

Abstract
In sepsis-induced acute kidney injury, kidney blood flow may increase despite decreased glomerular filtration. Normally, angiotensin-II reduces kidney blood flow to maintain filtration. We hypothesized that sepsis reduces angiotensin type-1 receptor (AT1R) expression to account for this observation and tested this hypothesis in a patient case-control study and studies in mice. Seventy-three mice underwent cecal ligation and puncture (a sepsis model) or sham operation. Additionally, 94 septic mice received losartan (selective AT1R antagonist), angiotensin II without or with losartan, or vehicle. Cumulative urine output, kidney blood flow, blood urea nitrogen, and creatinine were measured. AT1R expression was assessed using ELISA, qPCR, and immunofluorescence. A blinded pathologist evaluated tissue for ischemic injury. AT1R expression was compared in autopsy tissue from seven patients with sepsis to that of the non-involved portion of kidney from ten individuals with kidney cancer and three non-infected but critically ill patients. By six hours post ligation/puncture, kidney blood flow doubled, blood urea nitrogen rose, and urine output fell. Concurrently, AT1R expression significantly fell 2-fold in arterioles and the macula densa. Creatinine significantly rose by 24 hours and sham operation did not alter measurements. Losartan significantly exacerbated ligation/puncture-induced changes in kidney blood flow, blood urea nitrogen, creatinine, and urine output. There was no histologic evidence of cortical ischemia. Significantly, angiotensin II prevented changes in kidney blood flow, creatinine, and urine output compared to vehicle. Co-administering losartan with angiotensin-II reversed this protection. Relative to both controls, patients with sepsis had low AT1R expression in arterioles and macula densa. Thus, murine cecal ligation/puncture and clinical sepsis decrease renal AT1R expression. Angiotensin II prevents functional changes while AT1R-blockade exacerbates them independent of ischemia in mice.
Keywords: acute kidney injury, angiotensin II, receptor, angiotensin, type 1, sepsis
Sepsis, defined as life-threatening organ dysfunction caused by a dysregulated host-response to infection,1 is the most common cause of acute kidney injury.2 Sepsis-induced acute kidney injury (SIAKI) carries a particularly poor acute and long-term prognosis.3 Specific therapies remain elusive.
SIAKI is thought to reflect pathophysiology distinct from other acute kidney injuries.4-6 Clinical sepsis and large mammal sepsis models demonstrate a distinct renal physiology, characterized by decreased glomerular filtration rate (GFR) and urine output but, with appropriate resuscitation, normal-to-elevated renal blood flow (RBF).7-10 In this setting, an impaired renal response to angiotensin II may dilate efferent arterioles. This change would increase RBF but also decrease GFR and urine output by reducing filtration fraction.10 Therefore, the unique renal pathophysiology observed in sepsis11 might result from attenuation of signal transduction initiated by the angiotensin II type-1 receptor (AT1R), which mediates efferent arteriolar constriction. This mechanism is consistent with previous work demonstrating that sepsis attenuates receptor-mediated cell signaling in both the endocrine and immune systems.12,13 Changes in renal angiotensin receptor signaling, however, have not been explored. Therefore, we used cecal ligation and puncture (CLP), an animal model that mimics many aspects of human sepsis, and human tissue samples to test the hypothesis that sepsis decreases AT1R expression. We further tested the hypothesis that angiotensin II administration attenuates CLP-induced acute kidney injury while AT1R blockade exacerbates it.
RESULTS
CLP induces changes in urine output, blood urea nitrogen, and creatinine
Urine output (Figure 1a) decreased within 6 hours of CLP (0.6 ml/kg/h vs. 2.17 ml/kg/h, 95% confidence interval [CI]: 0.29–2.78, P = 0.011). This decrease persisted at later time points and was uniformly <0.5 ml/kg/h at 18 hours post-CLP, indicating Stage II acute kidney injury by Kidney Disease improving Global Outcomes (KDIGO) criteria.14 Serum blood urea nitrogen levels were significantly elevated by 6 hours after CLP (54 mg/dl vs. 16 mg/dl; 95% CI for difference: 7–69 mg/dl, P < 0.0001) (Figure 1b). Serum creatinine also increased (P = 0.018) following CLP (Figure 1c) and was still elevated at 48 hours. These changes were not observed postsham operation.
Figure 1∣. Cecal ligation and puncture (CLP) induces oliguric renal dysfunction and a high-flow, low-resistance hemodynamic state.
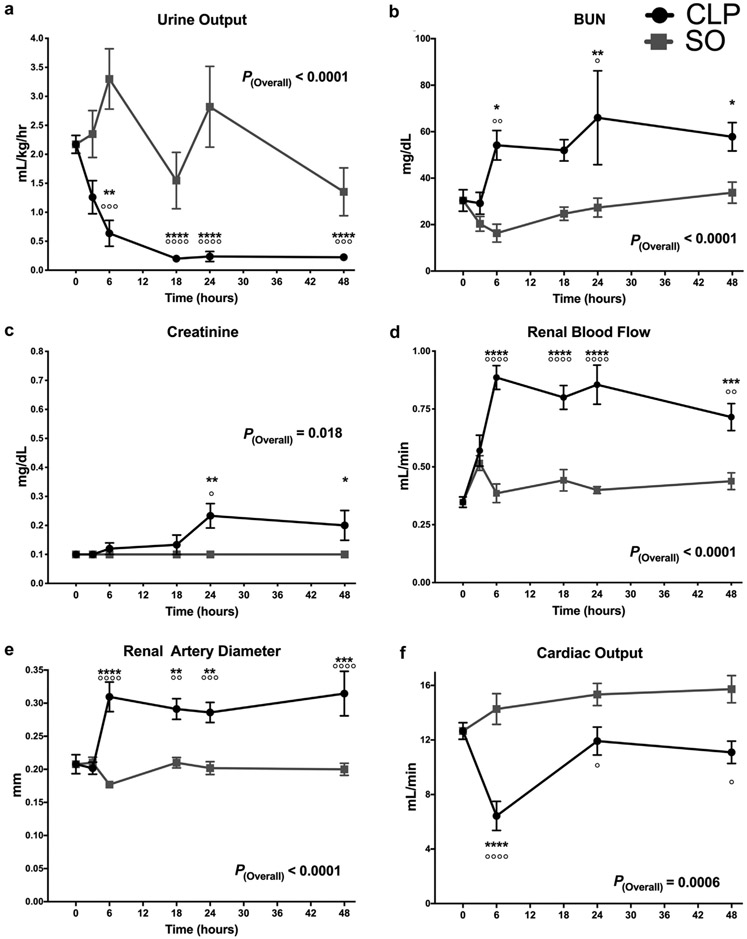
CLP versus sham operation (SO). Data as mean ± SD. p(overall) indicates the overall P value for the difference between CLP and SO groups. P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus T0 controls. °P < 0.05, °°P < 0.01, °°°P < 0.001, °°°°P < 0.0001 for CLP versus SO at that time point. P values calculated using 2-way analysis of variance with Sidak’s post hoc correction for multiple comparisons. Data in (a) violated the homoscedastic error assumption and were subjected to natural-logarithm transformation. The data in (a) were transformed for P value calculation but untransformed values were graphed to provide clinically relevant values for visual interpretation. (a) Urine output (UOP). (b) Blood urea nitrogen (BUN). (c) Creatinine. (d) Renal blood flow. (e) Renal artery diameter. (f) Cardiac output.
