

Abstract
Progressive supranuclear palsy (PSP) cases frequently have argyrophilic grain disease (AGD). However, the PSP‐like tau pathology in AGD cases has not been fully clarified. To address this, we examined tau pathologies in the subcortical nuclei and frontal cortex in 19 AGD cases that did not meet the pathological criteria of PSP or corticobasal degeneration, nine PSP cases and 20 Braak NFT stage‐matched controls. Of the 19 AGD cases, five (26.3%) had a few Gallyas‐positive tau‐positive tufted astrocytes (TAs) and Gallyas‐negative tau‐positive TA‐like astrocytic inclusions (TAIs), and six (31.6%) had only TAIs in the striatum and/or frontal cortex. Subcortical tau pathology was sequentially and significantly greater in AGD cases lacking these tau‐positive astrocytic lesions, AGD cases having them, and PSP cases than in controls. There was a significant correlation between three histologic factors, including the AGD stage and the quantities of subcortical neuronal and astrocytic tau pathologies. Tau immunoblotting demonstrated 68‐ and 64‐kDa bands and 33‐kDa low‐molecular mass tau fragments in PSP cases, and although with lesser intensity, in AGD cases with and without TAs and TAIs also. Given these findings, the progression of AGD may be associated with development of the neuronal and astrocytic tau pathologies characteristic of PSP.
Keywords: argyrophilic grain disease, progressive supranuclear palsy, subcortical nuclei, tau, tufted astrocyte
INTRODUCTION
Argyrophilic grain disease (AGD) is pathologically defined by the presence of argyrophilic grains in the limbic system and neocortical regions 6, 7. Pathological studies have demonstrated that the frequency of AGD increases with age, and that approximately 10% of autopsy cases aged 70 years or older have various quantities of argyrophilic grains 8, 28. Argyrophilic grains are spindle‐shaped Gallyas‐positive structures in which four‐repeat (4R) tau rather than three‐repeat (3R) tau is accumulated 6, 7, 13, 45. In addition, coiled bodies, mild degrees of neurofibrillary tangles (NFTs) and pretangles are found in the limbic regions and adjacent neocortex 7, 8, 19, 46, 47. Argyrophilic grains initially appear in the ambient gyrus, and spread to adjacent amygdala, hippocampus and temporofrontal cortex 38. Conversely, argyrophilic grains are hardly observed in the basal ganglia, brain stem nuclei and cerebellum. Cases having argyrophilic grains with an extensive distribution in the neocortex frequently show dementia in life 38.
Like AGD, progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD) are 4R tauopathies that have distinct disease‐specific glial inclusions 9. Glial lesions characteristic of PSP are Gallyas‐ and tau‐positive tufted astrocytes (TAs) 17, 23, 35, 36, 51. Predilection sites for TAs are the frontal cortex and striatum 22, 31, 44. In addition, NFTs associated with neuronal loss are observed in the basal ganglia, brain stem nuclei and dentate nucleus in the cerebellum in PSP cases 16, 41. The pathological diagnosis of PSP is based on the distribution and quantity of NFTs in the subcortical nuclei and the presence of TAs 16. That is, while AGD and PSP cases are characterized by the accumulation of 4R tau in disease‐specific neuronal and glial lesions, the morphological features and topographical distribution of the pathological hallmarks of AGD and PSP are different.
Although it has been reported that PSP cases often have various quantities of argyrophilic grains 19, 28, 45, a recent study demonstrated that eight of 30 PSP cases (26.7%) had AGD 42. Conversely, several reports demonstrated that some AGD cases had a few TAs characteristic of PSP 25, 26, 40 or a small number of NFTs in the basal ganglia and brain stem nuclei with a distribution similar to that observed in PSP brains 18, 20, 21, 25, 26, 29, 32, 40. These previous findings led us to speculate that there may be a pathophysiological relationship between AGD and PSP, but detailed histopathological characteristics of the tau pathology associated with PSP in AGD cases, including their quantity, frequency and morphological features, have not been fully explored. To address these issues, we prepared four groups that were defined by the presence or absence of argyrophilic grains in the limbic region and neocortex and NFTs and tau‐positive astrocytic lesions in the subcortical nuclei and frontal cortex, and then compared the histopathological features of these groups.
MATERIALS AND METHODS
Subjects
To examine the relationship between AGD and PSP pathologies, we selected 20 AGD cases that were pathologically diagnosed as lacking other degenerative diseases, including PSP and CBD, based on routine neuropathological examination. We first reexamined these cases using the Gallyas method and tau (AT8) imunotohistochemistry to screen for the presence or absence of astrocytic plaques in the frontal cortex and subcortical nuclei. As the result, one case had a very small number of morphologically typical astrocytic plaques in the frontal cortex, striatum and globus pallidus with minimal threads. This case was excluded from the following examination. The remaining 19 AGD cases lacked NFTs severe enough to meet the pathological criteria of PSP 16. The 19 AGD cases, nine pathologically‐confirmed PSP cases and 20 control cases were included in the further evaluation.
Then, we determined the presence or absence of Gallyas‐positive tau‐positive TAs and Gallyas‐negative but tau‐positive astrocytic inclusions morphologically similar to TAs (TAIs) in the subcortical nuclei (ie, the basal ganglia, brain stem nuclei, and dentate nucleus of the cerebellum) and frontal cortex in the 19 cases having argyrophilic grains using the Gallyas method and tau (AT8) immunohistochemistry. As a result, five cases (26.3%) had at least one Gallyas‐positive tau‐positive TA, six cases (31.6%) had at least one TAI alone, and the remaining eight cases (42.1%) lacked TAs or TAIs in any regions. Based on these preliminary results, we defined the following four pathological groups:
-
The AGD group (N = 8): AGD was defined by (i) the presence of argyrophilic grains (Figure 1a–d), (ii) the absence of TAs and TAIs in the frontal cortex, basal ganglia and brain stem nuclei, and (iii) the quantity of NFTs that did not meet the pathological criteria of PSP 16.
Figure 1.
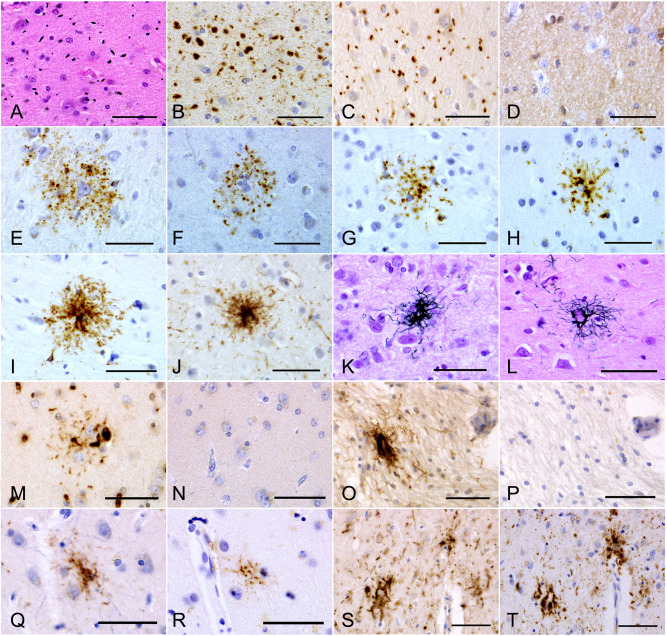 A–D. Argyrophilic grains in the amygdala in an AGD‐TA case. Argyrophilic grains are (A) Gallyas‐positive, (B) AT8‐positive, (C) RD4‐positive, but (D) RD3‐negative. E–G. AT8‐positive TAIs in the frontal cortex in AGD‐TA cases lacking Gallyas‐positive TAs in any region. Fine dot‐like granular phosphorylated tau accumulations surround the nuclei of astrocytes. H, I. AT8‐positive astrocytic lesions in the frontal cortex in AGD‐TA cases having Gallyas‐positive TAs. Phosphorylated tau tends to be densely accumulated around the nuclei of astrocytes. J. An AT8‐positive astrocytic lesion in a PSP case. Fibrous rather than dot‐like structures are densely distributed in the central portion. Their morphological features tended to be similar to those in AGD‐TA cases having Gallyas‐positive TAs (H, I) rather than in AGD‐TA cases lacking them (E, F, G). K, L. Gallyas‐positive TAs in the frontal cortex in (K) AGD‐TA and (L) PSP cases. They could not be distinguished morphologically. M, N. (M) An RD4‐positive TAI in an AGD‐TA case. (N) The lesion lacked RD3 immunoreactivity. Serial sections from the caudate nucleus. This case lacked Gallyas‐positive structures in this region. O, P. (O) An RD4‐positive astrocytic lesion in a PSP case. (P) The lesion lacked RD3 immunoreactivity. Serial sections from the putamen. Q, R. (Q) An RD4‐positive astrocytic lesion in the frontal cortex in an AGD‐TA case having Gallyas‐positive TAs. (R) The lesion also showed AT8 immunoreactivity. The identical region in mirror serial sections is shown after reversal. S, T. (S) RD4‐positive astrocytic lesions in the frontal cortex in a PSP case. S. The lesions also showed AT8 immunoreactivity. T. Mirror serial section of the identical region. All scale bars = 50 μm.
A–D. Argyrophilic grains in the amygdala in an AGD‐TA case. Argyrophilic grains are (A) Gallyas‐positive, (B) AT8‐positive, (C) RD4‐positive, but (D) RD3‐negative. E–G. AT8‐positive TAIs in the frontal cortex in AGD‐TA cases lacking Gallyas‐positive TAs in any region. Fine dot‐like granular phosphorylated tau accumulations surround the nuclei of astrocytes. H, I. AT8‐positive astrocytic lesions in the frontal cortex in AGD‐TA cases having Gallyas‐positive TAs. Phosphorylated tau tends to be densely accumulated around the nuclei of astrocytes. J. An AT8‐positive astrocytic lesion in a PSP case. Fibrous rather than dot‐like structures are densely distributed in the central portion. Their morphological features tended to be similar to those in AGD‐TA cases having Gallyas‐positive TAs (H, I) rather than in AGD‐TA cases lacking them (E, F, G). K, L. Gallyas‐positive TAs in the frontal cortex in (K) AGD‐TA and (L) PSP cases. They could not be distinguished morphologically. M, N. (M) An RD4‐positive TAI in an AGD‐TA case. (N) The lesion lacked RD3 immunoreactivity. Serial sections from the caudate nucleus. This case lacked Gallyas‐positive structures in this region. O, P. (O) An RD4‐positive astrocytic lesion in a PSP case. (P) The lesion lacked RD3 immunoreactivity. Serial sections from the putamen. Q, R. (Q) An RD4‐positive astrocytic lesion in the frontal cortex in an AGD‐TA case having Gallyas‐positive TAs. (R) The lesion also showed AT8 immunoreactivity. The identical region in mirror serial sections is shown after reversal. S, T. (S) RD4‐positive astrocytic lesions in the frontal cortex in a PSP case. S. The lesions also showed AT8 immunoreactivity. T. Mirror serial section of the identical region. All scale bars = 50 μm. The AGD with TAs or TAIs group (AGD‐TA: N = 11): AGD‐TA was defined by (i) the presence of argyrophilic grains, (ii) the presence of one or more TAs (Figure 1k,l) and/or TAIs (Figure 1e–g) in at least one region of the frontal cortex and subcortical nuclei and (iii) a quantity of NFTs that did not meet the pathological criteria of PSP 16.
