

Abstract
The majority of glioblastomas develop rapidly with a short clinical history (primary glioblastoma IDH wild‐type), whereas secondary glioblastomas progress from diffuse astrocytoma or anaplastic astrocytoma. IDH mutations are the genetic hallmark of secondary glioblastomas. Gliosarcomas and giant cell glioblastomas are rare histological glioblastoma variants, which usually develop rapidly. We determined the genetic patterns of 36 gliosarcomas and 19 giant cell glioblastomas. IDH1 and IDH2 mutations were absent in all 36 gliosarcomas and in 18 of 19 giant cell glioblastomas analyzed, indicating that they are histological variants of primary glioblastoma. Furthermore, LOH 10q (88%) and TERT promoter mutations (83%) were frequent in gliosarcomas. Copy number profiling using the 450k methylome array in 5 gliosarcomas revealed CDKN2A homozygous deletion (3 cases), trisomy chromosome 7 (2 cases), and monosomy chromosome 10 (2 cases). Giant cell glioblastomas had LOH 10q in 50% and LOH 19q in 42% of cases. ATRX loss was detected immunohistochemically in 19% of giant cell glioblastomas, but absent in 17 gliosarcomas. These and previous results suggest that gliosarcomas are a variant of, and genetically similar to, primary glioblastomas, except for a lack of EGFR amplification, while giant cell glioblastoma occupies a hybrid position between primary and secondary glioblastomas.
Keywords: gliosarcoma, giant cell glioblastoma, IDH mutation, TERT mutation, 19q, 10q
Introduction
Glioblastoma (WHO grade IV) is the most frequent and malignant glioma. The majority of glioblastomas (>90%) develop rapidly after a short clinical history, without evidence of a less malignant precursor lesion (primary glioblastoma). In contrast, secondary glioblastomas develop through progression from diffuse astrocytoma (WHO grade II) or anaplastic astrocytoma (WHO grade III)24, 27, 28. The decisive genetic alterations that reliably distinguish between primary and secondary glioblastoma are IDH mutations3, 24, 28, 35, 37 which constitute a reliable genetic marker of secondary glioblastoma. In a large population‐based study, IDH1 mutation status corresponded to the respective clinical diagnosis in 95% of cases24, 28.
Gliosarcoma and giant cell glioblastoma are rare histological variants of glioblastoma16. On clinical grounds, they are considered variants of primary glioblastoma, but genetic data that would allow an unambiguous classification are still scant. Gliosarcomas constitute approximately 2% of all glioblastomas16, and are characterized by a biphasic tissue pattern with alternating areas displaying glial or mesenchymal differentiation16. Despite these two distinct histological components, gliosarcomas are considered monoclonal, since glial and mesenchymal tumor areas were usually found to be genetically similar4, 31. It has been reported that PTEN mutations (37%–45%) and p16INK4a homozygous deletion (37%) are common, TP53 mutations (24%–26%) less frequent, and EGFR amplification rare or absent (0%–8%)1, 31.
Giant cell glioblastomas constitute up to 5% of all glioblastomas, and are characterized by a predominance of bizarre, multinucleated giant cells with an occasionally abundant reticulin network16. TP53 mutations are frequent (78%–90%) and PTEN mutations are common (33%), but EGFR amplification and p16INK4a homozygous deletion have been reported to be rare17, 18, 30.
The objective of the present study was to further genetically characterize these glioblastoma variants. We screened 36 gliosarcomas and 19 giant cell glioblastomas for IDH1 and IDH2 mutations (genetic hallmarks of secondary glioblastoma), and various additional genetic alterations known to be operative in diffuse gliomas.
Materials and Methods
Tumor samples
Formalin‐fixed and paraffin embedded tissue samples of 36 gliosarcomas were obtained from the Neurological Institute (Edinger Institute), University Hospital Frankfurt, Germany, the Institute of Neuropathology, University Hospital Munster, Germany, the Department of Neuropathology, University Hospital Rome, Italy, the Institute of Neuroscience, Bordeaux, France, and the Department of Neuropathology, University Hospital Zurich, Switzerland. Formalin‐fixed tissue samples of 19 giant cell glioblastomas, as well as a frozen sample of a giant cell glioblastoma were obtained from the Department of Neuropathology, University Hospital Zurich, Switzerland, the Institute of Neuropathology, University Hospital Munster, Germany, and the Departments of Neuropathology and Neurosurgery, University Hospital Essen, Germany.