CLP is not associated with acute tubular necrosis
Periodic acid–Schiff staining revealed mild focal tubular distention beginning at 24 hours post-CLP. Otherwise, there was minimal evidence of structural or inflammatory injury (Supplementary Figure S1). Terminal deoxynucleotidyl transferase–mediated dUTP nick end-labeling staining demonstrated a small number of apoptotic bodies in both baseline and CLP mice (Supplementary Figure S2).
CLP induces globally elevated RBF and low vascular resistance
At 6 hours post-CLP, RBF increased 2-fold versus baseline (0.89 ml/min vs. 0.35 ml/min, 95% CI: 0.28–0.80 ml/min) and sham operation (0.89 ml/min vs. 0.39 ml/min, 95% CI:0.28–0.72 ml/min). This difference persisted through 48 hours (Figure 1d) (P < 0.0001) and was accompanied by a 50% increase in arterial diameter (Figure 1e). In addition, the morphology of the renal artery Doppler waveform became progressively less pulsatile after CLP (Figure 2). By 6 hours post-CLP, log-renovascular resistance decreased 2-fold from baseline (Supplementary Figure S3). This change was sustained through 48h post-CLP. In contrast to RBF, cardiac output initially fell post-CLP (P < 0.0001) but returned to preoperative levels by 24 hours post-CLP (Figure 1f).
Figure 2 ∣. Cecal ligation and puncture (CLP) alters renal arterial pulsewave Doppler waveform morphology.

Representative images of pulsewave Doppler recording used to measure renal arterial velocities. Blue indicates flow away from the ultrasound probe (toward the kidney) and red toward the probe (away from the kidney). Waveforms show arterial velocity on the Y-axis and time on the X-axis. (a) T0 mouse. (b) Mouse 48 hours after CLP.
CLP induces kidney injury molecule-1 expression in the kidney
Enzyme-linked immunosorbent assay (ELISA) revealed markedly elevated kidney injury molecule-1 (KIM-1) in kidneys taken from mice sacrificed 18 hours post-CLP and at later time points (P < 0.0001) (Figure 3b). However, there was no KIM-1 elevation at 6 hours post-CLP. Mice did not have elevated KIM-1 at any time after sham operation.
Figure 3 ∣. Cecal ligation and puncture (CLP) induces early downregulation of renal angiotensin II type-1 receptor (AT1R) and kidney injury molecule 1 (KIM-1) elevation.
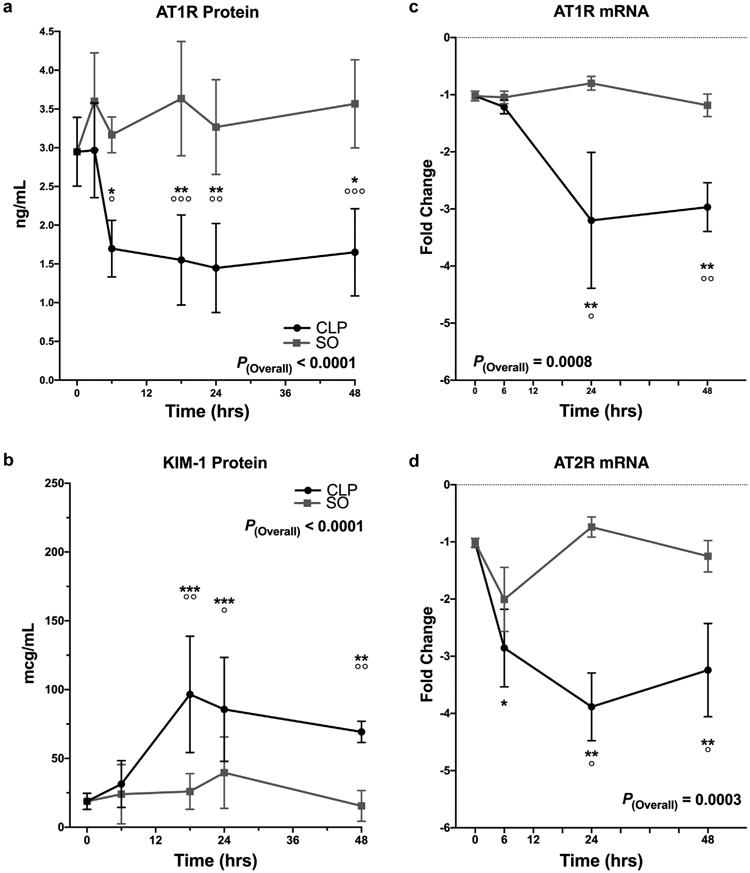
Concentration of specific proteins and mRNA in whole kidney homogenate determined using enzyme-linked immunosorbent assay and quantitative polymerase chain reaction, respectively. Data as mean ± SD. P(overall) indicates the overall p-value for the difference between CLP and sham operation (SO) groups. P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus T0 controls. °P < 0.05, °°P < 0.01, °°°P < 0.001, °°°°P < 0.0001 for CLP versus SO at that time point. The 95% confidence intervals (CIs) of group differences and P values are determined using 2-way analysis of variance with Sidak’s post hoc correction for multiple comparisons. (a,b) Black circles = CLP; gray squares = SO. (a) AT1R protein in CLP versus SO. The 95% CIs and P values for CLP versus SO at each time point were as follows: 3 hours: −1.8 to 0.6 ng/ml, P = 0.6; 6 hours: −2.7 to −0.2 ng/ml, P = 0.013; 18 hours: −3.3 to −0.9 ng/ml, P < 0.001; 24 hours: −3.1 to −0.6 ng/ml, P = 0.002; 48 hours: −3.1 to −0.7 ng/ml; P = 0.001. (b) KIM-1 protein in CLP versus SO. The 95% CIs and P values at each time point were as follows: 6 hours: −34 to 49 mcg/ml, P = 0.99; 18 hours: 22 to 119 mcg/ml, P = 0.002; 24 hours: 4 to 88 mcg/ml, P = 0.025; 48 hours: 14 to 93 mcg/ml, P = 0.004. (c) AT1R mRNA in CLP versus SO. Y-axis shows fold-change relative to the glyceraldehyde-3-phosphate dehydrogenase housekeeping mRNA. 6 hours: −1.5 to 1.2 relative units, P = 0.99; 24 hours: −4.5 to −0.3 relative units, P = 0.021; 48 hours: −3.1 to −0.5 relative units, P = 0.004. (d) AT2R mRNA in CLP versus SO; 6 hours: −2.7 to 1.0 relative units, P = 0.66; 24 hours: −5.7 to −0.6 relative units, P = 0.011; 48 hours: −3.7 to −0.3 relative units, P = 0.013. AT2R, angiotensin II type 2 receptor.
