

Abstract
Hippocampal sclerosis (HS) refers to loss of hippocampal neurons and astrogliosis. In temporal lobe epilepsy (TLE), HS is a key factor for pharmacoresistance, even though the mechanisms are not quite understood. While experimental TLE models are available, there is lack of models reflecting the natural HS development. Among domestic animals, cats may present with TLE‐like seizures in natural and experimental settings. With this study on the prevalence, segmental pattern and clinicopathological correlates of feline HS, we evaluated the translational value for human research. Evaluation schemes for human brains were applied to epileptic cats. The loss of neurons was morphometrically assessed and the degree of gliosis was recorded. Hippocampal changes resembling human HS were seen in about one third of epileptic cats. Most of these were associated with infiltrative diseases such as limbic encephalitis. Irrespective of the etiology and semiology of seizures, total hippocampal sclerosis was the most prevalent form seen in epileptic animals. Other HS types also occur at varying frequencies. Segmental differences to human HS can be explained by species‐specific synaptic connectivities and a different spectrum of etiologies. All these variables require consideration when translating results from feline studies regarding seizure‐associated changes of the temporal lobe and especially HS.
Keywords: feline, hippocampus temporal lobe epilepsy, mesial temporal lobe sclerosis, MTLS, seizure, TLE
Introduction
Hippocampal sclerosis (HS) is a major concern in progressive and therapy‐resistant epileptic disorders in humans 42. The term refers to the combination of neuronal loss in the pyramidal cell band of the hippocampus and reactive astrogliosis 11. The earliest description of HS dates back as far as 1825, which also was the first notification of hippocampal pathology in epileptic patients in general 13. The regional vulnerability to excitotoxic damage has been documented manifold ever since. However, the hippocampal contribution to epileptogenesis and disease progression still is far from being understood. Instead, the hippocampus has been considered being a target of convergent tissue changes in the course of seizures, as much as it may serve as primary epileptogenic zone 18.
Post‐mortem investigations in human patients affected by various epileptic syndromes identified the anterior part of the human hippocampus as the region that shows the most extensive degenerative changes in terms of HS when being examined along its longitudinal axis 45. This hippocampal head belongs to the temporal lobe and may explain the specific association of therapy‐resistance in temporal lobe epilepsy (TLE) and HS, which accounts for 10% of all human epilepsy cases 41. Compliant with the concept of HS providing an anatomical base of epileptogenesis, TLE patients represent nearly 70% of all epilepsy patients subjected to tailored brain resection 41 and 80% of TLE patients benefit from seizure freedom for at least 2 years post‐surgery 9.
With regard to the understanding of the pathobiology of TLE and elaboration of tissue‐sparing strategies, animal models of human TLE are indispensable. Even though highly standardized kindling 16 and chemoconvulsant 14 models are available, a natural disease model, reflecting the spontaneous development of HS, would be a valuable addendum for translational studies. Recent papers already highlight the feasibility of canine epilepsy as large animal model for human research 32. The authors state a “striking similarity in aetiology, clinical manifestation and disease course in between epileptic humans and dogs” 32. With regard to TLE, the dog model, however, does not appear to reflect the human situation sufficiently 15, 17.
Cats with limbic seizures with or without orofacial involvement may help to overcome this gap, as they appear to have epilepsy of temporal lobe origin 24, 29, 31, 49. Hence, research may benefit from comparative studies in this domestic animal that is broadly available through neurological veterinary practices. Moreover, epileptic cats of various seizure types also may exhibit resistance to antiepileptic drugs (AED), which may offer another relevant focus of research. As to whether this may be caused by anatomical changes or nonspecific multidrug resistance requires further investigations.
As a first step toward the evaluation of the feasibility of a feline epilepsy model for comparative research in temporal lobe epilepsy, in general, and HS, in particular, we assessed the hippocampal and extrahippocampal pathology in a cohort of cats suffering from various seizure types and etiologies. We addressed in particular the prevalence and segmental pattern of HS with regard to the subtypes reported in humans 9, 11. Thereby, we obtained the relative risk (RR) of HS development sorted for signalment, semiology and nosology.
Materials and Methods
Case selection
This study enrolled cats with epilepsies of different origins. The definitions of epilepsy and seizures were used in accordance with those proposed to and by the International League Against Epilepsy (ILAE) 5, 6, 22. Medical records and pathology reports from cats submitted to the neuropathology laboratory, between 1997 and 2011, were retrieved from the archives.
Cats with a documented history of recurrent seizures and/or status epilepticus (SE) were included if they underwent post‐mortem examination and if they had a full clinical workup consisting of a detailed description of out of clinic fits by the owners, documentation of seizure events witnessed by the neurology team, complete neurological examination, treatment and epicrisis, results from blood analyses and, possibly, magnetic imaging studies and cerebrospinal fluid (CSF) analyses. The group of epileptic cats was compared with an age‐ and sex‐matched control group.
Medical record analysis
Medical records were studied with regard to the signalment and the clinical presentation. Major determinants were, in particular, the reliability of seizure diagnosis, the time course and semiology of seizures, possible treatment schemes, the laboratory workup and the absence/presence of concurrent disorders.
Regarding seizure classification, focal seizures were defined as unilaterally involving parts of the body and occurring with or without loss of consciousness. Generalized seizures were defined as seizures involving the entire body accompanied by loss of consciousness. If generalized seizures developed out of primarily focal seizures, they were termed secondary generalized seizures. The term status epilepticus referred to generalized seizures sustained for longer than 5 minutes or cluster seizures without recovering consciousness between seizures.. The event was defined as primary SE, if it occurred without any prior seizures, while secondary SE was defined as being preceded by seizures of any type.
Tissue sampling and processing
All brains were fixed through immersion in 10% neutral‐buffered formalin for at least 3 days before external inspection and trimming as described earlier 19. For the standardized hippocampal examination, the brains were cut transversely at the level of the temporo‐ventral body of the hippocampus. This candidate area was chosen as it resembles the feline counterpart to the human mesial temporal lobe, namely, the center of the hippocampal head 33, which is the region most commonly affected by HS in humans 45, and because of its frequent involvement in feline seizures 29. Moreover, sampling can be easily reproduced by independent investigators and all segments of the cornu ammonis (CA1 through CA4 26) and dentate gyrus can be reliably identified in subsequent histological specimens.
The bihemispheric brain slides passed an ascending ethanol series and xylene treatment in an automatic tissue processor. Upon paraplast® (Leica Biosystems, Wetzlar, Germany) embedding, sections were taken at 5 μm slice thickness and stained with hematoxylin and eosin for histopathological examination. Further stains were elected case‐based, depending on the character of a particular lesion. These included Nissl's cresyl violet, luxol fast blue, periodic acid Schiff and immunohistochemical staining for feline infective agents (feline corona virus, feline leukemia virus, feline immunodeficiency virus, borna disease virus, Toxoplasma gondii) or specific cell markers (glial fibrillary acidic protein, S‐100, CD3, CD20, CD79a, lysozyme, vimentin).
Neuropathological examination
Standard algorithms were applied for general assessment of brain pathologies. Histological investigation was carried out using a Zeiss Axiophot® (Zeiss, Jena, Germany) equipped with a CCD camera (Leica) with optic magnification ranging from ×5 to ×1000.
