

Abstract
The paradigmatic relationship between aging and atherosclerotic cardiovascular events does not apply to all patient populations. Though trisomy 21 (T21) and its phenotypic expression, Down syndrome (DS), are conditions that involve premature aging, the cardiovascular system of adults with DS appears to be particularly spared from this early senescence. Despite a higher prevalence of some classic cardiovascular risk factors in adults with DS than in the general population, such as dyslipidemia, obesity, or sedentarism, these individuals do not develop hypertension or suffer major cardiovascular events as they age. The protective factors that prevent the development of hypertension in T21 are not well established. Genes like RCAN1 and DYRK1A, both on chromosome 21 and over‐expressed in adults with DS, appear to play a major role in cardiovascular prevention. Their regulation of the renin‐angiotensin‐aldosterone system (RAAS) and neprilysin synthesis could underlie the constitutive protection against arterial hypertension in adults with DS and explain the absence of increased arterial stiffness in this population. A better understanding of these molecular pathways could have enormous implications for the clinical management of adults with DS and might foster the development of novel therapeutic targets in cardiovascular prevention for the general population.
Keywords: cardiovascular risk, down syndrome, hypertension
1. INTRODUCTION
Atherosclerotic cardiovascular disease is the leading cause of mortality in Western countries and is one of the most important causes of morbidity worldwide. 1 Its clinical impact grows as the population ages and the prevalence of classic cardiovascular risk factors increases. 2 A pro‐inflammatory state occurs in the aging endothelium and in the arterial extracellular matrix leading to an increase in the stiffness of the vascular wall and finally, to the development of hypertension. 3 However, the paradigmatic association between aging, hypertension, and increased cardiovascular risk does not occur in all patient populations.
Although people with trisomy of chromosome 21 (T21), or Down syndrome (DS),1 present premature aging in most of their organs and tissues, 4 these individuals do not develop atherosclerosis with age. 5 In fact, despite having a higher prevalence than individuals without DS of some cardiovascular risk factors, the appearance of atherosclerotic cardiovascular events in this population is anecdotal. 6 It has been postulated that this cardiovascular protection is mediated by low constitutional blood pressure (BP). Understanding the molecular mechanisms underlying this phenomenon would allow a more accurate estimation of the real cardiovascular risk of individuals with DS and thus improve their clinical management. Equally important, it could also open the door to the study of novel therapeutic targets of great impact for the general population. This article reviews the particular relationship between T21 and cardiovascular disease, placing special emphasis on the possible molecular mechanisms that could regulate BP in subjects with DS.
2. THE AGING PROCESS IN DOWN SYNDROME
T21 is the most frequent chromosomal anomaly in live newborns and the most frequent cause of congenital intellectual disability, with a global incidence of approximately 1/1000 live newborns. 7 , 8 In recent decades, there has been a considerable, progressive increase in the life expectancy of people with DS. 9 However, there is still a significant knowledge gap about the aging process of these individuals as adults with DS.
This gradual increase in life expectancy occurs in a genetic context that promotes premature tissue aging through several mechanisms. An increase in tissular oxidative stress mediated by the enzymes superoxide dismutase and cystathionine‐β synthase, whose genes are located on chromosome 21, has been described. 10 Their over‐expression has been associated with elevated levels of pro‐inflammatory cytokines, such as leptin, interleukin‐6, and tumor necrosis factor alpha (TNF‐α). 11
As a consequence of tissue aging and the increase in life expectancy, a growing number of individuals face a life‐stage unbeknownst to previous generations of adults with DS. The studies carried out to date agree that these subjects present specific clinical problems that differ from those of the general population. 12 Although the development of premature Alzheimer's disease, early onset of cataracts, skin aging, immune system deterioration, or degenerative joint involvement are characteristic of this population, not all body systems exhibit signs of early senescence. In fact, these individuals have, for example, a remarkably lower incidence in the majority of the of solid tumors, hypertension, and atherosclerotic cardiovascular disease than the general population. 13
3. AN ATYPICAL MODEL OF CARDIOVASCULAR PROTECTION
In 1977, a team led by JC Murdoch described the differences in the cardiovascular risk factors profile of 70 institutionalized subjects with DS compared to 70 institutionalized controls with intellectual disability without DS matched by age and gender. 5 During the study period, necropsies were performed in 5 individuals who died in each group. The most striking finding of this work was the complete absence of histological changes suggestive of atherosclerosis in any of the main branches of the arterial system of the subjects with DS, while the controls presented atherosclerotic plaques in variable degrees. These results were replicated a decade later by Ylä‐Herttuala and colleagues, 14 which compared the postmortem findings of adults with DS with those of subjects with intellectual disability without DS and with controls without intellectual disability. Since then, DS has been considered an atheroma‐free biological model.
