

SUMMARY
Individually, dysfunction of both the endoplasmic reticulum (ER) and mitochondria has been linked to aging, but how communication between these organelles might be targeted to promote longevity is unclear. Here, we provide evidence that, in Caenorhabditis elegans, inhibition of the conserved unfolded protein response (UPRER) mediator, activating transcription factor (atf)-6, increases lifespan by modulating calcium homeostasis and signaling to mitochondria. Atf-6 loss confers longevity via downregulation of the ER calcium buffer, calreticulin. ER calcium release via the inositol triphosphate receptor (IP3R/itr-1) is required for longevity, while IP3R/itr-1 gain of function is sufficient to extend lifespan. Highlighting coordination between organelles, the mitochondrial calcium import channel mcu-1 is also required for atf-6 longevity. IP3R inhibition leads to impaired mitochondrial bioenergetics and hyperfusion, which is sufficient to suppress long life in atf-6 mutants. This study reveals the importance of organellar calcium handling as a critical output for the UPRER in determining the quality of aging.
In Brief
Burkewitz et al. show that modulating subcellular calcium compartmentalization and signaling is a mechanism of both aging and longevity. The loss of ATF-6, a conserved mediator of the unfolded protein response, disrupts calcium retention in the ER; subsequently, ER calcium release triggers lifespan extension by stimulating mitochondrial dynamics and function.
Graphical Abstract
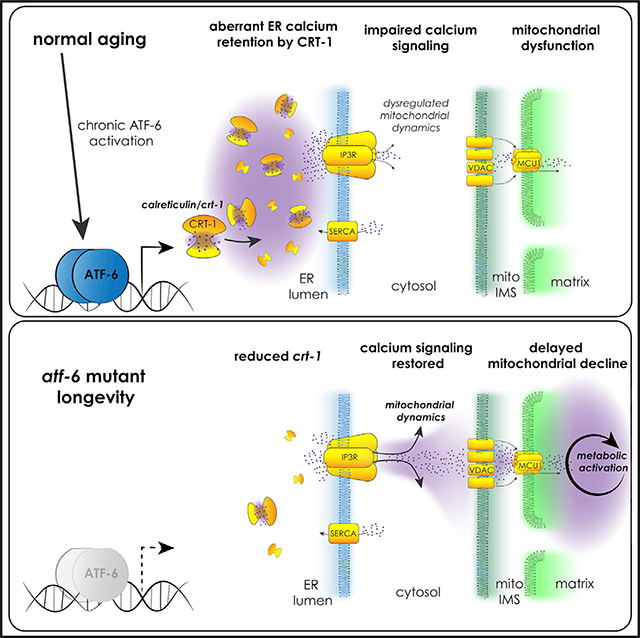
INTRODUCTION
Aging is associated with failures in the ability to maintain homeostasis at the molecular and subcellular levels, and understanding how to prevent these age-related changes is essential to developing therapies for a variety of age-onset diseases. The largest organelle system in cells, the endoplasmic reticulum (ER), serves as a hub of metabolism through a variety of critical roles in the cell. These roles include housing the secretory pathway and associated proteostasis machineries, providing storage and timely release of intracellular calcium, and acting as the primary site of the synthesis of triacylglycerols and membrane lipids (Bravo et al., 2013). Perturbations in any of these processes can trigger cell dysfunction and lead to disease, necessitating a robust homeostatic network to maintain the health and function of the ER. The unfolded protein response (UPR) serves as this network, and as its name suggests, historical insights into the UPR tend to center around its activation by unfolded proteins in the secretory pathway (Ron and Walter, 2007).