Specific examination of the hippocampus was carried out in agreement with a modified classification system for mesial temporal sclerosis and granule cell pathology (GCP) in humans 9. Histological evaluation was accomplished by quantitative assessment of neuronal density within CA‐subfields 1 through 4 and the straight inner and external limbs of the dentate gyrus (DG) using digital image analysis of non‐overlapping photomicrographs via modified Delaunay's triangulation plug‐in for Fiji image® (open source platform; http://fiji.sc/Fiji) processing package (Version 1.47 i). Conclusions on neuronal density were drawn from the surrogate parameter interneurononuclear distance (INND). Distances were therefore calculated between the geometrical centers of neighboring nerve cell nuclei. In the DG, measurements were accomplished by obtainment of the mean diameter of the granule cell band from three areas of the limbs.
Identification of HS
In agreement with consensus statements 9, 10, we defined HS as a significant dropout of pyramidal cells from any segment of the hippocampal cell band (CA1 through CA4) in combination with reactive astroglial proliferation. Changes to the neuronal density were calculated from the segmental INND (see above) while GFAP immunohistochemistry was launched to facilitate detection of astroglial changes. An INND increase exceeding twice the standard deviation (2 × SD) of the INND of the non‐neurological control population was considered abnormal. Mean values between 2 × SD and fourfold standard deviation (4 × SD) were graded as mildly increased. A moderate INND increase was defined as values between 4 × SD and sixfold standard deviation (6 × SD). INND exceeding 6 × SD was considered as a severe deviation 9 (see Figure 1).
Figure 1.
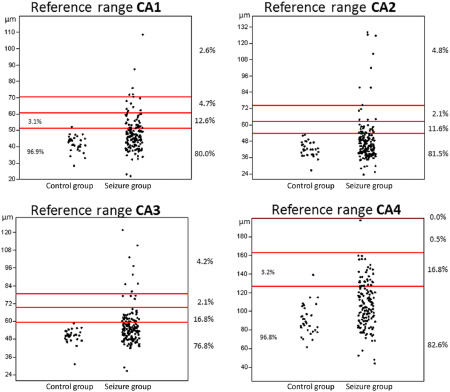
Segment‐specific INND values of the control group and the seizure group. The red lines indicate the cut‐off for mild (SD × 2), moderate (SD × 4) and severe (SD × 6) INND increase. Values lying within the defined boundaries are expressed as percentages for both groups, respectively.
Data analyses
All measurements per subfield were transferred into Excel® spreadsheets. Statistical analyses were conducted and illustrated using PAST® (http://palaeo‐electronica.org/2001_1/past/issue1_01.htm) and GraphPad Prism® (STATCON, Witzenhausen, Germany) statistic softwares. Group and subgroup characteristics were summarized as univariate analysis. Shapiro–Wilk algorithms gave way for parametric discriminative and correlation analyses. Thereby, single comparisons were carried out via Student's t‐test while multiple comparisons employed analysis of variance (ANOVA) with Tukey's post hoc modification. Correlation analyses were based on Spearman's rank sum test for nonparametric assays and Pearson's test for parametric studies. Categorical parameters were evaluated via chi square or Fisher's exact test that also assisted with the obtainment of the relative HS risk. Data analysis was accomplished by calculation of the confidence interval (CI95). P‐values ≤ 0.05 were accepted as indicating significance.
Results
Demographics, semiology and clinical course
One hundred eleven cats were included in the study. Ninety‐three of these had a history of seizures (Table 1), while the remaining 18 animals resembled non‐epileptic controls.
Table 1.
Included cases
| HS | Cohort | |||||
|---|---|---|---|---|---|---|
| (n) | (%) | CI95 | (n) | (%) | CI95 | |
| Animals (total) | 31 | 33.3 | 23.9–43.9 | 93 | 100 | |
| Mean age ± SD | 8.2 ± 4.9 | 8.0 ± 5.8 | ||||
| Sex distribution | ||||||
| Males | 15 | 48.4 | 30.2–66.9 | 38 | 40.9 | 30.8–51.5 |
| Females | 16 | 51.6 | 33.1–69.8 | 55 | 59.1 | 48.5–69.2 |
| Seizure type | ||||||
| Focal | 1 | 3.2 | 0.08–16.7 | 6 | 6.5 | 2.4–13.5 |
| Focal + generalized | 2 | 6.4 | 0.8–21.4 | 4 | 4.3 | 1.2–10.6 |
| Generalized | 13 | 41.9 | 24.5–60.9 | 51 | 54.8 | 44.2–65.2 |
| Secondary SE | 7 | 22.6 | 9.6–41.1 | 10 | 10.7 | 5.3–18.9 |
| Primary SE | 3 | 9.7 | 2.0–25.7 | 12 | 12.9 | 6.8–21.5 |
| Not defined | 5 | 16.2 | 5.5–33.7 | 10 | 10.8 | 5.3–18.9 |
| Seizure etiology | ||||||
| IC mass | 5 | 16.1 | 5.5–33.7 | 12 | 12.9 | 6.8–21.5 |
| Vascular | 1 | 3.2 | 0.08–16.7 | 8 | 8.6 | 3.8–16.2 |
| Metabolic | 3 | 9.7 | 2.0–25.7 | 8 | 8.6 | 3.8–16.3 |
| Inflammatory | 15 | 48.4 | 3 0.2–66.9 | 28 | 30.1 | 21.0–40.5 |
| Etiol. unknown | 3 | 9.7 | 2.0–25‐7 | 24 | 25.8 | 16.0–35.5 |
| Combined | 4 | 12.9 | 3.6–29.8 | 13 | 14.0 | 7.7–22.7 |
| Time course | ||||||
| <3 weeks | 22 | 71.0 | 52.0–85.8 | 72 | 77.4 | 65.6–85.4 |
| >3 weeks | 9 | 29.0 | 14.2–48.0 | 21 | 22.6 | 14.6–32.4 |
Case numbers, percentages and 95% confidence intervals of all epileptic cats are listed in the right column and of the HS‐positive cats in the left column. Please note, that for better distinction of the etiological groups, the animals with combined pathologies were listed as a separate entity and excluded from the other groups.
Corresponding to regional breed preferences, European shorthair cats were overrepresented in the epileptic group (83/93), followed by 4 Persian cats, 2 British shorthair and 1 Burmese, Birman, Siamese and Charteux each. In none of the cases, the owners/breeders indicated a litter or pedigree‐related neurological problem.
The mean age of epileptic cats was 8.0 ± 5.7 years. Concerning gender distribution, 59.1% (CI95 48.5–69.2) of the cats were females, 40.9% (CI95 30.8–51.5) were males.
Twenty‐two cats (22/93) had a history of status epilepticus (SE). In 10 animals, (10/22) the SE resulted from an aggravation of generalized seizures (secondary SE). The other 12 cats were presented to the practitioner or clinic because of SE without antecedent seizure events (primary SE). In eighteen (18/22) cases, SE resulted in early sudden death or euthanasia. Eight SE cats died during a seizure event. The other 10 animals were euthanized because of incomplete recovery (3/10), poor prognosis (3/10) or lack of owner's compliance (4/10). Only four animals with secondary SE survived more than 3 weeks after disease onset.
Within the non‐SE group, 51 animals (54.8%, CI95 44.2–65.2) had primarily generalized seizures. Ten cats (10.7%, CI95 5.3–18.9) were admitted because of focal seizures. Two out of these (2/10) were affected by simple focal seizures without loss of consciousness, four (4/10) had complex focal seizures with loss of consciousness and another four (4/10) presented with secondary generalized seizures. A typical TLE‐like seizure pattern with orofacial manifestation had been observed in seven of the animals presenting with focal seizures (7/10). In the remaining 10 animals (10/71), the clinical records did not reliably allow for classification of the seizure episodes, apart from an exclusion of SE.
