, striatum, temporal and occipital cortices, hippocampus, thalamus, hypothalamus and medulla oblongata. β‐Actin is used as a loading control. B. Distribution patterns of NBR1 and p62 in the sagittal section of mice brains. NBR1 or p62 levels are normalized by β‐actin. The circles represent the size of the expression level. C. NBR1 immunostaining in Tg and Tg/KO mice (9 weeks of age, n = 6 per group). NBR1 immunoreactivity is mainly detected in neurons of the thalamus of both Tg and Tg/KO mice. Note the increased intensity of NBR1 immunoreactivity in Tg/KO mice compared with Tg mice. Bar = 30 μm.
Discussion
p62 is an inducible protein that easily aggregates under pathological conditions, such as oxidative stress and disrupted proteolysis, and it is localized in cytoplasmic inclusions in LBD and other neurodegenerative diseases, suggesting that p62 contributes to inclusion formation. Moreover, p62‐ and ubiquitin‐positive inclusions in the neurons of brain‐specific Atg7‐deficient mice disappear with the loss of p62 17. Based on these findings, we initially predicted that p62 deficiency would lead to a decrease in the number of inclusions in Tg mice that overexpressed α‐synuclein. However, our data suggest that p62 deficiency results in an exaggeration of α‐synuclein pathology with regard to P‐syn staining intensity and inclusion number. Consistent with our findings, Doi et al demonstrated that a loss of p62 exacerbated neuropathological outcomes 5 in a mouse model of spinal and bulbar muscular atrophy, which is one of polyglutamine diseases. Our pathological data showed that the number of P‐syn‐positive inclusion increased by 1.5‐fold in Tg/KO mice compared with Tg mice. Consistently, this was supported by Western blot analyses showing that P‐syn level was higher in Tg/KO mice than Tg mice using two kinds of antibodies against P‐syn. Considering that increased P‐syn is mainly resistant to detergent of Triton X‐100, it is possible that biochemical property of α‐synuclein is altered and leads to more aggregation in Tg/KO mice. Although it remains controversial whether the formation of cytoplasmic inclusions exerts a beneficial or toxic effect on cells, our findings strengthen the idea that p62 can modulate α‐synuclein aggregation and the pathogenesis of diseases.
Consistent with previous results 28, 37, a p62 deficiency resulted in mature‐onset obesity in mice. Recent evidence indicates that hyperphagia is the primary cause of obesity in p62‐deficient mice due to the disruption of leptin signaling 9. Accordingly, p62 is highly expressed in hypothalamic neurons, including proopiomelanocortin (POMC) neurons in the arcuate nucleus 3, 9 that are responsible for the control of appetite and energy intake. Interestingly, lack of autophagic activity in POMC neurons caused higher post‐weaning body weight and p62/ubiquitin aggregation 4, 32. Furthermore, leptin signaling is also disrupted in these mice. This may have broad implications for the pathophysiology of p62 KO mice. Because p62 helps shuttle insoluble and ubiquitinated proteins into autophagosomes, disruption of autophagic flux or loss of p62 gives rise to the accumulation of p62 target molecules. Accordingly, we revealed that P‐syn level is increased in Triton X‐100 insoluble fraction of Tg/KO mice compared with Tg mice. Thus, p62 dysfunction observed in autophagy‐deficient POMC neurons or p62 KO mice might also affect intercellular environment through disturbance of p62 binding partners or substrates. One of p62 binding partners is known to be dopamine receptor 15. Because dopamine is widely involved in physiological conditions such as mood, cognition and motor control, it is possible that p62 modulates dopamine system, and p62 dysfunction may cause pathogenesis of PD.
Our immunoblot results confirmed that the hypothalamus is one of the regions with the highest p62 expression level. The hypothalamus is known to regulate various physiological functions, particularly the hypothalamus‐pituitary‐adrenal axis, which coordinates emotional, neuroendocrine and autonomic inputs in response to stress. Regarding behavioral abnormalities, we could not distinguish Tg mice from Tg/KO mice; however, p62‐deficient mice exhibited less activity and depression‐like behavior in the Morris water maze and forced swim test. This is consistent with previous results 33. It is conceivable that p62 deficiency affects the hypothalamus‐pituitary‐adrenal axis, leading to behavioral abnormalities in response to stress. The immobility rate of Tg/KO mice was comparable with that of normal control mice. Considering previous reports that mice overexpressing α‐synuclein are hyperactive 8, 30, 44, we speculate that the degree of immobility in Tg/KO mice is recovered because of the hyperactivity of Tg mice. Taken together, p62 plays an important role in modulating multiple physiological responses, including nutritional, oxidative and water stressors.