Gliosarcomas and giant cell glioblastomas were diagnosed according to the 2007 WHO Classification16. Histologically, gliosarcomas showed the typical biphasic pattern with alternating areas of glial and mesenchymal differentiation (Figure 1A). The glial area was composed of anaplastic glial cells with GFAP expression, and the mesenchymal component demonstrated bundles of spindle cells with malignant transformation and abundant connective tissue stained by reticulin, without GFAP expression (Figure 1B). For selection of giant cell glioblastomas, care was taken to include only typical cases showing a predominance of multinucleated, GFAP positive tumor cells in at least one large area of the biopsy specimen. All gliosarcomas and 18 giant cell glioblastomas were located in cerebral hemispheres; one giant cell glioblastoma was in the thalamus. This study was approved by the International Agency for Research on Cancer Ethics Committee.
Figure 1.
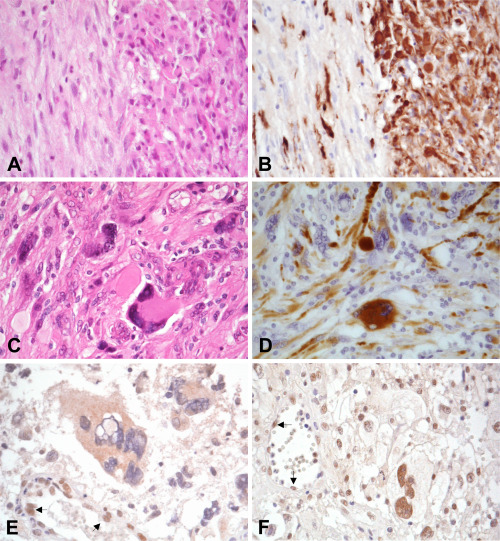
Gliosarcomas showing the typical biphasic pattern with alternating areas of glial and mesenchymal differentiation (A). Note that the glial area was composed of anaplastic glial cells with GFAP expression, while mesenchymal component lacks GFAP expression (B). Giant cell glioblastoma with multinucleated giant cells (C). GFAP is expressed at different levels in multinucleated giant cells in giant cell glioblastoma (D). Multinucleated giant cells with loss of nuclear ATRX expression in giant cell glioblastoma (E). Another giant cell glioblastoma showing nuclear immunoreactivity to ATRX (F). Arrows indicate nuclear ATRX expression in endothelium served as an internal positive control.
DNA extraction
Genomic DNA was extracted from typical tumor areas that were scraped from formalin‐fixed and paraffin‐embedded tissue slides or cryostat section from a frozen sample as previously described23, 29. DNA concentration and purity were measured by a ND8000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA).
IDH mutations
Screenings for the IDH1 and IDH2 mutations were performed in 36 gliosarcomas and 19 giant cell glioblastomas by Sanger sequencing as described previously24, 35.
TERT promoter mutations
Sequences covering the mutational hotspots (C228T and C250T) in the TERT core promoter were amplified by nested PCR. Primer sequences and detailed protocols were reported previously25. We considered as TERT mutations only cases when the height of the mutated allele was >20% of that of the wild‐type allele on sequencing data, as previously described25.
LOH 1p, 19q, 10q
Quantitative PCR was performed to assess LOH 1p, 19q, and 10q in 17 gliosarcomas and 12 giant cell glioblastomas. Microsatellite markers were D1S214, D1S468, and D1S2736 at 1p36.32‐1p36.22, D19S408, D19S596, and D19S867 at 19q13.31‐19q13.33, D10S536 at 10q23.3, and D10S1683 at 10q24.222, 26. We interpreted as LOH when 2 or 3 markers for each chromosome suggested loss, as described previously22, 26.