With reference to the time course of seizure events, 17 of the non‐SE cases had their first seizure earlier than 3 weeks prior to the post‐mortem examination (range 3 months to 3 years). The other 54 cats had their first seizures less than 3 weeks prior to death.
Six of the 71 non‐SE cases died during a seizure event and 18 cats died unobserved by the owners. In none of these, other causes of death were identified on post‐mortem examination or by revision of clinical data. In the remaining 47 cases, the owners elected euthanasia.
Medication records revealed antiepileptic treatment in 27 cats. Twelve of those received benzodiazepines with (4/12) or without a loading dose of phenobarbital (8/12) just a few hours prior to death. Four animals died under emergency treatment. In the other eight, the procedure did not immediately abolish seizure activity. Hence, the owners declined further therapies. Nine animals (9/27) died (2/9) or were euthanized (7/9) for the same reason while receiving acute treatment with phenobarbital. In another 6/27 cats, treated with phenobarbital for periods between 3 weeks and 3 years, seizures were not sufficiently suppressed (3/6) or recurred (3/6), thereby, leading to the overall decision for euthanasia.
Seizure nosology
Systematic brain inspection did not reveal specific pathologies (other than HS) in 24 of 93 epileptic cats (25.8%, CI95 16.0–35.5; Table 1). Lack of evidence from clinical, imaging and laboratory records is prerequisite for clinically defined epilepsy of unknown cause (EUC) 3, 36, 50. In this study, the absence of unequivocally primary brain lesions in combination with lacking clinical and laboratory indications of seizure origin was referred to as epilepsy of unclear etiology. The remaining 69 cats (74.2%, CI95 62.9–81.8) presented with structural forebrain lesions—independent of HS—that were epileptogenic or that, respectively, reflected systemic disturbances with an epileptogenic potential.
Among these structural epilepsies, inflammatory conditions comprised the largest group (36/70). An infectious etiology was identified in 12 of these cases, resembling the CNS variant of feline infectious peritonitis (4/36), bacterial infections (4/36), migrating nematodes (2/36), T. gondii (1/36) and one undetermined protozoal encephalitis (1/36). In two other cases (2/36), the infiltration pattern and cytopathic features suggested a viral origin. In nine of the 36 brains with inflammatory lesions, the changes were consistent with the feline limbic encephalitis‐hippocampal necrosis complex (LEHN) 29, 30. In the remaining 13/36 cats, clinical and pathological data did not allow for identification of the underlying trigger or target of inflammation. Independent of the spatial characteristics, the inflammatory brain diseases were not statistically linked to a certain type or course of seizures.
Vascular and vasogenic abnormalities were seen in 20 epileptic cats (28.6%, CI95 13.5–30.9). Among these, eight cats showed hypertensive lesions, one of which also exhibited hyalin microthrombi (shock bodies) throughout the microvessels of the forebrain. Other vasogenic/vascular features comprised three cases with multiple ischemic forebrain infarcts (3/20), another three cases with parenchymal hemorrhage of undetermined etiology (3/20), and two cats each presenting with subtotal middle cerebral artery thrombosis (2/20), necrotizing vasculitis (2/20) and lymphocytic vasculitis (2/20).
The third largest group of epileptic animals (16/70) exhibited intracranial neoplasia (14/16), a choroid plexus cyst (1/16) or diffuse bihemispheric enlargement (1/16). Thereby, focal compression and/or shifting of the forebrain were seen in five cases with WHO grade I meningioma, one pituitary macroadenoma and the choroid plexus cyst.
Seven tumors (three secondary lymphomas, two primary CNS lymphomas, one undifferentiated round cell sarcoma, one meningeal sarcoma) caused disruption of the brain tissue because of parenchymal invasion. Another case presented with angioinvasive features caused by CNS manifestation of vasotropic lymphoma.
In 11 (11/70) cases, the brain tissue reflected metabolic‐toxic or degenerative conditions. One case was consistent with thiamine deficiency and two with uremic encephalopathy. In the remaining cats, it proved impossible to identify a specific etiology. One animal, however, presented with abundant Alzheimer type II cells without clinical or pathological evidence of liver disease. A group of five cats showed a uniform symmetric spongy encephalopathy indicative of a hitherto unreported neurodegenerative disorder.
Of the animals with structural epilepsy, 56 cats were affected by one single type of disease. In the remaining 13 cases, multiple modalities were encountered. Notably, in all but one of these cases (12/13), a vascular or vasogenic pathology was involved (Figure 2).
Figure 2.
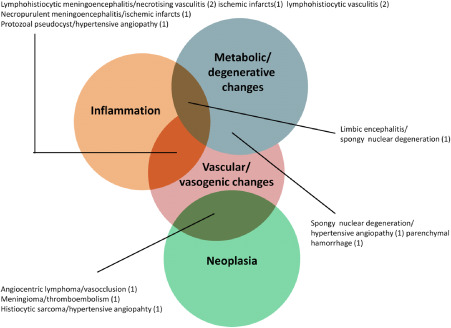
Combined pathologies in epileptic cats.
Pyramidal cell density, hippocampal sclerosis and mode of nerve cell decay
Univariate interneurononuclear distance values from the individual CA segments in control and epileptic cats are depicted in Table 2.
Table 2.
INND obtained from the different CA segments
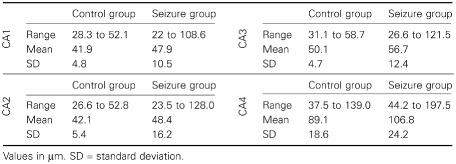
In control cats, the mean INND was 41.9 μm in CA1, 42.1 μm in CA2, 50.1 μm in CA3 and 89.1 μm CA4. The INND of CA1, CA2 and CA3 correlated positively to each other (P ≤ 6.6 10−6; r: 0.55 to 0.7), whereas the neuronal density of CA4 appeared to be independent to those of the other segments (P ≥ 0.3). In the control group, only two values exceeded 2 × SD: one in the CA1 segment and another one in the CA4 segment. Correlation analysis revealed a significant (P ≤ 0.03, r: 0.38 to 0.45) impact of the age on the INND throughout CA1 to CA3 (Figure 3). No such interdependence was seen in the endfolium and hilus (CA4; P = 0.24; r = 0.22).
Figure 3.
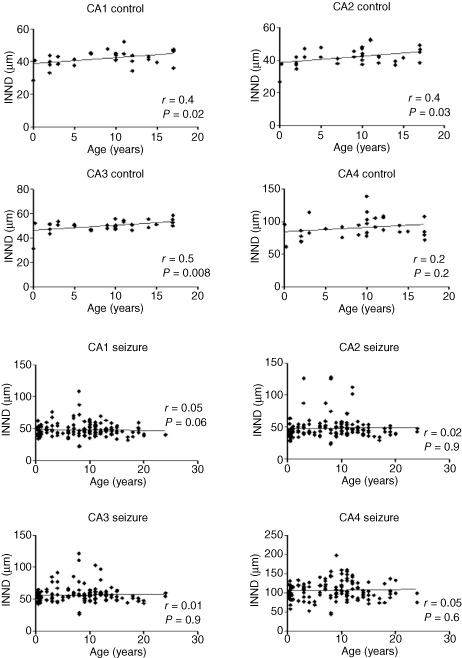
A positive correlation between age and INND is seen in control cats, while the interdependence is lost in hippocampi of epileptic cats.