CLP induces an early reduction in the expression of AT1R
ELISA measurements showed CLP reduced AT1R protein expression 2-fold within 6 hours of surgery. This decrease persisted to 48 hours (P < 0.0001) (Figure 3a). No change was observed with sham operation.
Quantitative polymerase chain reaction (qPCR) showed that CLP did not alter AT1R mRNA expression at 6 hours (Figure 3c). However, by 24 and 48 hours post-CLP AT1R mRNA expression had fallen 3-fold (P = 0.0008). No change occurred postsham operation.
Immunofluorescence microscopy (Figure 4) demonstrated that mean AT1R fluorescence intensity fell more than 2-fold at 6, 24, and 48 hours after CLP. This change was noted within periglomerular arterioles, macula densa, and large vessel smooth muscle cells (P < 0.0001 for all). In contrast, glomerular AT1R expression was unchanged through 24 hours and was only decreased after 48 hours. AT1R mean intensity determined at 24 hours post-CLP was also significantly lower in all regions than similar measurements made 48 hours posthemorrhagic shock.
Figure 4 ∣. Cecal ligation and puncture (CLP) decreases renal angiotensin II type-1 receptor (AT1R) expression in different regions of the renal cortex.

Representative fluorescence microscopic images of fixed kidney sections at original magnification ×20. Different structures labeled with different fluorescent tags. Green = AT1R; red = smooth muscle α-actin (smooth muscle); magenta = Na-K-Cl cotransporter (macula densa); blue = 4′,6-diamidino-2-phenylindole (nuclei). (a) T0 control. (b) Hemorrhagic shock (HS) control—kidney harvested 48 hours post–40% blood loss. (c) Six hours post-CLP. (d) Twenty-four hours post-CLP. (e) Forty-eight hours post-CLP. (f) Graphic depiction of quantification of AT1R fluorescence intensity in different renal structures at T0, at 48 hours post-HS or at 48 hours post-CLP. Bars indicate mean ± SD. P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns = P > 0.05 for the group difference. Brackets indicate the 2 groups being compared. Red = arterioles; magenta = macula densa; gray = arterial smooth muscle cells; blue = glomeruli. (g) Graphic depiction of quantification of AT1R fluorescence intensity in different renal structures at different time points post-CLP. Point and error bar indicate mean ± SD. Colors represent region noted above. Asterisks indicate P value levels compared with T0 controls as above. SMCs, smooth muscle cells. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
AT1R blockade exacerbates CLP-induced kidney injury without evidence of global renal hypoperfusion
We treated mice with losartan, a selective AT1R antagonist, to determine whether AT1R inhibition exacerbated abnormalities identified post-CLP. Decreases in urine output appeared at earlier post-CLP time points and were more severe in losartan-treated mice than mice subjected to CLP and vehicle (95% CI: 0.04–0.26 ml/kg/h lower, P = 0.0002) (Figure 5a). Specifically, oliguria was noted within 6 hours of CLP, and despite a second fluid bolus, anuria was often present by the 48th post-CLP hour. By 24 hours post-CLP, blood urea nitrogen was significantly higher in losartan-treated mice than in their vehicle-treated counterparts (101 mg/dl versus 75 mg/dl, 95% CI: 4–48 mg/dl, P = 0.028) (Figure 5b). Losartan treatment also accelerated the CLP-induced increase in creatinine (mean 48-hour creatinine: 0.80 mg/dl versus 0.27 mg/dl, 95% CI: 0.27–0.79 mg/dl, P < 0.0001) (Figure 5c). In contrast to vehicle-treated mice, the initial fall in cardiac output did not recover to T0 levels by 48 hours post-CLP in losartan-treated mice (P = 0.027 versus T0) (Figure 6).
Figure 5 ∣. Cecal ligation and puncture (CLP)-induced changes in renal hemodynamics and function are mediated by angiotensin II type-1 receptor (AT1R)-dependent angiotensin II signaling.
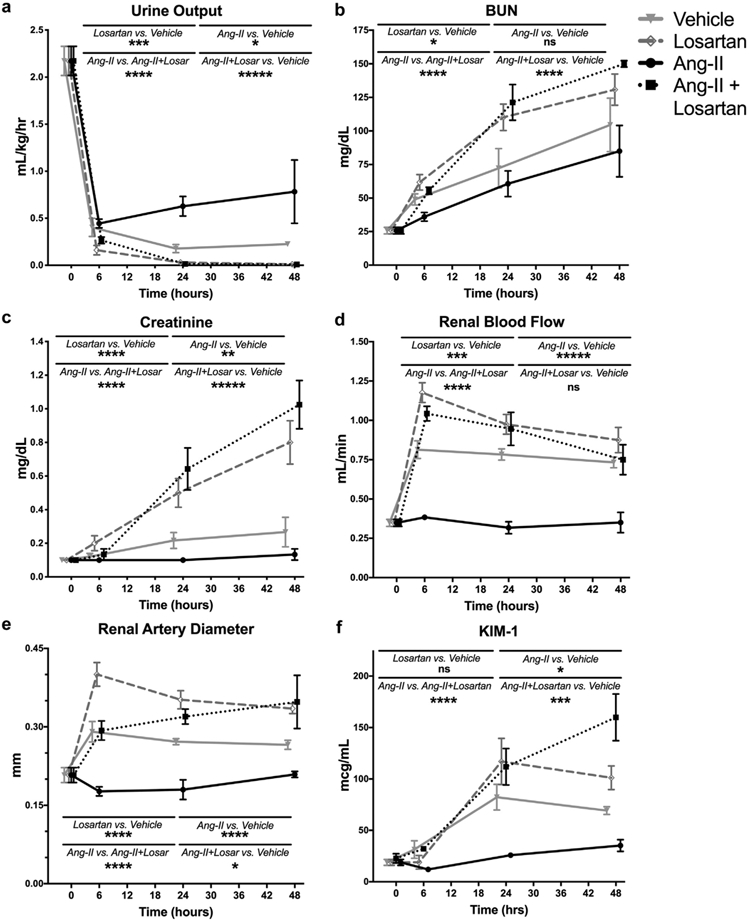
Data as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns = P > 0.05 for group difference. P values calculated using 2-way analysis of variance with Sidak’s post hoc correction for multiple comparisons. Gray circles/solid gray line = CLP + vehicle; black circles/solid black line = CLP + Ang-II, gray diamonds/dashed gray line = CLP + losartan; black boxes/dashed black line = CLP + Ang-II + losartan. (a) Urine output. (b) Blood urea nitrogen (BUN). (c) Creatinine. (d) Renal blood flow. (e) Renal artery diameter. (f) KIM-1.