ATRX immunohistochemistry
ATRX immunohistochemistry was carried out in 17 gliosarcomas and in 16 giant cell glioblastomas. Paraffin sections were deparaffinized in xylene and 95% ethanol for 5 min. After inactivation of endogenous peroxidases with 3% of H2O2 in methanol for 30 min, sections were incubated in epitope retrieval solution (diluted 1:10, H‐3300, Low pH 6.0, Vector Laboratories, inc. Burlingame, CA, USA) for 20 min at 95°C–99°C. Sections were cooled for 40 min at room temperature, and were incubated with anti‐ATRX antibody (1:400, HPA001906, rabbit polyclonal, Sigma‐Aldrich, St. Louis, MO, USA), diluted in antibody diluent (S3022, Dako, Les Ulis, France) at 4°C overnight. Sections were then washed in phosphate buffered saline (PBS) and incubated with the second antibody (EnVision+Single Reagents HRP rabbit, K4002, Dako, Les Ulis, France) for 30 min. Visualization was performed using Vector DAB Substrate Kit (SK‐4100; Vector Laboratories) for 4 min. After washing in PBS, sections were counterstained with hematoxylin.
For optimization of immunohistochemistry protocols, normal human brain tissue was used as a positive control, and human liver tissue was used as a negative control. For interpretation of ATRX immunoreactivity, vascular endothelial cells within tumor tissues on histological slides were used as internal positive controls. Cases with more than 10% immunoreactive tumor cells were considered as positive as previously described36.
Methylome and copy number profiling
The Illumina Infinium HumanMethylation450 (450k) array was used in 5 gliosarcomas, for which sufficient DNA from paraffin sections were available, to obtain the DNA methylation status of 482,421 CpG sites (Illumina, San Diego, CA, USA), according to the manufacturer's instructions at the Core Facility of the DKFZ. The methylation level of each CpG site was represented by beta‐values, which ranged from 0 (unmethylated) to 1 (methylated). Genome‐wide copy‐number profiles were calculated using the intensity measures of the aforementioned methylation probe loci throughout the genome. Chromosomal copy‐number alterations were visualized by the IdeogramBrowser tool19.
Statistical analysis
Fisher exact test was performed to evaluate the significance of difference in various genetic alterations. The significance level chosen was P < 0.05. Statistical analyses were carried out using Stat View J‐5.0 software (Abacus Concepts, Berkeley, CA, USA).
Results
IDH mutations
IDH1 mutations were absent in all 36 gliosarcomas and in 18 out of 19 giant cell glioblastomas analyzed. One giant cell glioblastoma with an IDH1 (R132S) mutation was diagnosed in a 44 year‐old male patient, who developed anaplastic astrocytoma one year earlier. TP53 mutation (R280T) was also detected in both anaplastic astrocytoma and giant cell glioblastoma. IDH2 mutations were absent in all gliosarcomas and giant cell glioblastomas analyzed.
TERT promoter mutations
TERT promoter mutations were detected in 30 of 36 (83%) gliosarcomas. Of these, 76% were C228T and 24% were C250T. This ratio was similar to that reported in primary glioblastomas25.
For the 20 selected gliosarcomas, we carried out screening for TERT promoter mutations separately in glial and mesenchymal components, and found identical results in 19 cases. In one gliosarcoma, TERT mutation was observed in only the mesenchymal component, but not in the glial component.
TERT promoter mutations (C228T) were found in 4 of 16 (25%) giant cell glioblastomas.
LOH 1p, 19q, 10q
Quantitative PCR revealed LOH 1p in 1/17 (6%) gliosarcomas and in 2 of 12 (17%) giant cell glioblastomas. LOH 19q was detected in 3 gliosarcomas (3/17; 18%) and 5 giant cell glioblastomas (5/12; 42%). LOH 10q was detected in 15/17 gliosarcomas (88%) and in 6 of 12 (50%) giant cell glioblastomas. Co‐deletion of 1p/19q was detected in 1/17 (6%) gliosarcomas and in 2/12 (17%) giant cell glioblastomas.
ATRX immunohistochemistry
Loss of nuclear ATRX expression was observed in 3 of 16 (19%) giant cell glioblastoma, but in none of the 17 gliosarcomas analyzed (Figure 1).
ATRX was expressed in vascular endothelial cells (internal positive control) in all histological sections. Sections without ATRX antibodies showed no immunoreactivity.
Copy number profiling
Copy number plots generated from 450k methylation data in 5 gliosarcomas revealed CDKN2A homozygous deletion (3 cases), monosomy chromosome 10 (2 cases), trisomy chromosome 7 (2 cases), gain at chromosome 7 (one case), and CDK4 amplification (one case).
Comparison of genetic alterations with primary and secondary glioblastomas
Genetic alterations of the gliosarcomas and giant cell glioblastomas which were obtained in the present study as well as those reported previously are summarized in Table 1. These data were compared with those of the primary glioblastomas (IDH‐wild‐type) and secondary glioblastomas (IDH mut) reported previously (Table 1)24, 25.