In epileptic cats, the mean INND was 47.9 μm in CA1, 48.4 μm in CA2, 56.7 μm in CA3 and 106.8 μm CA4. An age dependency was not evident (P ≥ 0.87; r: −0.04 to 0.04; Figure 3). However, 52 epileptic cats exceeded the INND reference range of control animals and categorical testing obtained a significant reduction of cellular density compared with non‐epileptics. Significant neuronal loss was identified in 19.5% of the CA1 segments, 18.5% of CA2 segments, 23.2% of CA3 segments and 17.4% of CA4 segments (Table 3). Thereby, a significant correlation was seen between the same CA segments of both hemispheres (P ≥ 0.027; r: 0.3 to 0.8). This association was independent of the topography of a possible extrahippocampal lesion.
Table 3.
Animals meeting the reference range of INND and outliers
| CA 1 | Control | Seizure | ||
|---|---|---|---|---|
| (n) | (%) | (n) | (%) | |
| Ref | 31 | 96.9 | 153 | 80.5 |
| Mild | 1 | 3.1 | 23 | 12.1 |
| Moderate | 0 | 0.0 | 9 | 4.7 |
| Severe | 0 | 0.0 | 5 | 2.6 |
| Total | 32 | 100.0 | 190 | 100.0 |
| CA 2 | Control | Seizure | ||
|---|---|---|---|---|
| (n) | (%) | (n) | (%) | |
| Ref | 32 | 100.0 | 154 | 81.5 |
| Mild | 0 | 0.0 | 22 | 11.6 |
| Moderate | 0 | 0.0 | 4 | 2.1 |
| Severe | 0 | 0.0 | 9 | 4.8 |
| Total | 32 | 100.0 | 189 | 100.0 |
| CA 3 | Control | Seizure | ||
|---|---|---|---|---|
| (n) | (%) | (n) | (%) | |
| Ref | 32 | 100.0 | 146 | 76.8 |
| Mild | 0 | 0.0 | 32 | 16.8 |
| Moderate | 0 | 0.0 | 4 | 2.1 |
| Severe | 0 | 0.0 | 8 | 4.2 |
| Total | 32 | 100.0 | 190 | 100.0 |
| CA 4 | Control | Seizure | ||
|---|---|---|---|---|
| (n) | (%) | (n) | (%) | |
| Ref | 30 | 96.8 | 153 | 83.2 |
| Mild | 1 | 3.2 | 30 | 16.3 |
| Moderate | 0 | 0.0 | 1 | 0.5 |
| Severe | 0 | 0.0 | 0 | 0.0 |
| Total | 31 | 100.0 | 184 | 100.0 |
Ref = reference range.
In contrast to the age‐associated neuronal loss seen in the control group, pyramidal cell decay in epileptic animals significantly correlated to and segregated with astrogliosis (P ≤ 0.0001; r: 0.38) (Figure 3) leading to a positive HS diagnosis of an INND exceeding the SD×2 accompanied by any degree of astrogliosis in at least one segment of the pyramidal cell band in 31 of 52 cats with INND deviation. Bihemispheric investigation identified bilateral HS in 10/31 and unilateral lesions in 21/31 (equaling 41 HS hippocampi in total). The pattern was bilaterally symmetrical in 4/10 hippocampi. The segments with mild increase in INND were affected by concurrent fibrillary astrogliosis in 50.5% of cases. The segments with moderate INND increase were associated with gliosis in 61.1% and finally the ones with severe nerve cell loss showed significant gliosis in 90.9% of cases (Figure 4).
Figure 4.
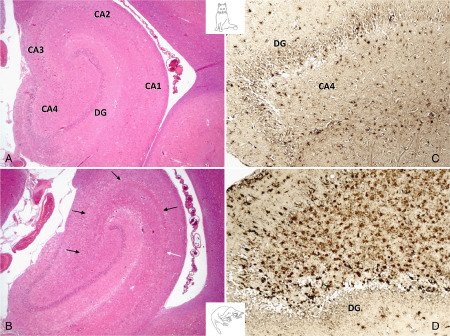
The left pictures show the physiological ( A ) and pathological ( B ) subgross appearance of the temporal hippocampus. B features the most common polysegmental HS with losses throughout all CA segments (black arrows) and a few residual pyramidal clusters (white arrows) in CA1. On the right, GFAP‐positive astrocytes are shown in the unaffected hilus (C) and this of a cat with severe HS (D).
Statistical testing revealed a significantly larger INND within the HS group compared with the general seizure cohort and control animals throughout all CA segments (P ≤ 2.2 × 10−5).
Actual nerve cell necroses, in far most cases, resembled excitotoxic phenotypes (pink neurons). In 2/9 LEHN cats and one case of viral encephalitis, lymphocytic attack and neuronophagia was widespread throughout all CA segments and the dentate gyrus, whereas two remaining LEHN cats showed isolated neuronophagic lesions in CA1, CA2 and CA3 (Figure 5). The latter and animals suffering from attenuated limbic encephalitis without neuronophagia showed occasional clusters and isolated neurons undergoing eosinophilic necroses. Highly active LEHN cases presented with massive neuronal necroses within and outside of infiltrated areas (Figure 5).
Figure 5.
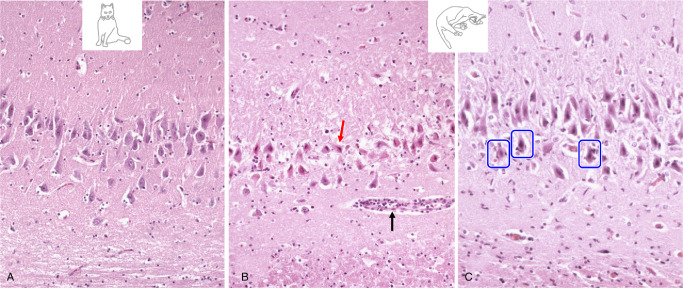
(A) Pyramidal cell degeneration in limbic encephalitis compared to a normal CA segment. (B) Neuronal loss shows morphologic features of excitotoxicity in terms of eosinophilic necrosis (red arrow). The black arrow indicates scanty perivascular inflammation. (C) Note the perineuronal infiltrates (blue frames).
Segmental characteristics of HS and INND in epileptic cats
Of the four CA segments, the CA3 region was most frequently involved comprising 26 out of 41 HS‐bearing hemispheres, followed by CA4 (22/41), CA1 and CA2 (20/41 and 18/41 respectively). Monosegmental HS was identified in 17 out of 41 hemispheres. Thereby, involvement of CA3 and CA4 was almost twice as frequent as that of CA1 and CA2 (5/17 and 6/17 vs. 3/17 for CA1 and CA2). The remaining 24 cases presented with polysegmental HS. All CA segments were affected in seven hemispheres. Among the remaining cases, the combinations CA3 + 4 and CA1 + 2 + 3 predominated with five hemispheres each. Other combinations (Figure 6) were seen in individual cases only. The only combinations missing were CA2 + 4 and CA1 + 3 + 4. A further insight into association of seizure type and nosology with INND in cats with and without HS is provided in Table 1.
Figure 6.
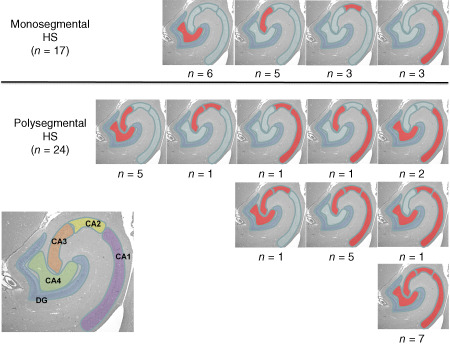
Segmental HS pattern seen in the hippocampi of epileptic cats from both sides. The picture in the left corner visualizes the borders of each CA segment.