The absence of atheromatous plaques in necropsies and the low prevalence of cardiovascular events in this population with a molecular milieu of increased oxidative stress and premature tissular aging are striking. Moreover, studies show that adults with DS have a different, but not improved, cardiovascular risk factor profile, than the general population. 15 Two of the most relevant risk factors in this population are weight disorders and sedentary lifestyle 13 (Table 1). It is estimated that approximately 25%‐40% of adults with DS are overweight and an additional 25%‐45% are obese according to the usual body mass index cutoff values. 16 However, using the waist‐hip ratio as a measure of abdominal obesity, up to 90% of the individuals in a Spanish cohort of adults with DS were diagnosed with abdominal obesity. 17 On the other hand, the prevalence of metabolic syndrome is surprisingly low in subjects with DS and oscillates around 10% even in the group with abdominal obesity. 17
TABLE 1.
Cardiovascular risk factor distribution in a population of adults with Down syndrome
| Risk factor | Total (n = 144) | Under 30 ( n = 54) | 30‐39 (n = 44) | 40‐49 (n = 23) | Over 50 (n = 23) | F 3, 140 | Pearson | P |
|---|---|---|---|---|---|---|---|---|
| Weight (Kg) | 65 ± 14 | 62 ± 13 | 72 ± 13 | 62 ± 11 | 60 ± 12 | 7,22 | < .001 | |
| Height (cm) | 152 ± 10 | 152 ± 10 | 155 ± 9 | 143 ± 15 | 146 ± 9 | 8,33 | < .001 | |
| BMI (Kg/m2) | 29.2 ± 6.4 | 27.4 ± 6.5 | 31.3 ± 6.1 | 31.0 ± 3.6 | 28.7 ± 6.8 | 4,03 | < .01 | |
| Systolic BP (mm Hg) | 104 ± 13 | 107 ± 10 | 104 ± 10 | 107 ± 26 | 98 ± 11 | 2,51 | Ns | |
| Diastolic BP (mm Hg) | 63 ± 16 | 65 ± 6 | 57 ± 22 | 71 ± 17 | 64 ± 14 | 4,58 | < .01 | |
| Heart rate (lpm) | 67 ± 12 | 68 ± 12 | 68 ± 9 | 67 ± 23 | 64 ± 9 | 0,57 | Ns | |
| Family history of cardiovascular disease | 12 (8%) | 3 (6%) | 5 (11%) | 3 (13%) | 1 (4%) | 2,22 | Ns | |
| Arterial hypertension | 0 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Dyslipidemia | 14 (10%) | 1 (2%) | 6 (14%) | 2 (9%) | 5 (21%) | 8,39 | < .05 | |
| Diabetes mellitus | 2 (1%) | 1 (2%) | 0 | 0 | 1 (4%) | 2,5 | Ns | |
| Metabolic syndrome | 7 (5%) | 2 (4%) | 2 (5%) | 1 (4%) | 2 (9%) | 0,91 | Ns | |
| Hyperuricemia | 3 (2%) | 1 (2%) | 0 | 2 (9%) | 0 | 6,37 | Ns | |
| Physical activity (h/semana) | 2.9 ± 2.2 | 3.4 ± 1.6 | 2.9 ± 2.6 | 2.7 ± 2.5 | 0.5 ± 0.6 | 11,38 | < .001 | |
| Tobacco/Alcohol abuse | 0/0 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
(Reproduced with permission from Real de Asúa et al Ref. 13).
BMI, body mass index; BP, blood pressure; ns, non significant.
However, there is yet another significant difference in the cardiovascular profile of adults with DS compared to the general population that could explain the low prevalence of atherosclerotic events: the absence of arterial hypertension. In a study conducted on 144 outpatient adults with DS, Real de Asúa et al did not find any individual with high blood pressure (HBP). 13 This finding, also observed by Murdoch et al, 5 has been replicated in other cohorts. 18 The absence of arterial hypertension could explain why the prevalence of asymptomatic organ damage as measured by carotid intima‐media thickness or by pulse wave velocity is basically absent in the population with DS regardless of the presence of other risk factors. To this particular, Draheim et al showed significantly lower values of systolic BP, diastolic BP, and carotid intima‐media thickness in adults with DS compared to age‐and‐sex‐matched controls. 19 Similarly, Rodrigues et al, 18 Roy‐Vallejo et al 20 and Parra et al 21 observed significantly lower peripheral and central BP values, in the DS population than in age‐matched controls without DS (Figure 1). Altogether the evidence points to a central role of the absence of hypertension in the cardiovascular protection of adults with DS. Since arterial hypertension is considered the main risk factor for cardiovascular mortality and morbidity in developed countries, the study of the mechanisms that regulate BP in DS could have enormous clinical and epidemiological relevance.