Figure 6 ∣. Angiotensin II type-1 receptor (AT1R) modulation alters the trajectory of cardiac output after cecal ligation and puncture (CLP).
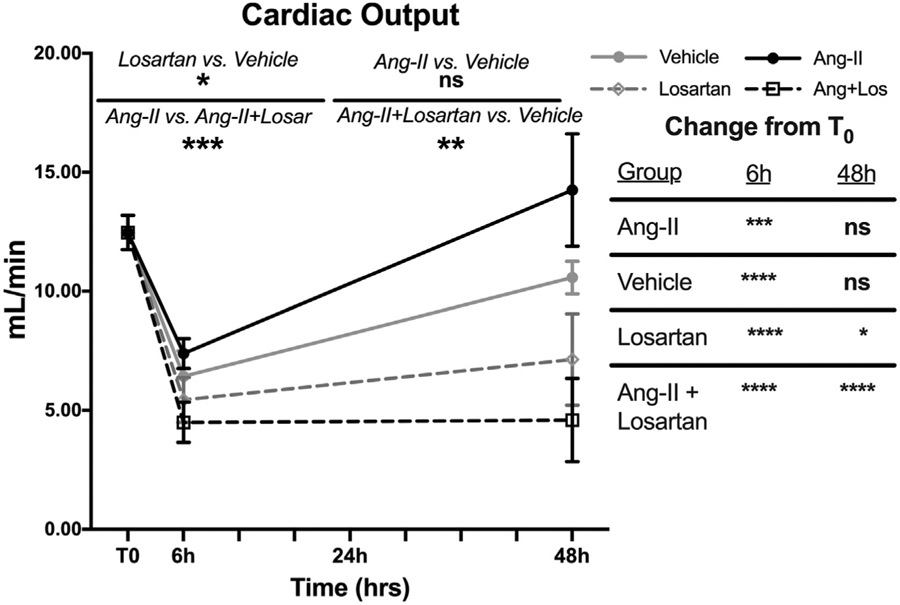
Data as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns = P > 0.05 for the group difference. P values calculated using 2-way analysis of variance with Sidak’s post hoc correction for multiple comparisons. Gray circles/solid gray line = CLP + vehicle; black circles/solid black line = CLP + Ang-II; gray diamonds/dashed gray line = CLP + losartan; black boxes/dashed black line = CLP + Ang-II + losartan. Table at right shows comparison of the treatment group at the indicated time to the T0 measurements.
Despite worsened renal function and reduced cardiac output with losartan treatment, sections stained with periodic acid–Schiff demonstrated that losartan treatment did not result in ischemic renal injury. Treatment with losartan significantly augmented the CLP-induced increases in RBF and arterial diameter (Figure 5d,e). CLP-induced decreases in total renovascular resistance were significantly more pronounced following losartan treatment (Supplemental Figure 4).
Angiotensin II reverses CLP-induced kidney injury by stimulating AT1R
In contrast to vehicle treatment, administration of angiotensin II prevented CLP-induced changes in RBF, renal artery diameter, and renovascular resistance (Figure 5, Supplementary Figure S4). Angiotensin II treatment also prevented CLP-induced creatinine elevation (P = 0.004 vs. vehicle) and attenuated oliguria (P = 0.015 vs. vehicle). KIM-1 levels were significantly lower in angiotensin II treated mice vs. vehicle (P = 0.021) (Figure 5). The cardiac output trajectory in angiotensin-treated mice post-CLP was similar to that in the vehicle-treated group (Figure 6).
Coadministering losartan with angiotensin II blocked the effects of angiotensin II alone. Compared with angiotensin II alone, mice undergoing CLP who were treated with both losartan and angiotensin II had greater elevations in blood urea nitrogen, creatinine, RBF, and renal artery diameter (P < 0.0001 for all), and greater decreases in urine output and total renovascular resistance (P < 0.0001 for both) (Figure 5, Supplementary Figure S4). These CLP-induced changes in mice treated with both losartan and angiotensin II were all of similar magnitude to those in mice treated with losartan alone. Following CLP, elevated KIM-1 levels in mice treated with both losartan and angiotensin II were significantly higher than in vehicle-treated (P = 0.028) or angiotensin II-treated (P < 0.0001) mice but were not significantly different from mice that were treated with losartan alone (P = 0.41) (Figure 5). Post-CLP cardiac output remained depressed through 48 hours in mice that received both drugs (P < 0.0001 vs. T0) (Figure 6).
Recently deceased sepsis patients show decreased renal AT1R expression
Relative to healthy and nonseptic critically ill controls, immunofluorescence imaging of kidney sections obtained from patients who recently died of sepsis showed markedly decreased AT1R fluorescence. Immunofluorescent colocalization indicated that these differences were most pronounced in the macula densa, arterioles, tubules, and glomeruli (Figure 7), as well as large resistance vessels (Supplemental Figure 5).
Figure 7 ∣. Recently deceased sepsis patients show decreased renal angiotensin II type-1 receptor (AT1R).

Representative immunofluorescence images of renal cortical tissue from sepsis patients versus healthy and noninfected critically ill controls. Fluorescence: Green = AT1R; red = smooth muscle α-actin (smooth muscle); magenta = Na-K-Cl cotransporter (macula densa); blue = 4′,6-diamidino-2-phenylindole (nuclei). Control (Ctrl) is a serial section in which the primary antibody cocktail was preincubated with free AT1R peptide to prevent anti-AT1R primary antibody from binding to target antigen in tissue. Green fluorescence in the sample that is absent in the adjacent control therefore confirms the specificity of the signal to AT1R. (a) Healthy tissue. (b) Serial section from healthy tissue where anti-AT1R antibody was preadsorbed with control peptide. (c) Tissue from a sepsis victim. (d) Serial section from a sepsis victim with preadsorbed anti-AT1R. (e) Tissue from a deceased noninfected critically ill control. (f) Serial section with preadsorbed anti-AT1R from a deceased noninfected critically ill control. ICU, intensive care unit; MD, macula densa. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
Periodic acid–Schiff sections showed minimal glomerular sclerosis and interstitial fibrosis in all 3 groups. There was no evidence of acute interstitial nephritis in any samples. All sepsis patients had some mild acute tubular injury, whereas none was observed in healthy controls (Supplemental Figure 6).
DISCUSSION
In controlled animal CLP experiments and in immunohistological studies of septic human kidneys, we found evidence of decreased AT1R expression. In CLP, decreased AT1R expression appeared to contribute to the development of acute kidney injury. In particular, AT1R protein expression throughout renal cortical and vascular tissues was significantly reduced within 6 hours of CLP and persisted to 48 hours. These molecular findings chronologically aligned with the development of characteristic physiology predicted by impaired AT1R signaling: oliguric renal dysfunction and high-flow, low-resistance renal hemodynamics. Selective AT1R antagonism exacerbated these physiologic changes, whereas angiotensin II infusion ameliorated them in an AT1R-dependent fashion. Finally, we found similar reduction in AT1R expression in renal tissue obtained from recently deceased sepsis patients.
Our findings suggest that renal AT1R receptor downregulation contributes to the pathophysiology of SIAKI. A decrease in renal AT1R was absent in both noninfected critically ill patients and mice subjected to hemorrhagic shock. In contrast to our observations, prior data report that AT1Rantagonism enhances the restoration of RBF after ischemic kidney injury.15 In this context, our study further indicates that some aspect of infection-induced critical illness, rather than critical illness alone, uniquely impairs AT1R-mediated angiotensin signaling.
Whereas renal AT1R protein decreased within 6 hours, mRNA did not decrease until 24 hours after CLP. These findings are compatible with early endocytosis-mediated downregulation and a later, CLP-induced suppression of transcription.16,17 This hypothesis is consistent with a prior study that found endotoxemia reduces expression of ARAP1, a protein that promotes AT1R cell-surface recycling, within 3 hours of insult.18
KIM-1 elevations were noted later in the disease course than oliguria and azotemia, suggesting that renal dysfunction occurs before tubular cell injury. Therefore, it is unlikely that tubular injury is responsible for the observed decreases in urine output and increases in urea and creatinine levels. In contrast, AT1R downregulation developed in parallel with renal dysfunction and before KIM-1 elevations. Angiotensin II treatment attenuated decreases in urine output and increases in urea and creatinine levels and prevented a rise in KIM-1. Taken together, these findings further implicate decreased renal AT1R expression and function in the pathogenesis of SIAKI.
Importantly, a blinded clinical renal pathologist reported no evidence of cortical ischemia or acute tubular necrosis in either untreated or losartan-treated mice. In fact, losartan-treated mice had higher RBF post-CLP than vehicle-treated mice despite lower cardiac output, suggesting CLP dissociates renovascular and systemic hemodynamics. Therefore, the exaggeration of CLP-induced renal dysfunction accompanying AT1R blockade cannot be logically explained by renal hypoperfusion or acute tubular necrosis.
CLP-induced changes in AT1R expression and the accompanying exacerbation of renal injury were consistent with findings in tissue from recently deceased sepsis patients. The human tissue also showed only mild acute tubular injury, consistent with prior histopathological studies of clinical and experimental SIAKI that repeatedly demonstrate only mild, nonspecific injury.19-23 Several investigations of clinical sepsis and large mammalian sepsis models documented increased RBF8,9,24 and restoration of normal blood flow with angiotensin II treatment,25-27 similar to our findings in mice. These concordant results provide evidence that, at least in the first 48 hours, SIAKI does not reflect global renal hypoperfusion or severe tubular injury but rather functional changes in glomerular hemodynamics.
Although our study was translational, we believe our results are clinically relevant. The low renal AT1R expression in septic patients is consistent with clinical literature reporting that hyperreninemia is common and prognostic in patients with sepsis-induced renal dysfunction.28,29 Compensatory increases in renin release and angiotensin formation should occur in response to decreased AT1R. This may explain the substantially elevated plasma renin and angiotensin levels present in patients with sepsis, and their previously observed correlation with adverse outcomes.30-33
Our findings may have clinical implications. First, identifying indirect evidence of impaired angiotensin signaling might provide diagnostic, prognostic, or predictive information. Recent data suggest that renin levels might reflect global perfusion in critically ill patients.32 High renin also identified a population of hyperresponders to angiotensin II treatment in the ATHOS-III trial.33 More importantly, angiotensin II may have value beyond its ability to reverse sepsis-induced hypotension. Angiotensin II is now an U.S. Food and Drug Administration–approved therapy for distributive shock.34 Our data suggest that angiotensin II might directly alter the pathophysiology that underlies SIAKI, and therefore attenuate or reverse renal dysfunction. Post hoc analysis of data from the recent ATHOS-III trial reported better outcomes in patients receiving renal replacement therapy at enrollment.35 Sepsis patients previously on chronic renin-angiotensin system blockade therapy are more likely to develop acute kidney injury, have more severe injury when kidney injury develops, and have worse outcomes.36-38 In contrast, studies that did not specify the cause of acute kidney injury found no association between prior renin-angiotensin system blockade and long-term proteinuria.39 Our results provide a plausible biochemical explanation for the above clinical findings in sepsis, support the need for a randomized trial of angiotensin II in the treatment of SIAKI, and suggest a need for future work exploring whether angiotensin-modulating approaches should be tailored to the cause of kidney injury.
We highlight several limitations. CLP has welldocumented deficiencies as a model of clinical sepsis.40,41 However, CLP has important advantages over many other sepsis models used in rodents, such as endotoxemia, and more reliably recapitulates many aspects of human diesease.42-44 Further, singly housing mice in metabolic cages likely recapitulated chronic daily stress, which is shown to be an important component of sepsis modeling in animals.45 Concerns regarding the use of mice in the study of sepsis also abound.46 Sepsis is a heterogenous clinical entity with respect to site, species, and response to infection, limiting the applicability of all standardized attempts to model the syndrome in vivo. Despite these limitations, our findings are pathophysiologically consistent with results from studies of large mammals and human patients, including our experiments using human tissue. Additionally, we could not conduct imaging and tissue collection in a blinded fashion. Concealment was hampered because losartan-treated mice became visibly sicker than untreated mice, while angiotensin II treated mice appeared patently less ill. Ascertainment bias could therefore impact hemodynamic measurements. Finally, we modulated the angiotensin system pharmacologically, which cannot rule out offtarget effects as rigorously as a selective genetic knockout model.
In conclusion, renal AT1R expression was reduced following CLP and in patients succumbing to sepsis. In mice, pharmacological modulation of AT1R-mediated angiotensin II signaling altered renal dysfunction in the early post-CLP period. These changes did not result from cortical ischemia or global renal hypoperfusion. These findings imply that AT1R-mediated angiotensin II signaling is important to protect from the development of septic kidney injury.
METHODS
Ethics statement regarding animal and human experiments
All animal studies were approved by the Institutional Animal Care and Use Committee at the Feinstein Institute for Medical Research and adhered to National Institutes of Health guidelines as well as Animal Research: Reporting of In Vivo Experiments criteria.47 The Northwell Health Institutional Review Board determined our experiments on human tissues were review exempt.
Mice
Male C57BL/6 mice aged 12 to 14 weeks (Jackson Labs, Farmington, CT) were acclimated and maintained in a conventional, light-cycled facility.
CLP versus sham
In the first cohort, mice were a priori randomized to an exposure (CLP vs. sham operation) and a terminal timepoint (0, 3, 6, 18, 24, and 48 hours postsurgery) at which surviving mice were sacrificed. In a second cohort, all mice underwent CLP but were randomized to a treatment and terminal timepoint (0, 6, 24, or 48 hours post-CLP). We allocated 5 mice to each combination of surgery, treatment, and time of euthanasia (for 48 hours, 2 additional mice were preallocated to CLP to account for early mortality). We used an additional 5 mice that did not undergo surgery as a baseline. A single author, blinded to time and treatment allocation, performed all CLP or sham surgeries as previously described.48 Sham involved laparotomy and cecal manipulation without ligation and puncture.48 Mice were resuscitated with subcutaneous 0.9% saline (50 ml/kg) immediately following surgery and again at 24 hours postintervention. We withheld antibiotics to avoid potentially confounding nephrotoxicity. To obtain serial echocardiographic measurements, 1 additional cohort underwent CLP (n = 13) or sham operation (n = 5) while another cohort underwent CLP with each combination of treatment (n = 7/group).
Study drugs
In the second cohort, we allocated mice to 4 treatments: losartan (a selective AT1R antagonist), angiotensin II, angiotensin II+losartan, or vehicle. We administered losartan (15 mg/kg i.p.) at the time of surgery and again 24 h later. Synthetic angiotensin II (LaJolla Pharmaceuticals, La Jolla, CA), was diluted in 0.9% saline and infused continuously (10 ng/kg/min at 1 μl/h)34 using i.p. microosmotic pumps (Alzet 1003D, ALZET Osmotic Pumps, Cupertino, CA) implanted at the time of CLP. Control mice had vehicle-infusing pumps implanted. Mice allocated to angiotensin II+losartan treatment received losartan at a higher dose (45 mg/kg) to ensure competition for AT1R would favor losartan over angiotensin II.
Post-CLP and terminal point procedures
We used metabolic cages to monitor post-operative urine output. We considered urine output <0.5 ml/kg/h to be oliguria.14 At the prespecified time for euthanasia, anesthetized mice (inhaled isoflourane) underwent sonography. Immediately after imaging, we performed in vivo bilateral nephrectomy to minimize potentially confounding ischemia. One kidney was immediately fixed in formalin for 24 h; the second was flash-frozen. Mice were then exsanguinated by cardiac puncture, with blood used for point-of-care assays.
In vivo imaging
Renal artery diameter and arterial velocities (measured 0.5 mm medial to the renal capsule) were measured using ultrasongraphy and pulsewave Doppler (Vevo 3100, Fujifilm Visual Sonics, Toronto, Canada). RBF was calculated with the formula:
We also calculated the renal arterial pulse-power and used Ohm’s law to estimate the total renovascular resistance (details in supplement) using the formula:
Echocardiography was performed (Vevo 3100, Fujifilm Visual Sonics, Toronto, Canada) by a single, blinded, experienced cardiologist (CC) according to a standardized image acquisition protocol. Images were obtained under isoflurane titrated to target a heart rate of 400 beats per minute to standardize anesthetic effects on cardiac function during imaging. Standard 2-dimensional (B-mode), Doppler and pulsed tissue Doppler studies were performed. The B-mode parasternal long axis view was used to measure end-diastolic volume, end-systolic volume, stroke volume, and cardiac output.
Assessment of renal injury
A clinical renal pathologist blinded to time, operation and/or treatment examined periodic acid–Schiff-stained kidney sections for evidence of ischemic/structural damage using a previously described grading system.19 We identified apoptosis with terminal deoxynucleotidyl transferase–mediated dUTP nick end-labeling staining and tubular injury using an ELISA kit to detect KIM-1 (Abcam, Cambridge, MA). Blood urea nitrogen and creatinine levels were measured from whole blood immediately upon collection using iStat Chem8+ cartridges, a point-of-care measurement device (Abbott Point of Care Inc., Princeton, NJ) that has excellent correlation (R2 = 0.99) with clinical core lab values.49
Regional and total AT1R expression
We coincubated tissue sections with rabbit anti-AT1R (Alomone Labs, Jerusalem, Israel), mouse anti-smooth muscle α-actin (to identify arterioles; Novus Biologicals, Centennial, CO), and goat anti-Na-K-Cl cotransporter antibodies (to identify macula densa; OriGene Technologies, Rockville, MD). We used fluorescently tagged secondary antibodies preadsorbed to have <5% species crossreactivity for detection. Images (20×) were obtained using identical acquisition settings for each experiment. A brightfield channel was also captured for each image to aid in structural confirmation. Mean AT1R fluorescence intensity within individual structures was determined using ZEN:Blue software (Zeiss International, North Chesterfield, VA). (For additional details, see the supplementary materials.)
As additional controls, we also determined fluorescence in sections taken from mice (n = 2) 48 h after hemorrhagic shock was induced by 40% blood loss (a gift from Joaquin Cagliani, MD; model as described50).
Total AT1R protein expression was determined by ELISA (MyBioSource, San Diego, CA). mRNA expression was determined by qPCR. (Detailed methods in the supplement; see validation assay in Supplementary Figure S7).
AT1R expression in recently deceased human sepsis patients
Human tissue experiments were conducted using a case-control study design. Renal tissue from critically ill sepsis patients (n = 7) fixed within an hour of death (a gift from Richard Hotchkiss, MD) was compared with both healthy tissue from patients undergoing nephrectomy for renal carcinoma (n = 10) and autopsy tissue obtained from noninfected critically ill patients (n = 3). Inclusion criteria and details of tissue collection and processing were described previously.19 Patients with any known prior chronic kidney disease were excluded from the healthy tissue group. For the noninfected critically ill group, inclusion criteria were admission to an intensive care unit for >24 hours with at least 1 form of organ support (defined: endotracheal intubation with mechanical ventilation, continuous vasopressor or inotrope infusion, continuous renal replacement therapy or acute dialysis, or extracorporeal membrane oxygenation), absence of any clinical evidence or suspicion of acute infection, and absence of prior history of end-stage renal disease. We performed staining and immunofluorescence assays as described earlier. (Additional details in supplement.)
Statistical analysis
Individual mice or patients were the unit of analysis. Two-way analysis of variance with Sidak corrections for multiple comparisons and generalized linear models, as appropriate, were used to identify differences between groups over time. Data distributions were assessed before statistical hypothesis testing, and an appropriate transformation was applied if found to violate assumptions.
Supplementary Material
Supplemental Methods Section. (1) Estimation of total renovascular resistance. (2) AT1R expression assessment. (3) AT1R protein expression assessment in tissue sections. (4) Validation of AT1R ELISA for whole tissue homogenate. (5) AT1R mRNA expression assessment by qPCR. (6) AT1R expression in human sepsis.
Figure S1. Untreated and losartan-treated mice with cecal ligation and puncture–induced renal dysfunction show mild, nonspecific renal histopathology.
Figure S2. Terminal deoxynucleotidyl transferase–mediated dUTP nick end-labeling (TUNEL) stain for apoptosis.
Figure S3. Estimated total renovascular resistance in cecal ligation and puncture (CLP) versus sham operation.
Figure S4. Estimated total renovascular resistance by treatment group over time following cecal ligation and puncture.
Figure S5. Recently deceased sepsis patients had low angiotensin II type-1 receptor (AT1R) in large renal resistance vessels.
Figure S6. Human sepsis patients display mild, nonspecific histopathologic injury.
Figure S7. Validation of the enzyme-linked immunosorbent assay (ELISA) for angiotensin II type-1 receptor (AT1R) concentration in whole kidney homogenate.
Translational Statement.
Sepsis-induced acute kidney injury (SIAKI) is the most common type of AKI. Here, we report decreased angiotensin II type-1 receptor (AT1R) expression in sepsis patients’ kidneys relative to healthy and noninfected critically ill controls. In mice, cecal-ligation and puncture (a sepsis model) reduced renal AT1R before tubular injury and simultaneously with physiologic changes that suggest decreased renal angiotensin II signaling. In mice, renal dysfunction/injury was exacerbated by AT1R antagonism, attenuated by angiotensin II, and dissociated from cardiac output. Decreased renal AT1R expression may play an important role in the pathophysiology of SIAKI.
ACKNOWLEDGMENTS
Human synthetic angiotensin II used in this study was generously provided by La Jolla Pharmaceutical Company. La Jolla Pharmaceutical Company had no access to any of the data in this study, was not involved in any part of its analysis or interpretation, and had no role in the drafting or editing of this manuscript.
We thank Richard Hotchkiss, MD, for providing human sepsis tissue specimens; Joaquin Cagliani, MD, for providing tissue samples from mice subjected to a model of hemorrhagic shock; and Lahkmir Chawla, MD, and John Kellum, MD, PhD, for their comments on preliminary results from these experiments. MDT received grant support (NIGMS K08GM132794), and CSD received grant support (R01GM121102).
Footnotes
DISCLOSURE
MDT receives grant support from the National Institutes of Health (NIH) National Institute of General Medical Sciences (NIGMS). CSD receives grant support from NIH NIGMS and is a consultant for Enlivex Therapeutics. Views expressed in this article do not necessarily represent the views of the NIH or NIGMS. All other authors declared no competing interests.
REFERENCES
- 1.Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315:801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ali T, Khan I, Simpson W, et al. Incidence and outcomes in acute kidney injury: a comprehensive population-based study. J Am Soc Nephrol. 2007;18:1292–1298. [DOI] [PubMed] [Google Scholar]
- 3.Bagshaw SM, George C, Bellomo R, Committee ADM. Early acute kidney injury and sepsis: a multicentre evaluation. Crit Care. 2008;12:R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kellum JA, Prowle JR. Paradigms of acute kidney injury in the intensive care setting. Nat Rev Nephrol. 2018;14:217–230. [DOI] [PubMed] [Google Scholar]
- 5.Gomez H, Kellum JA. Sepsis-induced acute kidney injury. Curr Opin Crit Care. 2016;22:546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellomo R, Kellum JA, Ronco C, et al. Acute kidney injury in sepsis. Intensive Care Med. 2017;43:816–828. [DOI] [PubMed] [Google Scholar]
- 7.Langenberg C, Bellomo R, May C, et al. Renal blood flow in sepsis. Crit Care. 2005;9:R363–R374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langenberg C, Wan L, Egi M, et al. Renal blood flow in experimental septic acute renal failure. Kidney Int. 2006;69(11):1996–2002. [DOI] [PubMed] [Google Scholar]
- 9.Correa TD, Vuda M, Takala J, et al. Increasing mean arterial blood pressure in sepsis: effects on fluid balance, vasopressor load and renal function. Crit Care. 2013;17:R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Post EH, Kellum JA, Bellomo R, Vincent JL. Renal perfusion in sepsis: from macro- to microcirculation. Kidney Int. 2017;91:45–60. [DOI] [PubMed] [Google Scholar]
- 11.Ma S, Evans RG, Iguchi N, et al. Sepsis-induced acute kidney injury: a disease of the microcirculation. Microcirculation. 2019;26:e12483. [DOI] [PubMed] [Google Scholar]
- 12.Ingels C, Gunst J, Van den Berghe G. Endocrine and metabolic alterations in sepsis and implications for treatment. Crit Care Clin. 2018;34:81–96. [DOI] [PubMed] [Google Scholar]
- 13.Deutschman CS, Tracey KJ. Sepsis: current dogma and new perspectives. Immunity. 2014;40:463–475. [DOI] [PubMed] [Google Scholar]
- 14.Kellum JA, Lameire N, Group KAGW. Diagnosis, evaluation, and management of acute kidney injury: a KDIGO summary (Part 1). Crit Care. 2013;17:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodríguez-Romo R, Benítez K, Barrera-Chimal J, et al. AT1 receptor antagonism before ischemia prevents the transition of acute kidney injury to chronic kidney disease. Kidney Int. 2016;89:363–373. [DOI] [PubMed] [Google Scholar]
- 16.Deutschman CS, De Maio A, Clemens MG. Sepsis-induced attenuation of glucagon and 8-BrcAMP modulation of the phosphoenolpyruvate carboxykinase gene. Am J Physiol. 1995;269(3 pt 2):R584–591. [DOI] [PubMed] [Google Scholar]
- 17.Abcejo AS, Andrejko KM, Raj NR, Deutschman CS. Failed interleukin-6 signal transduction in murine sepsis: attenuation of hepatic glycoprotein 130 phosphorylation. Crit Care Med. 2009;37:1729–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mederle K, Schweda F, Kattler V, et al. The angiotensin II AT1 receptorassociated protein Arap1 is involved in sepsis-induced hypotension. Crit Care. 2013;17:R130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takasu O, Gaut JP, Watanabe E, et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am J Respir Crit Care Med. 2013;187:509–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kosaka J, Lankadeva YR, May CN, Bellomo R. Histopathology of septic acute kidney injury: a systematic review of experimental data. Crit Care Med. 2016;44:e897–e903. [DOI] [PubMed] [Google Scholar]
- 21.Langenberg C, Gobe G, Hood S, et al. Renal histopathology during experimental septic acute kidney injury and recovery. Crit Care Med. 2014;42:e58–e67. [DOI] [PubMed] [Google Scholar]
- 22.Arulkumaran N, Sixma ML, Jentho E, et al. Sequential analysis of a panel of biomarkers and pathologic findings in a resuscitated rat model of sepsis and recovery. Crit Care Med. 2017;45:e821–e830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arulkumaran N, Pollen S, Greco E, et al. Renal tubular cell mitochondrial dysfunction occurs despite preserved renal oxygen delivery in experimental septic acute kidney injury. Crit Care Med. 2018;46:e318–e325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lankadeva YR, Kosaka J, Iguchi N, et al. Effects of fluid bolus therapy on renal perfusion, oxygenation, and function in early experimental septic kidney injury. Crit Care Med. 2019;47:e36–e43. [DOI] [PubMed] [Google Scholar]