The PSP group (N = 9) was composed of cases having AT8‐positive NFTs whose quantity and distribution met the pathological criteria of either typical or atypical PSP 16. The regions examined for assessment of NFTs were the caudate nucleus, putamen, globus pallidus, subthalamic nucleus, oculomotor nucleus, substantia nigra, pontine nucleus, inferior olivary nucleus and dentate nucleus in the cerebellum. Of these nine PSP cases, eight cases had TAs in at least the striatum and frontal cortex, and one had TAIs in these regions. Two of nine PSP cases had stage III argyrophilic grains. The remaining seven PSP cases lacked argyrophilic grains. The distribution and quantities of NFTs, TAs and TAIs in PSP cases with and without argyrophilic grains were comparable (data not shown).
The control group (N = 20) was composed of cases that lacked abnormal tau accumulation except for mild to moderate NFTs. The Braak NFT stage in the control group was matched to those in AGD and AGD‐TA groups, respectively.
None of the subjects had a high level of Alzheimer's disease (AD) neuropathologic changes with Braak NFT stages V–VI 5, 34, CBD 11, Pick's disease, white matter tauopathy with globular glial inclusions 24, primary TDP‐43 proteinopathies, or the NFTs of senile dementia 50. All subjects were selected from an autopsy case series registered with the Department of Neuropsychiatry, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences. Seven of eight AGD cases, 10 of 11 AGD‐TA cases, two of nine PSP cases, and all control cases died in psychiatric hospitals, and the remaining cases died in general hospitals. Autopsies were performed after informed consent was obtained from family members, and all experiments in this study were approved by the ethics committees of the Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, the National Hospital Organization Minami‐Okayama Medical Center, Zikei Institute of Psychiatry, and Tokyo Metropolitan Institute of Medical Science. A standardized neuropathological evaluation was done on all of these subjects. Brain tissue samples were fixed post‐mortemwith 10% formaldehyde and embedded in paraffin. Ten‐micrometer‐thick sections from the frontal, temporal, parietal, occipital, insular, and cingulate cortices, hippocampus, amygdala, basal ganglia, midbrain, pons, medulla oblongata and cerebellum were prepared. For the standardized neuropathological assessment, sections were stained with hematoxylin‐eosin and Klüver‐Barrera stains, and selected regions with modified Bielschowsky silver and Gallyas methods, and anti‐tau, Aβ, α‐synuclein, and TDP‐43 antibodies.
Immunohistochemistry
Paraffin sections were cut at 10 μm thickness and included the frontal, temporal, parietal, and occipital cortices, basal ganglia, brain stem and cerebellum from all AGD, PSP and control cases. Immunochemistry was performed by the avidin‐biotin‐peroxidase complex (ABC) method using a Vectastain Elite ABC kit (Vector Laboratories, Inc., Burlingame, CA, USA) as reported previously 52. Antibodies used were against tau phosphorylated at Ser 202 (AT8, mouse, monoclonal, 1:1000, Innogenetics, Ghent, Belgium), tau phosphorylated at Thr212 and Ser 214 (AT100, mouse, monoclonal, 1:100, Innogenetics), tau phosphorylated at Thr 231 (AT180, mouse, monoclonal, 1:40; Innogenetics), tau phosphorylated at Thr 181 (AT270, mouse, monoclonal, 1:500, Innogenetics), tau epitope at aa404‐441 (T46, mouse, monoclonal, 1:100, Invitrogen, Camarillo, CA, USA), 3R tau (RD3, mouse, monoclonal, 1:2000, Millipore, Temecula, CA, USA), 4R tau (RD4, mouse, monoclonal, 1:100, Millipore), 4R tau (anti‐4R tau, rabbit, polyclonal, 1:2000, Cosmo Bio Co., Tokyo, Japan), Aβ11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28 (12B2, mouse, monoclonal, 1:4000, IBL, Fujioka, Japan), phosphorylated α‐synuclein (psyn#64, mouse, monoclonal, 1:1000, Wako Co. Ltd., Osaka, Japan), and phosphorylated TDP‐43 (pS409/410‐2, rabbit, polyclonal, 1:5000, Cosmo Bio). When using anti‐Aβ antibodies, sections were pretreated with 98% formic acid for 1 minute for antigen retrieval. When using psyn#64 and pS409/410‐2, sections were autoclaved for 10 minutes in 10 mM sodium citrate buffer at 120°C. When using RD4, RD3, and anti‐4R tau, sections were pretreated with 98% formic acid for 1 minute and autoclaved for 10 minutes in 10 mM sodium citrate buffer at 120°C 45. Recently, it was also reported that RD3 immunoreactivity was strongly enhanced by additional treatment with 10 μg/mL proteinase K for 30 minutes after autoclaving and formic acid treatment 15. Therefore, this procedure was also applied to confirm the absence of 3R tau immunoreactivity in TAs and TAIs. The peroxidase labeling was visualized with 0.2% 3,3′‐diaminobenzidine (DAB) as the chromogen. Sections were lightly counterstained with hematoxylin.
Whether both epitopes in the same astrocytic lesions were recognized by AT8 and RD4 was examined using mirror sections. Two 10‐μm‐thick serial paraffin sections were obtained with the cut surfaces facing each other. Each section was stained by AT8 or RD4 immunohistochemistry, respectively, and then the distribution of AT8‐positive inclusions was compared that of RD4‐positive inclusions.
In addition, to distinguish TAIs from TAs, sections from the frontal cortex and putamen in representative cases of PSP and AGD‐TA having Gallyas‐positive TAs were stained by the Gallyas method, followed by immunostaining with AT8. The peroxidase labeling was visualized with DAB or Vector NovaRED (Vector Laboratories, Inc., Burlingame, CA, USA) as the chromogen. Sections were lightly counterstained with hematoxylin.
Semiquantitative assessment of tau‐positive NFTs, Gallyas‐positive TAs, tau‐positive astrocytic lesions and neuronal loss in subcortical nuclei and frontal cortex
NFTs in the superior frontal gyrus, caudate nucleus, putamen, globus pallidus, subthalamic nucleus, substantia nigra, oculomotor nucleus, pontine nucleus, inferior olivary nucleus and dentate nucleus in the cerebellum in all subjects were semiquantitatively assessed by the following staging system using AT8 immunohistochemistry by a reviewer blinded to any clinical information: stage 0, no lesion; stage 1, one or more thread‐like structures without NFT in the anatomical region; stage 2, one NFT in the anatomical region; stage 3, two to four NFTs in the anatomical lesion but fewer than one NFT per ×200 visual field; stage 4, one NFT per ×200 visual field; stage 5, 2 to 10 NFTs per ×200 visual field; stage 6, 11 to 20 NFTs per ×200 visual field; stage 7, over 20 NFTs per ×200 visual field. For the pathological diagnosis of PSP 16, at least our stage 4 for the diagnosis of “atypical PSP” or our stage 5 for the diagnosis of “typical PSP” in prescribed subcortical regions examined in the present study was necessary.
In the same regions in AGD‐TA cases, TAs were also semiquantitatively assessed on sections stained with the Gallyas method using the following grading system: stage 0, no lesion; stage 1, one TA in the anatomical region; stage 2, two to four TAs in the anatomical lesion but less than one TA per ×200 visual field; stage 3, one TA per ×200 visual field; stage 4, 2 to 10 TAs per ×200 visual field; stage 5, 11 to 20 TAs per ×200 visual field; stage 6, over 20 TAs per ×200 visual field.
In AGD‐TA cases, the number of tau‐positive astrocytic lesions was also examined using the same grading system for TAs on sections stained with AT8, because AT8 immunoreactivity was stronger than those of AT100, AT180, AT270, T46, anti‐4R tau and RD4 in some cases. In AGD‐TA cases without Gallyas‐positive TAs, the number of tau‐positive astrocytic lesions is considered the same as the number of TAIs. Conversely, in AGD‐TA cases with Gallyas‐positive TAs, we counted “tau‐positive astrocytic lesions” without distinguishing between TAIs and TAs because the boundary between morphological features of TAIs and TAs could not be sharply demarcated on AT8‐immunostained sections (Figure 1e–i). Therefore, in this article, we use the term “tau‐positive astrocytic lesion” when the absence of Gallyas‐positive structures was not confirmed in the region or the lesion, and used the term “TAI” only when it was confirmed. When a case had Aβ deposits in the frontal cortex, these tau‐positive astrocytic lesions in the site were not assessed, because dystrophic neurites of neuritic plaques were often morphologically similar to these astrocytic lesions. None of the AGD or AGD‐TA cases had neuritic plaques in the basal ganglia, brains stem nuclei, or dentate nucleus in the cerebellum.
The degree of neuronal loss in the caudate nucleus, putamen, globus pallidus, subthalamic nucleus, substantia nigra and oculomotor nucleus in all subjects was semiquantitatively assessed on sections stained by hematoxylin‐eosin and Klüver‐Barrera methods according to the grading system used previously52, 53, 54: stage 0, no neuronal loss; stage 1, mild neuronal loss with gliosis; stage 2, moderate neuronal loss with gliosis, but no tissue rarefaction; stage 3, severe neuronal loss with severe gliosis and tissue rarefaction.
Assessment of argyrophilic grains and other lesions
The presence of argyrophilic grains was evaluated using the Gallyas method and AT8 immunohistochemistry. In cases having argyrophilic grains, the accumulation of 4R tau and absence of 3R tau in the lesions were confirmed using RD4 and RD3 immunohistochemistry. The distribution of argyrophilic grains was classified into four stages (stage 0–III) using a system proposed by Saito et al 38. Briefly, argyrophilic grains initially develop in the ambient gyrus and amygdala (stage I), subsequently in the entorhinal cortex, hippocampus and occipitotemporal cortex (stage II), and finally in the cingulate and insular cortices and gyri recti (stage III).
The distribution of NFTs as AD pathology in the limbic system and neocortex was evaluated by Braak stage 5. The stage was first determined using AT8 immunohistochemistry and RD3 immunohistochemistry, respectively, because it is difficult to differentiate NFTs related to AD 14 from neuronal tau accumulation related to four‐repeat tauopathies (ie, AGD and PSP) only using AT8 immunohistochemistry. As a result, in our subjects, the number of Braak stage of RD3‐positive NFTs tended to be lower than that of the Braak stage of AT8‐positive NFTs, and the numbers of Braak stage of RD3‐positive NFTs, as well as the Braak stage of AT8‐positive NFTs, did not significantly differ between the four experimental groups (ie, AGD, AGD‐TA, PSP, and control groups). Therefore, only the Braak stage of RD3‐positive NFTs, which may not be affected by tau accumulation as AGD or PSP pathology, is shown in Tables 1, 2, 3.
Table 1.
