

Abstract
Genome‐wide association studies have pointed to clusterin (apolipoprotein J) as being linked to the occurrence of Alzheimer's disease (AD); studies have identified the protein as a possible biomarker. The association between clusterin and senile plaques in AD brain is well known, and clusterin levels in AD brain are 40% higher than that in control subjects. The present study investigates, immunohistochemically, the association between clusterin and Aβ peptides in AD and control cortex. A unique and specific association between clusterin and Aβ40 was observed in plaques in the cerebral cortex from AD subjects in that only plaques that contained Aβ40 showed clusterin immunoreactivity, while the many plaques with Aβ42 alone lacked clusterin labeling. Cerebrovascular Aβ in AD brain generally lacked Aβ42 but was positively labeled by both the Aβ40 and the clusterin antibodies. In control subjects, however, Aβ40 was absent from plaques, although very occasional plaques were found to be labeled by both the Aβ42 and the clusterin antibodies. Overall, in AD, but not aged control brain, clusterin was associated specifically with the Aβ40 form of Aβ in the brain. The lack of clusterin in association with Aβ42 may be a significant feature in neuronal loss and neurodegeneration in the disease state.
Keywords: Alzheimer's disease, amyloid beta‐protein, apolipoprotein J, biomarker, clusterin
Introduction
A prerequisite for a therapeutic agent directed at halting disease progression in Alzheimer's disease (AD) would be the ability to detect the disease at an earlier enough stage to maintain an acceptable quality of life. While the Braak staging systems provide for post‐mortem identification of tau pathology in AD, diagnostic initiatives, such as CERAD (Consortium to Establish a Registry for Alzheimer's Disease), have indicated that pre‐mortem diagnosis is possible, although definite diagnosis remains neuropathological 2, 18. Coupled with this, efforts have been directed toward the discovery of biomarkers, specifically plasma markers, that could direct physicians toward early and accurate diagnosis of the disease and guide translational medicine studies of new compounds 14.
A link between the multifunctional glycoprotein clusterin (apolipoprotein J) and AD has been described in genome‐wide association studies 6, 13. Moreover, we have previously shown clusterin to be associated with rapid clinical progression in AD and have identified the protein as a likely candidate for a biomarker 24. Furthermore, we reported that an elevated concentration of plasma clusterin 10 years earlier was predictive of a greater level of fibrillar amyloid beta‐protein (Aβ) in the medial temporal lobe, when determined by positron emission tomography (PET). Clusterin has also been shown to be associated with Aβ plaques in AD brain and amyloid precursor protein (APP) transgenic mice 16, although the precise nature of the plaque–clusterin association is not clear. Furthermore, in preliminary studies, we found that clusterin was increased in both the plasma and the brain of an APP transgenic mouse line 24.
Despite these findings, the relationship between plasma and brain clusterin concentration and that of Aβ is far from clear. We now report that in AD brain, clusterin shows a specific association with the 40, but not the 42, amino acid form of the Aβ molecule, a finding that may have profound implications for disease pathogenesis.
Materials and Methods
Alzheimer's disease brain tissue
Tissue was supplied (7‐μm wax sections), with ethics approval, by the Thomas Willis Oxford Brain Collection and the London Neurodegenerative Diseases Brain Bank, both part of the Brains for Dementia Research network. Samples from prefrontal cortex (BA 9), primary visual cortex (BA 17), temporal lobe neocortex with the superior and middle temporal gyrus (BA 21, 22), and inferior parietal lobe neocortex (BA 39, 40) were studied. In total, 12 AD subjects (age 74–90 years), 8 aged controls (age 62–90 years) and 4 young controls (age 20–35 years) were studied. All brains were sampled, assessed and diagnosed according to standard neuropathological criteria by a consultant neuropathologist. For a summary, see Table 1.
Table 1.
Details of Alzheimer's disease (AD) and control subjects. Abbreviations: CERAD = Consortium to Establish a Registry for Alzheimer's Disease; PMD = post‐mortem delay.