FIGURE 1.
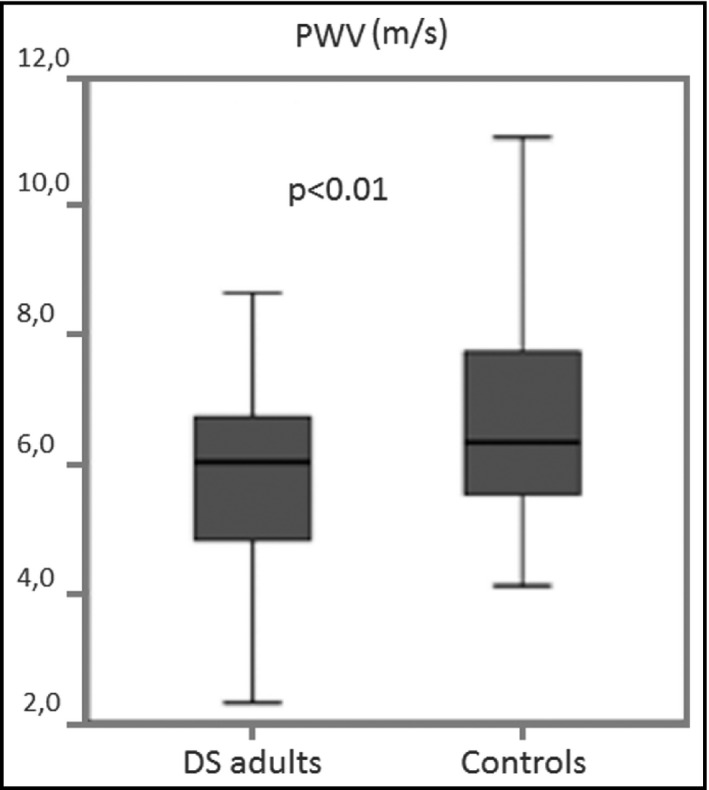
Differences in pulse wave velocity between subject with and without Down syndrome. (Reproduced with permission from Parra et al Ref. 21)
4. MOLECULAR MECHANISMS OF BLOOD PRESSURE CONTROL IN DOWN SYNDROME
The molecular mechanisms underlying BP control in the general population are polygenic and of complex characterization. Nonetheless, two genes located on chromosome 21 and over‐expressed in DS seem to play an essential role. These genes are dual specificity Yak1‐related kinase 1A (DYRK1A) and regulator of calcineurin 1 (RCAN1, formerly known as Down syndrome critical region 1 or DSCR1), which are also responsible for many of the phenotypic characteristics of people with DS. 22 There are three isoforms of RCAN1 (RCAN1.1L, RCAN1.1S, and RCAN1.4), which all act in a concentration‐dependent manner, albeit with a variable effect and possible differing actions, on the transcription factor nuclear factor of activated T cells (NFAT). 23 DYRK1A participates in the regulation of cell proliferation and is known for its crucial role in neuronal development and the onset of Alzheimer's disease in people with DS. 24 Through complementary pathways, both genes have a key influence in the regulation of the three main systems involved in the development of arterial hypertension: the renin‐angiotensin‐aldosterone system (RAAS), the sympathetic autonomic nervous system, and the endothelial function (Figure 2).
FIGURE 2.