Demographic data of all subjects.
| AGD group* | AGD‐TA groupb | PSP groupc | Control groupd | P value | |
|---|---|---|---|---|---|
| N | 8 | 11 | 9 | 20 | |
| Female (%) | 4 (50.0) | 7 (63.6) | 2 (22.2) | 6 (30.0) | 0.1860 |
| Age at onsete (y, mean ± SD) | 50.4 ± 24.7e | 65.1 ± 24.2e | 59.3 ± 9.0 | 41.4 ± 21.9e | |
| Age at death (y, mean ± SD) | 74.0 ± 8.4 | 82.0 ± 6.8f | 67.0 ± 7.2 | 71.1 ± 7.9 | 0.0018 |
| Brain weight (g, mean ± SD) | 1253 ± 130 | 1111 ± 198.3 | 1155 ± 190 | 1229 ± 180 | 0.1653 |
| Braak NFT stageg | |||||
| Median (25‐75 percentile) | 2.0 (1.0–2.0) | 2.0 (2.0–2.0) | 1.0 (1.0–2.0) | 2.0 (1.0–2.0) | 0.4951 |
| Braak stage 0 [N, (%)] | 1 (12.5) | 0 (0.0) | 2 (22.2) | 2 (10.0) | |
| Braak stage I [N, (%)] | 2 (25.0) | 2 (18.2) | 3 (33.3) | 7 (35.0) | |
| Braak stage II [N, (%)] | 5 (62.5) | 8 (72.7) | 3 (33.3) | 7 (35.0) | |
| Braak stage III [N, (%)] | 0 (0.0) | 0 (0.0) | 0 (0.0) | 3 (15.0) | |
| Braak stage IV [N, (%)] | 0 (0.0) | 1 (9.1) | 1 (11.1) | 1 (5.0) | |
| Braak stage V [N, (%)] | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Braak stage VI [N, (%)] | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Thal Aβ phase [N, (%)] | |||||
| Median (25‐75 percentile) | 0.0 (0.0–3.0) | 1.0 (0.0–3.0) | 0 (0.0–1.0) | 0.0 (0.0–1.0) | 0.6629 |
| Phase 0 [N, (%)] | 5 (62.5) | 5 (45.5) | 5 (55.6) | 13 (65.0) | |
| Phase 1 [N, (%)] | 0 (0.0) | 1 (9.1) | 2 (22.2) | 3 (15.0) | |
| Phase 2 [N, (%)] | 1 (12.5) | 0 (0.0) | 1 (11.1) | 1 (5.0) | |
| Phase 3 [N, (%)] | 0 (0.0) | 4 (36.4) | 1 (11.1) | 1 (5.0) | |
| Phase 4 [N, (%)] | 1 (12.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Phase 5 [N, (%)] | 1 (12.5) | 1 (9.1) | 0 (0.0) | 2 (10.0) | |
| AGD stage | |||||
| Median (25–75 percentile) | 0 (1.0–2.0)h | 2 (1.0–3.0)i | 0 (0–0) | 0 (0–0) | <0.0001 |
| Stage 0 [N, (%)] | 0 (0.0) | 0 (0.0) | 7 (77.8) | 20 (100.0) | |
| Stage I [N, (%)] | 4 (50.0) | 4 (36.4) | 0 (0.0) | 0 (0.0) | |
| Stage II [N, (%)] | 4 (50.0) | 2 (18.2) | 0 (0.0) | 0 (0.0) | |
| Stage III [N, (%)] | 0 (0.0) | 5 (45.5) | 2 (22.2) | 0 (0.0) | |
AGD cases had argyrophilic grains but lacked AT8‐positive astrocytic lesions and sufficient NFTs that met the pathological criteria of PSP 16.
AGD‐TA cases had argyrophilic grains and a few Gallyas‐positive tau‐positive TAs and/or Gallyas‐negative tau‐positive TAIs but no NFTs that met the pathological criteria of PSP 16.
PSP cases had both TAs/TAIs and NFTs that meet the pathological criteria of PSP 16.
The Braak NFT stage in a control group was matched with those in both AGD and AGD‐TA groups, respectively.
The mean age at onset of psychiatric disorders is shown.
The age at death in the AGD‐TA group was significantly higher than that in the PSP group (P = 0.0064) and control group (P = 0.0081).
Braak NFT stage determined with RD3 immunohistochemistry was shown.
The AGD stage in the AGD group was significantly higher than that in the control group (P < 0.0001).
The AGD stage in the AGD‐TA group was significantly higher than that in the PSP group (P = 0.0451) and control group (P < 0.0001). N = number of cases; y = years; SD = standard deviation.
Table 2.
Distribution of AT8‐positive NFTs in all AGD cases.
| Neurofibrillary tangles | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Braak NFT stage*/Thal Aβ phase | Lewy body disease subtype | Argyrophilic grain disease stage | TDP‐43 pathology | Superior frontal cortexb | Caudate nucleus | Putamen | Globus pallidus | Subthalamic nucleus | Substantia nigra | Oculomotor nucleus | Pontine nucleus | Inferior olivary nucleus | Dentate nucleus in cerebellum | |
| AGD 1 | II/5 | 0 | I | 0 | n.e. | − | − | − | − | − | − | − | − | − |
| AGD 2 | II/0 | 0 | I | 0 | − | − | − | − | − | − | − | − | − | − |
| AGD 3 | I/0 | Limbic type | I | 0 | + | − | − | − | − | − | − | − | − | − |
| AGD 4 | I/0 | 0 | II | 0 | + | − | − | − | +++ | +++ | +++ | − | − | + |
| AGD 5 | II/0 | 0 | II | 0 | − | − | +++ | +++ | ++++ | ++ | +++++ | − | − | − |
| AGD 6 | II/4 | 0 | II | 0 | n.e. | +++ | +++ | +++++ | ++++ | ++++ | +++++ | +++ | − | − |
| AGD 7 | II/2 | 0 | I | Temporal type | +++ | +++ | +++ | +++ | n.a. | +++ | ++ | +++ | − | − |
| AGD 8 | 0/0 | 0 | II | 0 | + | ++++ | ++++ | ++++ | n.a. | ++++ | +++++ | ++++ | − | − |
| (%) | 66.7 | 37.5 | 50.0 | 50.0 | 50.0 | 62.5 | 62.5 | 37.5 | 0.0 | 12.5 | ||||
Braak NFT stage determined on sections stained with RD3 is shown.
The number of NFTs in the superior frontal cortex was assessed when the case did not have Aβ deposits in the region. Semiqunatitative NFT stages 0 to 7 correspond to – to +++++++ (see text). n.e. = not examined; n.a. = not available.
Table 3.
Distribution of AT8‐positive NFTs in all AGD‐TA cases.
| Neurofibrillary tangles | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Braak NFTstage*/Thal Aβ phase | Lewy body disease subtype | Argyrophilic grain disease stage | TDP‐43 pathology | Superior frontal cortexb | Caudate nucleus | Putamen | Globus pallidus | Subthalamic nucleus | Substantia nigra | Oculomotor nucleus | Pontine nucleus | Inferior olivary nucleus | Dentate nucleus in cerebellum | ||
| AGD‐TA 1 | I/3 | Brain stem type | I | 0 | − | ++++ | − | − | − | + | − | − | − | − | |
| AGD‐TA 2 | II/0 | 0 | I | 0 | +++ | − | ++++ | +++ | n.a. | + | +++++ | + | − | − | |
| AGD‐TA 3 | II/3 | 0 | II | 0 | n.e. | +++ | +++ | + | n.a. | + | +++ | +++ | +++ | − | |
| AGD‐TA 4 | II/0 | 0 | I | 0 | +++ | − | ++ | +++ | n.a. | + | − | ++ | + | + | |
| AGD‐TA 5 | II/0 | 0 | I | 0 | +++ | +++ | +++ | + | ++ | ++ | ++ | ++ | +++ | + | |
| AGD‐TA 6 | II/5 | 0 | III | 0 | n.e. | +++ | ++++ | + | +++++ | +++++ | +++ | +++ | − | +++ | |
| AGD‐TA 7 | II/3 | 0 | II | 0 | n.e. | +++ | +++ | +++ | +++ | ++++ | +++++ | +++ | +++ | +++ | |
| AGD‐TA 8 | II/0 | 0 | III | 0 | +++ | +++++ | +++++ | ++++ | +++++ | +++++ | +++++ | + | + | ++ | |
| AGD‐TA 9 | I/1 | 0 | III | Amygadala type | ++++ | +++ | + | +++ | ++ | + | − | +++ | +++ | +++ | |
| AGD‐TA 10 | IV/3 | Limbic type | III | Limbic type | n.e. | +++++ | ++++ | ++++ | ++++ | ++++ | +++ | +++ | ++ | +++ | |
| AGD‐TA 11 | II/0 | 0 | III | Amygdala type | ++++ | +++++ | ++++ | +++ | +++++ | +++++ | ++++++ | +++ | ++ | +++ | |
| (%) | 85.7 | 81.8 | 90.9 | 90.9 | 87.5 | 100.0 | 72.7 | 90.9 | 72.7 | 72.7 | |||||
Braak NFT stage determined with RD3 immunohistochemistry is shown.
The number of NFTs in the superior frontal cortex was assessed when the case did not have Aβ deposits in the region. Semiqunatitative NFT stages 0 to 7 correspond to – to +++++++ (see text). n.e. = not examined; n.a. = not available.
Aβ deposits 43, α‐synuclein‐positive Lewy body‐related pathology 33, 48, and TDP‐43‐positive lesions 2, 27, 52 were assessed using established criteria, respectively.
Confocal laser scanning microscopy
Double‐labeling immunofluorescence was performed with the combination of phosphorylation dependent anti‐tau antibody (AT8, mouse, monoclonal, 1:500) and anti‐GFAP antibody (GFAP, rabbit, polyclonal, 1:100, Dako, Glostrup, Denmark) to examine the immunohistochemical features of tau‐positive astrocytic lesions. Sections from the frontal cortex in AGD‐TA and PSP cases were autoclaved for 10 minutes in 10 mM sodium citrate buffer at 120°C. Following washing in phosphate buffered saline (PBS), nonspecific antibody binding was blocked with a 5% skim‐milk buffer, and sections were incubated overnight with a mixture of the two primary antibodies at 4°C. After washing in PBS, sections were incubated with fluorescence‐labeled secondary antibodies (AlexaFluor 488 anti‐mouse IgG (1:200) and AlexaFluor 594 anti‐rabbit IgG (1:200), Molecular Probes, Inc., Eugene, OR) for 1 h. To quench (lipofuscin) autofluorescence, sections were incubated in 0.1% Sudan Black B for 10 minutes at room temperature and washed with 0.5% Tx‐PBS for 30 minutes. Sections were coverslipped with FluoromountTM (Diagnostic Biosystems, Pleasanton, CA, USA). Images were collected using a confocal microscope LSM780 (Carl Zeiss, Jena, Germany).