| Case # | Age | Sex | PMD | Braak and Braak tau stage*; CERAD diagnostic group** |
|---|---|---|---|---|
| AD | ||||
| 1 | 88 | M | 13 | Braak VI; definite AD |
| 2 | 82 | F | 64 | Braak VI; definite AD |
| 3 | 90 | F | 70 | Braak V; definite AD |
| 4 | 82 | M | 20 | Braak VI; definite AD |
| 5 | 87 | F | 42 | Braak VI; definite AD |
| 6 | 79 | M | 45 | Braak VI; definite AD |
| 7 | 78 | M | 45 | Braak VI; definite AD |
| 8 | 81 | M | ? | Braak VI; definite AD |
| 9 | 74 | M | 66 | Braak VI; definite AD |
| 10 | 76 | M | 21 | Braak VI; definite AD |
| 11 | 87 | F | 42 | Braak VI; definite AD |
| 12 | 81 | F | 36 | Braak VI; definite AD |
| Aged control | ||||
| 13 | 84 | F | 9 | Braak II; no evidence of AD |
| 14 | 62 | M | 65 | None; no evidence of AD |
| 15 | ? | M | 42 | None; no evidence of AD |
| 16 | 62 | M | 30 | None; no evidence of AD |
| 17 | 70 | M | 72 | Braak I; no evidence of AD |
| 18 | 72 | M | 72 | Braak I; no evidence of AD |
| 19 | 70 | M | 88 | None; no evidence of AD |
| 20 | 90 | F | 50 | Braak II; possible mild AD |
| Young control | ||||
| 21 | 25 | M | 18 | None; no evidence of AD |
| 22 | 20 | M | 48 | None; no evidence of AD |
| 23 | 35 | M | 96 | None; no evidence of AD |
| 24 | 32 | M | ? | None; no evidence of AD |
Immunohistochemistry
Labeling of Aβ40 and Aβ42 was generally undertaken as previously described 8. Antigen retrieval for Aβ species was carried out by immersion in 98% formic acid for 15 minutes. In certain studies, as described in the Results section, formic acid treatment was omitted. Sections being labeled with clusterin antibodies received an additional antigen retrieval step in that following dewaxing, they were microwaved (from cold, 1 × 10 minutes at 900 W followed by 1 × 10 min at 300 W) in 10 mM citrate buffer, pH 6.0 [as described in 7], prior to formic acid treatment. For Aβ40/42–clusterin association studies, sections received citric acid antigen retrieval followed by 15 minutes immersion in 98% formic acid prior to overnight incubation at 4°C with pairs of antibodies (raised in different species). Sections being investigated for clusterin–phosphotau association only received citric acid antigen retrieval prior to primary antibody exposure (formic acid pretreatment had no effect on clusterin or phosphotau immunoreactivity—data not shown). Subsequent development was with appropriate Alexafluor‐coupled secondary antibodies (Alexa Fluor™ 488, goat anti‐mouse IgG and Alexa Fluor™ 568 goat anti‐rabbit IgG, both from Invitrogen, Life Technologies Ltd., Paisley, UK). Autofluorescence was quenched with Sudan black (0.1% in 70% ethanol). Fluorescent images are presented in pairs, showing green and red channel data. Quantification of plaques was undertaken with Fuji‐Image J, with outline images being utilized in order to demonstrate green and red channel labeling of the same plaque (see Supporting Information Figure S1). A minimum of 10 images were captured, at random, across each section. In some cases, additional sections of the same subjects were labeled and quantified in order to assess the reproducibility of the image analysis. In all cases, images were captured on a Leica DMRB microscope (Leica Microsystems, Milton Keynes, UK) equipped with DFC420 camera (Leica Microsystems).
A range of Aβ antibodies was supplied by GlaxoSmithKline, Stevenage, UK [see 8]: mouse monoclonal 20G10 (0.28 mg/mL) raised against the Aβ35–42 fragment and selected for its C‐terminal Aβ42 specificity; rabbit antiserum G30 (3 mg/mL) raised against Aβ35–40 for Aβ40; and mouse monoclonal 11C5 (2.35 mg/mL) raised against the Aβ27–40 fragment and selected for its C‐terminal Aβ40 specificity. Additionally, a rabbit polyclonal antibody to Aβ42 was utilized (#44–344, Invitrogen). Other antibodies were a rabbit polyclonal to human clusterin (ab42673, Abcam, Cambridge, UK) and mouse anti‐PHF tau (clone AT8; MN1020, Thermo Scientific, Fisher Scientific UK Ltd., Loughborough, UK).
Results
Aβ species and clusterin in AD brain
Focal deposits of clusterin were observed, to varying extents, in all AD brain regions studied, although not in all AD subjects. The clusterin deposits were usually positive for Aβ40; clusterin labeling was not observed in the absence of Aβ40, although some plaques were positive for both Aβ40 and Aβ42 (Figure 1A–F; white arrows). Plaques labeled by an Aβ42, but not an Aβ40 antibody (Figure 1D–F,J–L,P–R; yellow arrows), did not show clusterin labeling. In general, Aβ42‐positive plaques greatly predominated in the AD brain parenchymal sections (Table 2). These were generally of a diffuse, ill‐defined nature and lacked Aβ40 labelling. A proportion of neuritic plaques, however, were positive for both Aβ40 and Aβ42; these also exhibited clusterin labeling (Figure 1; white arrows). Occasional neuritic plaques were Aβ40‐positive but lacked any apparent substantial Aβ42 (Figure 1; blue arrows). The proportion of Aβ40 to Aβ42 plaques varied between regions and subjects, although within any single subject, the ratio of Aβ40 to Aβ42 plaques was fairly consistent (Table 2). Double labelling with Aβ40 (11C5) and clusterin antibodies showed a highly significant correlation between the number and area of plaques (Table 3).
Figure 1.
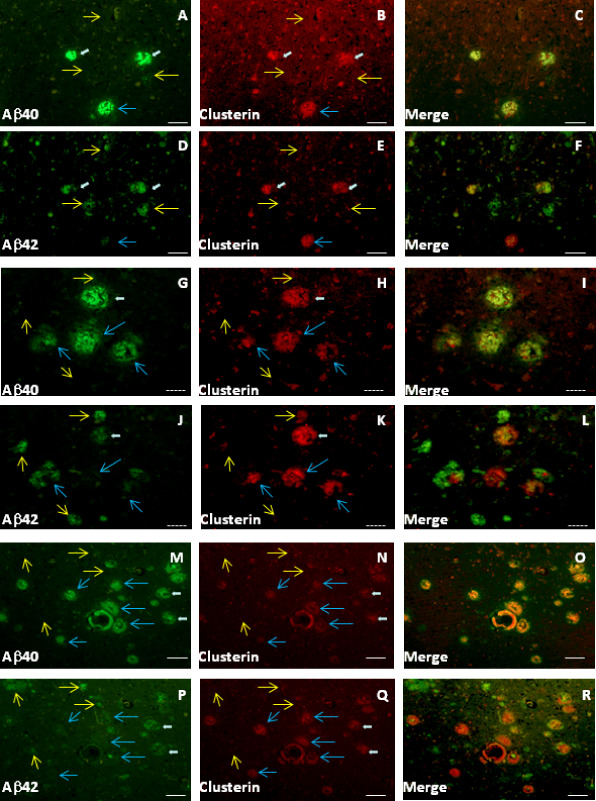
Clusterin is consistently co‐localized with Aβ40 in Alzheimer's disease (AD) plaques. Aβ40, Aβ42 and clusterin labeling in BA21,22 from AD patients. Adjacent sections were used for labeling with antibodies to either Aβ40 (antibody 11C5) plus clusterin or Aβ42 (20G10) plus clusterin. Aβ40 and Aβ42 are green channel; red channel is always clusterin. Subjects: AD#6 (A–F); AD#5 (G–R). Yellow arrows indicate plaques positive for Aβ42 but largely negative for Aβ40 and clusterin. White arrows represent plaques with Aβ40, Aβ42 and clusterin. Blue arrows point to plaques largely negative for Aβ42 but positive for Aβ40 and clusterin. Scale bars are either 50 μm (solid bars, A–F and M–R) or 25 μm (dashed bars, G–L).