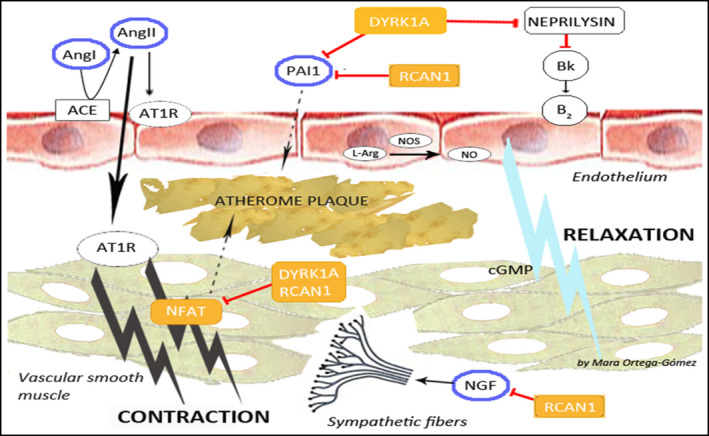
Molecular mechanisms of blood pressure control in subjects with Down syndrome ACE, angiotensin‐converting enzyme; Bk, bradykinin; B2, bradykinin receptor B2; NO, nitric oxide; NOS, nitric oxide syntase; L‐Arg, L‐arginine. Yellow square: genes in the chromosome 21; Blue circle: their action affect blood pressure and atherosclerosis; Red line: inhibition; Black line: activation; Dashed line: relation
4.1. Influence on the renin‐angiotensin‐aldosterone system
RAAS is the widest‐studied BP‐regulating system. Angiotensin II (AngII), the main effector of RAAS, exerts its function through AT1R and AT2R receptors, triggering the contraction or relaxation, respectively, of vascular smooth muscle cells (VSMCs). 25 In addition, the intracellular signaling produced by AngII‐AT1R interaction is implicated not only in the pathogenesis of hypertension, but also in vascular remodeling and the development of atherosclerosis. 26 The relationship between an over‐expressed RCAN1 and the control of AngII‐dependent BP is still unclear. While some authors have shown in cellular models that RCAN1‐mediated inhibition of NFAT decreases the appearance of AngII‐dependent cardiac hypertrophy, 27 others have reported that AngII, through NFAT, promotes the expression of RCAN1.4, which, in turn, is necessary for VSMCs migration and vascular remodeling in models of vascular restenosis and aortic aneurysm. 28 RCAN1.4 has also been implicated in the development of atheromatous plaques, endothelial activation, and angiogenesis. 29
DYRK1A is also an NFAT inhibitor, 22 although its role in the regulation of RAAS has not been well characterized. However, DYRK1A has recently been postulated to decrease in neprilysin levels in fibroblasts of subjects with DS. 30 Neprilysin is an endopeptidase responsible for the degradation of natriuretic peptides and vasodilators, whose inhibition is related to beta‐amyloid accumulation and the development of Alzheimer's disease in adults with DS. 31 Therefore, the inhibition of neprilysin in DS subjects could be one putative link between Alzheimer's disease and cardiovascular protection in this population. So far, the function of RAAS has not been evaluated in subjects with DS, but these clinical parallels suggests that a deeper analysis of the influence of the different RCAN1 and DYRK1A isoforms on the regulation of AngII and neprilysin may be an essential starting point.
4.2. Influence on the sympathetic autonomic nervous system
The sympathetic nervous system regulates vascular tone by controlling cardiac output and peripheral vascular resistances. Different clinical findings have led to suggest that people with DS have defects in their peripheral sympathetic innervation. First, people with DS have a lower baseline heart rate than the general population. 32 In addition, several studies have shown a poorer adaptation of heart rate and BP to moderate efforts in subjects with DS. 33 , 34
This sympathetic dysautonomy also seems to be mediated by RCAN1 and DYRK1A, through their inhibition of neural growth factor (NGF). NGF is one of the factors involved in the development of the nervous system and its inhibition leads to a decrease in peripheral sympathetic innervation and loss of sympathetic neurons. 35 A recent work by Patel et al 36 showed that individuals with DS have a lower sympathetic innervation of some organs than controls without DS and that over‐expression of RCAN1 would be able to block the development of sympathetic neurons through the inhibition of NGF in a murine model of DS. Likewise, Stefos et al 37 demonstrated that both RCAN1 and DYRK1A inhibit NGF function in the brain. In sum, these results point to a hypertensive role of NGF mediated through the sympathetic system that would be reduced by the over‐expression of RCAN1 and DYRK1A in the population with DS.
4.3. Influence on endothelial function
The third determinant that regulates blood pressure and can contribute to the development of hypertension is endothelial function. RCAN1 and DYRK1A might critically regulate endothelial function exerting their influence on three interrelated pathways: the plasminogen activator inhibitor‐1 (PAI‐1) pathway, the balance between leptin and adiponectin, and the oxidation of LDL cholesterol.
Plasminogen activator inhibitor‐1 (PAI‐1) is a crucial factor in the progression of endothelial damage, and its concentration correlates with the development of arterial hypertension and with an increase in cardiovascular mortality. 38 This factor may be also involved in endothelial senescence and increased arterial stiffness. 39 Stefos et al demonstrated that RCAN1 and DYRK1A negatively regulate PAI‐1, whose circulating levels are decreased in people with DS. 37 , 40 Likewise, DYRK1A activates sirtuin‐1 (SIRT‐1), an enzyme that inhibits the deleterious effects of PAI‐1 through epigenetic changes. 41
Additionally, adiponectin exerts its beneficial effect on endothelial function through nitric oxide‐dependent arterial vasodilation, by increasing the expression of endothelial nitric oxide synthase 42 and decreasing the expression of adhesion molecules. 43 Conversely, leptin is related to VSMCs hypertrophy and, therefore, to arterial hypertension. 44 Kim et al 45 observed that NFAT negatively regulates the expression of adiponectin, and Soudani et al 44 showed that the synthesis and secretion of leptin is activated by NFAT. There are no studies that directly link RCAN1 or DYRK1A to the regulation of the leptin/ adiponectin balance, but taking into account that both genes inhibit NFAT, it seems plausible to suggest that this might be another mechanism by which adults with DS have a more favorable adiponectin/leptin ratio. 11
The oxidation of LDL cholesterol molecules (oxLDL) occurs early in the process of atherogenesis. A paradoxical increase in both circulating oxLDL and anti‐LDL antibodies has been observed in the DS population, 46 which, however, do not cause an increased risk of atherosclerosis in these people. Although elevated levels of oxLDL produce an increase in RCAN1.4 that promotes VSMCs migration, 47 it seems that the key to the absence of atherosclerotic plaque formation in subjects with DS could be the inhibition of macrophage‐mediated migration by adiponectin, 11 which further increases the interest in clarifying the role of RCAN1 and DYRK1A in atherogenesis.