We screened multiple protein and mRNA levels to study the molecular mechanisms associated with the loss of p62 in Tg mice. We found that NBR1 was significantly increased in Tg/KO mice compared with Tg mice at both the mRNA and protein levels. p62 and NBR1 contain an N‐terminal PB1 domain, an intermediate LC3 binding region, and a C‐terminal UBA domain, and they function as cargo adapters for the autophagic degradation of ubiquitinated substrates 10, 12, 29. Intriguingly, our immunoblotting results suggest that these molecules are similarly distributed in distinct regions of the mouse brain. This spatial pattern and functional similarity raise the possibility that NBR1 levels can be up‐regulated to compensate for the loss of p62 protein. Therefore, the functional redundancy of NBR1 may mask the anticipated abnormalities of p62‐deficient mice.
In conclusion, we have provided evidence that p62 is unnecessary for the formation of inclusions in an animal model that overexpresses α‐synuclein. In addition, p62 deficiency enhanced α‐synuclein pathology based on the number of inclusions and staining intensity of P‐syn. In support of this finding, it is likely that p62 indirectly helps sequester abnormal molecules through its own oligomerization 35. Further analyses at the molecular level suggest that NBR1 plays a compensatory role for p62 in the central nervous system. NBR1 and p62 double KO mice would be a useful tool to test this hypothesis.
Supporting information
Video Clip S1. The Morris water maze test. A wild‐type mouse successfully reaches the platform within 30 s on the fourth day of training course.
Video Clip S2. The Morris water maze test. A p62‐knockout mouse does not take an action within 60 s, and end up failing on the fourth day of training course.
Acknowledgments
This work was supported by JSPS KAKENHI grant numbers 26430050 (K.T.), 26860655 (Y.M.), 26430049 (F.M.), 24300131 (K.W.); Priority Research Grant for Young Scientists designated by the president of Hirosaki University (K.T., J.M.); Hirosaki University Institutional Research Grant (K.I., K.W.); grants‐in‐aid from the Research Committee for Ataxic Disease from the Ministry of Health, Labor and Welfare, Japan (K.W.); and an Intramural Research Grant (24‐5) for Neurological and Psychiatric Disorders of NCNP (K.W.). The authors wish to express their gratitude to K. Saruta, T. Kon, M. Nakata and A. Ono for their technical assistance.
References
- 1. Bartlett BJ, Isakson P, Lewerenz J, Sanchez H, Kotzebue RW, Cumming RC et al (2011) p62, Ref(2)P and ubiquitinated proteins are conserved markers of neuronal aging, aggregate formation and progressive autophagic defects. Autophagy 7:572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bedford L, Hay D, Devoy A, Paine S, Powe DG, Seth R et al (2008) Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy‐like inclusions resembling human pale bodies. J Neurosci 28:8189–8198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Braak H, Thal DR, Del Tredici K (2011) Nerve cells immunoreactive for p62 in select hypothalamic and brainstem nuclei of controls and Parkinson's disease cases. J Neural Transm 118:809–819. [DOI] [PubMed] [Google Scholar]
- 4. Coupe B, Ishii Y, Dietrich MO, Komatsu M, Horvath TL, Bouret SG (2012) Loss of autophagy in pro‐opiomelanocortin neurons perturbs axon growth and causes metabolic dysregulation. Cell Metab 15:247–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Doi H, Adachi H, Katsuno M, Minamiyama M, Matsumoto S, Kondo N et al (2013) p62/SQSTM1 differentially removes the toxic mutant androgen receptor via autophagy and inclusion formation in a spinal and bulbar muscular atrophy mouse model. J Neurosci 33:7710–7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ebrahimi‐Fakhari D, Cantuti‐Castelvetri I, Fan Z, Rockenstein E, Masliah E, Hyman BT et al (2011) Distinct roles in vivo for the ubiquitin‐proteasome system and the autophagy‐lysosomal pathway in the degradation of alpha‐synuclein. J Neurosci 31:14508–14520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM (2002) Neuronal alpha‐synucleinopathy with severe movement disorder in mice expressing A53T human alpha‐synuclein. Neuron 34:521–533. [DOI] [PubMed] [Google Scholar]