Impact of seizure course, semiology and antiepileptic treatment on INND and HS
In the HS group, the known duration of seizure disease was longer than in the remaining epilepsy group. Thus, 29% (CI95 14.2–48) of animals had a disease course lasting more than 3 weeks, compared with only 21% (CI95 14.6–32.4) of all animals with seizures. Risk analysis obtained a 1.4 times higher RR of developing HS for animals with longer seizure disease, than for animals with shorter seizure histories.
Compared with both the persistent focal seizure cases and the primary generalized seizure group, focal‐onset seizures with secondary generalization showed a significantly higher INND throughout all segments (P ≤ 0.008). Further testing of generalized seizures against SE showed, that primary SE cases did not behave significantly different (P ≥ 0.4). Comparison between primary SE and secondary SE, rendered significant differences for CA1, in that INND (P = 0.05) was significantly higher in secondary SE. Animals presenting with secondary SE furthermore were at a 2.4 times higher risk of developing HS, than with any other seizure type (P = 0.014). Cats affected by focal seizures, in contrast, had a RR of only 0.5. The INND scatter‐plots sorted for the seizure types are depicted in Figure 7.
Figure 7.
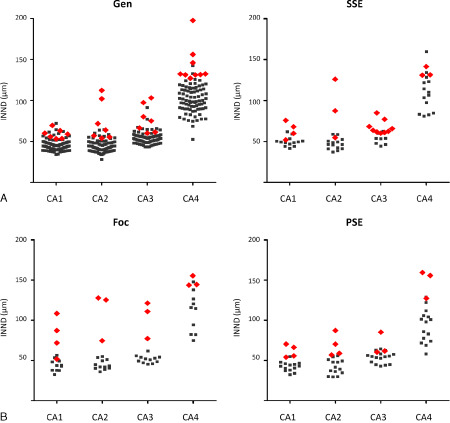
INND values sorted for the individual seizure type. The red rhombi represent the values that were accompanied by reactive astrogliosis and therefore are consistent with HS. Note that clinical seizure types did not predict reduction of neuronal cell density in certain CA segments in cats with or without HS.
Gen = generalized seizures; SSE = secondary status epilepticus; Foc = focal seizures; PSE = primary status epilepticus.
Prior to euthanasia, all cats with bilateral HS presented with generalized seizures or SE. The history of one single animal with bilateral HS revealed complex focal seizures at previous presentations. All other cats with bilateral HS were affected by primary or secondary SE prior to the final presentation.
Among the epilepsy group, cats submitted with a concise history of anticonvulsive treatment had a very high INND in CA2 (P = 0.005) and CA3 (P = 0.005). Thereby, no significant differences were seen between the emergency treatments and acute or chronic medication schedules (P ≥ 0.41).
Association of INND and HS with type of brain pathology
Concerning the individual disease categories (Figure 8), significant reduction of nerve cell densities were detected throughout all CA segments in the inflammatory disease group (P ≤ 7.2 × 10−5) and the group of combined pathologies (P ≤ 0.03). Intracranial mass lesion showed differences in all sectors but CA1 [P (CA1) = 0.08, P (other segments) ≤ 0.02].
Figure 8.
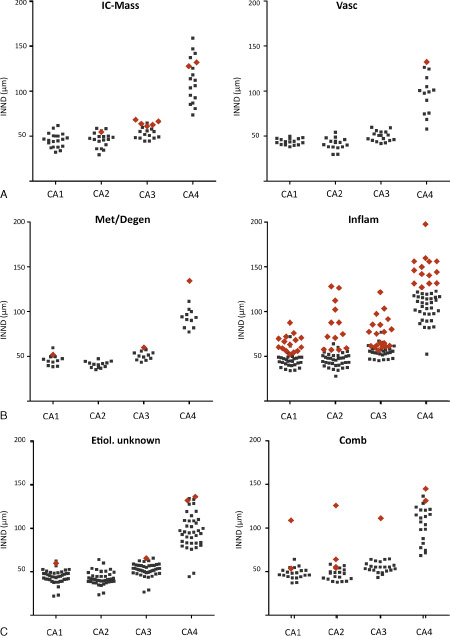
INND values sorted for the etiological category. There is no significant interdependence between seizure nosology and the segmental pattern of HS. Red rombi resemble HS‐affected hippocampi.
Comb = combined pathologies; IC mass = intracranial mass effects; Inflam = inflammatory; Met = metabolic/degenerative changes; Vasc = vascular/vasogenic changes.
Vascular encephalopathies and epilepsies of unclear etiology were not associated with a significant increase in INND if compared with non‐epileptic controls. The risk of HS was found to be highest for inflammatory diseases with a RR of 2.1 (P = 0.02). While the animals with epilepsy of unclear etiology, metabolic and vascular encephalopathies both had a RR below 0.3 (P = 0.02). The cats affected by intracranial masses only had a slightly elevated risk for HS compared with latter groups (RR 1.3; P = 0.7).
Segregation vs. dissociation, and laterality of HS and concurrent brain pathologies
In 79 hemispheres, structural lesions were extrahippocampal and noncontiguous to the hippocampus and parahippocampus. These extrahippocampal lesions were associated with ipsilateral HS in eight hemispheres and with contralateral HS in one case.
In 49 hemispheres, the hippocampus itself was affected by possibly epileptogenic structural lesions. These all were parenchymal neoplastic (12.5%) or inflammatory (87.5%) cell infiltrates. In 25 of these hippocampi (51.0%), the pathology was consistent with HS.
On direct comparison, the INND of affected hippocampi (72.1 μm ± 14.45) and those ipsilateral to a lesion (62.9 μm ± 17.7) were significantly higher than the INND of the control group throughout all segments (P ≤ 0.04). No significant difference (P ≥ 0.16) was detected between the corresponding ipsilateral and contralateral hippocampal segments in epileptic cats. Animals with hippocampal lesions showed a 6.9 times elevated RR with regard to HS (P < 0.0001).
Notably, all 10 animals affected by bilateral HS suffered from segregating infiltrative disorders, namely, one angiocentric B cell lymphoma and nine inflammatory pathologies.
Comparative temporal characteristics of seizure history and structural brain lesions
Special attention was directed at the match‐mismatch of seizure history and the age/stage of an epileptogenic lesion. In general, there was a high degree of congruence. As expected on the basis of the disease development and progression, neoplastic diseases had a rather short seizure history (mean: 8.6 days), whereas in metabolic and degenerative disorders, the onset of seizures dated further back (mean: 103.2 days). In LEHN, the time courses were explicitly variable, ranging from a few days only to several years. Notably, across all etiologies, the time course was neither predictive for the neuronal density nor for the occurrence of HS. Hence, a significantly increased INND was also seen in very acute courses with one fatal primary SE or even primary generalized seizures dating back a few hours. Similarly, the time span from seizure onset to death in HS cats ranged from 1 day to over 3 years.
Granule cell dispersion in HS
On quantitative assessment, granule cell band thickness was significantly increased in epileptic cats with HS as compared with those without HS and those spared from seizures (P ≤ 2.72 × 10–5). INND and DG morphology were consistent with GC dispersion in 14.6% of HS‐affected epileptics while only 0.6% epileptic animals of the non‐HS group showed this subtype of GCP type II. No such changes were seen in the single HS case of the non‐epileptic group. Notably, 7/14 cats with TLE presented with HS plus GC dispersion.