5. POTENTIAL PATHWAYS FOR THE PREVENTION OF ARTERIAL STIFFNESS AND ATHEROSCLEROSIS IN DOWN SYNDROME
As mentioned above, subjects with DS have lower wave pulse velocity and carotid intima‐media thickness than controls without DS. These findings could mean that DS entails a special protection against the development of arterial stiffness and atherosclerosis, too. However, there is an absolute paucity of data on whether the absence of arterial hypertension in individuals with DS is sufficient to avoid the development of arterial stiffness or atherosclerosis. Indeed, some mechanisms proposed such as the regulation of endothelial function (adiponectin/leptin balance, oxLDL, etc) may play equally relevant roles.
6. LIMITATIONS OF THIS PROPOSAL
Many of the mechanisms put forth in this review are based on cellular and murine models. The evidence to support the possible mechanisms leading to the low prevalence of hypertension in DS patients is indirect and most of these findings have not yet been accurately translated to the clinical field, our conclusions may need to be taken cautiously. Additionally, many of the clinical studies on this topic in adults with DS are small in sample size and their findings might not be generalizable. There is a significant paucity of data on whether our key argument (arterial hypertension is absent in individuals with DS) is sufficient to avoid the development of vascular damage. Indeed, we propose other concomitant mechanisms, such as an altered adiponectin/ leptin balance, or differences in oxLDL metabolism that may play equally relevant roles in the prevention of hypertension and atherosclerosis.
7. CONCLUSIONS
Adults with DS show a constitutional cardiovascular protection, by virtue of which they do not seem to develop arterial hypertension or arterial stiffness despite the early tissular aging and the increased prevalence of important classic vascular risk factors, such as obesity, a sedentary lifestyle, or dyslipidemia. RCAN1 and DYRK1A, two genes located on chromosome 21 and over‐expressed in DS, are probably involved in the regulation of RAAS, the autonomic nervous system, and endothelial function, and seem to be responsible for this protection. A deeper understanding of these two genes and their signaling pathways could help improve both the management of cardiovascular risk in adults with DS and have an enormous impact on cardiovascular health for the general population, which could lead to the investigation of new potential therapeutic targets.
8. Funding sources
The research team is supported by grants from the Sociedad Española de Medicina Interna (Premio a la Investigación Clínica Dr Prof. Miguel Vilardell 2017), from the Sociedad de Medicina Interna de Madrid y Castilla La Mancha (Beca al mejor Proyecto de Investigación 2018) and from the Jerôme Lejeune Foundation (Cycle2017B/Project #1711). ERV is supported by a scholarship from the Instituto de Salud Carlos III (Contrato Río Hortega;European Fund for Regional Development‐EFRD). DRA is partially supported by the Fondo de Investigaciones Sanitarias (FIS grant PI19/00634, European Fund for Regional Development—EFRD).
DISCLOSURE STATEMENT
The authors have no conflicts of interest to disclose.
AUTHOR CONTRIBUTIONS
ERV and JMGR substantially contributed to the conception of the present manuscript. ERV and DRA drafted the work and reviewed it critically for important intellectual content. ERV, JMGR, DRA, and FM approved the final version to be published. All authors agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately exposed and resolved.
ACKNOWLEDGEMENTS
The authors would like to thank Mr Jesús Coronado Hinojosa for his support of the Adult Down Syndrome Unit at the Hospital Universitario de La Princesa and its research program, and Mara Ortega, Pedro Parra and Manuel Gómez for their help elaborating this manuscript.
Roy‐Vallejo E, Galván‐Román JM, Moldenhauer F, Real de Asúa D. Adults with Down syndrome challenge another paradigm: When aging no longer entails arterial hypertension. J Clin Hypertens. 2020;22:1127–1133. 10.1111/jch.13930
Footnotes
Although they are not exactly equivalent, we will use the terms T21, which refers to a genotype, and DS, its phenotypic expression, interchangeably throughout this review.