Table 2.
Aβ40 and 42 plaque composition in Alzheimer's disease (AD) cortex.
| Subject | Amyloid plaques (per mm2) | %Aβ42 | ||
|---|---|---|---|---|
| Aβ42 plaques | Aβ40 plaques | |||
| BA39,40 | AD#1 | 297 | 50.0 | 85.6 |
| AD#2 | 332.7 | 40.7 | 89.1 | |
| AD#3 | 90 | 0.0 | 100.0 | |
| AD#4 | 224.5 | 0.0 | 100.0 | |
| AD#5 | 162 | 7.5 | 95.6 | |
| AD#6 | 180 | 9.7 | 94.9 | |
| AD#7 | 85 | 7.5 | 91.9 | |
| AD#8 | 138.5 | 8.5 | 94.2 | |
| AD#9 | 108 | 1.0 | 99.1 | |
| AD#10 | 156.5 | 0.0 | 100.0 | |
| AD#11 | 125 | 0.0 | 100.0 | |
| AD#12 | 191 | 1 | 99.5 | |
| 174.2 ± 21.9 | 10.5 ± 4.7 | |||
| BA17 | AD#5 | 72.5 | 1.5 | 98.0 |
| AD#6 | 243 | 14.0 | 94.6 | |
| AD#7 | 90 | 5.0 | 94.7 | |
| AD#8 | 155.5 | 0.0 | 100.0 | |
| AD#9 | 273 | 2.0 | 99.3 | |
| AD#10 | 107 | 0.0 | 100.0 | |
| AD#11 | 90 | 0.0 | 100.0 | |
| 147.3 ± 30.6 | 3.2 ± 1.9 | |||
| BA9 | AD#6 | 121.5 | 8.0 | 93.8 |
| AD#9 | 242 | 0.0 | 100.0 | |
| AD#10 | 136.5 | 0.0 | 100.0 | |
| AD#12 | 310 | 2.0 | 99.4 | |
| 202.5 ± 44.8 | 2.5 ± 1.9 | |||
| BA21,22 | AD#1 | 247 | 29.0 | 89.5 |
| AD#2 | 144.5 | 15.5 | 90.3 | |
| AD#3 | 36 | 3.0 | 92.3 | |
| AD#4 | 46 | 0.0 | 100.0 | |
| 118.4 ± 49.4 | 11.9 ± 6.6 | |||
Sections were double‐labeled with antibodies for Aβ42 (20G10) and Aβ40 (G30). Ten images were captured for each section and plaque counts determined as described in the Materials and Methods section, where each image constituted an area of 0.8 mm2. Mean counts (from the number of subjects shown) ± SEM are presented for each cortical region.
Table 3.
Comparison of Aβ40 and clusterin labelling in areas of AD brain.
| Subject region | Aβ40 (11C5) | Clusterin | ||
|---|---|---|---|---|
| Plaque counts | Plaque area | Plaque counts | Plaque area | |
| AD#1 BA39,40 | 37 | 0.058 | 43 | 0.062 |
| AD#2 BA39,40 | 85 | 0.090 | 102 | 0.097 |
| AD#2 BA21 | 42 | 0.051 | 64 | 0.070 |
| AD#6 BA9 | 74 | 0.087 | 62 | 0.072 |
| AD#6 BA17 | 4 | 0.004 | 4 | 0.003 |
| AD#6 BA39,40 | 26 | 0.041 | 25 | 0.049 |
| AD#7 BA9 | 11 | 0.018 | 7 | 0.010 |
| AD#7 BA17 | 39 | 0.073 | 36 | 0.046 |
| AD#7 BA39,40 | 10 | 0.019 | 9 | 0.008 |
| Total | 328 | 0.441 | 352 | 0.416 |
Sections were double‐labeled with antibodies for Aβ40 (11C5) and clusterin. Up to 10 images were captured for each section and plaque counts and area (mm2) determined as described in the Materials and Methods section, where each image constituted an area of 0.8 mm2. Pearson correlation (r 2) for both plaque counts and plaque area between Aβ40 and clusterin data = 0.900 (P < 0.0001). Subjects and brain regions with Aβ42 but lacking Aβ40 (Table 2) did not show any clusterin labeling (data not shown).