Discussion
Early chemoconvulsant kindling models in cats have brought some insight on epilepsy‐related hippocampal pathology in this species 24, 37, 38, 39, 49. Less information has been gathered from natural cases, which is surprising, concerning the prevalence of feline epilepsy in neurological practice, the unique occurrence of TLE among domestic animals and the anecdotal evidence of HS. Even though HS proved to contribute to progression and therapy resistance of epilepsy in people 11, the occurrence and possible impact of HS on the clinical course of feline seizures has not yet been explored.
With this first study dedicated to the prevalence and pattern of HS in cats, we found ILAE‐conforming HS subtypes in one‐third of a nonselected population of epileptic cats suffering from various types of seizures.
According to observations made in humans, three different onsets of HS are to be distinguished. Most commonly, HS develops as a consequence of ongoing seizure activity with or without initiating precipitating injury (known as secondary HS) 7, 47, 51. Alternatively, primary or idiopathic HS appears to coincide with the onset of seizures, which is particularly evident in human TLE 7, 47, 51. Another HS variant can be seen in non‐epileptic human patients admitted caused by cognitive dysfunction, often associated with Alzheimer's disease 52.
Compared with non‐epileptogenic HS that rarely goes with dentate gyrus pathology 46, about 50% of human TLE patients present with granule cell dispersion 8. Corresponding dentate gyrus changes were evident in 14.6% of the HS‐affected feline epileptics of this study, containing TLE and non‐TLE type of seizures. Concerning TLE cats only, granule cell dispersion also affected 50% of the animals while a total of 64.3% of TLE cats presented with HS.
Non‐epileptogenic HS cannot be excluded on scientific grounds in the remaining cats. On the other hand, feline HS was restricted to epileptic animals with exception of one single control cat being unilaterally affected by mild monosegmental HS. The remaining HS cases all were seen in epilepsy cases. Even though age is a major determinant of neuronal cell density in the feline hippocampus, this association was lost in epileptic cats and therefore did not influence the grade of confirmed HS. Other associative factors are hitherto unknown in cats and require specific exploration. A distinction of primary vs. secondary HS could essentially benefit from surgical samples and prospective studies, including serial magnetic resonance imaging (MRI).
Most human studies on the pattern and epileptogenic role of HS have been conducted on surgical samples obtained from patients suffering from long‐standing, therapy‐resistant TLE 9, 43 while only a few studies document HS in non‐epileptic patients and in epilepsies of extra‐temporal and extra(para)hippocampal origin 45. This preselection renders particular HS types and “HS‐only” pathologies more prevalent, than in the nonselected cat population. With an increased awareness of feline HS, future studies may improve the comparative value for human research prospectively by entity‐based selection criteria involving multiple referral centers.
This feline post‐mortem study, on the other hand, allowed for the evaluation of a plethora of seizure nosologies and the association with hippocampal vs. extrahippocampal structural pathologies. Downside of the diagnostic yield and resolution, the data obtained here require careful consideration of a selection bias. Most demographic and epidemiologic data obtained match the findings of previous studies 3, 31, 36 such as the 25.8% EUC cases 3, 36, 50. Yet, it has to be taken into account that in a post‐mortem setting, benign courses are underrepresented. In the same vein, animals affected by focal seizures resembled 10.7% of the investigated collective only. In feline neurological practice, they range up to about 52% of clinical presentations 36. With an expected loss of further mildly affected animals of an outhouse population, because of lack of owners' compliance regarding autopsy and nondetection of focal seizures, the caseload of this study is dominated by severe clinical presentations.
With 23.7% of feline HS cases being associated with epileptogenic extrahippocampal forebrain lesions, the causal relationship between HS and the concurrent pathology was further investigated. Brains with extrahippocampal lesions were sorted for etiological categories and compared within and across these groups. Not surprisingly, the vast majority of feline HS (76.3%) was found with pathologies disrupting the hippocampal architecture. Among these, inflammatory diseases were most prevalent (48.4%) and caused the most severe loss of pyramidal cells across all CA segments, either via local/remote excitotoxic events or via direct autoimmune attack. Lymphocytic invasion and neuronophagia indeed were most prominent in the feline anologon of human limbic encephalitis 21, 29, 35. Limbic encephalitis has been reported to account for up to 50% of HS‐positive human TLE cases 7. Among our post‐mortem cohort, we only identified nine LEHN cats, which may be caused by benign character of this disorder 29 and caused by the difficulty to identify non‐infiltrative stages histologically.
Now, that serological tests have been established for the voltage‐gated potassium channel (VGKC)‐subtype in cats, intravital diagnosis will facilitate comparative research on limbic encephalitis 30 while other paraneoplastic epileptic syndromes surely will flag‐up soon 1.
Apart from LEHN, HS was also seen in bacterial, parasitic and viral encephalitis involving the hippocampus. In loco production of proinflammatory cytokines such as interleukin‐1β 2 and interleukin‐6 as 20 well as shedding of bacterial lipopolysaccharides and parasitic toxins 34 cause damage to the blood–brain barrier 27 and may decrease the depolarization threshold in exposed neurons 48. Hence, disruptive and chemical changes of the microenvironment as well as specific cytotoxic actions all may contribute to the extensive histopathological sequelae triggering secondary HS. Only slightly less powerful, intractable seizures and HS were evoked by neoplasms with locally destructive growth.
It is noteworthy that all feline cases of bilateral HS were caused by either neoplastic or inflammatory infiltrates invading both hippocampi even though the lesions were symmetrical in only 40% of affected animals. This observation may be of predictive value for cats diagnosed with bilateral HS via MRI. All other entities lead to a unilateral HS, which also resembles the predominant manifestation of primary and secondary HS in humans 28, 47, 52.
HS contralateral to a structural brain lesion is extremely rare and in our cats just affected animals with hippocampal pathologies (on the non‐HS side). In particular, HS with ipsilateral lesions may be explained by changes to neuronal circuits, vascular supply or focal effects on the seizuring neurons. Unilateral HS with systemic disturbances such as metabolic disease still remains unclear. On the other hand, thorough revision of the slides revealed subsclerotic hippocampal changes in more than 50% of cases, which presented as either solitary INND increase or as pure astrogliosis.
Only three cats presented with unilateral “HS‐only pathology” compatible with idiopathic HS 7, 47, 51. As to whether HS resembles a perpetuating trigger of feline epilepsy remains to be elucidated via EEG mapping and lobectomy/lobotomy studies.
Concerning the clinical presentation, a clear ranking could be identified for the extent of neuronal loss: focal seizures were associated with least damage followed by primary SE, primary generalized seizures, secondary SE and, finally, focal seizures that progressed into generalized seizures had the worst impact on hippocampal cell density. Hence, seizures that progress into more severe clinical presentations also have advanced tissue changes. In human TLE, a similar scene set has been described. People with progression from focal to generalized seizures tend to have reduced hippocampal volumes on MRI when compared with those with focal seizures 40. In addition, there is a positive association between the occurrence of secondary generalizing tonic clonic seizures in TLE and the manifestation of HS 12.
In human TLE, the pattern of hippocampal affection is divided into four groups: (i) the most common classic HS, affecting mainly CA1 and CA4; (ii) the total or severe HS, affecting all segments to a similar degree; and (iii) the less CA1‐predominant; and (iv) endfolium sclerosis, affecting only the CA4 subfield 9. Apart from being anatomically different, the segmental pattern of HS appears to influence the degree of cognitive impairment 4 and postsurgical outcome as CA1‐predominant HS patients tend to have a less favorable prognosis and patients with endfolium sclerosis have a better postsurgical performance 44.
Even though all human HS types are represented in the epileptic cats of this study, the most common pattern is consistent with the severe or total HS phenotype (29.2%) that affects between 24% 44 and 53% 9 of human TLE patients only, depending on the respective study design.