Table 1.
Clinical and genetic profile of the gliosarcoma and the giant cell glioblastoma, in comparison with primary and secondary glioblastoma.
| Primary glioblastoma (IDH wild‐type) | Gliosarcoma | Giant cell glioblastoma | Secondary glioblastoma (IDH mutant) | |
|---|---|---|---|---|
| Age at GBM diagnosis | 59 years [Link] , [Link] , [Link] , [Link] | 56 years 5 | 44 years [Link] , [Link] | 43 years [Link] , [Link] , [Link] , [Link] |
| Sex ratio M/F | 1.4 [Link] , [Link] | 1.4 5 | 1.6 6 | 1.0 [Link] , [Link] |
| Clinical history | 3.9 months 1 | 3.0 months 5 | 1.6 months 6 | 15.2 months 1 |
| IDH1/2 mutation | 0% [Link] , [Link] , [Link] | 0% * | 5% * | 100% [Link] , [Link] , [Link] |
| PTEN mutation | 24% [Link] , [Link] , [Link] | 41% [Link] , [Link] | 33% 6 | 5% [Link] , [Link] , [Link] |
| ATRX expression loss | 0% 11 | 0% * | 19% * | 100% 11 |
| TERT mutation | 72% [Link] , [Link] , [Link] | 83% * | 25% * | 26% [Link] , [Link] , [Link] |
| TP53 mutation | 23% [Link] , [Link] , [Link] | 25% [Link] , [Link] | 84% [Link] , [Link] , [Link] | 74% [Link] , [Link] , [Link] |
| LOH 19q | 4% 1 | 18% * | 42% * | 32% 1 |
| EGFR amplification | 42% [Link] , [Link] , [Link] | 5% 5,10 | 6% [Link] , [Link] , [Link] | 4% [Link] , [Link] , [Link] |
| p16INK4a deletion | 45% [Link] , [Link] , [Link] | 37% 5 | 3% [Link] , [Link] | 30% [Link] , [Link] , [Link] |
| LOH 1p | 15% 1 | 6% * | 17% * | 24% 1 |
| LOH 1p/19q | 2% 1 | 6% * | 17% * | 11% 1 |
| LOH 10q | 67% 1 | 88% * | 50% * | 73% 1 |
PTEN mutations in gliosarcomas (41%) were significantly more frequent than those in primary glioblastomas (24%; P = 0.0190) and in secondary glioblastomas (5%; P = 0.0001). Loss of ATRX expression was less frequent in gliosarcomas than secondary glioblastomas (P = 0.0009). TERT promoter mutations in gliosarcomas were more frequent (83%) than those in secondary glioblastomas (26%; P < 0.0001). LOH 19q was more frequent in gliosarcomas (18%) than those in primary glioblastomas (4%; P = 0.0406). LOH 10q was more frequent in gliosarcomas (88%) than those in primary glioblastomas (67%; P = 0.0452).
PTEN mutations in giant cell glioblastomas (33%) were significantly more frequent than those in secondary glioblastomas (5%; P = 0.0126). Loss of ATRX expression was less frequent in giant cell glioblastomas than in secondary glioblastomas (P = 0.0206). TP53 mutations were more frequent in giant cell glioblastomas (84%) than in primary glioblastomas (23%; P < 0.0001). LOH 19q was more frequent in giant cell glioblastomas (42%) than in primary glioblastomas (4%; P = 0.0002; Table 1).
Discussion
IDH1 mutations are the definitive molecular marker of secondary glioblastoma24, 28. Gliosarcomas and giant cell glioblastomas usually develop rapidly without less malignant precursor lesions31, and have thus been considered to be primary glioblastomas. There have been few studies on IDH mutations. Lee et al 13 screened 26 gliosarcomas (21 primary gliosarcomas, 2 progressed from grade II or grade III astrocytomas, 2 developed from glioblastomas; one developed 9 years after radiotherapy for meningioma) for IDH1 mutations. IDH1 mutations were detected in one primary gliosarcoma and in a secondary gliosarcoma that progressed from a grade III astrocytoma13. Balss et al 3 reported that 2 of 8 giant cell glioblastomas but none of 5 gliosarcomas carried IDH1 mutations. Lotsch et al 15 reported the absence of IDH1 mutations in one gliosarcoma and one giant cell glioblastoma.