On occasion, labeling and quantification was undertaken on additional sections from the same subject to assess intersection variability. Parietal lobe sections from case AD#6 labeled with 20G10 and G30 (n = 3 for each) gave plaque counts of 180 ± 10.8 and 9.7 ± 5.2, respectively. Similar sections from case AD#2 gave counts of 332.7 ± 48.2 and 40.7 ± 8.4 (n = 3). Confirmation of the identity of the Aβ40 and Aβ42 labeling was achieved using an alternative pair of antibodies. Virtually identical images were observed with antibodies #44–344 (for Aβ42) and 11C5 (for Aβ40) (Figure 2A–F) to those observed with antibodies 20G10 (for Aβ42) and G30 (for Aβ40). Cerebrovascular amyloid deposits were usually negative for Aβ42 but were positive for Aβ40, although in some cases, a penumbra of parenchymal Aβ42 labeling encircled the Aβ40‐positive vessel wall (Figure 2A–F). Formic acid pretreatment of the sections was essential for demonstrating maximum Aβ42 immunoreactivity (Figure 3C,F); labeling with Aβ40 and clusterin antibodies was not dependent on formic acid treatment (Figure 3A,B,D,E).
Figure 2.
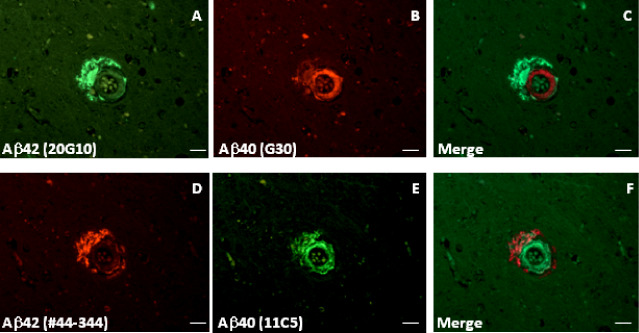
Aβ40 and Aβ42 antibodies label perivascular and cerebrovascular amyloid in the primary visual cortex (BA17) of an Alzheimer's disease (AD) subject. Aβ42 was labeled with either monoclonal antibody 20G10 (A) or polyclonal antibody #44–344 (D). Aβ40 was labeled with either polyclonal antibody G30 (B) or monoclonal antibody 11C5 (E). C and F show merged images. Subject is AD#10. Scale bars are 25 μm.
Figure 3.
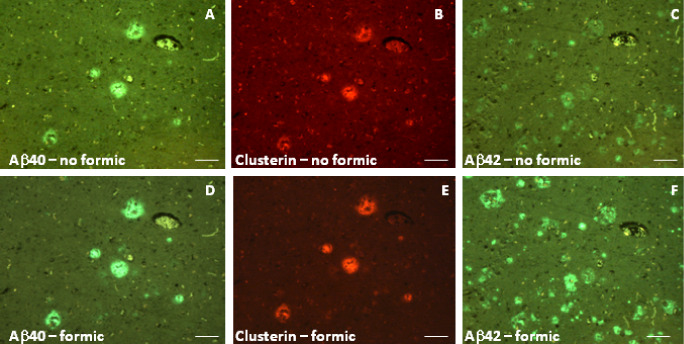
Formic acid pretreatment of sections has different effects on Aβ40, Aβ42 and clusterin immunolabeling of BA39,40 from an Alzheimer's disease (AD) subject. Adjacent sections (AD#6) were either treated with 98% formic acid (D–F) (as in the Materials and Methods section) or stood in PBS (A–C) for the same period prior to blocking and primary antibody incubation and were then lined up for image capture based on plaques, cerebral blood vessels or other landmark features. Aβ40 and Aβ42 were labeled with antibodies G30 and 20G10, respectively. Scale bars are 50 μm.
Layer V pyramidal neurons were also, on occasion, found to be clusterin‐positive; these neurons did not show any evidence of Aβ labeling (Figure 4A–F). Furthermore, the clusterin‐positive neurons were not labeled by an antibody (AT8) recognizing phosphorylated tau, and hyperphosphorylated tau‐positive neurofibrillary tangles, which were found in various neuronal layers of the AD brain sections, were clusterin negative (Figure 4G–I).
Figure 4.
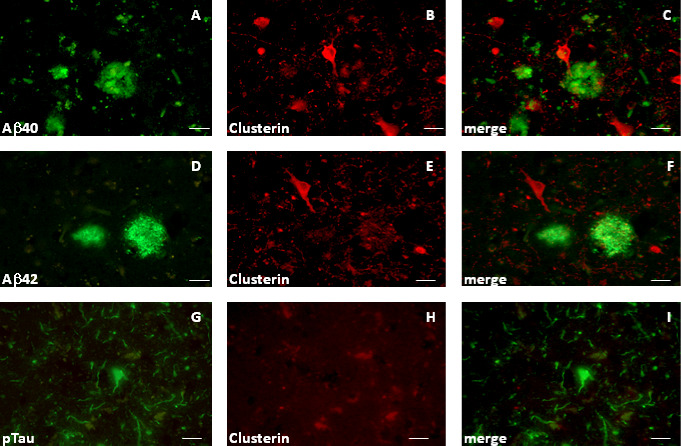
Labeling of BA21,22 neurons in Alzheimer's disease (AD) subjects (Braak V/VI) with a clusterin antibody is not associated with Aβ or phosphotau labeling. Sections were labeled with combinations of antibodies to clusterin and Aβ40 or Aβ42 (antibodies 11C5 and 20G10). Images are presented in pairs. For A and B and D and E, the green channel is Aβ40 and Aβ42, respectively; red is clusterin. For G and H, the green channel is phosphotau; red is clusterin. Merged images (C, F and I). Subjects: AD#2 (A–F); AD#1 (G–I). Scale bars are 50 μm.