REFERENCES
- 1. Piepoli MF, Hoes AW, Agewall S, et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts)Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Europ Heart J. 2016;37(29):2315‐2381. 10.1093/eurheartj/ehw106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mathers C, Stevens G, Hogan D, Mahanani WR, Ho J.Global and Regional Causes of Death: Patterns and Trends, 2000–15. In: Jamison DT, Gelband H, Horton S, et al., Disease control priorities: Improving health and reducing poverty. 3rd ed. Washington (DC): The International Bank for Reconstruction and Development / The World Bank; 2017. http://www.ncbi.nlm.nih.gov/books/NBK525280/. Accessed March 1, 2019. [Google Scholar]
- 3. Wang M, Monticone RE, McGraw KR. Proinflammatory arterial stiffness syndrome: A signature of large arterial aging. J Vasc Res. 2018;55(4):210‐223. 10.1159/000490244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Horvath S, Garagnani P, Bacalini MG, et al. Accelerated epigenetic aging in Down syndrome. Aging Cell. 2015;14(3):491‐495. 10.1111/acel.12325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Murdoch JC, Rodger JC, Rao SS, Fletcher CD, Dunnigan MG. Down’s syndrome: an atheroma‐free model? Br Med J. 1977;2(6081):226‐228. 10.1136/bmj.2.6081.226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sobey CG, Judkins CP, Sundararajan V, Phan TG, Drummond GR, Srikanth VK. Risk of major cardiovascular events in people with down syndrome. PLoS One. 2015;10(9):e0137093. 10.1371/journal.pone.0137093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Roizen NJ, Patterson D. Down’s syndrome. The Lancet. 2003;361(9365):1281‐1289. 10.1016/S0140-6736(03)12987-X [DOI] [PubMed] [Google Scholar]
- 8. Alexander M, Ding Y, Foskett N, Petri H, Wandel C, Khwaja O. Population prevalence of Down’s syndrome in the United Kingdom. J Intellect Disabil Res. 2016;60(9):874‐878. 10.1111/jir.12277 [DOI] [PubMed] [Google Scholar]
- 9. Glasson EJ, Sullivan SG, Hussain R, Petterson BA, Montgomery PD, Bittles AH. The changing survival profile of people with Down’s syndrome: implications for genetic counselling. Clin Genet. 2002;62(5):390‐393. 10.1034/j.1399-0004.2002.620506.x [DOI] [PubMed] [Google Scholar]
- 10. Campos C, Guzmán R, López‐Fernández E, Casado Á. Evaluation of urinary biomarkers of oxidative/nitrosative stress in children with Down syndrome. Life Sci. 2011;89(17–18):655‐661. 10.1016/j.lfs.2011.08.006 [DOI] [PubMed] [Google Scholar]
- 11. Corsi MM, Dogliotti G, Pedroni F, et al. Adipocytokines in Down’s syndrome, an atheroma‐free model: Role of adiponectin. Arch Gerontol Geriatrics. 2009;48(1):106‐109. 10.1016/j.archger.2007.10.011 [DOI] [PubMed] [Google Scholar]
- 12. Kerins G, Petrovic K, Bruder MB, Gruman C. Medical conditions and medication use in adults with Down syndrome: A descriptive analysis. Downs Syndr Res Pract. 2008;12(2):141‐147. 10.3104/reports.2009 [DOI] [PubMed] [Google Scholar]
- 13. Real de Asua D, Quero M, Moldenhauer F, Suarez C. Clinical profile and main comorbidities of Spanish adults with Down syndrome. Europ J Int Med. 2015;26(6):385‐391. 10.1016/j.ejim.2015.05.003 [DOI] [PubMed] [Google Scholar]
- 14. Ylä‐Herttuala S, Luoma J, Nikkari T, Kivimäki T. Down’s syndrome and atherosclerosis. Atherosclerosis. 1989;76(2–3):269‐272. 10.1016/0021-9150(89)90110-X [DOI] [PubMed] [Google Scholar]
- 15. Real de Asua D, Parra P, Costa R, Moldenhauer F, Suarez C. A Cross‐sectional study of the phenotypes of obesity and insulin resistance in adults with down syndrome. Diabet Metab J. 2014;38(6):464. 10.4093/dmj.2014.38.6.464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Melville CA, Cooper S‐A, McGrother CW, Thorp CF, Collacott R. Obesity in adults with Down syndrome: A case‐control study. J Intellect Disabil Res. 2005;49(Pt 2):125‐133. 10.1111/j.1365-2788.2004.00616.x [DOI] [PubMed] [Google Scholar]