Notably, polysegmental HS in cats was seen in 80% of hippocampal brain lesions and in a minority of extrahippocampal lesions. Most of extrahippocampal pathologies and incidental HS affected individual segments without systematic involvement of certain zones.
Concerning the individual segments, CA3 and CA4 were most commonly affected in feline patients, being involved in 63.4% and 53.6% of all HS cases. In contrast to humans 9, 10, 11, CA2 does not appear to be spared from nerve cell decay and gliosis as much as in humans. Moreover, the classic human HS ILAE type I is rarely seen in cats. Hence, CA1 does not represent the most vulnerable part of the feline pyramidal cell band.
Some dissimilarities to the appearance in humans may be explained by the unique situation that in cats, the entorhinal cortex layer III projects into CA2 and CA3, while in other mammalian species, they address the subiculum and CA1 25. A particular knowledge of the species‐specific neuronal circuits is mandatory for the correct interpretation and translational considerations.
Lacking longitudinal monitoring tools, in this retrospective study, we could not make concise conclusions about the development of HS over time. Seizure onset of our epileptic cats with HS, ranged back between 1 day and more than 3 years. It remains unclear, whether HS may have been present already in a subconvulsive state, or if seizures had gone unrecognized by the owners or were lasting longer than the clinical data suggested. Also, in humans, it has not been sufficiently elucidated when and how exactly HS is supposed to be kicked off. This is due to the fact that in TLE patients, surgery is performed only if seizure control with conservative therapy fails and the seizures have an extremely negative impact on life quality. Only a few studies report on acute HS that can occur as early as 3 months after onset of toxically induced seizures 23.
In order to obtain stringent conclusions about the development, progression, therapeutic options, prognosis, predictive and comparative value of feline HS, an intravital diagnosis of this condition will be necessary. The use of high‐resolution MRI for sequential examination of clinical cats could provide information about the epileptogenicity of HS in this species, the spatiotemporal development of the changes and the relationship between HS and treatment response in felids. The proof of principle regarding the role for epileptogenesis and seizure perpetuation by HS requires to be established via tailored hippocampectomy. More data will be required to strengthen specific aspects of this rather preliminary study. Even then, translation of animal data into human neurology requires careful consideration of feline particularities, some of which became apparent already during this pilot investigation.
References
- 1. Anderson NE, Barber PA (2008) Limbic encephalitis—a review. J Clin Neurosci 15:961–971. [DOI] [PubMed] [Google Scholar]
- 2. Balosso S, Maroso M, Sanchez‐Alavez M, Ravizza T, Frasca A, Bartfai T, Vezzani A (2008) A novel non‐transcriptional pathway mediates the proconvulsive effects of interleukin‐1beta. Brain 131(Pt 12):3256–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barnes HL, Chrisman CL, Mariani CL, Sims M, Alleman AR (2004) Clinical signs, underlying cause, and outcome in cats with seizures: 17 cases (1997–2002). J Am Vet Med Assoc 225:1723–1726. [DOI] [PubMed] [Google Scholar]
- 4. Baxendale SA, Van Paesschen W, Thompson PJ, Duncan JS, Harkness WF, Shorvon SD (1998) Hippocampal cell loss and gliosis: relationship to preoperative and postoperative memory function. Neuropsychiatry Neuropsychol Behav Neurol 11:12–21. [PubMed] [Google Scholar]
- 5. Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W et al (2010) Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 51:676–685. [DOI] [PubMed] [Google Scholar]
- 6. Berg AT, Scheffer IE (2011) New concepts in classification of the epilepsies: entering the 21st century. Epilepsia 52:1058–1062. [DOI] [PubMed] [Google Scholar]
- 7. Bien CG, Urbach H, Schramm J, Soeder BM, Becker AJ, Voltz R et al (2007) Limbic encephalitis as a precipitating event in adult‐onset temporal lobe epilepsy. Neurology 69:1236–1244. [DOI] [PubMed] [Google Scholar]
- 8. Blumcke I, Kistner I, Clusmann H, Schramm J, Becker AJ, Elger CE et al (2009) Towards a clinico‐pathological classification of granule cell dispersion in human mesial temporal lobe epilepsies. Acta Neuropathol 117:535–544. [DOI] [PubMed] [Google Scholar]
- 9. Blumcke I, Pauli E, Clusmann H, Schramm J, Becker A, Elger C et al (2007) A new clinico‐pathological classification system for mesial temporal sclerosis. Acta Neuropathol 113:235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Blumcke I, Thom M, Aronica E, Armstrong DD, Bartolomei F, Bernasconi A et al (2013) International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: a Task Force report from the ILAE Commission on Diagnostic Methods. Epilepsia 54:1315–1329. [DOI] [PubMed] [Google Scholar]
- 11. Blumcke I, Thom M, Wiestler OD (2002) Ammon's horn sclerosis: a maldevelopmental disorder associated with temporal lobe epilepsy. Brain Pathol 12:199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bone B, Fogarasi A, Schulz R, Gyimesi C, Kalmar Z, Kovacs N et al (2012) Secondarily generalized seizures in temporal lobe epilepsy. Epilepsia 53:817–824. [DOI] [PubMed] [Google Scholar]
- 13. Bouchet C, Cazauvieilh CA (1825) De l'épilepsie considerée dans ses rapports avec l'aliénation mentale. Recherche sur la nature et le siège de ces deux maladies. Arch Gen Med 9:510–542. [Google Scholar]
- 14. Buckmaster PS, Dudek FE (1997) Neuron loss, granule cell axon reorganization, and functional changes in the dentate gyrus of epileptic kainate‐treated rats. J Comp Neurol 385:385–404. [PubMed] [Google Scholar]
- 15. Buckmaster PS, Smith MO, Buckmaster CL, LeCouteur RA, Dudek FE (2002) Absence of temporal lobe epilepsy pathology in dogs with medically intractable epilepsy. J Vet Intern Med 16:95–99. [DOI] [PubMed] [Google Scholar]
- 16. Cardoso A, Lukoyanova EA, Madeira MD, Lukoyanov NV (2011) Seizure‐induced structural and functional changes in the rat hippocampal formation: comparison between brief seizures and status epilepticus. Behav Brain Res 225:538–546. [DOI] [PubMed] [Google Scholar]
- 17. Chandler K (2006) Canine epilepsy: what can we learn from human seizure disorders? Vet J 172:207–217. [DOI] [PubMed] [Google Scholar]
- 18. de Lanerolle NC, Lee TS (2005) New facets of the neuropathology and molecular profile of human temporal lobe epilepsy. Epilepsy Behav 7:190–203. [DOI] [PubMed] [Google Scholar]
- 19. De Risio L, Brown R, Tennant B, Sparkes A, Matiasek L, de Stefani A et al (2012) Slowly progressive lymphohistiocytic meningoencephalomyelitis in 21 adult cats presenting with peculiar neurological signs. J Feline Med Surg 14:250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. De Sarro G, Russo E, Ferreri G, Giuseppe B, Flocco MA, Di Paola ED, De Sarro A (2004) Seizure susceptibility to various convulsant stimuli of knockout interleukin‐6 mice. Pharmacol Biochem Behav 77:761–766. [DOI] [PubMed] [Google Scholar]