Aβ species and clusterin in control human brain
Brain sections from a number of cognitively normal, aged controls and four young controls (aged 25–35 years) were also investigated (Table 1). In two of the aged controls, occasional diffuse plaques were noted positive for Aβ42 and weakly for Aβ40 but not clusterin (Figure 5A–C,D–F), or were positive for Aβ42 and clusterin but not Aβ40 (Figure 5G–I). Overall, labeling of brain parenchyma of the aged control subjects with the Aβ40 antibodies was minimal (Table 4). In contrast with cerebrovascular amyloid in AD subjects, in some aged controls, vessels were labeled by Aβ40 and Aβ42 but not by clusterin antibodies (Figure 5J–L), while in others, clusterin labeling was observed in the absence of either Aβ40 or Aβ42 (Figure 5M–O). No parenchymal or cerebrovascular deposits (Aβ or clusterin‐positive) were observed in any of the younger controls (not shown).
Figure 5.
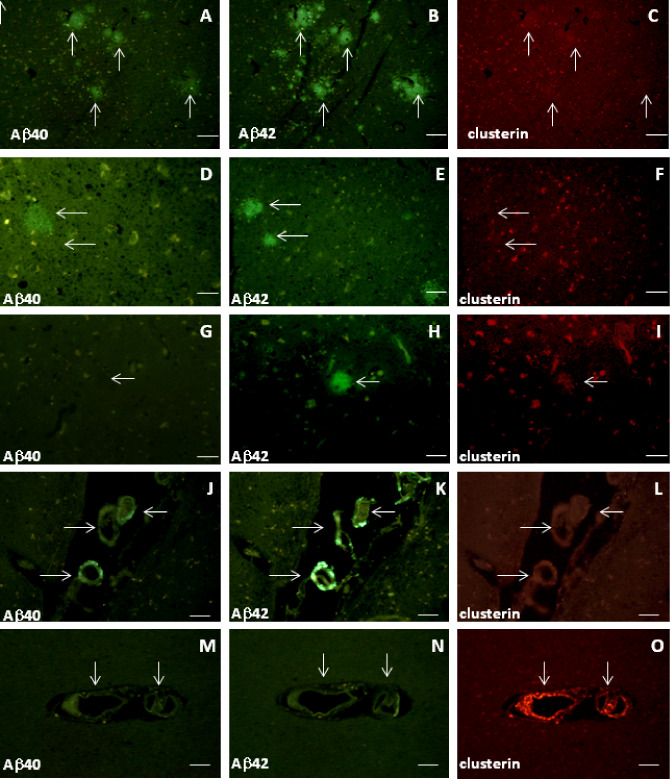
Clusterin does not have specific associations with Aβ40 or Aβ42 in brain parenchyma or cerebral blood vessels from aged control subjects. Adjacent slides of subjects were labeled with antibodies to either 20G10 and clusterin or 11C5 and clusterin as follows: A–C (Con#20, BA21,22); D–F (Con#16, BA17); G–I (Con#15, BA21,22); J–L (Con#20, BA21,22); M–O (Con#14, BA39,40). Features such as plaques and/or blood vessels were used to identify identical areas of the pairs of slides. Arrows indicate matched plaques/blood vessels (or positions of unlabeled structures). Scale bars are 50 μm.
Table 4.
Clusterin does not have a consistent association with sparse Aβ40 and Aβ42 deposits in parenchyma and cerebral blood vessels of aged control subjects.
| Subject | Region | Plaque Aβ40 | Plaque Aβ42 | Plaque clusterin | Cerebrovasculature |
|---|---|---|---|---|---|
| Con#14 | BA39,40 | ✓ | ✓✓ | 0 | Aβ40/Clusterin ✓ |
| Aβ42 none | |||||
| Con#15 | BA21,22 | 0 | ✓ | ✓ | 0 |
| Con#16 | BA17 | ✓ | ✓ | 0 | 0 |
| Con#18 | BA21,22 | 0 | 0 | 0 | 0 |
| Con#19 | BA21,22 | 0 | 0 | 0 | 0 |
| Con#20 | BA21,22 | ✓ | ✓✓ | 0 | Clusterin none |
| Aβ40/42 ✓ |
Sections were double‐labeled with antibodies for either Aβ40 (11C5) and clusterin or Aβ42 (20G10) and clusterin. A semiquantitative assessment of plaque labeling was made by eye with a score of ✓✓ representing an average of one plaque per two to four 0.8 mm images and ✓ representing very occasional (one or two) plaques per 10 images. 0 indicates that no plaques were observed in any of the 10 images.
Discussion
We have demonstrated a specific association between Aβ40 (but not Aβ42) and clusterin in AD brain. In several cortical areas of a number of AD subjects, plaques were labeled to varying degrees by antibodies to Aβ40, Aβ42 and clusterin. The largest proportion of plaque labeling was for Aβ42; neuritic plaques that were positive for Aβ42 were also positive for Aβ40 and clusterin. Many plaques, generally of a diffuse appearance, however, only showed Aβ42 immunoreactivity. Plaques lacking Aβ40 also lacked clusterin. Hence, the apparent association between Aβ40 and clusterin may relate to the species of Aβ or to the type of plaque in which Aβ40 occurs (ie, neuritic). The number of plaques labeled varied across the regions studied, with BA9 showing the most Aβ42, and BA21,22 having the most Aβ40. There were no significant differences between areas (statistics not shown), however, as there was large intersubject variation. Within any one subject, however, there was some degree of consistency in plaque count across areas. Although this fact is obviously limited by the brain areas available for study, it can be seen, for instance, that cases AD#1 and AD#2 had considerably greater plaque counts, particularly for Aβ40, in both BA21,22 and BA39,40 than the other subjects.