- 8. Graham DR, Sidhu A (2010) Mice expressing the A53T mutant form of human alpha‐synuclein exhibit hyperactivity and reduced anxiety‐like behavior. J Neurosci Res 88:1777–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harada H, Warabi E, Matsuki T, Yanagawa T, Okada K, Uwayama J et al (2013) Deficiency of p62/Sequestosome 1 causes hyperphagia due to leptin resistance in the brain. J Neurosci 33:14767–14777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T et al (2008) Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem 283:22847–22857. [DOI] [PubMed] [Google Scholar]
- 11. Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y et al (2000) Transcription factor Nrf2 coordinately regulates a group of oxidative stress‐inducible genes in macrophages. J Biol Chem 275:16023–16029. [DOI] [PubMed] [Google Scholar]
- 12. Johansen T, Lamark T (2011) Selective autophagy mediated by autophagic adapter proteins. Autophagy 7:279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kabeya Y, Mizushima N, Yamamoto A, Oshitani‐Okamoto S, Ohsumi Y, Yoshimori T (2004) LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form‐II formation. J Cell Sci 117:2805–2812. [DOI] [PubMed] [Google Scholar]
- 14. Kahle PJ (2008) alpha‐Synucleinopathy models and human neuropathology: similarities and differences. Acta Neuropathol 115:87–95. [DOI] [PubMed] [Google Scholar]
- 15. Kim OJ, Ariano MA, Namkung Y, Marinec P, Kim E, Han J, Sibley DR (2008) D2 dopamine receptor expression and trafficking is regulated through direct interactions with ZIP. J Neurochem 106:83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I et al (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441:880–884. [DOI] [PubMed] [Google Scholar]
- 17. Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T et al (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy‐deficient mice. Cell 131:1149–1163. [DOI] [PubMed] [Google Scholar]
- 18. Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y et al (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12:213–223. [DOI] [PubMed] [Google Scholar]
- 19. Kramer ML, Schulz‐Schaeffer WJ (2007) Presynaptic alpha‐synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci 27:1405–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S et al (1998) Ala30Pro mutation in the gene encoding alpha‐synuclein in Parkinson's disease. Nat Genet 18:106–108. [DOI] [PubMed] [Google Scholar]
- 21. Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS et al (2002) Human alpha‐synuclein‐harboring familial Parkinson's disease‐linked Ala‐53 –> Thr mutation causes neurodegenerative disease with alpha‐synuclein aggregation in transgenic mice. Proc Natl Acad Sci U S A 99:8968–8973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mak SK, McCormack AL, Manning‐Bog AB, Cuervo AM, Di Monte DA (2010) Lysosomal degradation of alpha‐synuclein in vivo. J Biol Chem 285:13621–13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A et al (2000) Dopaminergic loss and inclusion body formation in alpha‐synuclein mice: implications for neurodegenerative disorders. Science 287:1265–1269. [DOI] [PubMed] [Google Scholar]
- 24. Matsuoka Y, Vila M, Lincoln S, McCormack A, Picciano M, LaFrancois J et al (2001) Lack of nigral pathology in transgenic mice expressing human alpha‐synuclein driven by the tyrosine hydroxylase promoter. Neurobiol Dis 8:535–539. [DOI] [PubMed] [Google Scholar]
- 25. Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA et al (2011) Gaucher disease glucocerebrosidase and alpha‐synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146:37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nezis IP, Simonsen A, Sagona AP, Finley K, Gaumer S, Contamine D et al (2008) Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J Cell Biol 180:1065–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Odagiri S, Tanji K, Mori F, Kakita A, Takahashi H, Wakabayashi K (2012) Autophagic adapter protein NBR1 is localized in Lewy bodies and glial cytoplasmic inclusions and is involved in aggregate formation in alpha‐synucleinopathy. Acta Neuropathol 124:173–186. [DOI] [PubMed] [Google Scholar]
- 28. Okada K, Yanagawa T, Warabi E, Yamastu K, Uwayama J, Takeda K et al (2009) The alpha‐glucosidase inhibitor acarbose prevents obesity and simple steatosis in sequestosome 1/A170/p62 deficient mice. Hepatol Res 39:490–500. [DOI] [PubMed] [Google Scholar]
- 29. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H et al (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282:24131–24145. [DOI] [PubMed] [Google Scholar]