Tau immunoblotting
Frozen brain tissue in one AGD, one AGD‐TA having Gallyas‐positive TAs, and four PSP cases was available. These samples were used for Western blotting according to methods described previously 10, 30. Brain samples (0.5 g) from patients with AGD and PSP were individually homogenized in 10 mL of homogenization buffer (HB: 10 mM Tris–HCl, pH7.5, containing 0.8 M NaCl, 1 mM EGTA, 10% sucrose). Sarkosyl was added to the lysates (final concentration: 2%), which were then incubated for 30 minutes at 37°C and centrifuged at 20 000 × g for 10 minutes at 25°C. The supernatant was divided into six tubes (each 1.3 mL) and centrifuged at 100 000 × g for 20 minutes at 25°C. The pellets were further washed with sterile saline (0.5 mL/tube) and centrifuged at 100 000 × g for 20 minutes. The resulting pellets were used as the sarkosyl‐insoluble fraction (ppt).The sarkosyl‐ppt was sonicated in 50 μL (/tube) of 30 mM Tris‐HCl (pH 7.5) and solubilized in 2× sample buffer. Samples were run on gradient 4% to 20% polyacrylamide gels and electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes. Residual protein‐binding sites were blocked by incubation with 3% gelatin (Wako) for 10 minutes at 37°C, followed by overnight incubation at room temperature with the primary antibody (T46, mouse, monoclonal, 1:2000). The membrane was incubated for 1 h at room temperature with anti‐mouse IgG (BA‐2000, Vector Lab), then incubated for 30 minutes with avidin‐horseradish peroxidase (Vector Lab), and the reaction product was visualized using 0.1% 3,3‐diaminobenzidine (DAB) and 0.2 mg/mL NiCl2 as the chromogen.
Genetic analysis
The H1/H2 haplotype of one AGD, one AGD‐TA having TAs, and four PSP cases for which frozen tissue samples were available was determined. Genomic DNA was extracted from frozen brain using the High Pure PCR Template Preparation kit (Roche, Mannheim, Germany). We selected a single nucleotide polymorphism of the microtubule‐associated protein tau gene, rs9468, which is used to determine whether a sample has the H1 or H2 haplotype 1, 39. Genotyping was performed using TaqMan technology on an ABI7500 Real Time PCR system (Applied Biosystems, Foster City, CA, USA), according to methods described previously 37.
Statistical analysis
Kruskal–Wallis and Steel tests were used when comparing the control group and the three disease groups (ie, AGD, AGD‐TA and PSP groups). When comparing AGD, AGD‐TA, PSP and control groups and when comparing the three AGD stage groups (stages I–III), Kruskal–Wallis and Steel‐Dwass tests were used. The Mann‐Whitney U test and Fisher's exact test were used to compare two groups. Spearman rank order correlation analysis was applied for univariate correlations between (i) the AGD stage, (ii) the quantity of tau (AT8)‐positive NFTs in the subcortical nuclei (ie, the caudate nucleus, putamen, globus pallidus, subthalamic nucleus and substantia nigra), (iii) the quantity of tau (AT8)‐positive astrocytic lesions in the subcortical nuclei (the same regions for AT8‐positive NFTs) and (iv) the Braak stage of RD3‐positive NFTs. The correlation between the quantity of AT8‐positive NFTs and AT8‐positive astrocytic lesions was examined in each subcortical nucleus, respectively. A P value <0.05 was accepted as significant. Statistical analysis was performed using Excel.
RESULTS
Demographic data of all subjects
he demographic data of all subjects are shown in Table 1. The sex ratio, brain weight, Braak NFT stage and Thal Aβ stage were not significantly different among the four groups (Fisher exact test, Kruskal–Wallis and Steel‐Dwass tests). The age at death was significantly higher in AGD‐TA cases than in PSP and control cases, respectively (P = 0.0064 and 0.0081, Kruskal–Wallis and Steel‐Dwass tests). Lewy bodies were found in one AGD (limbic type), two AGD‐TA (brain stem type and limbic type), one PSP (brain stem type), and five control cases (brain stem type [N = 4], limbic type [N = 1]). Mild TDP‐43 pathology was found in the limbic region with or without it in the adjacent temporal cortex in one AGD, three AGD‐TA, and three PSP cases. Clinical diagnoses in AGD cases were senile‐onset psychosis (N = 4), schizophrenia (N = 2), depression (N = 1), and dementia (N = 1), and those in AGD‐TA cases were senile‐onset psychosis (N = 4), dementia (N = 3), schizophrenia (N = 2), cerebral infarction (N = 1) and gait disturbance (N = 1). Clinical diagnoses in pathologically confirmed PSP cases were PSP (N = 3), CBD (N = 3), parkinsonian syndrome (N = 2) and dementia (N = 1). Clinical diagnoses in control cases were schizophrenia (N = 11), bipolar disorder (N = 3), depression (N = 2), cerebrovascular disease (N = 2), senile‐onset psychosis (N = 1), and dementia with Lewy bodies (N = 1).
Distribution of NFTs in subcortical nuclei and frontal cortex in AGD, AGD‐TA, PSP and control cases
The distributions of tau (AT8)‐positive NFTs in AGD and AGD‐TA groups are shown in Tables 2 and 3, respectively. In both groups, the region most frequently affected by NFTs was the substantia nigra, followed by the putamen, globus pallidus, subthalamic nucleus and pontine nucleus. The frontal cortex was also frequently affected by NFTs in both groups. NFTs in the medulla oblongata and cerebellum were rare in AGD cases but frequent in AGD‐TA cases.
In comparisons of the quantities of NFTs in AGD, AGD‐TA and PSP groups to that in the Braak NFT stage‐matched control group, NFTs in all regions in the PSP group were significantly more frequent than those in the control group (P < 0.0001, respectively, Kruskal–Wallis and Steel tests; Figure 2). Likewise, NFTs in all regions except for the substantia nigra in the AGD‐TA group were significantly more frequent than those in the control group (P < 0.05, respectively). Conversely, NFTs were significantly more frequent in the globus pallidus and pontine nucleus in the AGD group compared with the control group (P <0.05, respectively).
Figure 2.
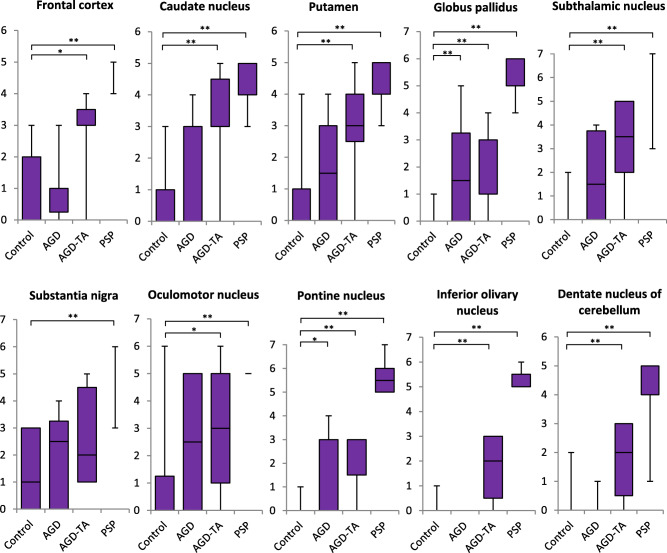
The quantity of tau (AT8)‐positive NFTs in AGD, AGD‐TA, PSP and control groups by anatomical region. The quantity of NFTs is shown on the vertical axis (see text for the definition of each stage). In all anatomical regions, NFTs were sequentially increased in number from control, AGD, AGD‐TA, to PSP groups. NFTs in all regions in the PSP group were significantly more severe than those in the control group. Likewise, NFTs in all regions except for the substantia nigra in the AGD‐TA group were significantly more severe than those in the control group. NFTs in the globus pallidus and pontine nucleus in the AGD group were significantly more severe than those in the control group. Kruskal–Wallis and Steel tests; *P < 0.05; **P < 0.01.
Severity of neuronal loss in subcortical nuclei in AGD, AGD‐TA, PSP and control cases
In all regions examined, the degree of neuronal loss in PSP cases was significantly more severe than that in control cases (P < 0.01, respectively, Kruskal–Wallis and Steel tests; Figure 3). In contrast, the degree of neuronal loss did not significantly differ between AGD‐TA and control groups in any region except the oculomotor nucleus (Figure 3). Likewise, there was no significant difference in the degree of neuronal loss in any region between the AGD and control groups (Figure 3).
Figure 3.
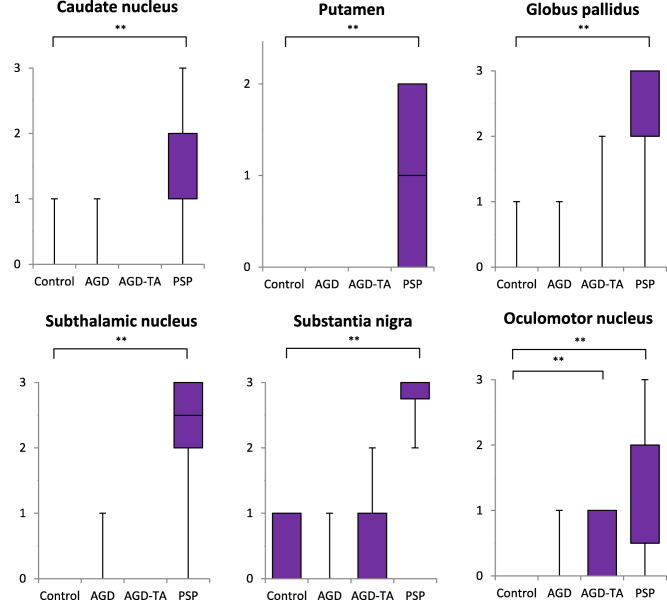
Severity of neuronal loss in AGD, AGD‐TA, PSP and control groups. The severity of neuronal loss is shown on the vertical axis (stage 0–3 means no, mild, moderate and severe). Neuronal loss in the caudate nucleus, putamen, globus pallidus, subthalamic nucleus, substantia nigra and oculomotor nucleus in the PSP group was significantly more severe than that in the control group. In contrast, no significant loss of neurons was found in any region in the AGD group compared with the control group. Likewise, significant loss of neurons was not found in any region except for the oculomotor nucleus in the AGD‐TA group compared with the control group. Kruskal–Wallis and Steel tests; **P < 0.01.
Distributions of Gallyas‐positive TAs and tau‐positive astrocytic lesions in AGD‐TA cases
The distributions of Gallyas‐positive TAs and tau (AT8)‐positive astrocytic lesions in AGD‐TA cases are shown in Table 4. The order of cases in this table is identical to that in Table 3. The Gallyas method demonstrated a few TAs in 5 of 11 AGD‐TA cases (AGD‐TA 7–11 in Table 4). The morphological features of the TAs in AGD‐TA cases (Figure 1k) could not be distinguished from those of TAs observed in PSP cases (Figure 1l). TAs were found in the frontal cortex and putamen in AGD‐TA cases that had a relatively large number of NFTs and tau‐positive astrocytic lesions in the subcortical nuclei (Tables 3 and 4).
Table 4.