Clusterin is a multifunctional heterodimeric glycoprotein, originally discovered in ram testis fluid and shown to have cell adhesion properties 4. In AD brain, it has been shown that anti‐clusterin antibodies label senile plaques, produce some punctate labeling of pyramidal neurons and, to a minor extent, label neurofibrillary tangles 16. While previous studies have shown an association with Aβ, the present study establishes that in cortical tissue from AD subjects, this association is specifically with senile plaques containing Aβ40; plaques lacking Aβ40 but showing Aβ42 labeling did not exhibit clusterin labeling. Cortical and hippocampal levels of clusterin are 40% greater in AD brain than that in control subjects 20. At least some of this must be attributable to the clusterin found in senile plaques. The association of clusterin with senile plaques and pyramidal neurons in the brains of AD subjects is confirmed in the present studies, although the latter did not appear to be disease‐state related as clusterin labeling of pyramidal neurons occurred in aged control subjects. Although a previous report suggested a minor degree of binding of a clusterin antibody to neurofibrillary tangles 16, in the present study, and in agreement with other data 5, there was no evidence of clusterin being associated with tangles or with dystrophic neurites. This may reflect the differing antibodies used, differing immunohistochemical procedures or collection/processing conditions of the brain material utilized in the different investigations.
Diffuse plaques in the brains of AD and Down syndrome subjects lack Aβ40 plaques 9, and overall, Aβ42‐positive deposits have been reported to exceed significantly the number of Aβ40‐positive ones 15. In the present study, very occasional plaques appeared to lack appreciable Aβ42 but were positive for Aβ40 and clusterin. It is not clear whether Aβ42 was, in fact, completely absent from the plaque or was undetectable, either caused by low levels of the Aβ42 protein or it exists in a conformation resistant to the 98% formic acid antigen retrieval treatment. Pretreatment with 98% formic acid for 72 h did not result in any further enhancement of Aβ42 immunoreactivity (data not shown). Diffuse and compact plaques in the brains of subjects with Down syndrome have been reported to be Aβ42‐positive but only the latter exhibited clusterin labeling 22. Although the diffuse plaques lack Aβ40 15, senile plaques in Down syndrome subject are labeled by Aβ40 antibodies 17. Data on the association of clusterin labeling with Aβ40 in Down syndrome subjects require further study in order to assess whether the observed association of the two proteins is specific for AD.
As commented earlier, diffuse plaques lack Aβ40 (and clusterin), whereas neuritic plaques often contained both proteins. Thus, clusterin may be specifically associated with Aβ40 or may be confined to neuritic plaques (where Aβ40 is found). However, in agreement with previous reports, Aβ within the wall of the cerebral vasculature was predominantly Aβ40 15, 23, associated with which was clusterin, supporting the idea that in AD brain, clusterin is specifically associated with the 40 amino acid forms of Aβ. There were no obvious differences in Aβ–clusterin associations between the different regions of the cortex included in the present study (data not shown).
In aged control brain, the number of plaques was much less, although in line with published data 26, Aβ‐positive plaques, mainly diffuse and very occasionally neuritic, were present in some subjects. In contrast with the AD subjects, there was fairly minimal plaque‐associated Aβ40. There was no consistent association pattern between clusterin and the Aβ species. Clusterin labeling of plaques was rarely observed, although, on occasion, it did show some association with Aβ42; however, not all Aβ42 plaques were clusterin‐positive. The AD temporal cortex differed from that of aged controls in that, in the former, a proportion of plaques contained Aβ40, and when this isoform was present, clusterin was also found. Moreover, when Aβ42 occurred without Aβ40, there was no clusterin associated with the plaques. In contrast, in aged control subjects, the plaques that were present were labeled by an Aβ42 antibody but lacked significant Aβ40 labelling, although clusterin was found to be associated with some of the Aβ42‐positive deposits. Furthermore, although Aβ40 and Aβ42 were occasionally found in the cerebral blood vessels of some aged control subjects in the absence of clusterin, in other subjects, clusterin labelling was noted in the absence of Aβ. Hence, the association observed between clusterin and Aβ40 or the lack of association of clusterin with Aβ42 in AD brain may be specific to the disease. Differences have been described between the conformational states of Aβ in control and AD brain 25 and may give rise to the differences in association properties described in the present study. A future investigation of the relationship between clusterin binding and conformational changes in Aβ in controls and subjects at various stages of AD pathology should aid our understanding of this phenomenon.
The origin of cerebrovascular Aβ is not clear—it may be in the process of being transported out of the brain, it may be crossing the blood–brain barrier into the brain, possibly caused by deficits in barrier integrity with aging 1 or it may simply be lining the vessel wall. It was, however, never observed in the young control subjects. Data from normal aged individuals showed that higher plasma clusterin was associated with a greater retention of 11C‐PiB in PET imaging studies (indicative of a fibrillar Aβ burden), suggesting that these clusterin measurements could be indicative of an early phase of the Aβ deposition process 24. As discussed in this latter report, the origins of plasma clusterin are not fully understood, although it was reported that in an APP transgenic mouse line, brain clusterin increased with age, more or less in parallel to Aβ content.