- 25.May CN, Ishikawa K, Wan L, et al. Renal bioenergetics during early gramnegative mammalian sepsis and angiotensin II infusion. Intensive Care Med. 2012;38:886–893. [DOI] [PubMed] [Google Scholar]
- 26.Wan L, Langenberg C, Bellomo R, May CN. Angiotensin II in experimental hyperdynamic sepsis. Crit Care. 2009;13:R190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lankadeva YR, Kosaka J, Evans RG, et al. Urinary oxygenation as a surrogate measure of medullary oxygenation during angiotensin II therapy in septic acute kidney injury. Crit Care Med. 2018;46:e41–e48. [DOI] [PubMed] [Google Scholar]
- 28.du Cheyron D, Lesage A, Daubin C, et al. Hyperreninemic hypoaldosteronism: a possible etiological factor of septic shock-induced acute renal failure. Intensive Care Med. 2003;29(10):1703–1709. [DOI] [PubMed] [Google Scholar]
- 29.Findling JW, Waters VO, Raff H. The dissociation of renin and aldosterone during critical illness. J Clin Endocrinol Metab. 1987;64:592–595. [DOI] [PubMed] [Google Scholar]
- 30.Doerschug KC, Delsing AS, Schmidt GA, Ashare A. Renin-angiotensin system activation correlates with microvascular dysfunction in a prospective cohort study of clinical sepsis. Crit Care. 2010;14:R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hilgenfeldt U, Kienapfel G, Kellermann W, et al. Renin-angiotensin system in sepsis. Clin Exp Hypertens A. 1987;9(8-9):1493–1504. [DOI] [PubMed] [Google Scholar]
- 32.Gleeson PJ, Crippa IA, Mongkolpun W, et al. Renin as a marker of tissueperfusion and prognosis in critically ill patients. Crit Care Med. 2019;47: 152–158. [DOI] [PubMed] [Google Scholar]
- 33.Bellomo R, Forni LG, Busse LW, et al. Renin and survival in patients given angiotensin II for catecholamine-resistant vasodilatory shock. Am J Respir Crit Care Med. 2020;202:1253–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khanna A, English SW, Wang XS, et al. Angiotensin II for the treatment of vasodilatory shock. N Engl J Med. 2017;377:419–430. [DOI] [PubMed] [Google Scholar]
- 35.Tumlin JA, Murugan R, Deane AM, et al. Outcomes in patients with vasodilatory shock and renal replacement therapy treated with intravenous angiotensin II. Crit Care Med. 2018;46:949–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Plataki M, Kashani K, Cabello-Garza J, et al. Predictors of acute kidney injury in septic shock patients: an observational cohort study. Clin J Am Soc Nephrol. 2011;6:1744–1751. [DOI] [PubMed] [Google Scholar]
- 37.Suh SH, Kim CS, Choi JS, et al. Acute kidney injury in patients with sepsis and septic shock: risk factors and clinical outcomes. Yonsei Med J. 2013;54:965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gayat E, Hollinger A, Cariou A, et al. Impact of angiotensin-converting enzyme inhibitors or receptor blockers on post-ICU discharge outcome in patients with acute kidney injury. Intensive Care Med. 2018;44:598–605. [DOI] [PubMed] [Google Scholar]
- 39.Parr SK, Matheny ME, Abdel-Kader K, et al. Acute kidney injury is a risk factor for subsequent proteinuria. Kidney Int. 2018;93:460–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iskander KN, Craciun FL, Stepien DM, et al. Cecal ligation and puncture-induced murine sepsis does not cause lung injury. Crit Care Med. 2013;41: 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zingarelli B, Coopersmith CM, Drechsler S, et al. Part I: Minimum Quality Threshold in Preclinical Sepsis Studies (MQTiPSS) for study design and humane modeling endpoints. Shock. 2019;51:10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dejager L, Pinheiro I, Dejonckheere E, Libert C. Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol. 2011;19:198–208. [DOI] [PubMed] [Google Scholar]
- 43.Libert C, Ayala A, Bauer M, et al. Part II: Minimum Quality Threshold in Preclinical Sepsis Studies (MQTiPSS) for types of infections and organ dysfunction endpoints. Shock. 2019;51:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Remick DG, Newcomb DE, Bolgos GL, Call DR. Comparison of the mortality and inflammatory response of two models of sepsis: lipopolysaccharide vs. cecal ligation and puncture. Shock. 2000;13:110–116. [DOI] [PubMed] [Google Scholar]
- 45.Stortz JA, Hollen MK, Nacionales DC, et al. Old mice demonstrate organ dysfunction as well as prolonged inflammation, immunosuppression, and weight loss in a modified surgical sepsis model. Crit Care Med. 2019;47:e919–e929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Osuchowski MF, Ayala A, Bahrami S, et al. Minimum Quality Threshold in Pre-Clinical Sepsis Studies (MQTiPSS): an international expert consensus initiative for improvement of animal modeling in sepsis. Shock. 2018;50:377–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kilkenny C, Browne W, Cuthill IC, et al. and the NC3Rs Reporting Guidelines Working Group. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Exp Physiol. 2010;95:842–844. [DOI] [PubMed] [Google Scholar]
- 48.Abraham MN, Jimenez DM, Fernandes TD, Deutschman CS. Cecal ligation and puncture alters glucocorticoid receptor expression. Crit Care Med. 2018;46:e797–e804. [DOI] [PubMed] [Google Scholar]
- 49.Gbinigie O, Price CP, Heneghan C, et al. Creatinine point-of-care testing for detection and monitoring of chronic kidney disease: primary care diagnostic technology update. Br J Gen Pract. 2015;65:608–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cagliani J, Yang WL, McGinn JT, et al. Anti-interferon-a receptor 1 antibodies attenuate inflammation and organ injury following hemorrhagic shock. J Trauma Acute Care Surg. 2019;86:881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Methods Section. (1) Estimation of total renovascular resistance. (2) AT1R expression assessment. (3) AT1R protein expression assessment in tissue sections. (4) Validation of AT1R ELISA for whole tissue homogenate. (5) AT1R mRNA expression assessment by qPCR. (6) AT1R expression in human sepsis.
Figure S1. Untreated and losartan-treated mice with cecal ligation and puncture–induced renal dysfunction show mild, nonspecific renal histopathology.
Figure S2. Terminal deoxynucleotidyl transferase–mediated dUTP nick end-labeling (TUNEL) stain for apoptosis.
Figure S3. Estimated total renovascular resistance in cecal ligation and puncture (CLP) versus sham operation.
Figure S4. Estimated total renovascular resistance by treatment group over time following cecal ligation and puncture.
Figure S5. Recently deceased sepsis patients had low angiotensin II type-1 receptor (AT1R) in large renal resistance vessels.
Figure S6. Human sepsis patients display mild, nonspecific histopathologic injury.
Figure S7. Validation of the enzyme-linked immunosorbent assay (ELISA) for angiotensin II type-1 receptor (AT1R) concentration in whole kidney homogenate.