- 30. Paumier KL, Sukoff Rizzo SJ, Berger Z, Chen Y, Gonzales C, Kaftan E et al (2013) Behavioral characterization of A53T mice reveals early and late stage deficits related to Parkinson's disease. PLoS ONE 8:e70274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A et al (1997) Mutation in the alpha‐synuclein gene identified in families with Parkinson's disease. Science 276:2045–2047. [DOI] [PubMed] [Google Scholar]
- 32. Quan W, Kim HK, Moon EY, Kim SS, Choi CS, Komatsu M et al (2012) Role of hypothalamic proopiomelanocortin neuron autophagy in the control of appetite and leptin response. Endocrinology 153:1817–1826. [DOI] [PubMed] [Google Scholar]
- 33. Ramesh Babu J, Lamar Seibenhener M, Peng J, Strom AL, Kemppainen R, Cox N et al (2008) Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem 106:107–120. [DOI] [PubMed] [Google Scholar]
- 34. Richfield EK, Thiruchelvam MJ, Cory‐Slechta DA, Wuertzer C, Gainetdinov RR, Caron MG et al (2002) Behavioral and neurochemical effects of wild‐type and mutated human alpha‐synuclein in transgenic mice. Exp Neurol 175:35–48. [DOI] [PubMed] [Google Scholar]
- 35. Riley BE, Kaiser SE, Shaler TA, Ng AC, Hara T, Hipp MS et al (2010) Ubiquitin accumulation in autophagy‐deficient mice is dependent on the Nrf2‐mediated stress response pathway: a potential role for protein aggregation in autophagic substrate selection. J Cell Biol 191:537–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E (2002) Differential neuropathological alterations in transgenic mice expressing alpha‐synuclein from the platelet‐derived growth factor and Thy‐1 promoters. J Neurosci Res 68:568–578. [DOI] [PubMed] [Google Scholar]
- 37. Rodriguez A, Duran A, Selloum M, Champy MF, Diez‐Guerra FJ, Flores JM et al (2006) Mature‐onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab 3:211–222. [DOI] [PubMed] [Google Scholar]
- 38. Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW (2004) Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol 24:8055–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S et al (2011) TFEB links autophagy to lysosomal biogenesis. Science 332:1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J et al (2003) alpha‐Synuclein locus triplication causes Parkinson's disease. Science 302:841. [DOI] [PubMed] [Google Scholar]
- 41. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha‐synuclein in Lewy bodies. Nature 388:839–840. [DOI] [PubMed] [Google Scholar]
- 42. Tanji K, Mori F, Mimura J, Itoh K, Kakita A, Takahashi H, Wakabayashi K (2010) Proteinase K‐resistant alpha‐synuclein is deposited in presynapses in human Lewy body disease and A53T alpha‐synuclein transgenic mice. Acta Neuropathol 120:145–154. [DOI] [PubMed] [Google Scholar]
- 43. Tanji K, Zhang HX, Mori F, Kakita A, Takahashi H, Wakabayashi K (2012) p62/sequestosome 1 binds to TDP‐43 in brains with frontotemporal lobar degeneration with TDP‐43 inclusions. J Neurosci Res 90:2034–2042. [DOI] [PubMed] [Google Scholar]
- 44. Unger EL, Eve DJ, Perez XA, Reichenbach DK, Xu Y, Lee MK, Andrews AM (2006) Locomotor hyperactivity and alterations in dopamine neurotransmission are associated with overexpression of A53T mutant human alpha‐synuclein in mice. Neurobiol Dis 21:431–443. [DOI] [PubMed] [Google Scholar]
- 45. van der Putten H, Wiederhold KH, Probst A, Barbieri S, Mistl C, Danner S et al (2000) Neuropathology in mice expressing human alpha‐synuclein. J Neurosci 20:6021–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC (2003) Alpha‐Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 278:25009–25013. [DOI] [PubMed] [Google Scholar]
- 47. Winder‐Rhodes SE, Garcia‐Reitbock P, Ban M, Evans JR, Jacques TS, Kemppinen A et al (2012) Genetic and pathological links between Parkinson's disease and the lysosomal disorder Sanfilippo syndrome. Mov Disord 27:312–315. [DOI] [PubMed] [Google Scholar]
- 48. Wooten MW, Geetha T, Babu JR, Seibenhener ML, Peng J, Cox N et al (2008) Essential role of sequestosome 1/p62 in regulating accumulation of Lys63‐ubiquitinated proteins. J Biol Chem 283:6783–6789. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video Clip S1. The Morris water maze test. A wild‐type mouse successfully reaches the platform within 30 s on the fourth day of training course.
Video Clip S2. The Morris water maze test. A p62‐knockout mouse does not take an action within 60 s, and end up failing on the fourth day of training course.