Formic acid pretreatment of histological sections has long been utilized to enhance antibody binding and it is believed that this treatment is necessary for the labeling of fibrillar Aβ 11. Diffuse plaques in the absence of formic acid pretreatment lack antibody binding; formic acid treatment renders the material immunoreactive to Aβ antibodies 27. Comparison of formic acid‐ and non–formic acid‐treated sections permits some degree of identification of the conformation of Aβ present. The lack of Aβ42 labeling in non–formic acid‐treated sections in the present study would suggest that most of the Aβ42 is in a fibrillar form. In contrast, Aβ40/clusterin‐positive plaques (and cerebrovascular labeling) were not dependent on formic acid pretreatment. A possible role for clusterin may, therefore, be to preserve or package the Aβ40 in a non‐fibrillar form suitable for removal from the brain. In fact, clusterin has been shown to facilitate both the aggregation and the disaggregation of Aβ1–40 in vitro 19 and a similar relationship may be suggested in AD brain by the present studies. Alternatively (or additionally), putative functions of clusterin with implications for AD pathology include inhibition of the complement attack complex 22, inhibition of apoptosis 12 and regulation of lipid transport 3. Thus, an absence of clusterin in Aβ42‐only plaques might support a neurodegenerative outcome and a lack of clearance by the low‐density lipoprotein receptor‐related protein 1 (LRP‐1) receptor 21 may aid the accumulation of the longer Aβ form. It is difficult to reconcile a theory proposing that a lack of clusterin in association with Aβ42 deposition is a causative factor in AD, with the observation of increased plasma clusterin in a subgroup of AD subjects 24 and the data from genome‐wide association studies 6, 13. Furthermore, pathogenic links between clusterin and AD development/progression have been proposed from data suggesting that clusterin upregulation promotes or supports Aβ neurotoxicity and facilitates tau phosphorylation through increases in DKK1 expression 10. The differences in the conformation of Aβ species between those detected in fixed post‐mortem tissue, those in plasma and the forms used in in vitro experiments may be a crucial factor in determining clusterin–Aβ interactions.
Clarity is clearly lacking on the significance of the Aβ40–clusterin association and, in particular, whether it is a phenomenon peculiar to AD. However, our previous data showing that plasma clusterin level indicates disease state in AD 24 do hint at a disease‐specific interaction. Furthermore, the presence of Aβ42‐positive plaques that lack clusterin does support the view that the predominance of Aβ42 in the brain parenchyma of AD subjects may be a consequence of impaired interaction with clusterin.
Conclusions
In conclusion, we observed that in cortical tissue from AD subjects, clusterin labeling of brain parenchyma was specifically associated with deposits containing the Aβ40 form of the Aβ. In contrast, Aβ42 labeling was only associated with clusterin when Aβ40 was also present in the deposit. The presence of Aβ42‐positive deposits lacking clusterin may be a significant pathogenic feature in the accumulation of Aβ in the AD brain.
Supporting information
Figure S1. A and B show an example of corresponding green (Aβ42) and red (Aβ40) channel images for subject AD#2 BA39. Plaques were converted to outline images (C and D) using Fiji‐ImageJ (http://rsbweb.nih.gov/ij/) setting a threshold to quantify plaques greater than 10 μm in diameter. E is an overlay of C and D. Plaques indicated by arrows were labeled by both Aβ40 and Aβ42 antibodies. Plaques indicated by * were only labeled by the Aβ40 antibody. Green outlines show plaques only positive for Aβ42. The “Analyse” function of the software was used to determine the plaque areas described in Table 2.
Acknowledgments
Human post‐mortem tissue was supplied by the London Neurodegenerative Diseases Brain Bank and the Thomas Willis Oxford Brain Collection (Brains for Dementia Research network). Professor Margaret Esiri and Drs Olaf Ansorge, Safa Al‐Sarraj, Istvan Bodi and Andrew King are thanked for neuropathological diagnosis of cases. This work was funded in part by AddNeuroMed, part of the FP6 InnoMed program. The authors thank Dr Claire Troakes for tissue supply and GlaxoSmithKline for the supply of Aβ antibodies.
The authors claim no conflict of interest.