- 17. Real de Asua D, Parra P, Costa R, Moldenhauer F, Suarez C. Evaluation of the impact of abdominal obesity on glucose and lipid metabolism disorders in adults with Down syndrome. Res Dev Disabil. 2014;35(11):2942‐2949. 10.1016/j.ridd.2014.07.038 [DOI] [PubMed] [Google Scholar]
- 18. Rodrigues A, Coelho GW. Stiffness of the large arteries in individuals with and without Down syndrome. Vasc Health Risk Manag. 2011:375‐381. 10.2147/VHRM.S21273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Draheim CC, Geijer JR, Dengel DR. Comparison of intima‐media thickness of the carotid artery and cardiovascular disease risk factors in adults with versus without the down syndrome. Am J Cardiol. 2010;106(10):1512‐1516. 10.1016/j.amjcard.2010.06.079 [DOI] [PubMed] [Google Scholar]
- 20. Roy‐Vallejo E, Alonso E, Galván‐Román JM, et al. Haemodynamic profile of Spanish adults with Down syndrome. Rev Clin Esp. 2019;220(5):275‐281. 10.1016/j.rce.2019.09.003 [DOI] [PubMed] [Google Scholar]
- 21. Parra P, Costa R, de Asúa DR, Moldenhauer F, Suárez C. Atherosclerotic surrogate markers in adults with down syndrome: A case‐control study. J Clin Hyp. 2017;19(2):205‐211. 10.1111/jch.12890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Arron JR, Winslow MM, Polleri A, et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature. 2006;441(7093):595‐600. 10.1038/nature04678 [DOI] [PubMed] [Google Scholar]
- 23. Shin S‐Y, Yang HW, Kim J‐R, Do Heo W, Cho K‐H. A hidden incoherent switch regulates RCAN1 in the calcineurin‐NFAT signaling network. J Cell Sci. 2011;124(1):82‐90. 10.1242/jcs.076034 [DOI] [PubMed] [Google Scholar]
- 24. Ryoo S‐R, Jeong HK, Radnaabazar C, et al. DYRK1A‐mediated Hyperphosphorylation of Tau: A functional link between down syndrome and alzheimer disease. J Biol Chem. 2007;282(48):34850‐34857. 10.1074/jbc.M707358200 [DOI] [PubMed] [Google Scholar]
- 25. Murphy TJ, Alexander RW, Griendling KK, Runge MS, Bernstein KE. Isolation of a cDNA encoding the vascular type‐1 angiotensin II receptor. Nature. 1991;351(6323):233‐236. 10.1038/351233a0 [DOI] [PubMed] [Google Scholar]
- 26. Nakashima H, Suzuki H, Ohtsu H, et al. Angiotensin II regulates vascular and endothelial dysfunction: Recent topics of angiotensin II type‐1 receptor signaling in the vasculature. Curr Vasc Pharmacol. 2006;4(1):67‐78. 10.2174/157016106775203126 [DOI] [PubMed] [Google Scholar]
- 27. Harris CD, Ermak G, Davies KJA. Multiple roles of the DSCR1 (Adapt78 or RCAN1) gene and its protein product Calcipressin 1 (or RCAN1) in disease. Cell Mol Life Sci. 2005;62(21):2477‐2486. 10.1007/s00018-005-5085-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Esteban V, Méndez‐Barbero N, Jesús Jiménez‐Borreguero L, et al. Regulator of calcineurin 1 mediates pathological vascular wall remodeling. J Exp Med. 2011;208(10):2125‐2139. 10.1084/jem.20110503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fuentes JJ. DSCR1, overexpressed in Down syndrome, is an inhibitor of calcineurin‐mediated signaling pathways. Human Mol Gen. 2000;9(11):1681‐1690. 10.1093/hmg/9.11.1681 [DOI] [PubMed] [Google Scholar]
- 30. Kawakubo T, Mori R, Shirotani K, Iwata N, Asai M. Neprilysin is suppressed by dual‐specificity tyrosine‐phosphorylation regulated kinase 1A (DYRK1A) in down‐syndrome‐derived fibroblasts. Biol Pharmac Bull. 2017;40(3):327‐333. 10.1248/bpb.b16-00825 [DOI] [PubMed] [Google Scholar]
- 31. Miners JS, Morris S, Love S, Kehoe PG. Accumulation of insoluble amyloid‐β in down’s syndrome is associated with increased BACE‐1 and neprilysin activities. J Alzheimer’s Dis. 2011;23(1):101‐108. 10.3233/JAD-2010-101395 [DOI] [PubMed] [Google Scholar]
- 32. Goulopoulou S, Baynard T, Collier S, et al. Cardiac autonomic control in individuals with down syndrome. Am J Ment Retard. 2006;111(1):8. 10.1352/0895-8017(2006)111[27:CACIIW]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- 33. de Carvalho T, Masseti T, da Silva TD, et al. Heart rate variability in individuals with Down syndrome–A systematic review and meta‐analysis. Auton Neurosci. 2018;213:23‐33. 10.1016/j.autneu.2018.05.006 [DOI] [PubMed] [Google Scholar]
- 34. Iellamo F, Galante A, Legramante JM, et al. Altered autonomic cardiac regulation in individuals with Down syndrome. American Journal of Physiology‐Heart and Circulatory Physiology. 2005;289(6):H2387‐H2391. 10.1152/ajpheart.00560.2005 [DOI] [PubMed] [Google Scholar]
- 35. Crowley C, Spencer SD, Nishimura MC, et al. Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell. 1994;76(6):1001‐1011. 10.1016/0092-8674(94)90378-6 [DOI] [PubMed] [Google Scholar]
- 36. Patel A, Yamashita N, Ascaño M, et al. RCAN1 links impaired neurotrophin trafficking to aberrant development of the sympathetic nervous system in Down syndrome. Nat Commun. 2015;6(1): 10.1038/ncomms10119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stefos GC, Soppa U, Dierssen M, Becker WNGF. Upregulates the plasminogen activation inhibitor‐1 in neurons via the calcineurin/NFAT pathway and the down syndrome‐related proteins DYRK1A and RCAN1 attenuate this. Effect. PLoS One. 2013;8(6):e67470. 10.1371/journal.pone.0067470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Samarakoon R, Higgins PJ. Integration of non‐SMAD and SMAD signaling in TGF‐β1‐induced plasminogen activator inhibitor type‐1 gene expression in vascular smooth muscle cells. Thromb Haemost. 2008;100(6):976‐983. [PMC free article] [PubMed] [Google Scholar]
- 39. Vaughan DE, Rai R, Khan SS, Eren M, Ghosh AK. PAI‐1 is a Marker and a mediator of senescence. Arterioscler Thromb Vasc Biol. 2017;37(8):1446‐1452. 10.1161/ATVBAHA.117.309451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hopkins WE, Fukagawa NK, Sobel BE, Schneider DJ. Plasminogen activator inhibitor type 1 in adults with Down syndrome and protection against macrovascular disease. Am J Cardiol. 2000;85(6):784‐786. 10.1016/S0002-9149(99)00864-4 [DOI] [PubMed] [Google Scholar]
- 41. Wan Y‐Z, Gao P, Zhou S, et al. SIRT1‐mediated epigenetic downregulation of plasminogen activator inhibitor‐1 prevents vascular endothelial replicative senescence. Aging Cell. 2014;13(5):890‐899. 10.1111/acel.12247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sena CM, Pereira A, Fernandes R, Letra L, Seiça RM. Adiponectin improves endothelial function in mesenteric arteries of rats fed a high‐fat diet: role of perivascular adipose tissue: Adiponectin improves endothelial dysfunction. British J Pharmacol. 2017;174(20):3514‐3526. 10.1111/bph.13756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Han X, Wu Y, Liu X, et al. Adiponectin improves coronary no‐reflow injury by protecting the endothelium in rats with type 2 diabetes mellitus. Biosci Rep. 2017;37(4):BSR20170282. 10.1042/BSR20170282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Soudani N, Ghantous CM, Farhat Z, Shebaby WN, Zibara K, Zeidan A. Calcineurin/NFAT activation‐dependence of Leptin synthesis and vascular growth in response to mechanical stretch. Front. Physiol.. 2016;7 10.3389/fphys.2016.00433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim HB, Kong M, Kim TM, et al. NFATc4 and ATF3 negatively regulate adiponectin gene expression in 3T3‐L1 adipocytes. Diabetes. 2006;55(5):1342‐1352. 10.2337/db05-1507 [DOI] [PubMed] [Google Scholar]
- 46. Licastro F, Dogliotti G, Goi G, Malavazos AE, Chiappelli M, Corsi MM. Oxidated low‐density lipoproteins (oxLDL) and peroxides in plasma of Down syndrome patients. Arch Gerontol Geriatr. 2007;44(Suppl 1):225‐232. 10.1016/j.archger.2007.01.031 [DOI] [PubMed] [Google Scholar]
- 47. Méndez‐Barbero N, Esteban V, Villahoz S, et al. A major role for RCAN1 in atherosclerosis progression: RCAN1 mediates atherosclerosis. EMBO Mol. Med.. 2013;5(12):1901‐1917. 10.1002/emmm.201302842 [DOI] [PMC free article] [PubMed] [Google Scholar]