This study provides evidence that gliosarcoma is indeed a histological variant of primary glioblastoma IDH wild‐type. IDH1 and IDH2 mutations were absent in all 36 gliosarcomas analyzed. TERT promoter mutations, which are frequent in primary glioblastomas2, 25, were detected in 83% of gliosarcomas (Table 1). Furthermore, LOH 10q was observed in 15/17 (88%) gliosarcomas. Monosomy of chromosome 10 was also observed in 2/5 gliosarcomas in the analysis calculated from 450k methylome array. Immunohistochemistry showed that ATRX is expressed in all gliosarcomas, suggesting the absence of ATRX mutations14, 36. Thus, gliosarcomas share clinical and genetic features with primary glioblastomas IDH wild‐type. The only major difference was EGFR amplification, which is common in primary glioblastomas and was considered to be a rare event in gliosarcomas1, 31 (Table 1).
Despite two distinct histological components, gliosarcomas are considered monoclonal, since glial and mesenchymal tumor areas were usually found to be genetically similar with respect to TP53 mutations, PTEN mutations, p16INK4a deletion, EGFR amplification, and 10q loss4, 31. The present study shows that this is also the case for TERT promoter mutations, as they were present in both glial and mesenchymal tumor areas in 95% (19/20) of gliosarcomas.
The molecular mechanisms involved in the mesenchymal differentiation in gliosarcomas are not yet fully understood. Expression of several transcription factors (eg, Slug, Twist) associated with epithelial‐mesenchymal transition (EMT), was found in mesenchymal tumor areas, suggesting that signalling pathways involved in EMT may play a role in the induction of mesenchymal differentiation21. In studies using conventional CGH, patterns of chromosomal imbalance were largely similar in glial and mesenchymal tumor areas, but there were also gains and losses at several loci unique to either glial or mesenchymal tumor areas1, 6. Using array CGH, we have shown that in a small fraction of gliosarcomas the gain at 13q13.3‐q14.1 and expression of several genes (STOML3, FREM2, LHFP) at this locus was restricted to the mesenchymal tumor area of gliosarcomas20.
Giant cell glioblastomas develop rapidly with a short clinical history, but they occur in young adults (mean, 44 years), similar to secondary glioblastomas IDH mut18, 30. We here provide genetic evidence that giant cell glioblastoma is a histological variant of primary glioblastoma IDH wild‐type as IDH1/2 mutations were absent in 18 of 19 giant cell glioblastomas analyzed. However, frequencies of TERT promoter mutations, TP53 mutations, LOH 19q, and EGFR amplification were similar to those of secondary IDH1 mut glioblastomas17, 18, 30 (Table 1). Thus, giant cell glioblastoma occupies a hybrid position, sharing with primary glioblastoma a short clinical history, the absence of less malignant lesions, absence of IDH1/2 mutations, frequent PTEN mutations and infrequent ATRX loss. In common with secondary glioblastomas, they have a younger patient age at manifestation, infrequent TERT promoter mutations, frequent TP53 mutations, frequent LOH 19q, and lack of EGFR amplification.
There was the exceptional case of a giant cell glioblastoma IDH1 mut (R132S) with clinical evidence suggesting progression from an anaplastic astrocytoma diagnosed a year earlier. This suggests that rarely, a secondary glioblastoma may show the histologic features of a giant cell glioblastoma.
The molecular mechanisms involved in the giant cell phenotype are not yet fully understood. Temme et al 34 reported that Aurora B expression is significantly higher in giant cell glioblastomas than in other glioblastomas, and that ectopic overexpression of Aurora B induced a significant increase in the proportion of multinucleated giant cells in TP53 mutant but not in TP53 wild‐type malignant glioma cells.
LOH 10q is a frequent genetic alteration in both primary and secondary glioblastomas, suggesting that this chromosomal region may contain tumor suppressor gene(s), essential for malignant glioblastoma phenotype27, 28. LOH 10q is also associated with shorter survival of glioblastoma patients in both population based24, 26 and hospital‐based studies7, 33. We here provide evidence that LOH 10q is the genetic hallmark of gliosarcomas (88%) and giant cell glioblastomas (50%), further suggesting that gene(s) at 10q play an important role in the pathogenesis of glioblastomas and their variants.
In summary, gliosarcomas and giant cell glioblastomas are both histological variants of primary glioblastoma IDH1 wild‐type. The genetic pattern of gliosarcoma is largely similar to that of primary glioblastoma except for a lack of EGFR amplification, while giant cell glioblastoma genetically occupies a hybrid position between primary and secondary glioblastomas.
References
- 1. Actor B, Cobbers JM, Buschges R, Wolter M, Knobbe CB, Reifenberger G, Weber RG (2002) Comprehensive analysis of genomic alterations in gliosarcoma and its two tissue components. Genes Chromosomes Cancer 34:416–427. [DOI] [PubMed] [Google Scholar]
- 2. Arita H, Narita Y, Fukushima S, Tateishi K, Matsushita Y, Yoshida A, Miyakita Y et al (2013) Upregulating mutations in the TERT promoter commonly occur in adult malignant gliomas and are strongly associated with total 1p19q loss. Acta Neuropathol 126:267–276. [DOI] [PubMed] [Google Scholar]
- 3. Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 116:597–602. [DOI] [PubMed] [Google Scholar]
- 4. Biernat W, Aguzzi A, Sure U, Grant JW, Kleihues P, Hegi ME (1995) Identical mutations of the p53 tumor suppressor gene in the gliomatous and the sarcomatous components of gliosarcomas suggest a common origin from glial cells. J Neuropathol Exp Neurol 54:651–656. [DOI] [PubMed] [Google Scholar]
- 5. Bleeker FE, Atai NA, Lamba S, Jonker A, Rijkeboer D, Bosch KS, Tigchelaar W et al (2010) The prognostic IDH1 (R132) mutation is associated with reduced NADP+‐dependent IDH activity in glioblastoma. Acta Neuropathol 119:487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boerman RH, Anderl K, Herath J, Borell T, Johnson N, Schaeffer‐Klein J, Kirchhof A et al (1996) The glial and mesenchymal elements of gliosarcomas share similar genetic alterations. J Neuropathol Exp Neurol 55:973–981. [DOI] [PubMed] [Google Scholar]
- 7. Brat DJ, Seiferheld WF, Perry A, Hammond EH, Murray KJ, Schulsinger AR, Mehta MP et al (2004) Analysis of 1p, 19q, 9p, and 10q as prognostic markers for high‐grade astrocytomas using fluorescence in situ hybridization on tissue microarrays from Radiation Therapy Oncology Group trials. Neuro Oncol 6:96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S et al (2013) The somatic genomic landscape of glioblastoma. Cell 155:462–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2:401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eckel‐Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, Pekmezci M et al (2015) Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med 372:2499–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6:l1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ichimura K, Pearson DM, Kocialkowski S, Backlund LM, Chan R, Jones DT, Collins VP (2009) IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol 11:341–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee D, Kang SY, Suh YL, Jeong JY, Lee JI, Nam DH (2012) Clinicopathologic and genomic features of gliosarcomas. J Neurooncol 107:643–650 [DOI] [PubMed] [Google Scholar]
- 14. Liu XY, Gerges N, Korshunov A, Sabha N, Khuong‐Quang DA, Fontebasso AM, Fleming A et al (2012) Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol 124:615–625. [DOI] [PubMed] [Google Scholar]
- 15. Lotsch D, Ghanim B, Laaber M, Wurm G, Weis S, Lenz S, Webersinke G et al (2013) Prognostic significance of telomerase‐associated parameters in glioblastoma: effect of patient age. Neuro Oncol 15:423–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, (eds) (2007) WHO Classification of Tumours of the Central Nervous System. IARC: Lyon: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martinez R, Roggendorf W, Baretton G, Klein R, Toedt G, Lichter P, Schackert G et al (2007) Cytogenetic and molecular genetic analyses of giant cell glioblastoma multiforme reveal distinct profiles in giant cell and non‐giant cell subpopulations. Cancer Genet Cytogenet 175:26–34 [DOI] [PubMed] [Google Scholar]
- 18. Meyer‐Puttlitz B, Hayashi Y, Waha A, Rollbrocker B, Bostrom J, Wiestler OD, Louis DN et al (1997) Molecular genetic analysis of giant cell glioblastomas. Am J Pathol 151:853–857 [PMC free article] [PubMed] [Google Scholar]