References
- 1. Bading JR, Yamada S, Mackic JB, Kirkman L, Miller C, Calero M et al (2002) Brain clearance of Alzheimer's amyloid‐beta 40 in the squirrel monkey: a SPECT study in a primate model of cerebral amyloid angiopathy. J Drug Target 10:359–368. [DOI] [PubMed] [Google Scholar]
- 2. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 3. Calero M, Tokuda T, Rostagno A, Kumar A, Zlokovic B, Frangione B et al (1999) Functional and structural properties of lipid‐associated apolipoprotein J (clusterin). Biochem J 344:375–383. [PMC free article] [PubMed] [Google Scholar]
- 4. Fritz IB, Burdzy K, Setchell B, Blasckuk O (1983) Ram rete testis fluid contains a protein (clusterin) which influences cell‐cell interaction in vitro . Biol Reprod 28:1173–1188. [DOI] [PubMed] [Google Scholar]
- 5. Giannakopoulos P, Kovari E, French LE, Viard I, Hof PR, Bouras C (1998) Possible neuroprotective role of clusterin in Alzheimer's disease: a quantitative immunocytochemical study. Acta Neuropathol 95:387–394. [DOI] [PubMed] [Google Scholar]
- 6. Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML et al (2009) Genome‐wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet 41:1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Howlett DR, Bowler K, Soden PE, Riddell D, Davis JB, Richardson JC et al (2008) Abeta deposition and related pathology in an APP x PS1 transgenic mouse model of Alzheimer's disease. Histol Histopathol 23:67–76. [DOI] [PubMed] [Google Scholar]
- 8. Howlett DR, Richardson JC, Austin A, Parsons AA, Bate ST, Davies DC et al (2004) Cognitive correlates of A beta deposition in male and female mice bearing amyloid precursor protein and presenilin‐1 mutant transgenes. Brain Res 1017:130–136. [DOI] [PubMed] [Google Scholar]
- 9. Iwatsubo T, Saido TC, Mann DMA, Lee VMY, Trojanowski JQ (1996) Full‐length amyloid‐beta(1‐42(43)) and amino‐terminally modified and truncated amyloid‐beta‐42(43) deposit in diffuse plaques. Am J Pathol 149:1823–1830. [PMC free article] [PubMed] [Google Scholar]
- 10. Killick R, Ribe EM, Al‐Shawi R, Malik B, Hooper C, Fernandes C et al (2013) Clusterin regulates b‐amyloid toxicity via Dickkopf‐1‐driven induction of the wnt‐PCP‐JNK pathway. Mol Psychiatry. doi: 10.1038/mp.2012.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kitamoto T, Ogomori K, Tateishi J, Prusiner SB (1987) Formic acid pretreatment enhances immunostaining of cerebral and systemic amyloids. Lab Invest 57:230–236. [PubMed] [Google Scholar]
- 12. Koch‐Brandt C, Morgans C (1996) Clusterin: a role in cell survival in the face of apoptosis? Prog Mol Subcell Biol 16:130–149. [DOI] [PubMed] [Google Scholar]
- 13. Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M et al (2009) Genome‐wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 41:1094–1099. [DOI] [PubMed] [Google Scholar]
- 14. Lovestone S, Guntert A, Hye A, Lynham S, Thambisetty M, Ward M (2007) Proteomics of Alzheimer's disease: understanding mechanisms and seeking biomarkers. Expert Rev Proteomics 4:227–238. [DOI] [PubMed] [Google Scholar]
- 15. Mann DM, Iwatsubo T, Ihara Y, Cairns NJ, Lantos PL, Bogdanovic N et al (1996) Predominant deposition of amyloid‐beta 42(43) in plaques in cases of Alzheimer's disease and hereditary cerebral hemorrhage associated with mutations in the amyloid precursor protein gene. Am J Pathol 148:1257–1266. [PMC free article] [PubMed] [Google Scholar]
- 16. McGeer PL, Kawamata T, Walker DG (1992) Distribution of clusterin in Alzheimer brain tissue. Brain Res 579:337–341. [DOI] [PubMed] [Google Scholar]
- 17. Miller DL, Potempska A, Wegiel J, Mehta PD (2011) High‐affinity rabbit monoclonal antibodies specific for amyloid peptides amyloid‐40 and amyloid‐42. J Alzheimer Dis 23:293–305. [DOI] [PubMed] [Google Scholar]
- 18. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM et al (1991) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41:479–486. [DOI] [PubMed] [Google Scholar]
- 19. Narayan P, Orte A, Clarke RW, Bolognesi B, Hook S, Ganzinger KA et al (2012) The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid‐beta(1‐40) peptide. Nat Struct Mol Biol 19:79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oda T, Pasinetti GM, Osterburg HH, Anderson C, Johnson SA, Finch CE (1994) Purification and characterization of brain clusterin. Biochem Biophys Res Commun 204:1131–1136. [DOI] [PubMed] [Google Scholar]
- 21. Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B et al (2000) Clearance of Alzheimer's amyloid‐beta(1‐40) peptide from brain by LDL receptor‐related protein‐1 at the blood‐brain barrier. J Clin Invest 106:1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stoltzner SE, Grenfell TJ, Mori C, Wisniewski KE, Wisniewski TM, Selkoe DJ et al (2000) Temporal accrual of complement proteins in amyloid plaques in Down's syndrome with Alzheimer's disease. Am J Pathol 156:489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Suzuki N, Iwatsubo T, Odaka A, Ishibashi Y, Kitada C, Ihara Y (1994) High tissue content of soluble beta 1‐40 is linked to cerebral amyloid angiopathy. Am J Pathol 145:452–460. [PMC free article] [PubMed] [Google Scholar]
- 24. Thambisetty M, Simmons A, Velayudhan L, Hye A, Campbell J, Zhang Y et al (2010) Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch Gen Psychiatry 67:739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tomic JL, Pensalfini A, Head E, Glabe CG (2009) Soluble fibrillar oligomer levels are elevated in Alzheimer's disease brain and correlate with cognitive dysfunction. Neurobiol Dis 35:352–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xuereb JH, Brayne C, Dufouil C, Gertz H, Wischik C, Harrington C et al (2000) Neuropathological findings in the very old. Results from the first 101 brains of a population‐based longitudinal study of dementing disorders. Annals N Y Acad Sci 903:490–496. [DOI] [PubMed] [Google Scholar]
- 27. Yamaguchi H, Hirai S, Morimatsu M, Shoji M, Harigaya Y (1988) Diffuse type of senile plaques in the brains of Alzheimer‐type dementia. Acta Neuropathol 77:113–119. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. A and B show an example of corresponding green (Aβ42) and red (Aβ40) channel images for subject AD#2 BA39. Plaques were converted to outline images (C and D) using Fiji‐ImageJ (http://rsbweb.nih.gov/ij/) setting a threshold to quantify plaques greater than 10 μm in diameter. E is an overlay of C and D. Plaques indicated by arrows were labeled by both Aβ40 and Aβ42 antibodies. Plaques indicated by * were only labeled by the Aβ40 antibody. Green outlines show plaques only positive for Aβ42. The “Analyse” function of the software was used to determine the plaque areas described in Table 2.