Distribution of TAs and tau‐positive astrocytic lesions in all AGD‐TA cases.
| Quantity of Gallyas‐positive TAs | Quantity of tau (AT8)‐positive astrocytic lesions* | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Frontal cortex † | Caudate nucleus | Putamen | Globus pallidus | Subthalamic nucleus | Substantia nigra | Oculomotor nucleus | Pontine nucleus | Inferior olivary nucleus | Dentate nucleus in cerebellum | Frontal cortex† | Putamen | Putamen | Globus pallidus | Subthalamic nucleus | Substantia nigra | Oculomotor nucleus | Pontine nucleus | Inferior olivary nucleus | Dentate nucleus in cerebellum | ||
| AGD‐TA | AGD‐TA 1 | − | − | − | − | − | − | − | − | − | − | − | ++ | − | − | − | − | − | − | − | − |
| having | AGD‐TA 2 | − | − | − | − | n.a. | − | − | − | − | − | ++ | − | ++ | ++ | n.a. | − | − | + | − | − |
| only | AGD‐TA 3 | n.e. | − | − | − | n.a. | − | − | − | − | − | n.e. | − | +++ | − | n.a. | − | + | − | − | − |
| TAIs | AGD‐TA 4 | − | − | − | − | n.a. | − | − | − | − | − | +++ | − | + | + | n.a. | − | − | − | − | − |
| AGD‐TA 5 | − | − | − | − | − | − | − | − | − | − | − | + | + | − | − | − | − | − | − | − | |
| AGD‐TA 6 | n.e. | − | − | − | − | − | − | − | − | − | n.e. | ++ | − | − | − | − | − | − | − | − | |
| % | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 50.0 | 50.0 | 66.7 | 33.3 | 0.0 | 0.0 | 16.7 | 16.7 | 0.0 | 0.0 | |
| AGD‐TA | AGD‐TA 7 | n.e. | − | ++ | − | − | − | − | − | +++ | − | n.e. | +++ | ++ | ++ | ++ | − | ++ | − | ++ | − |
| having | AGD‐TA 8 | ++ | ++++ | ++++ | − | − | − | − | − | + | − | ++ | ++++ | ++++ | − | − | − | +++ | − | + | − |
| both TAs | AGD‐TA 9 | +++ | − | ++ | − | − | − | − | − | − | − | +++ | ++ | ++ | ++ | ++ | − | − | − | − | − |
| and TAIs | AGD‐TA 10 | n.e. | − | − | − | − | − | − | − | ++ | − | n.e. | +++ | +++ | ++ | ++ | − | ++ | ++ | ++ | − |
| AGD‐TA 11 | ++ | − | − | − | − | − | − | − | − | − | +++ | +++ | +++ | − | +++ | ++ | + | + | ++ | − | |
| % | 100 | 20.0 | 60.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 60.0 | 0.0 | 100 | 100 | 100 | 60.0 | 80.0 | 20.0 | 80.0 | 40.0 | 80.0 | 0.0 | |
In AGD‐TA 1–6, the number of AT8‐positive astrocytic lesions shown in this table is that of TAIs, because these cases lacked TAs. In AGD‐TA 7–11, AT8‐positive astrocytic lesions may include both TAIs and TAs because these lesions cannot be completely differentiated by AT8 immunohistochemistry. n.e. = not examined; n.a. = not available. Semiquanatitative TA stages 0 to 6 correspond to – to ++++++ (see text).
†The number of TAs and tau‐positive astrocytic lesions in the superior frontal cortex was assessed when the case did not have Aβ deposits in the region. None of subjects had neuritic plaques in other regions examined.
The remaining six AGD‐TA cases (ie, AGD‐TA 1–6 in Table 4) had a small number of tau‐positive astrocytic lesions on AT8 immunostained sections but lacked TAs in any region on sections stained with the Gallyas method; therefore, all of the tau‐positive astrocytic lesions were considered to be Gallyas‐negative TAIs. TAIs preferentially occurred in the putamen, followed by the frontal cortex, caudate nucleus and globus pallidus (Table 4). Thus, regions frequently affected by TAIs in these six AGD‐TA cases (AGD‐TA 1–6) were almost the same as those of TAs observed in the previous five AGD‐TA cases (AGD‐TA 7–11).
Morphological and immunohistochemical features of Gallyas‐positive tau‐positive TAs and Gallyas‐negative tau‐positive TAIs in AGD‐TA and PSP cases
On AT8 immunostained sections, in TAIs in AGD‐TA cases lacking Gallyas‐positive TAs, fine granular tau accumulations were scattered or often radially arranged around the nuclei of astrocytes, but dense accumulations of tau protein were hardly found in the proximal portion of astrocytic processes and cytoplasm (Figure 1e–g).
Conversely, in tau‐positive astrocytic lesions in AGD‐TA cases having Gallyas‐positive TAs, phosphorylated tau tended to be densely accumulated in the proximal portion of astrocytic processes and cytoplasm, and tau‐positive small granules radially surrounded the astrocytic nuclei (Figure 1h,i). These features tended to be more prominent in tau‐positive astrocytic lesions in PSP cases (Figure 1j). However, it was difficult to distinguish TAIs from TAs on AT8‐stained sections.
When double staining with AT8 immunohistochemistry and the Gallyas method, Gallyas‐positive TAs were clearly distinguished from Gallyas‐negative TAIs. In AGD‐TA cases having Gallyas‐positive TAs, both TAIs (Figure 4a) and TAs (Figure 4b–d) were found. The quantity of Gallyas‐positive structures in TAs varied between TAs (Figure 4b–d). In some TAs, a small number of fine Gallyas‐positive thread‐like structures were observed only in the distal portion of each lesion (Figure 4c). In PSP cases also, not only TAs but also TAIs were observed (Figure 4e–h). The quantity of argyrophilic thread‐like structures tended to be larger than that of TAs in AGD‐TA cases.
Figure 4.
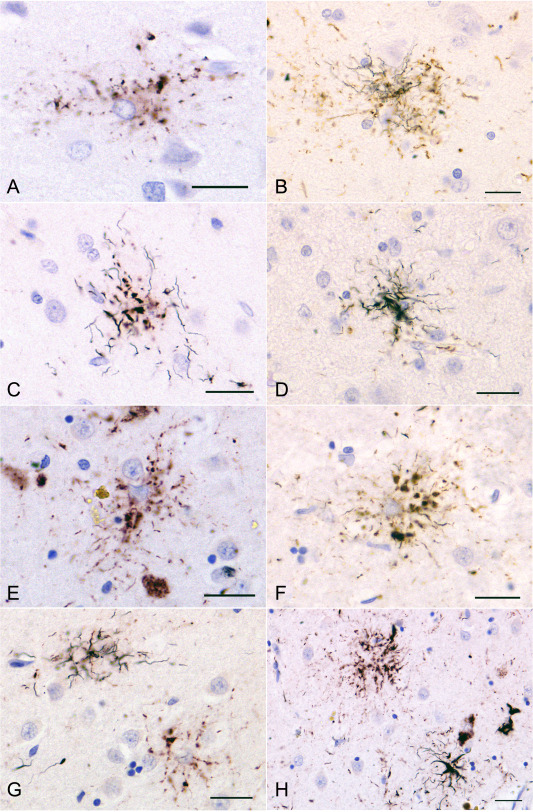
Double staining of AT8 immunohistochemistry and Gallyas method in an AGD‐TA case having Gallyas‐positive TAs and a PSP case. A, B, C, D. TAIs (A) and TAs (B, C, D) in the superior frontal cortex in an AGD‐TA case (AGD‐TA9 in Table 4). The proportion of Gallyas‐positive structures varied between lesions. In some TAs, fine argyrophilic thread‐like structures were found only in the distal portion of each astrocytic lesion (C). E, F, G, H. TAIs and TAs in the putamen in a PSP case. (E) As in AGD‐TA cases, TAIs had fine granular tau accumulations that were radially arranged. It is hard to morphologically distinguish TAIs in PSP cases (E) from TAIs in AGD‐TA cases (A). F. When the number of Gallyas‐positive thread‐like structures was small, they were often found in the distal portion of each astrocytic lesion. G. The lesion on the right lacks a Gallyas‐positive structure (ie, TAI), while the one on the left has Gallyas‐positive thread‐like structures (ie, TAs). H. A right‐hand lesion has Gallyas‐positive thread‐like structures (ie, TA), while the left‐hand one lacks Gallyas‐positive structures (ie, TAIs). The peroxidase labeling was visualized with DAB (B, D, F) or Vector NovaRED (A, C, E, G, H). All scale bars = 20 μm.
RD4 and RD3 immunohistochemistry demonstrated that only 4R tau was accumulated in both TAIs in AGD‐TA cases (Figure 1m,n) and tau‐positive astrocytic lesions in PSP cases (Figure 1o,p). Tau immunohistochemistry using mirror sections demonstrated that tau‐positive astrocytic lesions in AGD‐TA cases (Figure 1q,r) and PSP cases (Figure 1s,t) were labeled with both RD4 and AT8.
Quantities of NFTs and tau‐positive astrocytic lesions by AGD stage in cases of AGD and AGD‐TA
In cases having argyrophilic grains (ie, AGD and AGD‐TA cases), tau (AT8)‐positive NFTs in the subcortical nuclei sequentially increased in number with the progression of AGD (Figure 5). The difference in the quantity of NFTs in the caudate nucleus, subthalamic nucleus, substantia nigra, oculomotor nucleus and dentate nucleus in the cerebellum reached statistical significance between groups with different AGD stages (Kruskal–Wallis and Steel‐Dwass tests).
Figure 5.
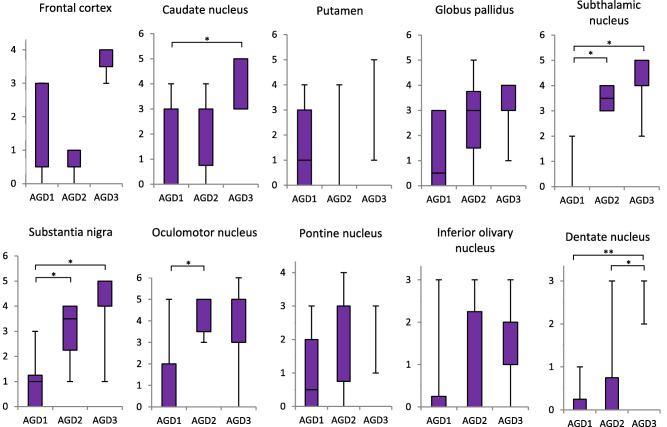
Quantity of tau (AT8)‐positive NFTs by AGD stage in the combined group of AGD and AGD‐TA. The quantity of NFTs is shown on the vertical axis (for definition of each stage, see text). In all regions except for the frontal cortex, the number of NFTs was sequentially increased with the progression of the AGD stage. In the caudate nucleus, subthalamic nucleus, substantia nigra, oculomotor nucleus and dentate nucleus in the cerebellum, the differences in the quantities of NFTs reached statistical significance between AGD stages. Kruskal–Wallis and Steel‐Dwass tests; *P < 0.05; **P < 0.01.
Likewise, tau (AT8)‐positive astrocytic lesions including TAs and TAIs successively increased in number with the progression of AGD in AGD‐TA cases (Figure 6). In the caudate nucleus, oculomotor nucleus and inferior olivary nucleus, the difference in the quantities of astrocytic lesions reached statistical significance between groups with different AGD stages (Kruskal–Wallis and Steel‐Dwass tests).
Figure 6.
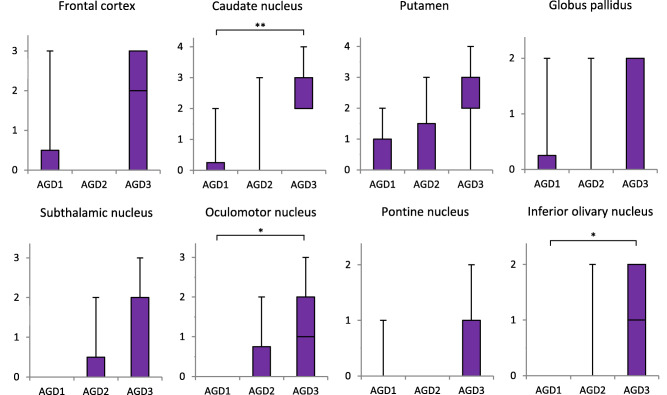
Quantity of tau (AT8)‐positive astrocytic lesions by AGD stage in the combined group of AGD and AGD‐TA. The number of astrocytic lesions is shown on the vertical axis (for definition of each stage, see text). The numbers of TAs and TAIs are included as tau‐positive astrocytic lesions because these two kinds of lesions cannot be assessed separately on AT8‐stained sections. In all regions, the number of tau‐positive astrocytic lesions was sequentially increased with the progression of AGD stage. In the caudate nucleus, oculomotor nucleus, and inferior olivary nucleus, the differences between the quantities of astrocytic lesions in different AGD stages reached statistical significance. Kruskal–Wallis and Steel‐Dwass tests; *P < 0.05; **P < 0.01.
Correlation between AGD stage, Braak stage of RD3‐positive NFTs and AT8‐positive NFTs and AT8‐positive astrocytic lesions in subcortical nuclei
The correlations between four histological factors, (i) the AGD stage, (ii) the Braak stage of RD3‐positive NFTs, (iii) the quantity of AT8‐positive NFTs in the subcortical nuclei and (iv) the quantity of tau (AT8)‐positive astrocytic lesions in the subcortical nuclei were examined (Figure 7). Regarding AT8‐positive NFTs and astrocytic lesions in the subcortical nuclei, the data on the caudate nucleus, putamen, globus pallidus, subthalamic nucleus and substantia nigra were used. Spearman rank order correlation analysis demonstrated that the AGD stage was significantly correlated with the quantity of NFTs in the caudate nucleus (ρ = 0.59, P = 0.007), putamen (ρ = 0.57, P = 0.011), globus pallidus (ρ = 0.50, P = 0.029), subthalamic nucleus (ρ = 0.84, P = 0.0002), and substantia nigra (ρ = 0.72, P = 0.0005). Likewise, the AGD stage was significantly correlated with the quantity of AT8‐positive astrocytic lesions in the caudate nucleus (ρ = 0.62, P = 0.0046), putamen (ρ = 0.47, P = 0.041) and subthalamic nucleus (ρ = 0.57, P = 0.034). In addition, the quantity of AT8‐positive NFTs was significantly correlated with that of AT8‐positive astrocytic lesions in the caudate nucleus (ρ = 0.74, P = 0.0003) and putamen (ρ = 0.54, P = 0.018). In contrast, there was no statistically significant correlation between the Braak stage of RD3‐positive NFTs and the other three factors, including the AGD stage and quantities of AT8‐positive NFTs and astrocytic lesions in the subcortical nuclei.
Figure 7.

Statistical analyses of the relationship between four histological factors, the AGD stage, AT8‐positivie NFTs in the subcortical nuclei, AT8‐positive astrocytic lesions in the subcortical nuclei and Braak stage of RD3‐positive NFTs, in the combined group of AGD and AGD‐TA. The data regarding the quantity of NFTs and AT8‐positive astrocytic lesions in the caudate nucleus, putamen, globus pallidus, subthalamic nucleus and substantia nigra were used in this correlation study. Solid lines represent statistically significant correlation (P < 0.05). N.S. = not significant. See details in the text.
Double‐labeling immunofluorescence of tau and GFAP in tau‐positive astrocytic lesions in frontal cortex of AGD‐TA and PSP cases
In AGD‐TA cases lacking Gallyas‐positive TAs, TAIs tended to have a small number of radially scattered AT8‐positive dot‐ or spindle‐shaped structures (Figure 8a). In these glial lesions, AT8 epitopes were only partially colocalized with GFAP epitopes (Figure 8b,c).
Figure 8.
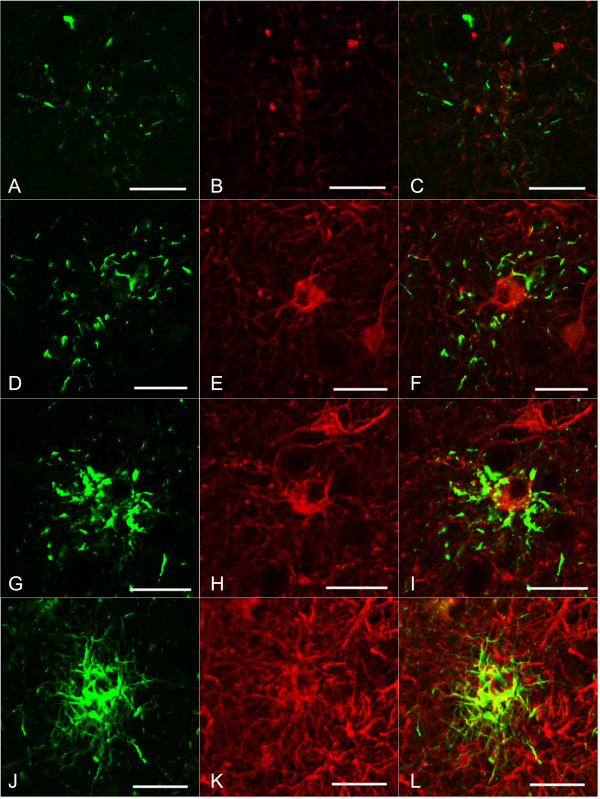
Confocal laser micrographs of double labeling with AT8 (A, D, G, J) and GFAP (B, E, H, K) of the frontal cortex in AGD‐TA and PSP cases. C, F, I, L. Merged images. Except for one PSP case, none of these cases had Aβ deposits in this region. A–C. A TAI in an AGD‐TA case without Gallyas‐positive TAs. An AT8 epitope (green) is only partially colocalized with a GFAP epitope (red) in a TAI. Short and fine AT8‐positive structures with an annular and radial pattern are shown. D–F and G–I. Tau‐positive astrocytic lesions observed in an AGD‐TA case having Gallyas‐positive TAs. An AT8 epitope (green) is colocalized with a GFAP epitope (red) in these lesions. Tau accumulation in the proximal portion in the latter tau‐positive astrocytic lesions (G) is more prominent than that in AGD‐TA cases lacking Gallyas‐positive TAs (A). J–L. A tau‐positive astrocytic lesion (probably a TA based on this morphology). AT8 epitopes (green) were frequently colocalized with GFAP epitopes (red). J. AT8‐positive structures in this lesion tended to be more densely packed and longer than those observed in AGD‐TA cases lacking Gallyas‐positive TAs (A) and AGD‐TA cases having Gallyas‐positive TAs (D, G). All scale bars = 20 μm.
In AGD‐TA cases having Gallyas‐positive TAs, tau‐positive astrocytic lesions tended to have a larger number of AT8‐positive dot‐ and spindle‐shaped structures that were more densely distributed in the paroxysmal portion of astrocytic processes (Figure 8d–i). Tau epitopes were more frequently colocalized with GFAP epitopes in AGD‐TA cases having Gallyas‐positive TAs (Figure 8a–c) than in AGD‐TA cases lacking them (Figure 8d–i).
In PSP cases, fibrillary tau aggregates densely packed in the proximal and distal portions in astrocytic processes were found (Figure 8j). AT8 epitopes were frequently colocalized with GFAP epitopes (Figure 8j–l).
Biochemical analyses of tau in PSP, AGD‐TA and AGD cases
Immunoblot analysis of the sarkosyl‐insoluble, urea‐soluble fraction with T46 demonstrated approximately 68‐ and 64‐kDa bands in the frontal cortex (Fr), striatum (St), pons (Po) and/or occipital cortex (Oc) in all typical PSP 16 (PSP1 and PSP2) and atypical PSP 16 (PSP3 and PSP4) cases (Figure 9A). All PSP cases showed the approximately 33‐kDa low‐molecular mass tau fragments characteristic of PSP. Some lanes of PSP1, PSP3 and PSP4 also showed a minor 60 kDa band that may reflect the coexistence of mild AD pathology.
Figure 9.
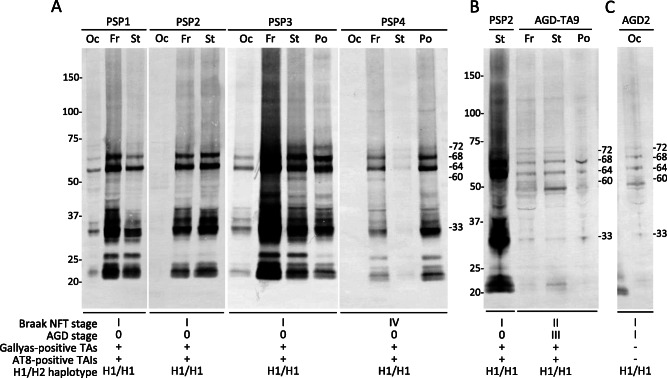
Immunoblot analysis of the sarkosyl‐insoluble, urea‐soluble fraction with T46 from frozen brain tissues of PSP A, B, AGD‐TA B and AGD C cases. A. Approximately 68‐ and 64‐kDa bands, as well as the 33‐kDa low‐molecular mass tau fragments characteristic of PSP, are seen in the frontal cortex, striatum, pons and/or occipital cortex in all PSP cases. In addition, a minor 60‐kDa band that may reflect the coexistence of AD pathology is seen in some lanes in PSP1, PSP3 and PSP4. B. Both 68‐ and 64‐kDa bands, as well as the 33‐kDa low‐molecular mass tau fragments characteristic of PSP, are found in not only a PSP case (PSP2) but also an AGD‐TA (AGD‐TA9) case having Gallyas‐positive TAs. C. Both 68‐ and 64‐kDa bands are seen in an AGD case. Although this case did not have any TAs or TAIs, a minor 33‐kDa band is also observed. Genetic analysis demonstrated that all cases had the H1/H1 haplotype. Fr = frontal cortex; St = striatum; Po = pons; Oc = occipital cortex; TAs = tufted astrocytes; TAIs = tufted astrocyte‐like astrocytic inclusions.
Tau immunoblotting was done in one AGD‐TA case (AGD‐TA9 in Figure 9B) and one AGD case (AGD2 in Figure 9C). Because 68‐ and 64‐kDa bands and only weakly reactive bands of 33‐kDa low‐molecular mass tau fragments were detected in a preliminary examination, reexamination was performed using a larger volume of samples (about twofold compared with PSP cases). As a result, 68‐ and 64‐kDa bands and minor 33‐kDa low‐molecular mass tau fragments were again observed in AGD‐TA and AGD cases (Figure 9B,C, respectively).
Genetic analysis demonstrated that all four PSP, one AGD‐TA, and one AGD cases examined had the H1/H1 haplotype (Figure 9).
DISCUSSION
This is the first study that comprehensively examined the relationship between the severities of AGD and PSP pathologies. The major findings in our 19 cases, which had argyrophilic grains but lacked NFTs whose quantity met the pathological criteria of PSP, were as follows: (i) five cases (26.3%) had at least one Gallyas‐positive tau‐positive TA, six cases (31.6%) had TAIs alone and eight cases (42.1%) lacked them in any region. TAs and TAIs commonly preferentially developed in the striatum and frontal cortex, and both lesions, especially TAs, were found in AGD cases that have relatively severe neuronal tau accumulation in the subcortical nuclei. (ii) Although completely lacking TAs or TAIs, AGD cases had significantly larger numbers of NFTs in the globus pallidus and pontine nucleus than Braak NFT stage‐matched control cases. (iii) AGD‐TA cases had significantly larger numbers of NFTs in more extensive subcortical regions and the frontal cortex than Braak NFT stage‐matched control cases. The distribution of NFTs in AGD‐TA cases was similar to that in PSP cases. (iv) There was a significant correlation between the three histological factors, AGD stage, quantity of NFTs in the subcortical nuclei, and quantity of tau‐positive astrocytic lesions in the subcortical nuclei. However, the Braak stage of RD3‐positive NFTs as AD pathology was not significantly correlated with these three factors. (v) Tau immunoblotting demonstrated 68‐ and 64‐kDa bands and 33‐kDa low‐molecular mass tau fragments in PSP cases, and with lesser intensity, in AGD and AGD‐TA cases also. These findings suggest that the pathogenic process of the formation of argyrophilic grains may be associated with tau accumulation in neurons and astrocytes in the basal ganglia, brain stem nuclei and frontal cortex independent of AD pathology.
Our findings regarding the distributions of NFTs, TAIs and TAs in the subcortical nuclei in cases having argyrophilic grains have several implications: NFTs may first develop in the substantia nigra, followed by the striatum, globus pallidus, subthalamic nucleus, oculomotor nucleus and pontine nucleus. Although with lesser frequency, the inferior olivary nucleus and dentate nucleus in the cerebellum also may be involved. Why the difference in the quantity of NFTs in the substantia nigra between the control group and AGD and AGD‐TA groups did not reach statistical significance is unclear. However, considering that the median (2.5 and 2.0) and max (4.0 and 5.0) in AGD and AGD‐TA groups were higher than the median (1.0) and max (3.0) in the control group (Figure 2), this might be explained by the small number of samples. In addition, as shown in Figure 2, a very small number of neurofibrillary changes was also found in the subcortical nuclei in some control cases. However, they may be very mild tau pathology as mild AD pathology, because no control case was excluded from this study because of the presence of TA or TAI (data not shown). Conversely, TAs and TAIs may develop first in the striatum and frontal cortex, and both lesions, especially TAs, were found in cases that had relatively severe neuronal tau accumulation in the subcortical nuclei. These findings suggest that some cases having argyrophilic grains may have various numbers of NFTs whose distribution is similar to that in PSP cases 16, 22, 49, and that along with the progression of NFT formation, first TAIs, and then TAs develop in the striatum and frontal cortex.
Several previous case reports and studies using a small number of samples demonstrated that some AGD cases may have a few NFTs or TAs in the basal ganglia and brain stem nuclei 18, 20, 21, 25, 26, 29, 40. Mattila et al 32 examined the number of NFTs in the subthalamic nucleus in AGD cases without PSP or CBD pathology, and demonstrated that the number of NFTs in the site in AGD cases was significantly larger than that in Braak NFT stage‐matched control cases. Our findings suggest that the occurrence of NFTs and tau‐positive astrocytic lesions in the subcortical nuclei and frontal cortex may be more common in AGD cases than believed previously. Because the lesions are often not very prominent, they might have been misidentified as age‐related mild neurofibrillary changes in the routine neuropathological examination.
The morphological features of Gallyas‐negative tau‐positive astrocytic lesions, TAIs, observed in some of our AGD cases were different from those of the thorn‐shaped astrocytes or astrocytic plaques characteristic to CBD. Several researchers reported that Gallyas‐negative tau‐positive astrocytic lesions were found in some AGD cases 4, 12, 26. Botez et al 4 reported Gallyas‐negative tau‐positive astrocytic lesions in the amygdala and entorhinal cortex in AGD cases, and called them “bush‐like astrocytes.” They noted that the lesions were distinguishable from TAs because they were Gallyas‐negative and distributed in the limbic region. However, in their study, the presence of the astrocytic lesions in the striatum or frontal cortex was not examined. Kovacs et al 26 also observed Gallyas‐negative tau‐positive astrocytic lesions in four AGD cases, and they called them diffuse tau immunoreactivity of astrocytic processes. Conversely, although Gallyas method was not employed, Ferrer et al 12 reported tau‐positive astrocytic lesions in PSP cases and elderly cases (“protoplasmic astrocytes” and “astrocytes with diffuse granular immunoreactivity of astrocytic processes”), whose morphological features were similar to those of TAIs. Whether these previously reported tau‐positive astrocytic lesions are identical to the TAIs observed in our cases having argyrophilic grains remains unclear, because, given our findings, the quantity, distribution and morphological features of tau‐positive astrocytic lesions in AGD cases can be affected by the progression of tau pathology including AGD and subcortical tau accumulation. However, considering that the predilection sites of TAIs were similar to those of TAs and TAs in AGD‐TA and PSP cases often had both Gallyas‐positive tau‐positive and Gallyas‐negative tau‐positive thread‐like structures in various proportions (Table 4, Figure 4), it may be natural to consider that there is a maturation process of tau‐positive astrocytic lesions in AGD and PSP cases, and that at least some TAIs in AGD cases may be early TAs.
In the present study, a significant correlation was found between the numbers of NFTs in the subcortical nuclei, the number of tau‐positive astrocytic lesions in the subcortical nuclei, and the progression of AGD, although the anatomical distributions of these pathologies are not identical: argyrophilic grains are hardly observed in the subcortical nuclei 8, 38. These findings led us to consider that a common pathological process may lie upstream of the occurrence of these lesions. This hypothesis appears to be supported by our biochemical finding that the 33‐kDa low‐molecular mass tau fragments characteristic to PSP 3 were observed not only in our PSP cases but also AGD‐TA and AGD cases, although with less intensity. The frequency of the 33‐kDa low‐molecular mass tau fragments in AGD cases should be examined by further studies using a larger number of samples.
Our finding that cases bearing AGD had neuronal and astrocytic tau pathologies with the morphological and distributional characteristics of PSP led us to speculate that the lesions might have evolved to severe PSP pathology if each case had lived longer. Because AGD was reported to be found in subjects who died after the age of 50 years in consecutive autopsy series 8, it is possible that AGD might develop prior to the formation of PSP pathology in some of the cases having both PSP and AGD.
There are some potential limitations in interpreting the findings presented in this study. First, the number of cases in each pathological subgroup is small. Biochemical examination was also done in only a small number of AGD‐TA and AGD cases. Second, almost all of our AGD and AGD‐TA cases were psychiatric hospital inpatients. Therefore, a case selection bias might influence the frequency, degree, and topographical distribution of tau pathology in the AGD and AGD‐TA cases presented in this article. Third, the clinicopathological correlation between mild tau pathology in the subcortical nuclei and clinical features could not be examined in the present study because detailed neurological data on our AGD and AGD‐TA cases were not available.
Several findings in AGD brains presented in this article also seem to include significant implications regarding a very early phase of the pathological process of PSP that may be difficult to explore in autopsy‐confirmed PSP cases having full‐blown pathology. For example, based on our results, the number of TAs in PSP cases also might be parallel to that of NFTs, and PSP cases with AGD might have more severe tau pathology in the subcortical nuclei and frontal cortex than PSP cases without AGD. In addition, if the regularity with which the neuronal tau accumulation in the subcortical nuclei, Gallyas‐negative tau‐positive TAIs and Gallyas‐positive tau‐positive TAs develop successively in AGD brains also exists in the pathological process in PSP brains, these three lesions should be evaluated in future pathological diagnosis of PSP; further, it should be considered that PSP pathology is comprehensively expressed as several levels based on the degree of these lesions, together with the presence or absence of argyrophilic grains.
Finally, the reason why our AGD cases frequently had early PSP pathology but hardly had early CBD pathology is discussed. As noted in the procedure of the case selection in the study, in the 20 AGD cases that were first selected, early CBD pathology was found only in one case (5%). Although the size of our sample was small, the low incidence of early CBD pathology contrasted with the high incidences of TAs and TAIs (26.3% and 57.9% of remaining the 19 AGD cases, respectively). Interestingly, a recent study demonstrated that the incidence of AGD in CBD cases was 100%, while that in PSP cases was only 26.7% 42. Based on these findings, argyrophilic grains may occur parallel to the progression of CBD pathology without exception in CBD brains, but it may be rare that CBD pathology is secondarily formed after the progression of AGD. These are in contrast to the fact that PSP pathology often occurs after the progression of AGD, and that PSP pathology can often occur independent of AGD. The potential difference in the significance of AGD in the pathological processes of PSP and CBD should be examined in the future.
ACKNOWLEDGMENTS
The authors thank Ms. M. Onbe for her technical assistance. This work was supported by grants from the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT KAKENHI Grant No. 23591708 and 15K09867 to OY and 10648231 to CI), Grants‐in‐Aid from the Research Committee of CNS Degenerative Diseases and Research on Dementia, the Ministry of Health, Labour and Welfare of Japan, an Intramural Research Grant 25‐7 for Neurological and Psychiatric Disorders of NCNP, and grants from Zikei Institute of Psychiatry.
Disclosure Statement: The authors declare that they have no conflict of interest.