- 19. Muller A, Holzmann K, Kestler HA (2007) Visualization of genomic aberrations using Affymetrix SNP arrays. Bioinformatics 23:496–497 [DOI] [PubMed] [Google Scholar]
- 20. Nagaishi M, Kim YH, Mittelbronn M, Giangaspero F, Paulus W, Brokinkel B, Vital A et al (2012) Amplification of the STOML3, FREM2, and LHFP genes is associated with mesenchymal differentiation in gliosarcoma. Am J Pathol 180:1816–1823 [DOI] [PubMed] [Google Scholar]
- 21. Nagaishi M, Paulus W, Brokinkel B, Vital A, Tanaka Y, Nakazato Y, Giangaspero F et al (2012) Transcriptional factors for epithelial‐mesenchymal transition are associated with mesenchymal differentiation in gliosarcoma. Brain Pathol 22:670–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nigro JM, Takahashi MA, Ginzinger DG, Law M, Passe S, Jenkins RB, Aldape K (2001) Detection of 1p and 19q loss in oligodendroglioma by quantitative microsatellite analysis, a real‐time quantitative polymerase chain reaction assay. Am J Pathol 158:1253–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nobusawa S, Lachuer J, Wierinckx A, Kim YH, Huang J, Legras C, Kleihues P et al (2010) Intratumoral patterns of genomic imbalance in glioblastomas. Brain Pathol 20:936–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nobusawa S, Watanabe T, Kleihues P, Ohgaki H (2009) IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin Cancer Res 15:6002–6007 [DOI] [PubMed] [Google Scholar]
- 25. Nonoguchi N, Ohta T, Oh JE, Kim YH, Kleihues P, Ohgaki H (2013) TERT promoter mutations in primary and secondary glioblastomas. Acta Neuropathol 126:931–937 [DOI] [PubMed] [Google Scholar]
- 26. Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL, Burkhard C et al (2004) Genetic pathways to glioblastoma: a population‐based study. Cancer Res 64:6892–6899 [DOI] [PubMed] [Google Scholar]
- 27. Ohgaki H, Kleihues P (2007) Genetic pathways to primary and secondary glioblastoma. Am J Pathol 170:1445–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ohgaki H, Kleihues P (2013) The definition of primary and secondary glioblastoma. Clin Cancer Res 19:764–772 [DOI] [PubMed] [Google Scholar]
- 29. Ohta T, Kim YH, Oh JE, Satomi K, Nonoguchi N, Keyvani K, Pierscianek D et al (2014) Alterations of the RRAS and ERCC1 genes at 19q13 in gemistocytic astrocytomas. J Neuropathol Exp Neurol 73:908–915 [DOI] [PubMed] [Google Scholar]
- 30. Peraud A, Watanabe K, Schwechheimer K, Yonekawa Y, Kleihues P, Ohgaki H (1999) Genetic profile of the giant cell glioblastoma. Lab Invest 79:123–129 [PubMed] [Google Scholar]
- 31. Reis RM, Konu‐Lebleblicioglu D, Lopes JM, Kleihues P, Ohgaki H (2000) Genetic profile of gliosarcomas. Am J Pathol 156:425–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reuss DE, Mamatjan Y, Schrimpf D, Capper D, Hovestadt V, Kratz A, Sahm F et al (2015) IDH mutant diffuse and anaplastic astrocytomas have similar age at presentation and little difference in survival: a grading problem for WHO. Acta Neuropathol 129:867–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schmidt MC, Antweiler S, Urban N, Mueller W, Kuklik A, Meyer‐Puttlitz B, Wiestler OD et al (2002) Impact of genotype and morphology on the prognosis of glioblastoma. J Neuropathol Exp Neurol 61:321–328 [DOI] [PubMed] [Google Scholar]
- 34. Temme A, Geiger KD, Wiedemuth R, Conseur K, Pietsch T, Felsberg J, Reifenberger G et al (2010) Giant cell glioblastoma is associated with altered aurora b expression and concomitant p53 mutation. J Neuropathol Exp Neurol 69:632–642 [DOI] [PubMed] [Google Scholar]
- 35. Watanabe T, Nobusawa S, Kleihues P, Ohgaki H (2009) IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 174:1149–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wiestler B, Capper D, Holland‐Letz T, Korshunov A, von DA, Pfister SM, Platten M et al (2013) ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol 126:443–451 [DOI] [PubMed] [Google Scholar]
- 37. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]