Three conserved branches mediate the UPR in metazoans, namely the inositol-requiring enzyme-1 (Ire1) branch, PKR-like ER kinase (PERK), and activating transcription factor 6 (atf-6). Previous studies of the UPR in mammalian models revealed that the loss of Ire1 or PERK results in lethality or severe metabolic pathology, respectively, in mice, demonstrating critical roles for these branches in both organelle and organismal homeostasis (Zhang et al., 2002, 2005). In contrast, the first Atf6α knockout models surprisingly did not result in overt phenotypes (Wu et al., 2007; Yamamoto et al., 2007). Upon chronic, non-physiological dosing of tunicamycin, an inhibitor of protein glycosylation and maturation in the ER, Atf6α−/− animals succumb to liver failure, revealing its importance in long-term adaptation to ER stress conditions (Wu et al., 2007; Yamamoto et al., 2007). Similar studies in Caenorhabditis elegans confirmed the hierarchical, relative importance of the three branches, revealing that ire-1/xbp-1 and PERK/pek-1 mutants exhibit the strongest phenotypes and are highly sensitive to tunicamycin. Unlike ire-1 and pek-1, atf-6 mutants present few baseline phenotypes and appear to exhibit normal, wild-type responses to tunicamycin (Bischof et al., 2008; Shen et al., 2005; Springer et al., 2005). These results suggest that (1) targeting atf-6 may be more tolerable than the alternative UPR branches and that (2) atf-6 may play a role in maintaining ER and cellular homeostasis beyond protein folding.
Despite the established and essential role of the UPR in maintaining cell homeostasis, fundamental questions regarding its roles in aging persist. First, while activation of the UPR is most commonly associated with cytoprotection, the outputs of the UPR are not universally beneficial. Chronic UPR activation can promote cell death and inflammation, ultimately contributing to pathology and, in some cases, limiting lifespan, through excessive and persistent activation later in life (Hotamisligil, 2010; Wang et al., 2015a). Depending on the context therefore, not only activation but also UPR inhibition may provide therapeutic value (e.g., Harnoss et al., 2019; Tufanli et al., 2017), suggesting that UPR inhibitors may be an underexplored strategy in combating age-associated diseases. Second, the importance of the proteostasis outputs of the ER and UPR are well established and critical in promoting healthy aging (Frakes and Dillin, 2017). As mentioned above, however, the ER performs myriad roles in parallel to proteostasis. While these functions are also influenced by the UPR, how they affect aging or longevity is not well understood.
Here, we focus on understanding the roles of the understudied atf-6 branch of the UPR in aging in C. elegans. atf-6 loss promotes longevity, and we find that the regulation of lifespan in this case does not occur through canonical proteostasis pathways but instead through the modulation of ER calcium homeostasis. Reduction in the expression of the ER calcium sink, calreticulin, mediates longevity in atf-6 mutants, and ER calcium efflux through the conserved inositol triphosphate receptor/itr-1 (IP3R/itr-1) channel is necessary and sufficient for lifespan extension in this pathway. Finally, we reveal mitochondrial reprogramming as a downstream consequence of modulating ER calcium release, highlighting interorganelle calcium signaling as a key factor in promoting healthier aging.
RESULTS AND DISCUSSION
Atf-6 Mutants Are Not Sensitive to Proteotoxic Stress and Exhibit Extended Lifespan
To define novel roles for ATF-6 in healthy aging, we used a C. elegans deletion mutant, atf-6(ok551). We confirmed that atf-6 mutants do not exhibit reduced survival in the presence of tunicamycin, an inhibitor of protein glycosylation and maturation in the ER lumen (Figure 1A). Indicating that ATF-6 still functions to regulate some aspect of ER homeostasis in the nematode, the loss of atf-6 results in synthetic lethality when combined with loss of the ire-1/xbp-1 pathway in C. elegans (Shen et al., 2005). Furthermore, the ER residency and activation of mammalian Atf6 are accomplished through a transmembrane domain and lumenal Golgi trafficking signals, which are conserved in the nematode (Shen et al., 2005).
Figure 1. Atf-6 Is Dispensable in Proteotoxic Stress, and Mutants Exhibit Extended Lifespans.

(A) Survival analysis of UPR mutant nematodes exposed chronically to 30 mg/mL tunicamycin starting on the first day of adulthood (n = 72 per condition).
(B) Lifespan analysis of atf-6 mutants (n = 100 per condition).
(C) Pharyngeal pumping rates as a measure of health span in 7-day old nematodes (means ± SDs of n = 31 combined over 2 independent repeats).
(D) Period of the defecation motor program, an ultradian behavioral rhythm, in aging nematodes (means ± SDs of n = 20–40 total periods measured from 4–5 animals on each day).
(E) