

Abstract
Cognitive impairment is a common and debilitating feature of multiple sclerosis (MS) that has only recent gained considerable attention. Clinical neuropsychological studies have made apparent the multifaceted nature of cognitive troubles often encountered in MS and continue to broaden our understanding of its complexity. Radiographic studies have started to decipher the neuroanatomic substrate of MS‐related cognitive impairment and have shed light onto its pathogenesis. Where radiographic studies have been limited by inadequate resolution or non‐specificity, pathological studies have come to the fore. This review aims to provide an overview of the nature of cognitive impairment typically seen in MS and to explore the literature on imaging and pathological studies relevant to its evolution. In particular, the relative contributions of gray (ie, cerebral cortex, hippocampus, thalamus and basal ganglia) and white matter to MS‐related cognitive impairment will be discussed and the importance of interconnectivity between structures highlighted. The pressing need for longitudinal studies combining standardized neuropsychometric, paraclinical and radiographic outcomes obtained during life with post‐mortem tissue analysis after death is presented.
Keywords: cognitive impairment, gray matter, imaging, multiple sclerosis, pathology, white matter
Introduction
Cognitive impairment in MS is complex and multifactorial, with both white (WM) and gray (GM) matter damage implicated in its pathogenesis 54. Neuropsychometric studies have mapped out the landscape of cognitive deficits encountered in the disease, which has helped the interpretation of imaging findings. Magnetic resonance imaging (MRI) studies have been monumental in teasing apart the relationship between the distribution and extent of pathology in the MS brain and how it relates to cognitive impairment. However, inherent limitations of current imaging modalities have made the study of MS post‐mortem tissue evermore topical and relevant.
In this review, we will present a broad overview of the nature of MS‐related cognitive impairment and highlight some of the neuropsychometric tools used in clinical practice and research settings. We will then explore the contribution of neuroimaging studies in dissecting out the relative impact of GM and WM pathology on MS cognitive impairment by focusing on cognitively relevant neuroanatomic structures. The same approach will be applied to a survey of the neuropathologic literature to aid in the interpretation of rapidly accumulating MRI findings. The emerging role of post‐mortem MRI studies in more precisely deciphering pathologic contributions (and their radiographic surrogates) to MS cognitive decline is discussed. The pressing need for longitudinal studies wherein serial neuropsychometric, neuroimaging and paraclinical data is accumulated in life and complemented with post‐mortem brain tissue analysis after death is outlined.
Cognitive Impairment in MS
Cognitive impairment is now recognized as a common and substantial consequence of MS. It occurs in 40%–70% of the MS patients, depending on the population studied and the diagnostic methodology and cognitive impairment is known to have a significant impact quality of life. MS patients who are cognitively impaired are more likely to report loss of self‐esteem, participation in fewer social activities and higher rates of divorce 107, 178. Both cross‐sectional studies and longitudinal studies indicate cognitive impairment is the strongest predictor of MS patients reducing work responsibilities or leaving the workforce 88, 157, 185.
Classically, the cognitive impairment associated with MS has been labeled as a “subcortical dementia.” Subcortical dementia has been previously described in other neurological conditions such as Huntington's disease or Parkinson's disease 205. The clinical presentation differs from cortical dementias such as Alzheimer's dementia mainly because of the lack of aphasia, apraxia and agnosia 61. In contrast, the subcortical dementias affect information processing speed, memory and executive function and often present as a profound slowing of cognition also referred to as bradyphrenia 61. Instead of cortical lesions or atrophy, the lesions responsible for subcortical dementia are thought to occur in the deep gray matter (basal ganglia and thalamus), brain stem and cerebellum 62. Additionally, psychiatric and personality changes are common with subcortical dementias, specifically cognitive inflexibility 177 and mood disorders such as depression and anxiety, apathy and irritability 1. Yet, as will be discussed below, the term subcortical dementia implies a disconnect between cortical and subcortical pathologies, which does not truly exist in MS. Further, there is a wide variability in the clinical presentation of cognitive impairment in MS and this presentation does not always fit the concept of subcortical dementia.
Cognitive impairment in MS most frequently presents as impaired information processing speed, as well as impaired immediate and delayed recall or memory 23, 177. Verbal fluency and executive function can also be involved 54. Although disease duration and physical disability do not predict the presence of cognitive impairment, there is a likely a relationship with disease type and time since diagnosis 2, 12. Feuillet et al found 80% of clinically isolated syndrome (CIS) patients who met McDonald's (2005) disseminated in space criteria on MRI were impaired on at least one cognitive domain, while 57% were impaired on two or more tests 86, 174. Similar patterns were found in other CIS and recently diagnosed (2–3 years) relapsing remitting (RR) MS patients 98, 180. Achiron et al in a large cross‐sectional study divided subjects in 5‐year cohorts by year of diagnosis and found a decrease in all cognitive domains after the fifth year after diagnosis 2. Cognitive impairment is known to be more frequent and more severe in progressive types of MS than RRMS 2, 3, 177; although studies vary when comparing primary progressive (PPMS) and secondary progressive (SPMS) MS. Comi et al reported a higher prevalence of cognitive impairment in SPMS than PPMS (57% vs. 7%); however, this was a small study with only 31 progressive patients included 59. Huijbregts found SPMS were more severely and more frequently impaired than PPMS subjects, who in turn were more impaired than the RRMS group 113. In contrast, larger population‐based studies have found 29%–87% of PPMS patients were cognitively impaired 42, 45, and in other studies, the degree of cognitive impairment between SP and PPMS is comparable 74, 91.
In contrast, all studies support the concept that cognitive impairment in MS is progressive with little evidence of improvement once apparent. Duque and colleagues evaluated 44 MS subjects of all types every 3 months over a 2‐year period 75. At baseline, 31% were considered impaired, and at 2 years, that increased to 41% with a decrease in verbal memory and processing speed being most prominent. Other short‐term (3 years or less) longitudinal studies have found similar results 11, 127. Only one long‐term study has been published by Amato et al 10. These investigators followed 45 MS subjects over 10 years, who were considered early in the disease as they were on average 1.5 years since diagnosis with Poser criteria. At baseline, 74% were cognitively normal, which decreased to 51% at 4 years and 44% at 10 years. These studies examined predictors of developing or worsening cognitive impairment over time; no characteristic was found to be predictive of worsening cognitive function other than the presence of cognitive impairment at baseline.
Although known to be insidiously progressive in nature, more recently, it has been demonstrated that acute inflammatory lesions, or relapses, can affect cognitive function. Morrow et al examined subjects receiving natalizumab monthly who also had the Symbol Digit Modalities Test (SDMT) administered at each infusion visit in the STRATA extension study 158. The authors retrospectively compared 53 subjects with documented relapses to 115 stable controls matched on age, gender and baseline SDMT values at time of study initiation and found SDMT change pre‐ to post‐relapse was significantly greater in the relapse group than matched stable controls, with a change of 4.0 on the SDMT found to be clinically meaningful. However, this study included all relapses documented in the phase IV study and was not specific to cognitive complaints. A prospective study by the same group examined MS patients with cognitive complaints and compared neuropsychological tests done before, during and after the relapse to matched controls without relapses. Again, a decline on the SDMT was noted during the acute inflammatory phase, on average a worsening of 3.5 points or approximately 6% 25. These studies confirm that acute inflammation also contributes to cognitive function in MS patients.
Tools to Measure Cognitive Decline in MS
It is important, when examining cognitive impairment clinically or for research protocols, to ensure the tests being used are appropriate, reliable and valid. In 1991, Rao developed and validated the Brief Repeatable Battery of Neuropsychological tests (BRB‐N) for MS, a comprehensive battery that could be administered in 1 h or less 30 (Table 1). The BRB‐N was found to have a sensitivity of 71% and specificity of 94% in discriminating cognitively impaired from cognitively intact MS patients 30. Some researchers noted this battery did not have a measure of visual/spatial ability or executive function; often other tests are added to the battery to assess other measures. In 2002, a panel of neuropsychologists and clinical psychologists with expertise in the field of cognitive impairment in MS developed a battery for what they determined to be a minimal neuropsychological examination to assessment the principal features noted in cognitive impairment in MS patients 24. This battery, named the Minimal Assessment of Cognitive Function in MS (MACFIMS), was similar to the BRB‐N but with additional tests ensuring a more comprehensive assessment but also increasing the time for administration to 1.5–2 h (Table 1). The panel also recommended, although not included in the battery per se, that premorbid cognitive ability, generalized fatigue and mood disorders, specifically depression, should also be measured as these factors can affect performance on objective cognitive measures. Impairment on one test was defined as worse than 1.5 standard deviations below the mean on that test and cognitive impairment was defined as impairment on two domains in the MACFIMS battery. Since that time, multiple studies have supported the lack of effect of generalized fatigue on objective measures of cognitive impairment, although fatigue has a significant impact on the subjective evaluation of cognitive function in MS; thus this symptom should still be considered when evaluating cognitive function. The MACFIMS was subsequently evaluated and found to be a valid and reliable measure of cognitive impairment in the MS population, demonstrating that all 11 neuropsychological outcomes included in the battery significantly discriminated between MS patients and normal controls, as well as between RRMS and SPMS patients 23.
Table 1.
Neuropsychometric batteries commonly used in multiple sclerosis. Abbreviations: BVMTR = Brief Visuospatial Memory Test—Revised; COWAT = Controlled Oral Word Association Test; CVLT2 = California Verbal Learning Test—2nd edition; D‐KEFS = Delis–Kaplan Executive Function System; JLO = Judgment of Line Orientation test; PASAT = Paced Auditory Serial Addition Test; SDMT = Symbol Digit Modalities Test; SPART = 10/36 Spatial Recall Test 16; SRT = Selective Reminding Test
| Domain | BRN‐B | MACFIMS | BiCAMS |
|---|---|---|---|
| Processing speed (auditory) and working memory | PASAT 103 | PASAT | |
| Processing speed (visual) | SDMT 194 | SDMT | SDMT |
| Verbal memory (learning and recall) | SRT 41 | CVLT2 65 | CLVT2* |
| Visual/spatial memory (learning and recall) | SPART 16 | BVMTR 22 | BVMT* |
| Verbal fluency | COWAT 36 | COWAT | |
| Spatial processing | JLO 28 | ||
| Executive function | D‐KEFS 66 |
*For the BiCAMS, only the immediate recall portion of the CVLT2 and BVMTR are included, while both the BRN‐B and MACFIMS include both the immediate and delayed recall test.
Both of the above batteries are comprehensive, and a recent study comparing the BRB‐N to the MACFIMS found both had similar discriminative validity to distinguish MS patients from healthy controls as well as an excellent prediction rate: 79% for the BRB‐N and 83% for the MACFIMS 198. However, the time it takes to administer the full battery remains a limiting step. Indeed, there is much ongoing research to develop either a screening test or a screening battery for cognitive impairment in MS. Many studies support the high validity, discriminative ability and test‐test reliability of the SDMT, one of the two measures of processing speed included in both comprehensive batteries 23, 73, 159, 175, 198, 217. Yet, there is concern that one test of only one domain will miss those MS patients who are impaired in other domains, specifically memory, which is also frequently affected in MS patients 23, 48, 160. A shorter battery based on the MACFIMS, which can be completed in 15 minutes, has been proposed by Langdon et al, the Brief International Cognitive Assessment for Multiple Sclerosis (BiCAMS) battery 131 (Table 1).
MRI and Cognitive Impairment in MS
MRI is currently the best biomarker available for both diagnosis and monitoring of disease activity in MS patients. Demyelinating plaques, both active and chronic, are correlated with high signal (hyperintensity) on T2‐weighted sequences on MRI 163, 190. Over time, new T2 hyperintense lesions may diminish in size as inflammation resolves, yet a permanent T2 lesion often remains. Overall T2 volume increases yearly by approximately 5%–10% 203. On T1‐weighted images, areas of hyperintensity on T2 often have corresponding low signal (hypointense) lesions. T1 hypointensities can resolve, as the T2 hyperintense lesion resolves. Permanent T1 hypointense lesions reflect irreversible axonal loss and/or permanent myelin damage 208. Gadolinium, a paramagnetic agent, causes a high signal intensity (hyperintensity) in areas of blood–brain barrier (BBB) breakdown on T1‐weighted images 104. Gadolinium‐enhancing lesions are correlated with pathologic evidence of BBB compromise, allowing infiltration of pro‐inflammatory T‐cells, and only appear when there is acute inflammation 141. These lesions are transient, lasting approximately 4 weeks, and may precede a new T2 lesion by hours to days 118, 149. New T2 lesions and contrast‐enhancing lesions correlate with active relapses 67, 111, 118, yet many new lesions appear on MRI that are clinically silent, and are estimated to occur 10× more frequently than clinically relevant relapses 111, 171, 203.
White matter
Inflammatory lesions and cognitive impairment
Hyperintense T2 lesions have been found to be related to cognitive impairment and correlate better with cognitive impairment better than T1 or gadolinium‐positive lesions 111, 179. Several studies have found a relationship between cognitively impaired MS subject and T2 lesion volumes 67, 171. Rossi et al found the overall anatomical distribution of T2 lesions in the brain similar between cognitively impaired and cognitively preserved but also found greater WM lesion volume in cognitively impaired vs. cognitively preserved MS patients. Further, the peak of lesion frequency was two times higher in the corpus callosum in the cognitively impaired group. The authors also found the SDMT was significantly correlated with WM lesions and low SDMT scores were associated with high lesion frequency in corpus callosum 183 (Table 2).
Table 2.
Radiographic and pathologic featured references by anatomic location
| Region | Subregion | Anatomic structure | Featured radiology references | Featured pathology references |
|---|---|---|---|---|
| White matter | Subcortical WM | N/A | 67, 111, 171, 179, 183, 187, 202 | 8, 35, 37, 82, 85, 92, 130, 135, 154, 186, 204, 206 |
| Gray matter | Cortical GM | Cerebral cortex | 9, 15, 43, 44, 50, 136, 155, 164, 191 | 32, 33, 39, 112, 130, 142, 144, 145, 172, 213 |
| Hippocampus | 121, 193 | 76, 77, 94, 168 | ||
| Deep GM | Thalamus | 17, 27, 56, 111 | 57, 96, 106, 209, 211 | |
| Basal ganglia | 17, 29, 38 | 106, 209, 211 |
Cortical gray matter
Cerebral cortex
Cerebral cortical lesions and cognitive impairmentt
Recently, advances in MRI technology, including new sequences and higher field strength, have led to the identification of cortical lesions in MS. Double Inversion recovery (DIR) sequence utilizes two inversion times to suppress the signal from WM and CSF allowing the images to show superior delineation of cortical GM. Cortical lesions can now be identified and cortical lesion volume has been found to be significantly higher in cognitively impaired MS patients 44. Calabrese et al (2009) reported cortical lesions in both cognitively impaired and cognitively preserved MS patients, similar to the pattern of demyelinating T2 lesions noted on conventional MRI, but noted that cognitively impaired MS patients had a higher cortical lesion volume and lower cortical volume overall. Regression analysis found both cortical volume and cortical lesion volume to be independent predictors of cognitive impairment 43. Studies using 3T MRI have demonstrated that T1 hypointense cortical lesions are common in MS patients and predict worse scores on neuropsychological tests of memory, processing speed and verbal fluency 15. Finally, 7T MRI imaging has further demonstrated that cortical lesions are consistently associated with cognitive impairment in MS patients 164. Post‐mortem MRI studies have demonstrated that MRI is not sensitive but highly specific in the detection of cortical lesions. Therefore, the true extent of cerebral cortical disease is likely significantly underrepresented using current imaging modalities 191.
Cerebral cortical atrophy and cognitive impairment
Imaging studies have consistently found that measures of brain atrophy on MRI correlate better with clinical disability than the number of volume of T2 lesions 87, 115. Studies have used brain parenchymal fraction (BPF), a measure of the ratio of brain parenchymal tissue volume within the total brain surface volume on MRI, to quantify the amount of atrophy, which has been found to be accurate and reliable with good reproducibility 87, 110, 184. Kassubek et al compared BPF measures of MS patients with healthy controls, controlling for age and gender 116. MS patients had significantly lower BPF measures than controls; BPF was also significantly correlated with disease duration (r = −0.6) and EDSS (r = −0.5), but not with relapse rate or lesion load measures 50. This data supports BPF, or atrophy, as a measure of degeneration or permanent damage. Although atrophy of both WM and GM are noted over time, MRI studies support GM atrophy as the driving force behind whole brain atrophy. Cross‐sectional studies comparing early MS patients to healthy controls confirm a significant brain volume reduction compared with healthy controls, but also find a greater reduction in GM than WM when controlling for age and gender, as well as a lack of correlation between brain atrophy measures and T2 lesion load 50, 202 (Figure 1). Early MS patients followed for 4 years were found to have GM atrophy occurring at a 3.4× greater rate in the group of first demyelinating event patients who converted to MS vs. those who did not. GM atrophy also occurred 8.1× greater in RRMS, 12.4× greater in RRMS converting to SPMS and 14× greater in SPMS patients. In contrast, WM atrophy occurred at a similar rate in all MS types (3× greater than normal controls) 27.
Figure 1.
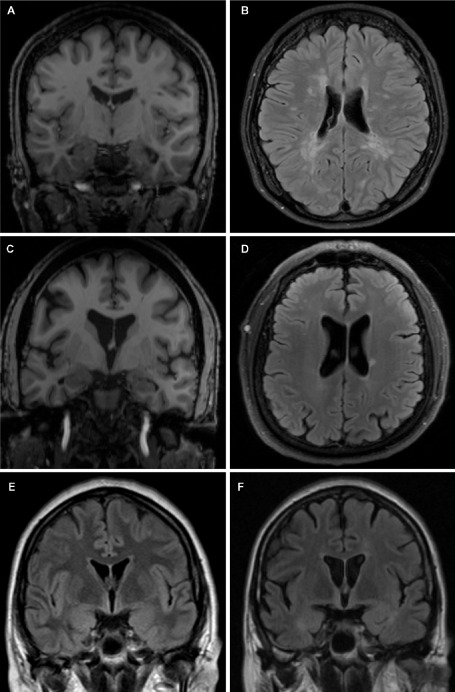
Cortical atrophy, lesion burden and cognitive impairment in MS. Coronal (A, C, E, F) and axial (B, D) views of MRI brain scans from RRMS patients without cognitive impairment (patient 1—A, B; patient 2—C, D) and with cognitive decline (patient 3—E, F). Patient 1 demonstrates relatively preserved brain parenchymal volume (A) despite moderately significant WM lesion burden (B). Conversely, patient 2 has evidence of significant globalized cerebral atrophy (C) in the absence of a significant WM lesion load (D). Patient 3 shows radiographic evidence of progressively worsening global cerebral atrophy over 18 months despite disease‐modifying therapy and relative stability of WM lesion burden. These images highlight the complex relationships between GM and WM pathology and cognitive impairment in MS. A, C. T1‐weighted sequences; B, D, E, F. T2‐FLAIR sequence.
A similar link has been described between atrophy measures and cognitive function. Whole brain atrophy has also been strongly linked with cognitive impairment in the MS population. MS patients who are cognitively impaired are found to have smaller overall brain volumes as well as GM 136 and WM volume 187. Cross‐sectional studies in MS patients have found performance on a test of processing speed to be correlated with BPF 27, 109, 188. Additionally, longitudinal studies also found that MS patients with greater brain atrophy worsened on cognitive tests over time more than MS patients with stable or improved cognitive function 155. Further studies have shown it is cortical volume that is most important in cognitive impairment. Amato et al examined 41 MS patients within 10 years of diagnosis and found MS patients had decreased neocortical volume compared with normal controls and MS patients with cognitive impairment had decreased neocortical volume compared with MS patients without cognitive impairment 9 (Table 2).
Hippocampus
There are also studies supporting the role of hippocampal atrophy in cognitive impairment. Sicotte et al examined the absolute volume of both hippocampi and its segments in early RRMS, SPMS and healthy controls compared with performance on memory tasks. Both types of MS had lower total hippocampal volume when compared with healthy controls, which was localized to the CA1 regions in RRMS and more extensively involved in SPMS. An inverse relationship was found between the number of trials needed to learn a new list of words and total hippocampal, CA1 and subiculum volume 193. This pattern of selectivity vulnerability in CA1 is reminiscent of that seen in anoxia, epilepsy and dementia (reviewed in 14). Koenig et al reported a 6%–7% smaller hippocampal volume in MS patients compared with normal controls, and a smaller hippocampal volume in MS patients who were impaired on measures of memory and processing speed 121 (Table 2).
Deep gray matter
The possibility that deep GM structures, as opposed to cortical GM, play a role in cognitive dysfunction has also been of interest. Deep GM matter consists of the thalamus, hippocampus, nucleus accumbens and basal ganglia, which encompass the caudate, putamen, globus pallidus and amygdala (see relevant pathology sections for further anatomic details).
Thalamus
Some studies have used third ventricular width (TVW) as a measure of whole brain atrophy. Larger ventricles, as measured by ventricular width, indicate a larger amount of cerebrospinal fluid (CSF) and less brain tissue. However, TVW could alternatively represent selective atrophy of the thalami, which border the third ventricle. When comparing MS patients with healthy controls, TVW was a strong predictor of impairment on tests of memory and processing speed 27. Additionally, central atrophy, measured as the percent of central CSF to total intracranial volume, had a strong correlation to cognitive performance 56. If the volume of the thalami is examined directly, it is lower in MS patients compared with normal controls and associated with impairment on tests of processing speed, working memory and visuo‐spatial memory 111. Finally, the thalamus was significantly smaller in MS patients with impaired processing speed, verbal fluency, verbal and visuo‐spatial learning and executive function 17 (Table 2).
Basal ganglia
Basal ganglia T1 hypointensity and T2 lesion volume were both found to be strong predictors of cognitive impairment in MS patients 38. Similarly, the bicaudate ratio, the ratio of the width of the ventricles between the two caudate to the overall brain width, was significantly greater in MS patients than healthy controls and was a strong predictor of impairment on tests of processing speed 29. Finally, impairment on tests of verbal fluency and verbal learning/memory was also significantly associated with lower volume in both caudate nuclei 17 (Table 2).
Cognitive reserve
Cognitive reserve is defined as “the hypothesized capacity of the mature adult brain to sustain the effects of disease or injury without clinical symptoms, but sufficient to cause clinical symptoms in an individual possessing less cognitive reserve” 197. It is the ability of the brain to tolerate pathology related to an underlying disease process without exhibiting overt signs or symptoms, allowing those with greater baseline cognitive ability to adapt or compensate for changes caused by either normal aging or damage caused by an underlying disease 117, 214. Thus, those with greater cognitive reserve would require more extensive pathology to cause the same degree of impairment, leading to the ability to cope with pathology better than those with less reserve. As mentioned previously, the onset of impairment does not correlate well with disease duration, can be present at initial diagnosis and does not necessarily follow the same pattern of severity as physical disability 12, 86, 98. The reason for this variability is unknown but may be partly explained by cognitive reserve. A case control study of 43 MS and normal controls matched on level of education, found that the low education MS group performed worse than controls on almost all tests, while the higher education MS group differed from controls only on measures of processing speed and attention. Additionally, the cognitive performance of MS patients was correlated with years of education while the control groups' performance did not differ based on education level 34. In a study of cognitive performance and third ventricular width (TVW) as a measure of brain atrophy, Benedict et al found that education level was correlated with each measure of atrophy 27. Sumowski and colleagues 199 investigated whether the adverse impact of brain atrophy on information processing speed and efficiency would be moderated by cognitive reserve. In 38 patients, cognitive function was assessed using the SDMT and Paced Auditory Serial Addition Test (PASAT), measures of processing speed, and measured brain atrophy using TVW. Regression models showed that the expected relationship between brain atrophy and cognitive processing was moderated by estimated pre‐morbid intelligence. The study also showed a significant interaction between atrophy and cognitive reserve such that the impact of atrophy on cognition was attenuated at higher levels of cognitive reserve. In other words, among patients with more severe brain atrophy, those with greater cognitive reserve had a better performance on formal cognitive testing. The same was not true of patients with minimal atrophy. Additionally, a longitudinal study examining 90 MS patients over a mean of 5 years, using education as an indicator of cognitive reserve, found those MS patients with more than 14 years of education (“high reserve”) showed no significant change on the SDMT over time, while those with low levels of education (<9 years, “low reserve”) declined significantly over time 26.
Pathology and Cognitive Impairment in MS
Pathology studies have played an important role in providing insight into the possible neuroanatomic substrate of cognitive impairment in MS and helping with the interpretation of MRI findings. From a pathologic point of view, MS lesions are heterogeneous in nature even within a given anatomic location and their distribution and size are highly variable between patients. Each MS lesion may have an impact on cognition through various mechanisms including chronic inflammation and/or demyelination, disruption of the neuronal/axonal compartments, oxidative stress, alterations in the BBB and cerebral metabolism and blood flow, and resultant changes in cortico‐cortical and cortico‐subcortical connectivity to name a few 114. That being said, the links between MS pathology and cognition remain poorly understood. This is largely because of frequent reports of neuroimaging abnormalities to which cognitive impairment has been attributed (eg, cortical, hippocampal, thalamic atrophy and subcortical WM pathology), also being found in “cognitively normal” MS sufferers. Further, the exploration of clinicopathological correlations in MS‐related cognitive impairment has been hampered by the application of inconsistent neuropsychometric protocol(s), variable definitions of cognitive impairment and varying MRI measures, as previously highlighted. The cross‐sectional nature of MRI studies and the lack of specificity of findings on the impact of surrogate markers of MS pathology are problematic. The impact of disease‐modifying therapies on the evolution of cognitive decline is not accounted for adequately adding yet another layer of complexity.
Pathology studies have filled some of these important gaps, though much work is needed. The confounding issues of lesion distribution/extent, cross‐sectional ascertainment and poor clinicopathologic correlations limit the ability of pathology studies to explain MS‐related cognitive impairment (Figure 2, Table 2).
Figure 2.
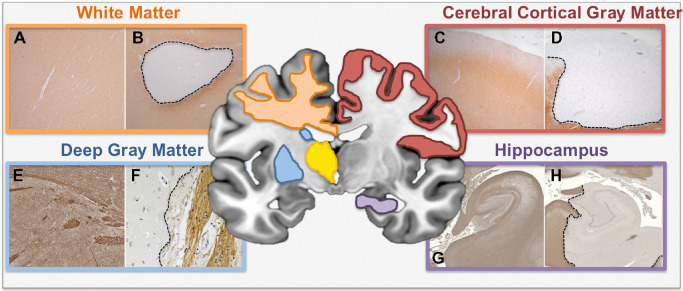
White and gray matter pathology in MS. Schematic diagram outlining the anatomical regions of WM and GM pathology covered in this review (white matter—orange box; deep gray matter—blue box; cerebral cortical GM—red box; hippocampus—purple box). Myelin immunolabeled sections illustrate normal or normal appearing WM/GM (left panel within each colored box—A, C, E, G) or demyelination (right panel in each colored box—B, D, F, H). Specifically, well‐demarcated subcortical WM (B), cerebral cortical subpial (D), caudate GM (F) and hippocampal mixed GM/WM (H) lesions are presented alongside MS subcortical NAWM (A), MS cerebral cortical NAGM (C), non‐neurologic control caudate nucleus (E) and non‐neurologic control hippocampus (H), respectively. A–D. Proteolipid protein immunolabeling. E–H. Myelin basic protein immunolabeling. E, F. Adapted from 208. G, H. Adapted from 166 and used with permission.
In the next section, we will highlight the neuropathology of key anatomic structures implicated in MS‐related cognitive impairment.
White matter pathology and cognitive impairment in MS
WM consists of glial cells that support aggregations of axons linked by common cortical and/or subcortical origins/destinations grouped into association, striatal, commissural and projection fiber bundles. We will next review the pathology of WM and its involvement in cognitive dysfunction in MS.
White matter demyelination and inflammation
There is a spectrum of WM abnormalities recognized in MS, ranging from lesional WM (acute and chronic), diffusely abnormal (or dirty appearing) WM (DAWM) and normal appearing WM (NAWM). WM lesions have long been considered the pathologic hallmark of MS and can be easily detected both in vivo by MRI and upon post‐mortem examination 132 (Figure 2). Acute inflammatory demyelination is widely accepted as the pathologic surrogate for clinical relapses, which demonstrate radiographic evidence of gadolinium enhancement indicating disruption of the BBB as outlined in the MRI section of this review. From a histologic perspective, these focal lesions are characterized by recruitment of monocytes and lymphocytes, which traverse the disrupted BBB into the perivascular space where ongoing immunological interactions take place 102. Early infiltration of macrophages/microglia engulf myelin sheaths with or without the deposition of complement or immunoglobulins 4, 40, 186. The distribution and extent of the microglial inflammatory reaction defines the “stage” of the lesion; active lesions are rich in macrophages at their lesional core, chronic (or border) active lesions show abundant macrophages at their margins, and chronic inactive lesions have minimal, if any, macrophage infiltration 72, 95.
CD8+T‐cells typically outnumber CD4+ T‐cells within the MS lesional milieu, with the latter being the predominant cell type in the perivascular space 35. Pro‐inflammatory IL‐17 expressing CD8+ and CD4+ cells (Th‐17 cells) are thought to play a pathogenic role given their abundance in acute lesions and relative paucity in more chronic lesions 206. B‐lymphocytes and/or plasma cells are also present, and become particularly prevalent in progressive disease stages 92, 144. Recent work has elegantly demonstrated the co‐localization of complement and immunoglobulin on oligodendrocytes and axons suggesting that complement‐mediated damage may be inciting factors in inflammatory demyelination, oligodendrocyte loss, axonal injury and damage, and reactive astrogliosis 186. Remyelination of WM lesions is highly variable between patients and within lesions of the same patient 170 and is inefficient compared with that, observed in cerebral cortical GM 122, 156. Myelin deposited from remyelination is susceptible to “second‐attack” demyelination, especially around lesional borders 37. The resulting disruption of WM fiber bundles has potential to have a deleterious impact on cognition.
While WM lesions can occur anywhere throughout the neuraxis, their distribution is certainly not random with periventricular, centrum semiovale and corpus callosal WM involvement being common 64, 89. Subcortical lesions stemming from small vessel disease can mimic the distribution of those seen in MS, and in fact, can share histopathologic features reminiscent of MS plaques. Some of these include alterations in BBB permeability, increased microglial activity, oligodendrocyte drop out and astroglial activation (reviewed by Wharton et al in this issue). The established cognitive manifestations of small vessel disease, including reduced information processing speed, and difficulties with attention and concentration, make it plausible that WM involvement in MS may have a similar clinical effect given the nature and overlapping distribution of lesions from both conditions 81.
Outside of lesions, myelin in MS WM typically displays a spectrum of pathologic change ranging from NAWM to DAWM. NAWM is different from WM in control tissue given evidence of diffuse parenchymal and perivascular T‐cell inflammation 6, 7, microglial activation with upregulation of major histocompatibility complex expression and gliosis 8, 99, 130. Abnormalities in endothelial tight junctions 120, 173, extracellular matrix proteins 196 and enhanced perivascular expression of matrix metalloproteinases 138, have been described, as has BBB permeability 120. Disruption of the BBB in NAWM allows leakage of large molecular weight proteins, such as fibrinogen, into the WM compartment triggering a deleterious inflammatory cascade 137. Davalos and colleagues recently demonstrated that BBB‐mediated perivascular fibrinogen deposition instigates a rapid and sustained microglial response resulting in release of reactive oxygen species that damages axons, even in the absence of overt demyelination 63. While pathology studies have not been in a position to compare specific pathologic changes to cognitive phenotypes, NAWM abnormalities as defined on MRI (as opposed to lesion burden) correlate with atrophy, disability and impaired cognition (reviewed in 153).
Diffusely abnormal WM (DAWM), otherwise known as “dirty appearing” WM, refers to WM myelin with hyperintensity on proton density and T2‐weighted imaging intermediate between that of NAWM and focal WM lesions 154. Typical locations of DAWM are adjacent to periventricular plaques and occipital WM, although they can affect any part of WM including areas distant to lesions 154). Pathologically, DAWM shows selective reduction of myelin phospholipids, as detected by Luxol Fast Blue and Weil myelin stains, with relative preservation of myelin proteins 154. Axonal loss is also present but does not feature to the same extent as the myelin phospholipid abnormalities, which are defining 134, 153. The relationships between DAWM and lesional WM, NAWM and neurodegeneration remain up for experimental discovery. Whether WM signal change (leukoaraiosis) 167 noted in ageing, subcortical vascular cognitive impairment 189 and Alzheimer's disease (AD) 101 share pathogenic mechanisms with DAWM evolution in MS warrants evaluation. If so, this could have important implications in the understanding of MS‐related cognitive impairment.
White matter axonal loss
Axonal injury and loss are established features of MS WM pathology and likely contribute significantly to the irreversible cognitive decline often seen in the disease 31, 201. Ferguson et al first described axonal injury in MS WM lesions via β‐APP immunohistochemistry, which refocused the field onto the neurodegenerative aspects of the disease 85. Several studies have replicated these findings and have asserted the relationship between acute axonal injury (and loss) and inflammation both within and outside areas of demyelination 13, 68, 140, 204. Axonal loss can be both an early and late feature, and its extent can reach 40%–80% 31, 69, 82. Small diameter axons are preferentially affected, with WM tracts relevant to cognition, such as the corpus callosum, commonly involved 69, 82, 130, 200.
The cause of axonal injury and loss is probably multifactorial. Axons, stripped of myelin and surrounding glial trophic support, are susceptible to degeneration especially when electrically active 161, 195, 216. CD4+ and CD8+ T‐cells in active WM lesions have been shown to be neurotoxic in vitro via direct cell contact‐dependent mechanisms 97. It is established that activated macrophages and microglia contribute to axonal damage in lesions 85 but the diversity of phenotypes of these cells 151 point to their involvement in neurotoxicity and neuroprotection 53. The observation that microglia can strip cortical neurons of inhibition synapses leading to neuronal survival is a recent example of the latter 52. The fact that peripherally derived macrophages compared with resident microglia can perform differing roles in driving lesion pathology and axonal loss further complicates matters 139. There are also longer standing consequences of inflammatory demyelination on the axonal unit, including redistribution of ion channels after demyelination 60, upregulation of the acid‐sensing ion channel‐1 with subsequent aberration of sodiumand calcium regulation 212, chronic mitochondrial injury with resultant oxidative stress and energy failure, and dysregulation of iron homeostasis 106, among others. The predisposition of remyelinated axons to be the target of “second‐hit” demyelination adds yet another layer of complexity to the pathogenesis of lesion‐associated axonal injury and loss 37.
The presence of axonal injury and loss in both NAWM and DAWM broadens the traditional plaque‐centric view of its pathogenesis 69, 82, 135. Where axonal loss does occur in NAWM and DAWM, it can be partly explained by secondary (Wallerian) degeneration stemming from injury from acute lesions in structures such as the corpus callosum 83. However, the lack of significant correlations between demyelination and axonal loss in other WM tracts draws attention to the possibilities that inflammation, diffusible cytotoxic factors 144, 145, glutamate excitotoxicity 126 and/or axon‐targeted immune‐mediated injury 147 rather than myelin loss per se, may be the culprit process(es) 13, 68, 112. One must also consider that axonal vulnerability to inflammatory demyelination may intrinsically differ between different WM tracts within the same patient 70. A better understanding of the contribution of these pathological processes to axonal demise in WM tracts important for cognition is urgently needed.
Cortical gray matter pathology and cognitive impairment in MS
Post‐mortem analysis of the MS brain has established that GM structures throughout the central nervous system, such as cerebral cortex, thalamus, hippocampus and basal ganglia, are commonly affected and have important consequences on cognitive function. The following sections will review the evidence for involvement of each of these GM structures in MS pathology and explore their potential impact on neuropsychometric outcomes.
Cerebral cortex
The cerebral cortex is an organized, laminated structure consisting of six layers defined by their predominant cell type. The cortex contains two types of neurons: principal (mostly pyramidal) neurons (80% of total population) and local interneurons (20% of the total population) that are surrounded by a complex network of glial cells and embedded in an intricate pattern of myelin and extracellular matrix proteins. Disruption of neuronal connections, glial support networks and myelination pattern are the substrate for several neuropsychiatric conditions, of which many have cognitive dysfunction as a core feature 21. It is feasible that similar processes are involved in MS‐related cognitive impairment.
Cerebral cortical demyelination
Involvement of cortical demyelination in MS has been a long recognized feature that has only gained particular attention over the last two decades. Descriptions of cerebral cortical demyelination were furnished in the late 19th and early 20th centuries by several eminent neuropathologists, including Dawson among others 64. In the early 1960s, Brownell and Hughes provided an authoritative addition to the literature through the examination of 22 post‐mortem MS cases in which they macroscopically evaluated the extent and distribution of cerebral lesions 39. Of the 1594 lesions identified, 80 (5%) involved the cortex and 265 (17%) were situated at the junction between cortex and WM. The authors astutely recognized that the extent of cortical GM demyelination was likely significantly under‐detected in their hands 39, a limitation that standard histochemical techniques and conventional in vivo imaging modalities would only partially address over subsequent decades. It was only since the relatively recent application of immunohistochemistry for myelin proteins in MS cortical tissue that the severe extent of cortical demyelination was appreciated. This, in combination with an increased awareness of cognitive dysfunction in MS patients and its relationship to GM structural change detected on imaging, dampened the WM‐centric view of MS pathology and brought to the fore the role of (cortical) GM pathology in the pathogenesis and phenotypic expression of the disease.
Cortical demyelination can occur at an early disease stage 142 and has been documented in every clinical phenotype, being most prevalent and extensive with advanced disease duration and in progressive disease 128, 130. Cortical regions that appear to be particularly prone to demyelination include the cingulate gyrus, temporal and frontal cortices, and the depths of sulci. These areas lie in regions of relative CSF stasis leading to the hypothesis that neurotoxic mediators in the CSF influence cortical demyelination 112. In contrast, the paracentral lobule, occipital lobe and primary motor cortex display comparatively smaller amounts of pathology 129. That said, no cortical region is universally spared from the MS disease process and indeed in progressive MS the majority of the cortical ribbon can be demyelinated 96, 128 although this can vary markedly between patients.
Several classification systems to describe the topography of cortical demyelination have been proposed 32, 119, 172. Four major lesion types in the MS cortex have been defined: type I (leukocortical) lesions span deeper GM layers with involvement of subcortical WM at the gray/white junction; type II (intracortical) lesions are limited to the cortex (sparing subpial and subjacent WM boundaries) and are typically small and centered around small veins; type III (subpial) lesions extend from the pial surface into superficial cortical layers; and type IV lesions originate from the pial surface and span the entire cortical width without involvement of underlying WM although extension into subcortical WM has been described 142. These lesion types not only differ in location but also in their extent of cortical area coverage with subpial lesions (types III and IV) being most extensive and intracortical lesions, although most numerous, being smallest. Both the location (cortical layer(s) and hemispheric position) and extent of cortical demyelination are likely to play critical roles in the evolution of cognitive impairment 51 (Figure 2).
The distribution and extent of cortical demyelination can vary markedly between MS patients, the reasons for which are unclear 129. It has recently been demonstrated that heterogeneity in cortical pathology in MS can be explained partly by genetic variation at the HLA‐DRB1*15 locus, with younger individuals positive for this allele having significantly more severe demyelination than those who are negative 215. The observation that cortical MS disease may be more severe in a discrete genotype group is supported by evidence of significantly greater cerebral hemispheric T2 lesion burden, decreased levels of N‐acetyl aspartate (a neuronal marker) and more severe cognitive impairment in MS patients carrying the HLA‐DRB1*15 allele 166. Given the limited resolution to detect cortical lesions, further imaging and pathological studies exploring the role of genetic factors on cortical lesion burden and topography and their relationship to cognitive dysfunction are warranted.
Cerebral cortical remyelination
Cortical demyelination is not a permanent outcome in MS. There is increasing evidence to suggest that cortical remyelination is frequent and can be extensive. In a comprehensive study by Albert et al using light and electron microscopy, cortical remyelination was detected in 28 of 29 (95%) cases and was more extensive compared with WM lesions from the same patients 5 but the investigators did not control for lesional age. To address this confound, Chang et al examined leukocortical lesions (where GM and WM involvement are presumably temporally congruent) and found that GM had significantly greater oligodendrocyte generation and remyelination compared with subjacent WM 49. Differences in the extent of astrogliosis and hyaluronon deposition, factors known to frustrate oligodendrocyte differentiation and myelination, were thought to account for these findings 49. An intriguing alternative possibility is that there may be factors in the cortex favoring oligodendrocyte reactions. In support of this view, some have argued that the commonly observed sparing of subcortical myelin in leukodystrophies may be secondary to repeatedly regenerated myelin of subcortical U‐fibers because of proximity to efficient myelin‐generating cortical oligodendrocytes rather than because of myelin sparing 156. Whether subpial (type III) demyelinating lesions demonstrate similar remyelination potential compared with the other lesional types remains unanswered and will only be elucidated when an accepted staging method for cortical lesions is devised 49.
The relationship between the extent of cerebral cortical remyelination and age remains contentious. Recent work by Yates et al and others have demonstrated that MS cases with longer disease duration and later age at death have more abundant cortical myelination 169, 170, 215. Given that actively remyelinating oligodendrocytes are commonly encountered in patients over 70 years of age with long‐standing disease duration 122, it is likely that remyelination extends into old age and may contribute to “cognitive reserve.” While experimental data suggests that oligodendrocyte remyelinating potential decreases with age 100, this phenomenon is undoubtedly influenced by the changing extent and quality of inflammatory activity associated with aging 165. This may subsequently impact the susceptibility of newly formed myelin from “second‐hit” demyelinating attack in older patients 37. The observed substantial interindividual variation in remyelinating capacity irrespective of age implicates underlying genetic and/or environmental factors 170, 182.
Cerebral cortical inflammation
Pathological studies evaluating inflammation in MS have traditionally focused on WM regions throughout the neuraxis 181. This WM exploratory bias has been fueled, in part, by the notion that GM structures, such as cerebral cortex, have a paucity of inflammation even in active disease stages. Recent data have challenged this view highlighting how inflammation within the cerebral cortex and overlying meninges can be severe and extensive, and contribute to other cortical pathological features likely important for cognitive function.
Cortical parenchymal inflammation within lesions and NAGM is variable but routinely less severe than underlying WM. Leukocortical lesions contain more pronounced inflammatory infiltrates compared with their intracortical and subpial counterparts 32, 33, 112, 123, 130, 142, 172, 211. In chronic MS, cortical lesions residing solely within the GM compartment show infrequent T‐cell lymphocytes, which when present, are typically restricted to the perivascular space. B‐cell infiltrates are rare. Varying degrees of activated microglia are also present, which typically take on a ramified appearance. Myelin‐laden macrophages are atypical. There is a widely held view that they are not associated with BBB breakdown or complement deposition 32, 172, 207. The ascertainment bias of post‐mortem tissue derived from end‐stage progressive MS cases demands consideration in the interpretation of these findings.
On the flip side of the ascertainment spectrum, a recent biopsy study has broadened the view of cortical demyelination in early MS 142. Cerebral cortex, sampled “en passant” of the stereotactic retrieval of WM lesions suspicious for tumor, revealed demyelination associated with pronounced CD3+ T‐cell infiltration and macrophages with myelin‐inclusions in the setting of radiographic evidence of BBB disruption. The presence of complement deposition was not studied. While the majority of patients studied later fulfilled criteria for a diagnosis of MS, there should be cautious extrapolation of these findings to understand the pathogenesis of the cortical MS lesion. The clinical and radiographic presentations justifying the need of biopsy suggest atypical MS presentations, and therefore, possibly less typical pathology. Further, the extent to which the cortical findings were influenced by the juxtaposed fulminant WM pathology that prompted tissue biopsy in the first place is not certain.
The contrasting features of cortical pathology in early and late disease stages leave unanswered important questions about the pathogenesis of the cortical lesion. Involvement of the BBB in MS cortical lesions is contentious despite the majority of these being centered on veins 152. Some investigators propose that CNS inflammation becomes compartmentalized behind a “closed” BBB once the disease transitions from a phenotype of focal inflammatory demyelination to that of more diffuse cortical disease as encountered in progressive disease 128, 130. The detection of marked endothelial tight junction abnormalities in cortical NAGM derived from SPMS and PPMS cases 137 and in cortical lesions despite minimal inflammation in a chronic murine model of the disease challenge this conceptual framework 80.
Microglial responses may prove a key culprit, independent of BBB disruption. Microglial activation can be severe and extensive in MS cortical lesions, with a recent report suggesting that its severity is influenced by HLA‐DRB1*15 status even in long‐standing cases 125, 215. The functional consequences of increased microglial burden are not certain however. Microglia respond to cytotoxic factors from the subarachnoid space (implicating neurovascular communication) 102 and show age‐related dysregulation 71, both of which may be relevant when weighing the role of these resident macrophages on cortical pathology and age‐related cognitive decline. Given that chronic disruption of the neurovascular unit and microglial activation contribute to both GM and WM pathology in diseases wherein progressive cognitive decline is a hallmark, such as AD (reviewed in 79, the possibility of similar parallel processes being operative in MS deserves attention. Complicating matters, recent work has demonstrated that activation of microglia in the cortex may confer neuronal protection via stripping of inhibitory synapses (see cerebral cortical neuronal and synaptic loss section) 52.
While features of cortical pathology appear to vary between early and late MS, meningeal inflammation is a unifying feature 68, 142, 145. Numerous studies have reported substantial meningeal immune cell infiltrates composed of T‐ and B‐cells and macrophages, and noted their relationship to underlying cortical GM pathology both in progressive MS 130, 144, 192 and early‐stage cortical biopsies 142, although these relationships have not been universally found 5, 123, 215. Organization of meningeal B‐cell inflammation in what resemble tertiary lymphoid‐like structures is a replicated feature 112, 144, 192. The extent of meningeal inflammation has been linked to underlying cortical demyelination, inflammation and a gradient of neuronal loss that is greatest in superficial cortical layers closest to meninges in both demyelinated and normal appearing GM (see section below) 145. These results point toward a cytotoxic milieu in the meningeal/CSF compartment, an idea supported by detection of enhanced meningeal gene expression of tumor necrosis factor and interferon‐gamma, cytokines known to induce subpial cortical pathology similar to that seen in MS 93.
Cerebral cortical neuronal and synaptic loss
Neuronal loss is a notable feature of the landscape of MS cortical pathology. In a seminal report, apoptotic neurons and transected neurites (ie, dendrites and axons) were observed in cortex, with the former concentrated in large, pyramidal neurons in cortical layers 3 and 5 and the extent of the latter associated with lesional inflammatory activity 172. Subsequent studies have built upon these findings demonstrating significant neuronal loss (approximately 10%–30%) occurs within lesions and in normal appearing GM 145, 210, 213 with specific neuronal subpopulations (ie, interneurons expressing parvalbumin in cortical layer 2) being particularly vulnerable 58. A gradient of neuronal loss has been observed, with most severe loss in superficial cortical layers especially in MS cases where overlying meningeal inflammatory lymphoid‐like follicles are present 145. In early biopsy‐derived MS tissue where pronounced cortical inflammatory demyelination features, neuronal and axonal injury is seen 142. Synaptic loss is another element of cortical pathology, with one study showing a 47% reduction in synaptophysin in lesional cortex 213.
Pathology of the neuronal‐axonal compartment is not restricted to cortical lesions. Several studies have now convincingly shown that neuronal mitochondrial dysfunction and cellular drop out is also detected in NAGM distant from lesions suggesting that myelin loss per se is not the driving substrate 47, 78, 145. Rather, mounting evidence suggests that parenchymal inflammatory cellular reactions and/or meningeal‐derived diffusible soluble cytotoxic factors play an important role in the disruption of the neuronal‐axonal unit.
The chronic inflammatory milieu in MS likely drives the observed deficiencies in oxidative phosphorylation and GABAergic neurotransmission. Gene and protein expression studies of normal appearing cortex have demonstrated defects in oxidative phosphorylation and functional reduction in mitochondrial respiratory chain complex I and III 78. In combination with the observation of significantly increased levels of mitochondrial DNA deletions in cortical neurons 46, the changes in oxidative phosphorylation are thought to set the stage for neuronal‐axonal energy failure leading to a neurodegenerative fate. In contrast, decreased inhibitory GABA transmission may confer neuroprotection through the upregulation of neuroprotective pathways 53. Studies have demonstrated that neuronal survival is enhanced by synaptic firing of NMDA receptors, a process facilitated by the inhibition or temporary disruption of GABA‐ergic inhibition 53. Recent elegant experiments by Trapp and colleagues have built upon this work establishing that activated microglia can migrate to and displace inhibitory GABA‐ergic synapses from cortical neurons. The resultant increased synchronized cortical neuronal firing increased expression of anti‐apoptotic and neurotrophic molecules and decreased cortical neuronal apoptosis following injury 52. These findings may be particularly relevant to cortical neurons in superficial layers I–III, which are involved in cortico‐cortical connections, given their vulnerability to subpial demyelination and meningeal‐derived cytotoxic soluble mediators as outlined above.
Cognitive implications of cerebral cortical pathology in MS
Direct clinicopathological evidence linking features of cortical pathology to MS‐related cognitive decline are lacking given insufficient clinical information and/or cognitive testing late in life typical of autopsy series. That being said, features of MS cortical pathology when married with what is known about cortical function provide insight into MS‐relevant cognitive phenotypes. The preferential affectation of prefrontal, middle temporal and cingulate cortices to demyelination likely contribute to deficiencies in executive function, learning, memory retrieval, and emotion formation and processing often encountered in the disease. The prevalence of demyelination, cytotoxic inflammation and neuronal loss in both superficial (I–III) and deep (V–VI) cortical layers is of particular relevance to cognitive disturbance given these layers are important for cortico‐thalamo‐cortical activity, cortical binding, synchronization and plasticity, which impact cognitive function. Chronic microglial activation, while likely a driver of free radical generation and excitotoxic neuronal injury, may confer cognitive protection by its anti‐apoptotic influence on neurons and promotion of cortical synchronization via stripping of GABAergic inhibitory synapses. The positive influence of microglia on amyloid deposition in the MS brain may additionally protect from Alzheimer's‐related cognitive decline.
Taken together, the complex interplay between the distribution, nature and extent of demyelination, inflammation, neuronal‐axonal injury and loss in the MS cerebral cortex, contribute to MS‐related cognitive decline and differentiate it from other degenerative dementia syndromes. While cerebral cortical atrophy is a recognized radiographic surrogate of cognitive decline in MS, so too is hippocampal atrophy (see MRI section above). The pathology of this archaeocortical structure will be reviewed next.
Hippocampus
The hippocampal formation is located in the medial temporal lobe and includes the dentate gyrus, the hippocampus proper (including CA1, CA2, CA3 and CA4 subfields) and the subiculum. These structures form part of a predominantly unidirectional glutaminergic excitatory pathway that is complemented by parallel and reciprocal connections within the hippocampus that form a functionally organized circuit 20. The hippocampal formation forms a circuit with neocortical structures subserving diverse function with memory formation, maintenance and retrieval being key elements. Emotional memory processing (via amygdala, orbitofrontal, medial prefrontal cortex) and visuospatial memory and navigation (via precuneus and posterior cingulate cortex) are additional features 20.
Hippocampal demyelination
Aside from incidental descriptions of hippocampal demyelination in a few atypical presentations of MS 90, 162, the first systematic assessment of hippocampal demyelination was carried out in 2007 94. In this study, a total of 37 lesions were found in or around the hippocampus in 15 of 19 (78.9%) MS cases sampled. Both intrahipppocampal lesions (involving dentate gyrus and/or ammon horns) and mixed GM and perihippocampal WM lesions featured, * with mixed GM/WM lesions covering larger areas and having a more prominent microglial reaction than ** pure GM lesions 94. Others have been documented similar findings with the proportion of MS cases exhibiting hippocampal demyelination ranging between 55% and 62% with around 30% of the total hippocampal area afflicted 76, 168. No hippocampal subregion appears to be spared 124, 168. Subpial and subependymal demyelination is commonly seen. A contribution of hippocampal demyelination to cognitive dysfunction is not certain although mixed hippocampal lesions have been shown to be overrepresented in MS cases with cognitive impairment compared with those without 94. Whether this finding points toward a site‐specific phenotypic consequence of demyelination or merely reflects a more severe global burden of disease is not known (Figure 2).
Hippocampal inflammation
Hippocampal inflammation is not prominent. In one series, the majority of demyelinated lesions were chronic inactive (25/35, 71.4%), with only 9/35 (25.7%) lesions demonstrating microglial activation at their borders. Only one acute lesion was identified, which demonstrated microglial activation and perivascular CD3 T‐cell infiltration. Microglial activation and T‐cell inflammation in non‐lesional areas is not significantly increased in MS compared with controls 168. CD3+ T‐cell and HLA class II+ cell infiltration is noted in the choroid plexus and meninges but its relationship to underlying subependymal and subpial lesions, respectively, is not certain. These findings suggest that chronic inflammation of the hippocampal parenchyma or overlying meninges/choroid plexus appears an unlikely candidate for progressive cognitive impairment in MS.
Hippocampal neuronal and synaptic loss
As noted above, GM structures appear susceptible to neuronal loss. However, the results on this aspect of hippocampal pathology have been conflicting. Papadopoulos et al reported that neuronal density was significantly decreased in CA1 (by 27%) and CA2‐3 (by 29.7%) in MS cases compared with controls with no differences between the groups noted in CA4 168. Neuronal atrophy in MS cases was restricted to the CA1 region, a finding not influenced by the extent of HLA class II+ cell expression. Correlations between global hippocampal cross‐sectional area and neuronal density and size independent of lesions have fueled the notion that hippocampal atrophy is secondary to neuronal loss and atrophy 168. These observations contrast with those of Dutta and colleagues who found variable, but nonsignificant, reductions (10%–20%) in neuronal densities in demyelinated CA1, CA3 and CA4 hippocampal subfields in MS cases compared with controls 76.
Findings on synaptic loss in the MS hippocampus have yielded more consistent results. In their investigation, Papadopoulos revealed a 46.1% reduction in synaptophysin immunolabeled neuropil in CA4 in MS cases compared with controls, without correlation to the extent of demyelination, HLA class II+ inflammation or neuronal loss 168. Dutta et al compared synaptophysin‐positive punctae in myelinated vs. demyelinated CA1 and dentate gyrus subregions. Loss of myelin was associated with significant reductions in synaptic density in both hippocampal areas (CA1—1.85‐fold reduction; dentate gyrus—2.4‐fold reduction). An elegant extension to this work revealed decreased expression of neuronal mRNA and proteins associated with axonal transport, glutamate neurotransmission and homeostasis, and memory and learning in demyelinated hippocampi 76, 77. Further experiments unraveled that demyelination altered neuronal gene expression and function by modulating levels of gene regulatory microRNAs in the hippocampus. Memory dysfunction in an animal model of hippocampal demyelination with similar consequent gene regulatory changes highlighted the functional significance of these findings though the mechanism by which this occurs is not fully understood 77. The influence of altered cholinergic metabolism, known to be dysfunctional in the MS hippocampus and tied to memory dysfunction, on these neuronal gene expression changes, is worthy of exploration 124.
In short, hippocampal pathology is common in MS. The available clinicopathologic and animal model data implicate MS‐related hippocampal damage as a factor leading to memory deficits. Although relationships between hippocampal pathology and cognitive dysfunction are already starting to emerge, much work is needed in understanding better the mechanism by which this structure is damaged and how this damage leads to cognitive troubles in MS. To this end, future studies should aim to implement more standardized methods to assess the nature, distribution and extent of hippocampal pathology in MS to not only facilitate useful comparisons between studies but also to allow meaningful correlations with cognitive measures obtained during life.
Deep gray matter and cognitive impairment in MS
Radiographic studies have provided unequivocal evidence that atrophy in deep GM structures is prominent and contributes to cognitive dysfunction in MS. In the next section of this review, we will shift attention to the pathology of deep GM structures and evaluate their contribution to MS‐related cognitive difficulties.
Thalamus
The thalamus is a highly integrated structure with connections to key cerebral cortical and subcortical structures critical for cognitive function, including hippocampus, amygdala, cingulate cortex, orbitofrontal cortex, retrosplenial cortex and inferior parietal lobule 18, 55, 150. It acts as an important gateway and has a multitude of reciprocal interactions between neocortical pyramidal, thalamic reticular nucleus and thalamocortical relay neurons central to normal oscillatory activity. Relevant to cognition, these diverse thalamic connections play a central role in arousal, executive function, emotional and episodic memory, spatial learning and memory, and recollective‐based and familiarity‐based recognition circuits 55. Therefore, damage to thalamic structures or their connections can potentially have a notable impact on cognitive function. As outlined above, radiographic studies have demonstrated that thalamic involvement is a feature of both early and late MS 108, 111 and contributes to cognitive disability in multiple domains. The neuropathologic features of MS‐related thalamic injury and the implications on MS cognition will be presented here.
Thalamic demyelination
Several pathologic studies confirm that demyelination in the thalamus is frequent with lesions commonly juxtaposed to the ependymal/CSF boundary 96, 106, 209, 211. In a study of 14 autopsy MS brains inclusive of RRMS and SPMS phenotypes, Vercellino et al observed lesions predominantly in periventricular areas of the thalamus involving cognitively relevant anterior and medial nuclei. Thalamic lesions affect both GM and WM structures to a similar degree, with mixed GM/WM lesions being relatively common 209. Where pure GM lesions arise, many respect the GM/WM boundary with lesional edges standing at the foot of where WM tracts and GM nuclei intersect 96, 209. While GM involvement in the thalamus is proportionally less than that observed in the spinal cord, cingulate and cerebellum 96, it can be extensive in a subset of cases 211. The predisposition GM thalamic lesions to extend from subependymal surface suggest that soluble CSF factors may a play a role in lesion formation here.
Thalamic inflammation
Inflammation in the thalamus is characterized by a diffuse microglial infiltrate that is more pronounced in NAWM compared with NAGM structures 106, 209. In non‐lesional matter, T‐ and B‐cell infiltrates are rare and BBB disruption evidenced by fibrinogen deposition is absent. In most series, thalamic GM lesions are typically chronic with the extent of T‐cell and macrophage/microglial infiltration relating to the stage of lesional activity 106, 209. Similar to other CNS regions, thalamic lesional WM is significantly more inflammatory than lesional GM, the former demonstrating more myelin‐laden macrophages than the latter where these are uncommon. Evidence of fibrinogen deposition in WM greater than GM lesional areas is observed suggesting differing grades of BBB disruption that may explain the noted differences in inflammation, especially in the perivascular region 209. In a pooled analysis of deep GM pathology inclusive of thalamus, the extent of inflammation within deep GM lesions is intermediate compared with WM and cerebral cortex 106. While lymphoid‐like structures have been demonstrated in the ventricular system of EAE 143, similar findings have not been demonstrated in human studies 144.
Thalamic neuronal loss
Neuronal loss and axonal injury are elements of MS thalamic pathology. Cifelli and colleagues demonstrated significant reductions in total (21%) and medial dorsal nuclear thalamic (21%) volumes attributed to a similar reduction in neuronal density (22%) in GM areas distant from focal lesions 57. Reduction in neuronal area has also been described, also likely contributing to the observed thalamic volume changes 209. The extent of neuronal atrophy and loss is exaggerated in lesional compared with non‐lesional matter and coincides with substantial glial cell loss found in a similar distribution. In contrast, no differences in synaptic density between lesional and non‐lesional matter were observed 209. These findings implicate direct injury from inflammatory demyelination supported by the presence of axonal spheroids in both GM and WM parts of lesions 85, 209. That being said, the pathogenesis of neuronal/axonal injury in the thalamic circuitry is likely to be more complex with evidence of size‐selective degenerative changes noted in a study exploring the anterior optic pathway and lateral geniculate nucleus (LGN). The selective neuronal size reduction in parvocellular, but not magnocellular, layers of the LGN, highlights how distinct areas of the thalamus may be differentially vulnerable to degeneration depending on their unique connectivity and cell composition 84. The relative contribution of retrograde degeneration, transynaptic reactive change, cytotoxic injury secondary to inflammatory cells or diffusible mediators to thalamic neuronal pathology remains unknown, although recent post‐mortem MRI studies have shed light on this issue 122.
Research into thalamus pathologic correlates of cognitive decline has thus far been limited. The complex anatomic architecture of the thalamus, lack of systematic study of cognitively relevant thalamic nuclei and unavailable clinical neuropsychology metrics further complicate the established biases of post‐mortem studies. Despite these limitations, the demyelinating, inflammatory and neurodegenerative profiles of the anterior and medial dorsal nuclei of the thalamus is well established. Other thalamic nuclei are likely implicated but human neuropathological evidence is lacking. Given that these thalamic nuclei relay to limbic and prefrontal cortical structures, their contribution to impaired processing speed, memory impairment and executive dysfunction is likely, a notion supported by the previously presented radiographic studies.
Basal ganglia
The basal ganglia consist of the striatum (composed of the caudate and putamen) and the globus pallidus 146. These deep GM nuclei receive inputs from intralaminar nuclei of the thalamus and several cortical regions, including frontal, inferotemporal and posterior parietal cortex to participate in parallel and partially segregated motor, oculomotor, cognitive and limbic circuits 19. There are several cortico‐basal ganglionic “loops,” subsets of which connect the basal ganglia with dorsolateral prefrontal cortex, lateral orbitofrontal cortex and anterior cingulate/medial orbitofrontal cortices 148. These frontal cortical regions are involved in executive function (eg, planning and attention), rule‐based learning and working memory, cognitive domains affected in MS. As noted previously, the relevance of basal ganglia on MS cognitive dysfunction is evidenced by strong correlations between MRI T2 hypointensities in caudate, globus pallidus and putamen and measures of cognitive dysfunction 38. Despite the anatomic relevance of the basal ganglia to cognitive function, there have been few pathological studies examining MS‐related pathology in this structure. We will present the available evidence here.
Basal ganglionic demyelination
Basal ganglionic structures demonstrate demyelination, but there is considerable variation in the extent to which they are affected. In several reports, the caudate nucleus appears most susceptible to demyelination, especially in periventricular areas 106, 209. This is in contrast to the putamen and global pallidus where demyelination is more restricted and typically centered on blood vessels 106, 209. In a combined analysis of several deep GM structures (inclusive of basal ganglia, thalamus and hypothalamus), the median percentage of deep GM demyelination was 3.1% (range 0%–26.8%) in acute MS, 14.5% (range 0.2%–31.6%) in RRMS, 6.0% (range 0%–46.4%) in secondary progressive MS and 9.9% (range 0%–91.3%) in primary progressive MS. Disease course and sex do not appear to influence the extent of demyelination in these deep GM structures 106. The majority of basal ganglionic lesions are mixed (ie, involving both GM and WM), with a greater proportion of the lesions in the caudate appearing “active” compared with putamen and globus pallidus in one series 209. Evidence of BBB disruption, inferred by fibrinogen deposition, is observed in both GM and WM components of deep GM lesions, being less pronounced in the former. The extent of deep GM demyelination appears to correlate with that found in the cerebral cortex in some studies 209 and appears to contribute to clinical deficits in an additive fashion when considered with cerebral cortical and WM lesion burden 106 (Figure 2).
Basal ganglionic inflammation
Consistent with other reports on deep GM, including thalamus, basal ganglia demonstrate an intermediate amount of inflammation in comparison to cerebral cortex and WM in both normal appearing and lesional tissue 106, 209. As typical of other brain regions, there is a gradient of T‐cell inflammation (perivascular and parenchymal) being most severe in active deep GM lesions, followed by inactive deep GM lesions, and normal appearing GM. Microglial and macrophage inflammation is most enhanced in active deep GM regions where coincident iNOS expression and severe signs of oxidative injury in neurons, axons and oligodendrocytes may be seen. Iron deposition, as visualized by Turnbull blue staining method, is primarily found on oligodendrocytes and myelin fibers in normal appearing GM, which on demyelination, is mostly concentrated within microglia at lesional borders. Deep GM lesional inflammatory activity appears to decline significantly with advancing age 106.
Basal ganglionic neuronal and synaptic loss
Neuronal loss and atrophy are features of basal ganglionic pathology in MS. In a comprehensive survey by Haider et al, abundant signs of neuronal/axonal injury as evidenced by neuronal cytoplasmic inclusive of oxidized phospholipids and β‐APP positive axonal spheroids, were found in non‐lesional areas of all deep GM nuclei. Neuronal loss was significantly reduced in normal appearing GM in MS (median 86.6 neurons/mm2; range 29.5–165.8 neurons/mm2) compared with matched controls (median 151.0 neurons/mm2; range 86.8–223.1 neurons/mm2; P < 0.001) 106. While this group did not find any statistically significant differences in neuronal density between lesional and non‐lesional deep GM, Vercellino et alfound a 35.5% reduction in neuronal density in demyelinated vs. non‐demyelinated caudate nucleus 209. This discrepancy between studies is not clear; however, differences in cohorts, sample size, counting methodology and precise deep GM regions studied may be contributory. No significant differences in synaptic density have been reported although further work in this area is required 209. Glial cells, implicated in neuronal trophic support, were also significantly reduced in the caudate by up to 70% 209. Neuronal density reductions in deep GM nuclei significantly associate with EDSS scores, with the impact on objective cognitive outcomes remaining unexplored 106.
Taken together, basal ganglionic pathology is a core feature of MS pathology. Given the intricate involvement of these deep GM nuclei in cortico‐basal ganglionic loops with cortical regions implicated in memory and executive function, it is likely these structures contribute to cognitive decline in MS. Future pathological studies evaluating the contribution of demyelination, inflammation and neuronal/axonal pathology in the MS basal ganglia to cognitive decline would fill an important knowledge gap.
Determinants of Cognitive Impairment: White Matter? Gray Matter? Or Both?
This review has thus far shown how MS pathology is multifaceted with cortical, deep GM and WM structures being significantly impacted. Given the intricate connectivity of these structures, it is likely that each contributes uniquely (and additively) to MS‐related cognitive impairment. Further, it is feasible that pathology of one structure may affect the pathology in the other. This concept was borne out in a recent post‐mortem imaging study investigating two thalamo‐cortical projection systems, namely lateral geniculate nucleus to primary visual cortex and mediodorsal nucleus to prefrontal cortex 122. The authors demonstrated that within the anatomically distinct thalamocortical projection systems, cerebral cortical thickness correlated significantly with myelination in the connected WM tract and neuronal density in the connected thalamic nucleus. Estimates of total lesion burden impinging on the connecting WM structures did not associate with the severity of pathology within each of the thalamo‐cortical projection systems studied. These results highlight that nerve cell or axonal loss in a specific brain area can lead to anterograde or retrograde degeneration in connected brain areas. The consequences of this “tract‐specific” pattern of MS neurodegenerative pathology are not clear. However, anterograde and retrograde neurodegeneration events are known to trigger microglial activation in areas distant to the initial insult (ie, focal inflammatory demyelination lesion). This can, in turn, propagate an inflammatory cascade leading to free radical generation and oxidative stress, factors known to compromise oligodendrocytes and the neuronal‐axonal unit 105, 133. In short, one can deduce that damage to interconnected GM and WM structures can lead to a tract‐specific neurodegenerative process with resultant inflammatory change that has the potential to further propagate neuronal‐axonal demise. Should these tracts be involved in cognition, such as the dorsomedial thalamic and prefrontal system, then cognitive decline would be expected to ensue. Pathology targeting either GM and/or WM structures appear important to this outcome (Figure 3).
Figure 3.
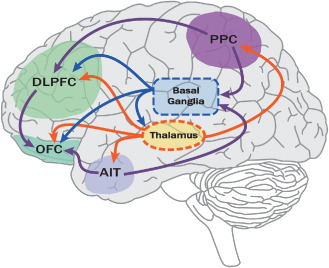
Connectivity in the brain and cognitive function. Cognitive function relies on complex interconnections between cortical (cerebral cortex and hippocampus) and deep gray matter structures (eg, thalamus and basal ganglia), with adjoining white matter tracts being important conduits. In the cortex, pyramidal neurons connect cortical structures (cortico‐cortical) or project to subcortical areas to form functionally distinct and temporally coordinated networks (purple lines). Various cortical regions are particularly relevant for cognition, including DLPFC (eg, working memory, planning, and cognitive flexibility), OFC (eg, decision making and adaptive learning), PPC (eg, spatial memory and attention) and AIT (eg, semantic memory) to name a few. The thalamus is a highly integrated structure with connections to several cerebral cortical and subcortical structures important for cognitive function (orange lines). The basal ganglia receive inputs from the intralaminar nuclei of the thalamus and several cortical regions, including frontal, inferotemporal and posterior parietal cortex and form several cortico‐basal ganglionic loops important for cognitive function, including executive function, rule‐based learning and working memory (blue lines). The hippocampal formation (not shown) forms a circuit with neocortical and subcortical structures and subserves diverse functions critical for cognition, including memory formation, maintenance and retrieval being key elements. Although shown with arrows, connections may be in both directions and produce feedforward cycles. AIT = anterior inferior temporal cortex; DLPFC = dorsolateral prefrontal cortex; OFC = orbitofrontal cortex; PPC = posterior parietal cortex.
How do these findings translate into a possible mechanism of cognitive impairment in MS? In early disease stages, where focal inflammatory demyelination is thought to predominate, GM and/or WM lesions may disrupt interconnected cognitive pathways (Figure 3). Given the bias toward WM pathology in early disease stages 130, this could lead to subtle changes in subcortical cognitive function often encountered early on, with psychomotor slowing and inattention being common manifestations. In later disease stages, diffuse cortical and WM pathology may overwhelm the modest contribution of focal inflammatory demyelinating lesions leading to cognitive deficits typical of degenerative dementias, such as aphasia, apraxia and agnosia 130, 176. This model parallels that, which is seen in mixed dementia syndromes where, for example, the cognitive manifestations secondary to subcortical small vessel disease are later dominated by AD pathology 81. Where cognitive dysfunction is a presenting or early clinical symptom of MS, early extensive cerebral cortical inflammation and demyelination and/or disruption of interconnected cognitive pathways remain leading suspects 142.
Conclusions
MS cognitive impairment is common, can occur at any disease stage and has a major impact on quality of life. Accordingly, cognitive batteries have become a staple of the clinical armamentarium used to evaluate patients with MS. Clinical neuropsychological studies have painted a clear picture of the range of cognitive deficits encountered in MS, and importantly, have laid the foundation to our understanding of the clinical consequences of disease gleaned from radiographic and pathologic studies. MRI studies have been monumental in dissecting the contribution of GM and WM pathology to MS cognitive impairment but have been limited by inadequate resolution to detect disease in brain areas critical for cognition, such as cerebral cortex. This void has been filled, in part, by pathological studies, which have systematically evaluated the nature, extent and distribution of GM and WM pathology although the standard biases of post‐mortem studies apply. The conflation of clinical, radiographic and pathologic data suggests that both GM and WM pathology play unique and likely additive roles in the pathogenesis of MS‐related cognitive impairment. The relative contributions of GM and WM pathology, the interplay of inflammation and AD‐like pathology, and the influence of genetic and environmental factors on MS cognitive decline are just a few of the many questions that warrant pursuit. Only through the study of prospective, longitudinally followed population‐based cohorts with standardized clinical, paraclinical, neuropsychometric and radiographic outcomes obtained during life and post‐mortem tissue available after death will meaningful answers to these questions surface.
Authors' Contributions
The specific authors were involved in the following aspects of the review: review design (GD and SM), data gathering (GD, RY, HB, SM), wrote the manuscript (pathology sections: GD, RY, and HB; clinical and radiology sections: SM), decided to publish the paper (GD and SM).
Acknowledgments
GD receives support from a Goodger Scholarship (University of Oxford), John Fell Award (University of Oxford) and the NIHR Biomedical Research Centre, Oxford. RY receives support from an MRC studentship.
References
- 1. Aarsland D, Karlsen K (1999) Neuropsychiatric aspects of Parkinson's disease. Curr Psychiatry Rep 1:61–68. [DOI] [PubMed] [Google Scholar]
- 2. Achiron A, Chapman J, Magalashvili D, Dolev M, Lavie M, Bercovich E et al (2013) Modeling of cognitive impairment by disease duration in multiple sclerosis: a cross‐sectional study. PLoS One 8:e71058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Achiron A, Polliack M, Rao SM, Barak Y, Lavie M, Appelboim N, Harel Y (2005) Cognitive patterns and progression in multiple sclerosis: construction and validation of percentile curves. J Neurol Neurosurg Psychiatry 76:744–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM (2011) Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci 14:1142–1149. [DOI] [PubMed] [Google Scholar]
- 5. Albert M, Antel J, Bruck W, Stadelmann C (2007) Extensive cortical remyelination in patients with chronic multiple sclerosis. Brain Pathol 17:129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Allen IV, Glover G, Anderson R (1981) Abnormalities in the macroscopically normal white matter in cases of mild or spinal multiple sclerosis (MS). Acta Neuropathol Suppl.7:176–178. [DOI] [PubMed] [Google Scholar]
- 7. Allen IV, McKeown SR (1979) A histological, histochemical and biochemical study of the macroscopically normal white matter in multiple sclerosis. J Neurol Sci 41:81–91. [DOI] [PubMed] [Google Scholar]
- 8. Allen IV, McQuaid S, Mirakhur M, Nevin G (2001) Pathological abnormalities in the normal‐appearing white matter in multiple sclerosis. Neurol Sci 22:141–144. [DOI] [PubMed] [Google Scholar]
- 9. Amato MP, Bartolozzi ML, Zipoli V, Portaccio E, Mortilla M, Guidi L et al (2004) Neocortical volume decrease in relapsing‐remitting MS patients with mild cognitive impairment. Neurology 63:89–93. [DOI] [PubMed] [Google Scholar]
- 10. Amato MP, Ponziani G, Siracusa G, Sorbi S (2001) Cognitive dysfunction in early‐onset multiple sclerosis: a reappraisal after 10 years. Arch Neurol 58:1602–1606. [DOI] [PubMed] [Google Scholar]
- 11. Amato MP, Portaccio E, Goretti B, Zipoli V, Iudice A, Della Pina D et al (2010) Relevance of cognitive deterioration in early relapsing‐remitting MS: a 3‐year follow‐up study. Mult Scler 16:1474–1482. [DOI] [PubMed] [Google Scholar]
- 12. Amato MP, Zipoli V, Goretti B, Portaccio E, De Caro MF, Ricchiuti L et al (2006) Benign multiple sclerosis: cognitive, psychological and social aspects in a clinical cohort. J Neurol 253:1054–1059. [DOI] [PubMed] [Google Scholar]
- 13. Androdias G, Reynolds R, Chanal M, Ritleng C, Confavreux C, Nataf S (2010) Meningeal T cells associate with diffuse axonal loss in multiple sclerosis spinal cords. Ann Neurol 68:465–476. [DOI] [PubMed] [Google Scholar]
- 14. Arendt T (2003) Synaptic plasticity and cell cycle activation in neurons are alternative effector pathways: the “Dr. Jekyll and Mr. Hyde concept” of Alzheimer's disease or the yin and yang of neuroplasticity. Prog Neurobiol 71:83–248. [DOI] [PubMed] [Google Scholar]
- 15. Bagnato F, Salman Z, Kane R, Auh S, Cantor FK, Ehrmantraut M et al (2010) T1 cortical hypointensities and their association with cognitive disability in multiple sclerosis. Mult Scler 16:1203–1212. [DOI] [PubMed] [Google Scholar]
- 16. Barbizet JCE (1968) Clinical and psychometric study of a patient with memory disturbances. Int J Neurol 7:44–54. [PubMed] [Google Scholar]
- 17. Batista S, Zivadinov R, Hoogs M, Bergsland N, Heininen‐Brown M, Dwyer MG et al (2012) Basal ganglia, thalamus and neocortical atrophy predicting slowed cognitive processing in multiple sclerosis. J Neurol 259:139–146. [DOI] [PubMed] [Google Scholar]
- 18. Benarroch EE (2008) The midline and intralaminar thalamic nuclei: anatomic and functional specificity and implications in neurologic disease. Neurology 71:944–949. [DOI] [PubMed] [Google Scholar]
- 19. Benarroch EE (2012) Effects of acetylcholine in the striatum. Recent insights and therapeutic implications. Neurology 79:274–281. [DOI] [PubMed] [Google Scholar]
- 20. Benarroch EE (2013) Adult neurogenesis in the dentate gyrus: general concepts and potential implications. Neurology 81:1443–1452. [DOI] [PubMed] [Google Scholar]
- 21. Benarroch EE (2013) Neocortical interneurons: functional diversity and clinical correlations. Neurology 81:273–280. [DOI] [PubMed] [Google Scholar]
- 22. Benedict R (1997) Brief Visuospatial Memory Test—Revised: Professional Manual. Psychological Assessment Resources: Odessa. [Google Scholar]
- 23. Benedict RH, Cookfair D, Gavett R, Gunther M, Munschauer F, Garg N et al (2006) Validity of the minimal assessment of cognitive function in multiple sclerosis (MACFIMS). J Int Neuropsychol Soc 12:549–558. [DOI] [PubMed] [Google Scholar]
- 24. Benedict RH, Fischer JS, Archibald CJ, Arnett PA, Beatty WW, Bobholz J et al (2002) Minimal neuropsychological assessment of MS patients: a consensus approach. Clin Neuropsychol 16:381–397. [DOI] [PubMed] [Google Scholar]
- 25. Benedict RH, Morrow S, Rodgers J, Hojnacki D, Bucello MA, Zivadinov R, Weinstock‐Guttman B (2014) Characterizing cognitive function during relapse in multiple sclerosis. Mult Scler 20:1745–1752. [DOI] [PubMed] [Google Scholar]
- 26. Benedict RH, Morrow SA, Weinstock Guttman B, Cookfair D, Schretlen DJ (2010) Cognitive reserve moderates decline in information processing speed in multiple sclerosis patients. J Int Neuropsychol Soc 16:829–835. [DOI] [PubMed] [Google Scholar]
- 27. Benedict RH, Weinstock‐Guttman B, Fishman I, Sharma J, Tjoa CW, Bakshi R (2004) Prediction of neuropsychological impairment in multiple sclerosis: comparison of conventional magnetic resonance imaging measures of atrophy and lesion burden. Arch Neurol 61:226–230. [DOI] [PubMed] [Google Scholar]
- 28. Benton AL (1994) Contributions to Neuropsychological Assessment, 2nd edn. Oxford University Press: New York. [Google Scholar]
- 29. Bermel RA, Bakshi R, Tjoa C, Puli SR, Jacobs L (2002) Bicaudate ratio as a magnetic resonance imaging marker of brain atrophy in multiple sclerosis. Arch Neurol 59:275–280. [DOI] [PubMed] [Google Scholar]
- 30. Bever CT Jr, Grattan L, Panitch HS, Johnson KP (1995) The brief repeatable battery of neuropsychological tests for multiple sclerosis: a preliminary serial study. Mult Scler 1:165–169. [DOI] [PubMed] [Google Scholar]
- 31. Bjartmar C, Kinkel RP, Kidd G, Rudick RA, Trapp BD (2001) Axonal loss in normal‐appearing white matter in a patient with acute MS. Neurology 57:1248–1252. [DOI] [PubMed] [Google Scholar]
- 32. Bo L, Vedeler CA, Nyland H, Trapp BD, Mork SJ (2003) Intracortical multiple sclerosis lesions are not associated with increased lymphocyte infiltration. Mult Scler 9:323–331. [DOI] [PubMed] [Google Scholar]
- 33. Bo L, Vedeler CA, Nyland HI, Trapp BD, Mork SJ (2003) Subpial demyelination in the cerebral cortex of multiple sclerosis patients. J Neuropathol Exp Neurol 62:723–732. [DOI] [PubMed] [Google Scholar]
- 34. Bonnet MC, Deloire MS, Salort E, Dousset V, Petry KG, Brochet B (2006) Evidence of cognitive compensation associated with educational level in early relapsing‐remitting multiple sclerosis. J Neurol Sci 251:23–28. [DOI] [PubMed] [Google Scholar]
- 35. Booss J, Esiri MM, Tourtellotte WW, Mason DY (1983) Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. J Neurol Sci 62:219–232. [DOI] [PubMed] [Google Scholar]
- 36. Borkowski JGBA, Spreen O (1967) Word fluency and brain damage. Neuropsychologia 5:135–140. [Google Scholar]
- 37. Bramow S, Frischer JM, Lassmann H, Koch‐Henriksen N, Lucchinetti CF, Sorensen PS, Laursen H (2010) Demyelination versus remyelination in progressive multiple sclerosis. Brain 133:2983–2998. [DOI] [PubMed] [Google Scholar]
- 38. Brass SD, Benedict RH, Weinstock‐Guttman B, Munschauer F, Bakshi R (2006) Cognitive impairment is associated with subcortical magnetic resonance imaging grey matter T2 hypointensity in multiple sclerosis. Mult Scler 12:437–444. [DOI] [PubMed] [Google Scholar]
- 39. Brownell B, Hughes JT (1962) The distribution of plaques in the cerebrum in multiple sclerosis. J Neurol Neurosurg Psychiatry 25:315–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bruck W, Porada P, Poser S, Rieckmann P, Hanefeld F, Kretzschmar HA, Lassmann H (1995) Monocyte/macrophage differentiation in early multiple sclerosis lesions. Ann Neurol 38:788–796. [DOI] [PubMed] [Google Scholar]
- 41. Buschke H, Fuld PA (1974) Evaluating storage, retention, and retrieval in disordered memory and learning. Neurology 24:1019–1025. [DOI] [PubMed] [Google Scholar]
- 42. Caceres F, Vanotti S, Rao S (2011) Epidemiological characteristics of cognitive impairment of multiple sclerosis patients in a Latin American country. J Clin Exp Neuropsychol 33:1094–1098. [DOI] [PubMed] [Google Scholar]
- 43. Calabrese M, Agosta F, Rinaldi F, Mattisi I, Grossi P, Favaretto A et al (2009) Cortical lesions and atrophy associated with cognitive impairment in relapsing‐remitting multiple sclerosis. Arch Neurol 66:1144–1150. [DOI] [PubMed] [Google Scholar]
- 44. Calabrese M, De Stefano N, Atzori M, Bernardi V, Mattisi I, Barachino L et al (2007) Detection of cortical inflammatory lesions by double inversion recovery magnetic resonance imaging in patients with multiple sclerosis. Arch Neurol 64:1416–1422. [DOI] [PubMed] [Google Scholar]
- 45. Camp SJ, Stevenson VL, Thompson AJ, Miller DH, Borras C, Auriacombe S et al (1999) Cognitive function in primary progressive and transitional progressive multiple sclerosis: a controlled study with MRI correlates. Brain 122(Pt 7):1341–1348. [DOI] [PubMed] [Google Scholar]
- 46. Campbell GR, Mahad DJ (2012) Clonal expansion of mitochondrial DNA deletions and the progression of multiple sclerosis. CNS Neurol Disord Drug Targets 11:589–597. [DOI] [PubMed] [Google Scholar]
- 47. Campbell GR, Ziabreva I, Reeve AK, Krishnan KJ, Reynolds R, Howell O et al (2011) Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann Neurol 69:481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Carone DA, Benedict RH, Munschauer FE 3rd, Fishman I, Weinstock‐Guttman B (2005) Interpreting patient/informant discrepancies of reported cognitive symptoms in MS. J Int Neuropsychol Soc 11:574–583. [DOI] [PubMed] [Google Scholar]
- 49. Chang A, Staugaitis SM, Dutta R, Batt CE, Easley KE, Chomyk AM, Yong VW, Fox RJ, Kidd GJ, Trapp BD (2012) Cortical remyelination: a new target for repair therapies in multiple sclerosis. Ann Neurol 72:918–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chard DT, Griffin CM, Parker GJ, Kapoor R, Thompson AJ, Miller DH (2002) Brain atrophy in clinically early relapsing‐remitting multiple sclerosis. Brain 125(Pt 2):327–337. [DOI] [PubMed] [Google Scholar]
- 51. Charil A, Zijdenbos AP, Taylor J, Boelman C, Worsley KJ, Evans AC, Dagher A (2003) Statistical mapping analysis of lesion location and neurological disability in multiple sclerosis: application to 452 patient data sets. Neuroimage 19:532–544. [DOI] [PubMed] [Google Scholar]
- 52. Chen Z, Jalabi W, Hu W, Park HJ, Gale JT, Kidd GJ et al (2014) Microglial displacement of inhibitory synapses provides neuroprotection in the adult brain. Nat Commun 5:4486–4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen Z, Jalabi W, Shpargel KB, Farabaugh KT, Dutta R, Yin X, Kidd GJ, Bergmann CC, Stohlman SA, Trapp BD (2012) Lipopolysaccharide‐induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J Neurosci 32:11706–11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chiaravalloti ND, DeLuca J (2008) Cognitive impairment in multiple sclerosis. Lancet Neurol 7:1139–1151. [DOI] [PubMed] [Google Scholar]
- 55. Child ND, Benarroch EE (2013) Anterior nucleus of the thalamus: functional organization and clinical implications. Neurology 81:1869–1876. [DOI] [PubMed] [Google Scholar]
- 56. Christodoulou C, Krupp LB, Liang Z, Huang W, Melville P, Roque C et al (2003) Cognitive performance and MR markers of cerebral injury in cognitively impaired MS patients. Neurology 60:1793–1798. [DOI] [PubMed] [Google Scholar]
- 57. Cifelli A, Arridge M, Jezzard P, Esiri MM, Palace J, Matthews PM (2002) Thalamic neurodegeneration in multiple sclerosis. Ann Neurol 52:650–653. [DOI] [PubMed] [Google Scholar]
- 58. Clements RJ, McDonough J, Freeman EJ (2008) Distribution of parvalbumin and calretinin immunoreactive interneurons in motor cortex from multiple sclerosis post‐mortem tissue. Exp Brain Res 187:459–465. [DOI] [PubMed] [Google Scholar]
- 59. Comi G, Filippi M, Martinelli V, Campi A, Rodegher M, Alberoni M et al (1995) Brain MRI correlates of cognitive impairment in primary and secondary progressive multiple sclerosis. J Neurol Sci 132:222–2227. [DOI] [PubMed] [Google Scholar]
- 60. Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG (2004) Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci U S A 101:8168–8173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cummings JL (1986) Subcortical dementia. Neuropsychology, neuropsychiatry, and pathophysiology. Br J Psychiatry 149:682–697. [DOI] [PubMed] [Google Scholar]
- 62. Darvesh S, Freedman M (1996) Subcortical dementia: a neurobehavioral approach. Brain Cogn 31:230–249. [DOI] [PubMed] [Google Scholar]
- 63. Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA et al (2012) Fibrinogen‐induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun 3:1227–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dawson J (1916) The histology of multiple sclerosis. Trans R Soc Edinburgh 50:517–740. [Google Scholar]
- 65. Delis DC (2000) California Verbal Learning Test: Adult Version. Psychological Corporation: San Antonio. [Google Scholar]
- 66. Delis DC, Kaplan E, Kramer JH (2001) Delis‐Kaplan Executive Funcion System. Psychological Corporation: San Antonio. [Google Scholar]
- 67. Deloire MS, Salort E, Bonnet M, Arimone Y, Boudineau M, Amieva H et al (2005) Cognitive impairment as marker of diffuse brain abnormalities in early relapsing remitting multiple sclerosis. J Neurol Neurosurg Psychiatry 76:519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. DeLuca GC, Alterman R, Martin JL, Mittal A, Blundell S, Bird S et al (2013) Casting light on multiple sclerosis heterogeneity: the role of HLA‐DRB1 on spinal cord pathology. Brain 136(Pt 4):1025–1034. [DOI] [PubMed] [Google Scholar]
- 69. DeLuca GC, Ebers GC, Esiri MM (2004) Axonal loss in multiple sclerosis: a pathological survey of the corticospinal and sensory tracts. Brain 127(Pt 5):1009–1018. [DOI] [PubMed] [Google Scholar]
- 70. DeLuca GC, Williams K, Evangelou N, Ebers GC, Esiri MM (2006) The contribution of demyelination to axonal loss in multiple sclerosis. Brain 129(Pt 6):1507–1516. [DOI] [PubMed] [Google Scholar]
- 71. Derecki NC, Katzmarski N, Kipnis J, Meyer‐Luehmann M (2014) Microglia as a critical player in both developmental and late‐life CNS pathologies. Acta Neuropathol 128:333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Diaz‐Sanchez M, Williams K, DeLuca GC, Esiri MM (2006) Protein co‐expression with axonal injury in multiple sclerosis plaques. Acta Neuropathol 111:289–299. [DOI] [PubMed] [Google Scholar]
- 73. Drake AS, Weinstock‐Guttman B, Morrow SA, Hojnacki D, Munschauer FE, Benedict RH (2010) Psychometrics and normative data for the Multiple Sclerosis Functional Composite: replacing the PASAT with the Symbol Digit Modalities Test. Mult Scler 16:228–237. [DOI] [PubMed] [Google Scholar]
- 74. Drew M, Tippett LJ, Starkey NJ, Isler RB (2008) Executive dysfunction and cognitive impairment in a large community‐based sample with Multiple Sclerosis from New Zealand: a descriptive study. Arch Clin Neuropsychol 23:1–19. [DOI] [PubMed] [Google Scholar]
- 75. Duque B, Sepulcre J, Bejarano B, Samaranch L, Pastor P, Villoslada P (2008) Memory decline evolves independently of disease activity in MS. Mult Scler 14:947–953. [DOI] [PubMed] [Google Scholar]
- 76. Dutta R, Chang A, Doud MK, Kidd GJ, Ribaudo MV, Young EA et al (2011) Demyelination causes synaptic alterations in hippocampi from multiple sclerosis patients. Ann Neurol 69:445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dutta R, Chomyk AM, Chang A, Ribaudo MV, Deckard SA, Doud MK et al (2013) Hippocampal demyelination and memory dysfunction are associated with increased levels of the neuronal microRNA miR‐124 and reduced AMPA receptors. Ann Neurol 73:637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dutta R, McDonough J, Yin X, Peterson J, Chang A, Torres T et al (2006) Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol 59:478–489. [DOI] [PubMed] [Google Scholar]
- 79. Erickson MA, Banks WA (2013) Blood‐brain barrier dysfunction as a cause and consequence of Alzheimer's disease. J Cereb Blood Flow Metab 33:1500–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Errede M, Girolamo F, Ferrara G, Strippoli M, Morando S, Boldrin V et al (2012) Blood‐brain barrier alterations in the cerebral cortex in experimental autoimmune encephalomyelitis. J Neuropathol Exp Neurol 71:840–854. [DOI] [PubMed] [Google Scholar]
- 81. Esiri MM, Joachim C, Sloan C, Christie S, Agacinski G, Bridges LR et al (2014) Cerebral subcortical small vessel disease in subjects with pathologically confirmed Alzheimer disease: a clinicopathologic study in the Oxford Project to Investigate Memory and Ageing (OPTIMA). Alzheimer Dis Assoc Disord 28:30–35. [DOI] [PubMed] [Google Scholar]
- 82. Evangelou N, Esiri MM, Smith S, Palace J, Matthews PM (2000) Quantitative pathological evidence for axonal loss in normal appearing white matter in multiple sclerosis. Ann Neurol 47:391–395. [PubMed] [Google Scholar]
- 83. Evangelou N, Konz D, Esiri MM, Smith S, Palace J, Matthews PM (2000) Regional axonal loss in the corpus callosum correlates with cerebral white matter lesion volume and distribution in multiple sclerosis. Brain 123(Pt 9):1845–1849. [DOI] [PubMed] [Google Scholar]
- 84. Evangelou N, Konz D, Esiri MM, Smith S, Palace J, Matthews PM (2001) Size‐selective neuronal changes in the anterior optic pathways suggest a differential susceptibility to injury in multiple sclerosis. Brain 124(Pt 9):1813–1820. [DOI] [PubMed] [Google Scholar]
- 85. Ferguson B, Matyszak MK, Esiri MM, Perry VH (1997) Axonal damage in acute multiple sclerosis lesions. Brain 120(Pt 3):393–399. [DOI] [PubMed] [Google Scholar]
- 86. Feuillet L, Reuter F, Audoin B, Malikova I, Barrau K, Cherif AA, Pelletier J (2007) Early cognitive impairment in patients with clinically isolated syndrome suggestive of multiple sclerosis. Mult Scler 13:124–127. [DOI] [PubMed] [Google Scholar]
- 87. Fisher E, Rudick RA, Cutter G, Baier M, Miller D, Weinstock‐Guttman B et al (2000) Relationship between brain atrophy and disability: an 8‐year follow‐up study of multiple sclerosis patients. Multiple Sclerosis 6:373–377. [DOI] [PubMed] [Google Scholar]
- 88. Flensner G, Landtblom AM, Soderhamn O, Ek AC (2013) Work capacity and health‐related quality of life among individuals with multiple sclerosis reduced by fatigue: a cross‐sectional study. BMC Public Health 13:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fog T (1965) The topography of plaques in multiple sclerosis with special reference to cerebral plaques. Acta Neurol Scand Suppl. 15:1–161. [PubMed] [Google Scholar]
- 90. Fontaine B, Seilhean D, Tourbah A, Daumas‐Duport C, Duyckaerts C, Benoit N et al (1994) Dementia in two histologically confirmed cases of multiple sclerosis: one case with isolated dementia and one case associated with psychiatric symptoms. J Neurol Neurosurg Psychiatry 57:353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Foong J, Rozewicz L, Chong WK, Thompson AJ, Miller DH, Ron MA (2000) A comparison of neuropsychological deficits in primary and secondary progressive multiple sclerosis. J Neurol 247:97–101. [DOI] [PubMed] [Google Scholar]
- 92. Frischer JM, Bramow S, Dal‐Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M et al (2009) The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 132(Pt 5):1175–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Gardner C, Magliozzi R, Durrenberger PF, Howell OW, Rundle J, Reynolds R (2013) Cortical grey matter demyelination can be induced by elevated pro‐inflammatory cytokines in the subarachnoid space of MOG‐immunized rats. Brain 136(Pt 12):3596–3608. [DOI] [PubMed] [Google Scholar]
- 94. Geurts JJ, Bo L, Roosendaal SD, Hazes T, Daniels R, Barkhof F et al (2007) Extensive hippocampal demyelination in multiple sclerosis. J Neuropathol Exp Neurol 66:819–827. [DOI] [PubMed] [Google Scholar]
- 95. Ghosh N, DeLuca GC, Esiri MM (2004) Evidence of axonal damage in human acute demyelinating diseases. J Neurol Sci 222:29–34. [DOI] [PubMed] [Google Scholar]
- 96. Gilmore CP, Donaldson I, Bo L, Owens T, Lowe J, Evangelou N (2009) Regional variations in the extent and pattern of grey matter demyelination in multiple sclerosis: a comparison between the cerebral cortex, cerebellar cortex, deep grey matter nuclei and the spinal cord. J Neurol Neurosurg Psychiatry 80:182–187. [DOI] [PubMed] [Google Scholar]
- 97. Giuliani F, Goodyer CG, Antel JP, Yong VW (2003) Vulnerability of human neurons to T cell‐mediated cytotoxicity. J Immunol 171:368–379. [DOI] [PubMed] [Google Scholar]
- 98. Glanz BI, Holland CM, Gauthier SA, Amunwa EL, Liptak Z, Houtchens MK et al (2007) Cognitive dysfunction in patients with clinically isolated syndromes or newly diagnosed multiple sclerosis. Mult Scler 13:1004–1010. [DOI] [PubMed] [Google Scholar]
- 99. Gobin SJ, Montagne L, Van Zutphen M, Van Der Valk P, Van Den Elsen PJ, De Groot CJ (2001) Upregulation of transcription factors controlling MHC expression in multiple sclerosis lesions. Glia 36:68–77. [DOI] [PubMed] [Google Scholar]
- 100. Goldschmidt T, Antel J, Konig FB, Bruck W, Kuhlmann T (2009) Remyelination capacity of the MS brain decreases with disease chronicity. Neurology 72:1914–1921. [DOI] [PubMed] [Google Scholar]
- 101. Gouw AA, Seewann A, Vrenken H, van der Flier WM, Rozemuller JM, Barkhof F et al (2008) Heterogeneity of white matter hyperintensities in Alzheimer's disease: post‐mortem quantitative MRI and neuropathology. Brain 131(Pt 12):3286–3298. [DOI] [PubMed] [Google Scholar]
- 102. Goverman J (2009) Autoimmune T cell responses in the central nervous system. Nat Rev Immunol 9:393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Gronwall DM (1977) Paced auditory serial‐addition task: a measure of recovery from concussion. Percept Mot Skills 44:367–373. [DOI] [PubMed] [Google Scholar]
- 104. Grossman RI, Yousem DM (2003) Neuroradiology, 2nd edn. Mosby: Philadelphia. [Google Scholar]
- 105. Haider L, Fischer MT, Frischer JM, Bauer J, Hoftberger R, Botond G et al (2011) Oxidative damage in multiple sclerosis lesions. Brain 134(Pt 7):1914–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Haider L, Simeonidou C, Steinberger G, Hametner S, Grigoriadis N, Deretzi G et al (2014) Multiple sclerosis deep grey matter: the relation between demyelination, neurodegeneration, inflammation and iron. J Neurol Neurosurg Psychiatry 85:1386–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hakim EA, Bakheit AM, Bryant TN, Roberts MW, McIntosh‐Michaelis SA, Spackman AJ et al (2000) The social impact of multiple sclerosis—a study of 305 patients and their relatives. Disabil Rehabil 22:288–293. [DOI] [PubMed] [Google Scholar]
- 108. Henry RG, Shieh M, Amirbekian B, Chung S, Okuda DT, Pelletier D (2009) Connecting white matter injury and thalamic atrophy in clinically isolated syndromes. J Neurol Sci 282:61–66. [DOI] [PubMed] [Google Scholar]
- 109. Hildebrandt H, Hahn HK, Kraus JA, Schulte‐Herbruggen A, Schwarze B, Schwendemann G (2006) Memory performance in multiple sclerosis patients correlates with central brain atrophy. Mult Scler 12:428–436. [DOI] [PubMed] [Google Scholar]
- 110. Horsfield MA, Rovaris M, Rocca MA, Rossi P, Benedict RH, Filippi M, Bakshi R (2003) Whole‐brain atrophy in multiple sclerosis measured by two segmentation processes from various MRI sequences. J Neurol Sci 216:169–177. [DOI] [PubMed] [Google Scholar]
- 111. Houtchens MK, Benedict RHB, Killiany R, Sharma J, Jaisani Z, Singh B et al (2007) Thalamic atrophy and cognition in multiple sclerosis. Neurology 69:113–123. [DOI] [PubMed] [Google Scholar]
- 112. Howell OW, Reeves CA, Nicholas R, Carassiti D, Radotra B, Gentleman SM et al (2011) Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 134(Pt 9):2755–2771. [DOI] [PubMed] [Google Scholar]
- 113. Huijbregts SC, Kalkers NF, de Sonneville LM, de Groot V, Reuling IE, Polman CH (2004) Differences in cognitive impairment of relapsing remitting, secondary, and primary progressive MS. Neurology 63:335–339. [DOI] [PubMed] [Google Scholar]
- 114. Iadecola C (2013) The pathobiology of vascular dementia. Neuron 80:844–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kalkers NF, Bergers E, Castelijns JA, van Walderveen MA, Bot JC, Ader HJ et al (2001) Optimizing the association between disability and biological markers in MS. Neurology 57:1253–1258. [DOI] [PubMed] [Google Scholar]
- 116. Kassubek J, Tumani H, Ecker D, Kurt A, Ludolph AC, Juengling FD (2003) Age‐related brain parenchymal fraction is significantly decreased in young multiple sclerosis patients: a quantitative MRI study. Neuroreport 14:427–430. [DOI] [PubMed] [Google Scholar]
- 117. Katzman R, Terry R, DeTeresa R, Brown T, Davies P, Fuld P et al (1988) Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol 23:138–144. [DOI] [PubMed] [Google Scholar]
- 118. Kermode AG, Tofts PS, Thompson AJ, MacManus DG, Rudge P, Kendall BE et al (1990) Heterogeneity of blood‐brain barrier changes in multiple sclerosis: an MRI study with gadolinium‐DTPA enhancement. Neurology 40:229–235. [DOI] [PubMed] [Google Scholar]
- 119. Kidd D, Barkhof F, McConnell R, Algra PR, Allen IV, Revesz T (1999) Cortical lesions in multiple sclerosis. Brain 122(Pt 1):17–26. [DOI] [PubMed] [Google Scholar]
- 120. Kirk J, Plumb J, Mirakhur M, McQuaid S (2003) Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood‐brain barrier leakage and active demyelination. J Pathol 201:319–327. [DOI] [PubMed] [Google Scholar]
- 121. Koenig KA, Sakaie KE, Lowe MJ, Lin J, Stone L, Bermel RA et al (2014) Hippocampal volume is related to cognitive decline and fornicial diffusion measures in multiple sclerosis. Magn Reson Imaging 32:354–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Kolasinski J, Stagg CJ, Chance SA, Deluca GC, Esiri MM, Chang EH et al (2012) A combined post‐mortem magnetic resonance imaging and quantitative histological study of multiple sclerosis pathology. Brain 135(Pt 10):2938–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Kooi EJ, Geurts JJ, van Horssen J, Bo L, van der Valk P (2009) Meningeal inflammation is not associated with cortical demyelination in chronic multiple sclerosis. J Neuropathol Exp Neurol 68:1021–1028. [DOI] [PubMed] [Google Scholar]
- 124. Kooi EJ, Prins M, Bajic N, Belien JA, Gerritsen WH, van Horssen J et al (2011) Cholinergic imbalance in the multiple sclerosis hippocampus. Acta Neuropathol 122:313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Kooi EJ, Strijbis EM, van der Valk P, Geurts JJ (2012) Heterogeneity of cortical lesions in multiple sclerosis: clinical and pathologic implications. Neurology 79:1369–1376. [DOI] [PubMed] [Google Scholar]
- 126. Kostic M, Zivkovic N, Stojanovic I (2013) Multiple sclerosis and glutamate excitotoxicity. Rev Neurosci 24:71–88. [DOI] [PubMed] [Google Scholar]
- 127. Kujala P, Portin R, Ruutiainen J (1997) The progress of cognitive decline in multiple sclerosis. A controlled 3‐year follow‐up. Brain 120(Pt 2):289–297. [DOI] [PubMed] [Google Scholar]
- 128. Kutzelnigg A, Faber‐Rod JC, Bauer J, Lucchinetti CF, Sorensen PS, Laursen H et al (2007) Widespread demyelination in the cerebellar cortex in multiple sclerosis. Brain Pathol 17:38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Kutzelnigg A, Lassmann H (2006) Cortical demyelination in multiple sclerosis: a substrate for cognitive deficits? J Neurol Sci 245:123–126. [DOI] [PubMed] [Google Scholar]
- 130. Kutzelnigg A, Lucchinetti CF, Stadelmann C, Bruck W, Rauschka H, Bergmann M et al (2005) Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 128(Pt 11):2705–2712. [DOI] [PubMed] [Google Scholar]
- 131. Langdon D, Amato M, Boringa J, Brochet B, Foley F, Fredrikson S et al (2012) Recommendations for a brief international cognitive assessment for multiple sclerosis (BICAMS). Mult Scler 18:891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Lassmann H (2011) Review: the architecture of inflammatory demyelinating lesions: implications for studies on pathogenesis. Neuropathol Appl Neurobiol 37:698–710. [DOI] [PubMed] [Google Scholar]
- 133. Lassmann H (2012) Cortical lesions in multiple sclerosis: inflammation versus neurodegeneration. Brain 135(Pt 10):2904–2905. [DOI] [PubMed] [Google Scholar]
- 134. Laule C, Pavlova V, Leung E, Zhao G, MacKay AL, Kozlowski P et al (2013) Diffusely abnormal white matter in multiple sclerosis: further histologic studies provide evidence for a primary lipid abnormality with neurodegeneration. J Neuropathol Exp Neurol 72:42–52. [DOI] [PubMed] [Google Scholar]
- 135. Laule C, Vavasour IM, Leung E, Li DK, Kozlowski P, Traboulsee AL et al (2011) Pathological basis of diffusely abnormal white matter: insights from magnetic resonance imaging and histology. Mult Scler 17:144–150. [DOI] [PubMed] [Google Scholar]
- 136. Lazeron RH, de Sonneville LM, Scheltens P, Polman CH, Barkhof F (2006) Cognitive slowing in multiple sclerosis is strongly associated with brain volume reduction. Mult Scler 12:760–768. [DOI] [PubMed] [Google Scholar]
- 137. Leech S, Kirk J, Plumb J, McQuaid S (2007) Persistent endothelial abnormalities and blood‐brain barrier leak in primary and secondary progressive multiple sclerosis. Neuropathol Appl Neurobiol 33:86–98. [DOI] [PubMed] [Google Scholar]
- 138. Lindberg RL, De Groot CJ, Montagne L, Freitag P, van der Valk P, Kappos L, Leppert D (2001) The expression profile of matrix metalloproteinases (MMPs) and their inhibitors (TIMPs) in lesions and normal appearing white matter of multiple sclerosis. Brain 124(Pt 9):1743–1753. [DOI] [PubMed] [Google Scholar]
- 139. London A, Cohen M, Schwartz M (2013) Microglia and monocyte‐derived macrophages: functionally distinct populations that act in concert in CNS plasticity and repair. Front Cell Neurosci 7:34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Lovas G, Szilagyi N, Majtenyi K, Palkovits M, Komoly S (2000) Axonal changes in chronic demyelinated cervical spinal cord plaques. Brain 123(Pt 2):308–317. [DOI] [PubMed] [Google Scholar]
- 141. Lublin FD (2007) The incomplete nature of multiple sclerosis relapse resolution. J Neurol Sci 256(Suppl. 1):S14‐S18. [DOI] [PubMed] [Google Scholar]
- 142. Lucchinetti CF, Popescu BF, Bunyan RF, Moll NM, Roemer SF, Lassmann H et al (2011) Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med 365:2188–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Magliozzi R, Columba‐Cabezas S, Serafini B, Aloisi F (2004) Intracerebral expression of CXCL13 and BAFF is accompanied by formation of lymphoid follicle‐like structures in the meninges of mice with relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol 148:11–23. [DOI] [PubMed] [Google Scholar]
- 144. Magliozzi R, Howell O, Vora A, Serafini B, Nicholas R, Puopolo M et al (2007) Meningeal B‐cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 130(Pt 4):1089–1104. [DOI] [PubMed] [Google Scholar]
- 145. Magliozzi R, Howell OW, Reeves C, Roncaroli F, Nicholas R, Serafini B et al (2010) A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol 68:477–493. [DOI] [PubMed] [Google Scholar]
- 146. Martin JH (2003) Neuroanatomy: Text and Atlas. McGraw‐Hill Medical: New York. [Google Scholar]
- 147. Mathey EK, Derfuss T, Storch MK, Williams KR, Hales K, Woolley DR et al (2007) Neurofascin as a novel target for autoantibody‐mediated axonal injury. J Exp Med 204:2363–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Middleton FA, Strick PL (2000) Basal ganglia output and cognition: evidence from anatomical, behavioral, and clinical studies. Brain Cogn 42:183–200. [DOI] [PubMed] [Google Scholar]
- 149. Miller DH, Rudge P, Johnson G, Kendall BE, Macmanus DG, Moseley IF et al (1988) Serial gadolinium enhanced magnetic resonance imaging in multiple sclerosis. Brain 111(Pt 4):927–939. [DOI] [PubMed] [Google Scholar]
- 150. Minagar A, Barnett MH, Benedict RH, Pelletier D, Pirko I, Sahraian MA et al (2013) The thalamus and multiple sclerosis: modern views on pathologic, imaging, and clinical aspects. Neurology 80:210–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL et al (2013) M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci 16:1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Mistry N, Dixon J, Tallantyre E, Tench C, Abdel‐Fahim R, Jaspan T et al (2013) Central veins in brain lesions visualized with high‐field magnetic resonance imaging: a pathologically specific diagnostic biomarker for inflammatory demyelination in the brain. JAMA Neurol 70:623–628. [DOI] [PubMed] [Google Scholar]
- 153. Moore GR, Laule C (2012) Neuropathologic correlates of magnetic resonance imaging in multiple sclerosis. J Neuropathol Exp Neurol 71:762–778. [DOI] [PubMed] [Google Scholar]
- 154. Moore GR, Laule C, Mackay A, Leung E, Li DK, Zhao G et al (2008) Dirty‐appearing white matter in multiple sclerosis: preliminary observations of myelin phospholipid and axonal loss. J Neurol 255:1802–1811, discussion 12. [DOI] [PubMed] [Google Scholar]
- 155. Morgen K, Sammer G, Courtney SM, Wolters T, Melchior H, Blecker CR et al (2006) Evidence for a direct association between cortical atrophy and cognitive impairment in relapsing‐remitting MS. Neuroimage 30:891–898. [DOI] [PubMed] [Google Scholar]
- 156. Morris CS, Esiri MM, Sprinkle TJ, Gregson N (1994) Oligodendrocyte reactions and cell proliferation markers in human demyelinating diseases. Neuropathol Appl Neurobiol 20:272–281. [DOI] [PubMed] [Google Scholar]
- 157. Morrow SA, Drake A, Zivadinov R, Munschauer F, Weinstock‐Guttman B, Benedict RH (2010) Predicting loss of employment over three years in multiple sclerosis: clinically meaningful cognitive decline. Clin Neuropsychol 24:1131–1145. [DOI] [PubMed] [Google Scholar]
- 158. Morrow SA, Jurgensen S, Forrestal F, Munchauer FE, Benedict RH (2011) Effects of acute relapses on neuropsychological status in multiple sclerosis patients. J Neurol 258:1603–1608. [DOI] [PubMed] [Google Scholar]
- 159. Morrow SA, O'Connor PW, Polman CH, Goodman AD, Kappos L, Lublin FD et al (2010) Evaluation of the symbol digit modalities test (SDMT) and MS neuropsychological screening questionnaire (MSNQ) in natalizumab‐treated MS patients over 48 weeks. Mult Scler 16:1385–1392. [DOI] [PubMed] [Google Scholar]
- 160. Morrow SA, Weinstock‐Guttman B, Munschauer FE, Hojnacki D, Benedict RH (2009) Subjective fatigue is not associated with cognitive impairment in multiple sclerosis: cross‐sectional and longitudinal analysis. Mult Scler 15:998–1005. [DOI] [PubMed] [Google Scholar]
- 161. Nave KA, Trapp BD (2008) Axon‐glial signaling and the glial support of axon function. Annu Rev Neurosci 31:535–561. [DOI] [PubMed] [Google Scholar]
- 162. Neumann PE, Mehler MF, Horoupian DS, Merriam AE (1988) Atypical psychosis with disseminated subpial demyelination. Arch Neurol 45:634–636. [DOI] [PubMed] [Google Scholar]
- 163. Newcombe J, Hawkins CP, Henderson CL, Patel HA, Woodroofe MN, Hayes GM et al (1991) Histopathology of multiple sclerosis lesions detected by magnetic resonance imaging in unfixed postmortem central nervous system tissue. Brain 114(Pt 2):1013–1023. [DOI] [PubMed] [Google Scholar]
- 164. Nielsen AS, Kinkel RP, Madigan N, Tinelli E, Benner T, Mainero C (2013) Contribution of cortical lesion subtypes at 7T MRI to physical and cognitive performance in MS. Neurology 81:641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Norden DM, Godbout JP (2013) Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol 39:19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Okuda DT, Srinivasan R, Oksenberg JR, Goodin DS, Baranzini SE, Beheshtian A et al (2009) Genotype‐Phenotype correlations in multiple sclerosis: HLA genes influence disease severity inferred by 1HMR spectroscopy and MRI measures. Brain 132(Pt 1):250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Pantoni L, Garcia JH (1997) Pathogenesis of leukoaraiosis: a review. Stroke 28:652–659. [DOI] [PubMed] [Google Scholar]
- 168. Papadopoulos D, Dukes S, Patel R, Nicholas R, Vora A, Reynolds R (2009) Substantial archaeocortical atrophy and neuronal loss in multiple sclerosis. Brain Pathol 19:238–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Patani R, Balaratnam M, Vora A, Reynolds R (2007) Remyelination can be extensive in multiple sclerosis despite a long disease course. Neuropathol Appl Neurobiol 33:277–287. [DOI] [PubMed] [Google Scholar]
- 170. Patrikios P, Stadelmann C, Kutzelnigg A, Rauschka H, Schmidbauer M, Laursen H et al (2006) Remyelination is extensive in a subset of multiple sclerosis patients. Brain 129(Pt 12):3165–3172. [DOI] [PubMed] [Google Scholar]
- 171. Patti F, Failla G, Ciancio MR, L'Episcopo MR, Reggio A (1998) Neuropsychological, neuroradiological and clinical findings in multiple sclerosis. A 3 year follow‐up study. Eur J Neurol 5:283–286. [DOI] [PubMed] [Google Scholar]
- 172. Peterson JW, Bo L, Mork S, Chang A, Trapp BD (2001) Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol 50:389–400. [DOI] [PubMed] [Google Scholar]
- 173. Plumb J, McQuaid S, Mirakhur M, Kirk J (2002) Abnormal endothelial tight junctions in active lesions and normal‐appearing white matter in multiple sclerosis. Brain Pathol 12:154–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Polman CH, Reingold SC, Edan G, Filippi M, Hartung HP, Kappos L et al (2005) Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol 58:840–846. [DOI] [PubMed] [Google Scholar]
- 175. Portaccio E, Goretti B, Zipoli V, Iudice A, Pina DD, Malentacchi GM et al (2010) Reliability, practice effects, and change indices for Rao's Brief Repeatable Battery. Mult Scler 16:611–617. [DOI] [PubMed] [Google Scholar]
- 176. Querfurth HW, LaFerla FM (2010) Alzheimer's disease. N Engl J Med 362:329–344. [DOI] [PubMed] [Google Scholar]
- 177. Rao SM, Leo GJ, Bernardin L, Unverzagt F (1991) Cognitive dysfunction in multiple sclerosis. I. Frequency, patterns, and prediction. Neurology 41:685–6891. [DOI] [PubMed] [Google Scholar]
- 178. Rao SM, Leo GJ, Ellington L, Nauertz T, Bernardin L, Unverzagt F (1991) Cognitive dysfunction in multiple sclerosis. II. Impact on employment and social functioning. Neurology 41:692–696. [DOI] [PubMed] [Google Scholar]
- 179. Rao SM, Leo GJ, Haughton VM, St Aubin‐Faubert P, Bernardin L (1989) Correlation of magnetic resonance imaging with neuropsychological testing in multiple sclerosis. Neurology 39(Pt 1):161–166. [DOI] [PubMed] [Google Scholar]
- 180. Reuter F, Zaaraoui W, Crespy L, Faivre A, Rico A, Malikova I et al (2011) Cognitive impairment at the onset of multiple sclerosis: relationship to lesion location. Mult Scler 17:755–758. [DOI] [PubMed] [Google Scholar]
- 181. Revesz T, Kidd D, Thompson AJ, Barnard RO, McDonald WI (1994) A comparison of the pathology of primary and secondary progressive multiple sclerosis. Brain 117(Pt 4):759–765. [DOI] [PubMed] [Google Scholar]
- 182. Rist JM, Franklin RJ (2008) Taking ageing into account in remyelination‐based therapies for multiple sclerosis. J Neurol Sci 274:64–67. [DOI] [PubMed] [Google Scholar]
- 183. Rossi F, Giorgio A, Battaglini M, Stromillo ML, Portaccio E, Goretti B et al (2012) Relevance of brain lesion location to cognition in relapsing multiple sclerosis. PLoS One 7:e44826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Rudick RA, Fisher E, Lee JC, Simon J, Jacobs L (1999) Use of the brain parenchymal fraction to measure whole brain atrophy in relapsing‐remitting MS. Multiple Sclerosis Collaborative Research Group. Neurology 53:1698–1704. [DOI] [PubMed] [Google Scholar]
- 185. Ruet A, Deloire M, Hamel D, Ouallet JC, Petry K, Brochet B (2013) Cognitive impairment, health‐related quality of life and vocational status at early stages of multiple sclerosis: a 7‐year longitudinal study. J Neurol 260:776–784. [DOI] [PubMed] [Google Scholar]
- 186. Sadaba MC, Tzartos J, Paino C, Garcia‐Villanueva M, Alvarez‐Cermeno JC, Villar LM, Esiri MM (2012) Axonal and oligodendrocyte‐localized IgM and IgG deposits in MS lesions. J Neuroimmunol 247:86–94. [DOI] [PubMed] [Google Scholar]
- 187. Sanfilipo MP, Benedict RH, Weinstock‐Guttman B, Bakshi R (2006) Gray and white matter brain atrophy and neuropsychological impairment in multiple sclerosis. Neurology 66:685–692. [DOI] [PubMed] [Google Scholar]
- 188. Sastre‐Garriga J, Arevalo MJ, Renom M, Alonso J, Gonzalez I, Galan I et al (2009) Brain volumetry counterparts of cognitive impairment in patients with multiple sclerosis. J Neurol Sci 282:120–124. [DOI] [PubMed] [Google Scholar]
- 189. Schmidt R, Enzinger C, Ropele S, Schmidt H, Fazekas F (2006) Subcortical vascular cognitive impairment: similarities and differences with multiple sclerosis. J Neurol Sci 245:3–7. [DOI] [PubMed] [Google Scholar]
- 190. Scotti G, Scialfa G, Biondi A, Landoni L, Caputo D, Cazzullo CL (1986) Magnetic resonance in multiple sclerosis. Neuroradiology 28:319–323. [DOI] [PubMed] [Google Scholar]
- 191. Seewann A, Kooi EJ, Roosendaal SD, Pouwels PJ, Wattjes MP, van der Valk P et al (2012) Postmortem verification of MS cortical lesion detection with 3D DIR. Neurology 78:302–308. [DOI] [PubMed] [Google Scholar]
- 192. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F (2004) Detection of ectopic B‐cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol 14:164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Sicotte NL, Kern KC, Giesser BS, Arshanapalli A, Schultz A, Montag M et al (2008) Regional hippocampal atrophy in multiple sclerosis. Brain 131(Pt 4):1134–1141. [DOI] [PubMed] [Google Scholar]
- 194. Smith A (1982) Symbol Digit Modalities Test: Manual. Western Psychological Services: Los Angeles. [Google Scholar]
- 195. Smith KJ, Kapoor R, Hall SM, Davies M (2001) Electrically active axons degenerate when exposed to nitric oxide. Ann Neurol 49:470–476. [PubMed] [Google Scholar]
- 196. Sobel RA, Ahmed AS (2001) White matter extracellular matrix chondroitin sulfate/dermatan sulfate proteoglycans in multiple sclerosis. J Neuropathol Exp Neurol 60:1198–1207. [DOI] [PubMed] [Google Scholar]
- 197. Starr JM, Lonie J (2007) The influence of pre‐morbid IQ on Mini‐Mental State Examination score at time of dementia presentation. Int J Geriatr Psychiatry 22:382–384. [DOI] [PubMed] [Google Scholar]
- 198. Strober LB, Christodoulou C, Benedict RH, Westervelt HJ, Melville P, Scherl WF et al (2011) Unemployment in multiple sclerosis: the contribution of personality and disease. Mult Scler 18:647–653 [DOI] [PubMed] [Google Scholar]
- 199. Sumowski JF, Chiaravalloti N, Wylie G, Deluca J (2009) Cognitive reserve moderates the negative effect of brain atrophy on cognitive effi ciency in multiple sclerosis. J Int Neuropsychol Soc 15:606–612. [DOI] [PubMed] [Google Scholar]
- 200. Tallantyre EC, Bo L, Al‐Rawashdeh O, Owens T, Polman CH, Lowe J, Evangelou N (2009) Greater loss of axons in primary progressive multiple sclerosis plaques compared to secondary progressive disease. Brain 132(Pt 5):1190–1199. [DOI] [PubMed] [Google Scholar]
- 201. Tallantyre EC, Bo L, Al‐Rawashdeh O, Owens T, Polman CH, Lowe JS, Evangelou N (2010) Clinico‐pathological evidence that axonal loss underlies disability in progressive multiple sclerosis. Mult Scler 16:406–411. [DOI] [PubMed] [Google Scholar]
- 202. Tiberio M, Chard DT, Altmann DR, Davies G, Griffin CM, Rashid W et al (2005) Gray and white matter volume changes in early RRMS: A 2‐year longitudinal study. Neurology 64:1001–1007. [DOI] [PubMed] [Google Scholar]
- 203. Traboulsee A (2007) MRI relapses have significant pathologic and clinical implications in multiple sclerosis. J Neurol Sci 256(Suppl. 1):S19–S22. [DOI] [PubMed] [Google Scholar]
- 204. Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L (1998) Axonal transection in the lesions of multiple sclerosis. N Engl J Med 338:278–285. [DOI] [PubMed] [Google Scholar]
- 205. Turner MA, Moran NF, Kopelman MD (2002) Subcortical dementia. Br J Psychiatry 180:148–151. [DOI] [PubMed] [Google Scholar]
- 206. Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L (2008) Interleukin‐17 production in central nervous system‐infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol 172:146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. van Horssen J, Brink BP, de Vries HE, van der Valk P, Bo L (2007) The blood‐brain barrier in cortical multiple sclerosis lesions. J Neuropath Exp Neur 66:321–328. [DOI] [PubMed] [Google Scholar]
- 208. van Walderveen MA, Kamphorst W, Scheltens P, van Waesberghe JH, Ravid R, Valk J et al (1998) Histopathologic correlate of hypointense lesions on T1‐weighted spin‐echo MRI in multiple sclerosis. Neurology 50:1282–1288. [DOI] [PubMed] [Google Scholar]
- 209. Vercellino M, Masera S, Lorenzatti M, Condello C, Merola A, Mattioda A et al (2009) Demyelination, inflammation, and neurodegeneration in multiple sclerosis deep gray matter. J Neuropathol Exp Neurol 68:489–502. [DOI] [PubMed] [Google Scholar]
- 210. Vercellino M, Merola A, Piacentino C, Votta B, Capello E, Mancardi GL et al (2007) Altered glutamate reuptake in relapsing‐remitting and secondary progressive multiple sclerosis cortex: correlation with microglia infiltration, demyelination, and neuronal and synaptic damage. J Neuropathol Exp Neurol 66:732–739. [DOI] [PubMed] [Google Scholar]
- 211. Vercellino M, Plano F, Votta B, Mutani R, Giordana MT, Cavalla P (2005) Grey matter pathology in multiple sclerosis. J Neuropathol Exp Neurol 64:1101–1107. [DOI] [PubMed] [Google Scholar]
- 212. Vergo S, Craner MJ, Etzensperger R, Attfield K, Friese MA, Newcombe J et al (2011) Acid‐sensing ion channel 1 is involved in both axonal injury and demyelination in multiple sclerosis and its animal model. Brain 134(Pt 2):571–584. [DOI] [PubMed] [Google Scholar]
- 213. Wegner C, Esiri MM, Chance SA, Palace J, Matthews PM (2006) Neocortical neuronal, synaptic, and glial loss in multiple sclerosis. Neurology 67:960–967. [DOI] [PubMed] [Google Scholar]
- 214. Whalley LJ, Deary IJ, Appleton CL, Starr JM (2004) Cognitive reserve and the neurobiology of cognitive aging. Ageing Res Rev 3:369–382. [DOI] [PubMed] [Google Scholar]
- 215. Yates RL, Esiri MM, Palace J, Mittal A, DeLuca GC (2014) The influence of HLA‐DRB1*15 on motor cortical pathology in multiple sclerosis. Neuropath Appl Neuro (in press). [DOI] [PubMed] [Google Scholar]
- 216. Yin X, Baek RC, Kirschner DA, Peterson A, Fujii Y, Nave KA et al (2006) Evolution of a neuroprotective function of central nervous system myelin. J Cell Biol 172:469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Zakzanis KK (2000) Distinct neurocognitive profiles in multiple sclerosis subtypes. Arch Clin Neuropsychol 15:115–1136. [PubMed] [Google Scholar]