REFERENCES
- 1. Abraham R, Sims R, Carroll L, Hollingworth P, O'Donovan MC, Williams J, Owen MJ (2009) An association study of common variation at the MAPT locus with late‐onset Alzheimer's disease. Am J Med Genet B Neuropsychiatr Genet 150:1152–1155. [DOI] [PubMed] [Google Scholar]
- 2. Amador‐Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, et al (2007) TDP‐43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol 61:435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arai T, Ikeda K, Akiyama H, Nonaka T, Hasegawa M, Ishiguro K, et al (2004) Identification of amino‐terminally cleaved tau fragments that distinguish progressive supranuclear palsy from corticobasal degeneration. Ann Neurol 55:72–79. 14705114 [Google Scholar]
- 4. Botez G, Probst A, Ipsen S, Tolnay M (1999) Astrocytes expressing hyperphosphorylated tau protein without glial fibrillary tangles in argyrophilic grain disease. Acta Neuropathol 98:251–256. [DOI] [PubMed] [Google Scholar]
- 5. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Braak H, Braak E (1987) Argyrophilic grains: characteristic pathology of cerebral cortex in cases of adult onset dementia without Alzheimer changes. Neurosci Lett 76:124–127. [DOI] [PubMed] [Google Scholar]
- 7. Braak H, Braak E (1989) Cortical and subcortical argyrophilic grains characterize a disease associated with adult onset dementia. Neuropathol Appl Neurobiol 15:13–26. [DOI] [PubMed] [Google Scholar]
- 8. Braak H, Braak E (1998) Argyrophilic grain disease: frequency of occurrence in different age categories and neuropathological diagnostic criteria. J Neural Transm 105:801–819. [DOI] [PubMed] [Google Scholar]
- 9. Buée L, Delacourte A (1999) Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP‐17 and Pick's disease. Brain Pathol 9:681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dan A, Takahashi M, Masuda‐Suzukake M, Kametani F, Nonaka T, Kondo H, et al (2013) Extensive deamidation at asparagine residue 279 accounts for weak immunoreactivity of tau with RD4 antibody in Alzheimer's disease brain. Acta Neuropathol Commun 1:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, et al (2002) Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 61:935–946. [DOI] [PubMed] [Google Scholar]
- 12. Ferrer I, López‐González I, Carmona M, Arregui L, Dalfó E, Torrejón‐Escribano B, et al (2014) Glial and neuronal tau pathology in tauopathies: characterization of disease‐specific phenotypes and tau pathology progression. J Neuropathol Exp Neurol 73:81–97. [DOI] [PubMed] [Google Scholar]
- 13. Fujino Y, Wang DS, Thomas N, Espinoza M, Davies P, Dickson DW (2005) Increased frequency of argyrophilic grain disease in Alzheimer disease with 4R tau‐specific immunohistochemistry. J Neuropathol Exp Neurol 64:209–214. [DOI] [PubMed] [Google Scholar]
- 14. Greenberg SG, Davies P (1990) A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA 87:5827–5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hasegawa M, Watanabe S, Kondo H, Akiyama H, Mann DM, Saito Y, Murayama S (2014) 3R and 4R tau isoforms in paired helical filaments in Alzheimer's disease. Acta Neuropathol 127:303–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hauw JJ, Daniel SE, Dickson D, Horoupian DS, Jellinger K, Lantos PL, et al (1994) Preliminary NINDS neuropathologic criteria for Steele‐Richardson‐Olszewski syndrome (progressive supranuclear palsy). Neurology 44:2015–2019. [DOI] [PubMed] [Google Scholar]
- 17. Hauw JJ, Verny M, Delaère P, Cervera P, He Y, Duyckaerts C (1990) Constant neurofibrillary changes in the neocortex in progressive supranuclear palsy. Basic differences with Alzheimer's disease and aging. Neurosci Lett 119:182–186. [DOI] [PubMed] [Google Scholar]
- 18. Hitoshi S, Mizutani T, Amano N, Bando M, Yamanouchi H (1994) Adult‐onset dementia with abundant neurofibrillary tangles resembling progressive supranuclear palsy. Rinsho Shinkeigaku 34:557–562. [PubMed] [Google Scholar]
- 19. Ikeda K, Akiyama H, Kondo H, Haga C (1995) A study of dementia with argyrophilic grains. Possible cytoskeletal abnormality in dendrospinal portion of neurons and oligodendroglia. Acta Neuropathol 89:409–414. [DOI] [PubMed] [Google Scholar]
- 20. Ishihara K, Araki S, Ihori N, Shiota J, Kawamura M, Yoshida M, et al (2005) Argyrophilic grain disease presenting with frontotemporal dementia: a neuropsychological and pathological study of an autopsied case with presenile onset. Neuropathology 25:165–170. [DOI] [PubMed] [Google Scholar]
- 21. Itagaki S, McGeer PL, Akiyama H, Beattie BL, Walker DG, Moore GR, McGeer EG (1989) A case of adult‐onset dementia with argyrophilic grains. Ann Neurol 26:685–689. [DOI] [PubMed] [Google Scholar]
- 22. Iwasaki Y, Yoshida M, Hattori M, Goto A, Aiba I, Hashizume Y, Sobue G (2004) Distribution of tuft‐shaped astrocytes in the cerebral cortex in progressive supranuclear palsy. Acta Neuropathol 108:399–405. [DOI] [PubMed] [Google Scholar]
- 23. Komori T, Arai N, Oda M, Nakayama H, Mori H, Yagishita S, et al (1998) Astrocytic plaques and tufts of abnormal fibers do not coexist in corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol 96:401–408. [DOI] [PubMed] [Google Scholar]
- 24. Kovacs GG, Majtenyi K, Spina S, Murrell JR, Gelpi E, Hoftberger R, et al (2008) White matter tauopathy with globular glial inclusions: a distinct sporadic frontotemporal lobar degeneration. J Neuropathol Exp Neurol 67:963–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kovacs GG, Milenkovic I, Wöhrer A, Höftberger R, Gelpi E, Haberler C, et al (2013) Non‐Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community‐based autopsy series. Acta Neuropathol 126:365–384. [DOI] [PubMed] [Google Scholar]
- 26. Kovacs GG, Molnár K, László L, Ströbel T, Botond G, Hönigschnabl S, et al (2011) A peculiar constellation of tau pathology defines a subset of dementia in the elderly. Acta Neuropathol 122:205–222. [DOI] [PubMed] [Google Scholar]
- 27. Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, et al (2011) A harmonized classification system for FTLD‐TDP pathology. Acta Neuropathol 122:111–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martinez‐Lage P, Munoz DG (1997) Prevalence and disease associations of argyrophilic grains of Braak. J Neuropathol Exp Neurol 56:157–164. [DOI] [PubMed] [Google Scholar]
- 29. Masliah E, Hansen LA, Quijada S, DeTeresa R, Alford M, Kauss J, Terry R (1991) Late onset dementia with argyrophilic grains and subcortical tangles or atypical progressive supranuclear palsy? Ann Neurol 29:389–396. [DOI] [PubMed] [Google Scholar]
- 30. Masuda‐Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, et al (2013) Prion‐like spreading of pathological α‐synuclein in brain. Brain 136:1128–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matsusaka H, Ikeda K, Akiyama H, Arai T, Inoue M, Yagishita S (1998) Astrocytic pathology in progressive supranuclear palsy: significance for neuropathological diagnosis. Acta Neuropathol 96:248–252. [DOI] [PubMed] [Google Scholar]
- 32. Mattila P, Togo T, Dickson DW (2002) The subthalamic nucleus has neurofibrillary tangles in argyrophilic grain disease and advanced Alzheimer's disease. Neurosci Lett 320:81–85. [DOI] [PubMed] [Google Scholar]
- 33. McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H, et al (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65:1863–1872. [DOI] [PubMed] [Google Scholar]
- 34. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al (2012) National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nishimura M, Namba Y, Ikeda K, Oda M (1992) Glial fibrillary tangles with straight tubules in the brains of patients with progressive supranuclear palsy. Neurosci Lett 143:35–38. [DOI] [PubMed] [Google Scholar]
- 36. Nishimura T, Ikeda K, Akiyama H, Kondo H, Kato M, Li F, et al (1995) Immunohistochemical investigation of tau‐positive structures in the cerebral cortex of patients with progressive supranuclear palsy. Neurosci Lett 201:123–126. [DOI] [PubMed] [Google Scholar]
- 37. Okahisa Y, Ujike H, Kunugi H, Ishihara T, Kodama M, Takaki M, et al (2010) Leukemia inhibitory factor gene is associated with schizophrenia and working memory function. Prog Neuropsychopharmacol Biol Psychiatry 34:172–176. [DOI] [PubMed] [Google Scholar]
- 38. Saito Y, Ruberu NN, Sawabe M, Arai T, Tanaka N, Kakuta Y, et al (2004) Staging of argyrophilic grains: an age‐associated tauopathy. J Neuropathol Exp Neurol 63:911–918. [DOI] [PubMed] [Google Scholar]
- 39. Santa‐Maria I, Haggiagi A, Liu X, Wasserscheid J, Nelson PT, Dewar K, et al (2012) The MAPT H1 haplotype is associated with tangle‐predominant dementia. Acta Neuropathol 124:693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Santpere G, Ferrer I (2009) Delineation of early changes in cases with progressive supranuclear palsy‐like pathology. Astrocytes in striatum are primary targets of tau phosphorylation and GFAP oxidation. Brain Pathol 19:177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Steele JC, Richardson JC, Olszewski J (1964) Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum white vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 10:333–359. [DOI] [PubMed] [Google Scholar]
- 42. Tatsumi S, Mimuro M, Iwasaki Y, Takahashi R, Kakita A, Takahashi H, Yoshida M (2014) Argyrophilic grains are reliable disease‐specific features of corticobasal degeneration. J Neuropathol Exp Neurol 73:30–38. [DOI] [PubMed] [Google Scholar]
- 43. Thal DR, Rüb U, Orantes M, Braak H (2002) Phases of A beta‐deposition in the human brain and its relevance for the development of AD. Neurology 58: 1791–800. [DOI] [PubMed] [Google Scholar]
- 44. Togo T, Dickson DW (2002) Tau accumulation in astrocytes in progressive supranuclear palsy is a degenerative rather than a reactive process. Acta Neuropathol 104:398–402. [DOI] [PubMed] [Google Scholar]
- 45. Togo T, Sahara N, Yen SH, Cookson N, Ishizawa T, Hutton M, et al (2002) Argyrophilic grain disease is a sporadic 4‐repeat tauopathy. J Neuropathol Exp Neurol 61:547–556. [DOI] [PubMed] [Google Scholar]
- 46. Tolnay M, Calhoun M, Pham HC, Egensperger R, Probst A (1999) Low amyloid (Abeta) plaque load and relative predominance of diffuse plaques distinguish argyrophilic grain disease from Alzheimer's disease. Neuropathol Appl Neurobiol 25:295–305. [DOI] [PubMed] [Google Scholar]
- 47. Tolnay M, Monsch AU, Probst A (2001) Argyrophilic grain disease. A frequent dementing disorder in aged patients. Adv Exp Med Biol 487:39–58. [PubMed] [Google Scholar]
- 48. Uchikado H, Lin WL, DeLucia MW, Dickson DW (2006) Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha‐synucleinopathy. J Neuropathol Exp Neurol 65:685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Williams DR, Holton JL, Strand C, Pittman A, de Silva R, Lees AJ, Revesz T (2007) Pathological tau burden and distribution distinguishes progressive supranuclear palsy‐parkinsonism from Richardson's syndrome. Brain 130:1566–1576. [DOI] [PubMed] [Google Scholar]
- 50. Yamada M (2003) Senile dementia of the neurofibrillary tangle type (tangle‐only dementia): neuropathological criteria and clinical guidelines for diagnosis. Neuropathology 23:311–317. [DOI] [PubMed] [Google Scholar]
- 51. Yamada T, McGeer PL, McGeer EG (1992) Appearance of paired nucleated, Tau‐positive glia in patients with progressive supranuclear palsy brain tissue. Neurosci Lett 135:99–102. [DOI] [PubMed] [Google Scholar]
- 52. Yokota O, Davidson Y, Bigio EH, Ishizu H, Terada S, Arai T, et al (2010) Phosphorylated TDP‐43 pathology and hippocampal sclerosis in progressive supranuclear palsy. Acta Neuropathol 120:55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yokota O, Tsuchiya K, Arai T, Yagishita S, Matsubara O, Mochizuki A, et al (2009) Clinicopathological characterization of Pick's disease versus frontotemporal lobar degeneration with ubiquitin/TDP‐43‐positive inclusions. Acta Neuropathol 117:429–444. [DOI] [PubMed] [Google Scholar]
- 54. Yokota O, Tsuchiya K, Terada S, Ishizu H, Uchikado H, Ikeda M, et al (2008) Basophilic inclusion body disease and neuronal intermediate filament inclusion disease: a comparative clinicopathological study. Acta Neuropathol 115:561–575. [DOI] [PubMed] [Google Scholar]