- 21. Fatzer R, Gandini G, Jaggy A, Doherr M, Vandevelde M (2000) Necrosis of hippocampus and piriform lobe in 38 domestic cats with seizures: a retrospective study on clinical and pathologic findings. J Vet Intern Med 14:100–104. [DOI] [PubMed] [Google Scholar]
- 22. Fisher RS, van Emde Boas W, Blume W, Elger C, Genton P, Lee P, Engel J Jr (2005) Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 46:470–472. [DOI] [PubMed] [Google Scholar]
- 23. Gardner H, Lawn N, Fatovich DM, Archer JS (2009) Acute hippocampal sclerosis following ecstasy ingestion. Neurology 73:567–569. [DOI] [PubMed] [Google Scholar]
- 24. Gastaut H, Naquet R, Meyer A, Cavanagh JB, Beck E (1959) Experimental psychomotor epilepsy in the cat; electro‐clinical and anatomo‐pathological correlations. J Neuropathol Exp Neurol 18:270–293. [DOI] [PubMed] [Google Scholar]
- 25. Hirama J, Shoumura K, Ichinohe N, You S, Yonekura H (1997) Cornu ammonis of the cat: lack of a separate field of CA2. J Hirnforsch 38:487–493. [PubMed] [Google Scholar]
- 26. Lorente de No R (1934) Studies on the structure of the cerebral cortex II. Contiuation of the study of the ammonic system. J Psychol Neurol 46:113–177. [Google Scholar]
- 27. Marchi N, Angelov L, Masaryk T, Fazio V, Granata T, Hernandez N et al (2007) Seizure‐promoting effect of blood‐brain barrier disruption. Epilepsia 48:732–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Margerison JH, Corsellis JA (1966) Epilepsy and the temporal lobes. A clinical, electroencephalographic and neuropathological study of the brain in epilepsy, with particular reference to the temporal lobes. Brain 89:499–530. [DOI] [PubMed] [Google Scholar]
- 29. Pakozdy A, Gruber A, Kneissl S, Leschnik M, Halasz P, Thalhammer JG (2011) Complex partial cluster seizures in cats with orofacial involvement. J Feline Med Surg 13:687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pakozdy A, Halasz P, Klang A, Bauer J, Leschnik M, Tichy A et al (2012) Suspected limbic encephalitis and seizure in cats associated with voltage‐gated potassium channel (VGKC) complex antibody. J Vet Intern Med 27:212–214. [DOI] [PubMed] [Google Scholar]
- 31. Pakozdy A, Leschnik M, Sarchahi AA, Tichy AG, Thalhammer JG (2010) Clinical comparison of primary versus secondary epilepsy in 125 cats. J Feline Med Surg 12:910–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Potschka H, Fischer A, von Ruden EL, Hulsmeyer V, Baumgartner W (2013) Canine epilepsy as a translational model? Epilepsia 54:571–579. [DOI] [PubMed] [Google Scholar]
- 33. Sasaki M, Tohyama K, Matsunaga S, Nakamura M, Tomizawa N, Inoue T et al (2004) MRI identification of dorsal hippocampus homologue in human brain. Neuroreport 15:2173–2176. [DOI] [PubMed] [Google Scholar]
- 34. Sayyah M, Javad‐Pour M, Ghazi‐Khansari M (2003) The bacterial endotoxin lipopolysaccharide enhances seizure susceptibility in mice: involvement of proinflammatory factors: nitric oxide and prostaglandins. Neuroscience 122:1073–1080. [DOI] [PubMed] [Google Scholar]
- 35. Schmied O, Scharf G, Hilbe M, Michal U, Tomsa K, Steffen F (2008) Magnetic resonance imaging of feline hippocampal necrosis. Vet Radiol Ultrasound 49:343–349. [DOI] [PubMed] [Google Scholar]
- 36. Schriefl S, Steinberg TA, Matiasek K, Ossig A, Fenske N, Fischer A (2008) Etiologic classification of seizures, signalment, clinical signs, and outcome in cats with seizure disorders: 91 cases (2000–2004). J Am Vet Med Assoc 233:1591–1597. [DOI] [PubMed] [Google Scholar]
- 37. Tanaka S, Tanaka T, Kondo S, Hori T, Fukuda H, Yonemasu Y et al (1993) Magnetic resonance imaging in kainic acid‐induced limbic seizure status in cats. Neurol Med Chir (Tokyo) 33:285–289. [DOI] [PubMed] [Google Scholar]
- 38. Tanaka T, Fujita T, Yamamoto K, Fukuda H, Yonemasu Y (1993) Experimental seizure‐induced brain damage: electrophysiological, metabolic and pathological correlation. Jpn J Psychiatry Neurol 47:239–244. [DOI] [PubMed] [Google Scholar]
- 39. Tanaka T, Kaijima M, Daita G, Ohgami S, Yonemasu Y, Riche D (1982) Electroclinical features of kainic acid‐induced status epilepticus in freely moving cats. Microinjection into the dorsal hippocampus. Electroencephalogr Clin Neurophysiol 54:288–300. [DOI] [PubMed] [Google Scholar]
- 40. Tasch E, Cendes F, Li LM, Dubeau F, Andermann F, Arnold DL (1999) Neuroimaging evidence of progressive neuronal loss and dysfunction in temporal lobe epilepsy. Ann Neurol 45:568–576. [DOI] [PubMed] [Google Scholar]
- 41. Tellez‐Zenteno JF, Hernandez‐Ronquillo L (2012) A review of the epidemiology of temporal lobe epilepsy. Epilepsy Res Treat 2012:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Thom M (2009) Hippocampal sclerosis: progress since Sommer. Brain Pathol 19:565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thom M, Eriksson S, Martinian L, Caboclo LO, McEvoy AW, Duncan JS, Sisodiya SM (2009) Temporal lobe sclerosis associated with hippocampal sclerosis in temporal lobe epilepsy: neuropathological features. J Neuropathol Exp Neurol 68:928–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Thom M, Liagkouras I, Elliot KJ, Martinian L, Harkness W, McEvoy A et al (2010) Reliability of patterns of hippocampal sclerosis as predictors of postsurgical outcome. Epilepsia 51:1801–1808. [DOI] [PubMed] [Google Scholar]
- 45. Thom M, Liagkouras I, Martinian L, Liu J, Catarino CB, Sisodiya SM (2012) Variability of sclerosis along the longitudinal hippocampal axis in epilepsy: a post mortem study. Epilepsy Res 102:45–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thom M, Martinian L, Catarino C, Yogarajah M, Koepp MJ, Caboclo L, Sisodiya SM (2009) Bilateral reorganization of the dentate gyrus in hippocampal sclerosis: a postmortem study. Neurology 73:1033–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thom M, Zhou J, Martinian L, Sisodiya S (2005) Quantitative post‐mortem study of the hippocampus in chronic epilepsy: seizures do not inevitably cause neuronal loss. Brain 128(Pt 6):1344–1357. [DOI] [PubMed] [Google Scholar]
- 48. Vezzani A, French J, Bartfai T, Baram TZ (2011) The role of inflammation in epilepsy. Nat Rev Neurol 7:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wada JA, Sata M (1974) Generalized convulsive seizures induced by daily electrical stimulation of the amygdala in cats. Correlative electrographic and behavioral features. Neurology 24:565–574. [DOI] [PubMed] [Google Scholar]
- 50. Wahle AM, Brühschwein A, Matiasek K, Wagner E, Putschbach K, Mueller RS, Fischer A (2013) Clinical characterization of epilepsy of unknown cause in cats. J Vet Intern Med 28:182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wieser HG (2004) ILAE Commission Report. Mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsia 45:695–714. [DOI] [PubMed] [Google Scholar]
- 52. Zarow C, Weiner MW, Ellis WG, Chui HC (2012) Prevalence, laterality, and comorbidity of hippocampal sclerosis in an autopsy sample. Brain Behav 2:435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]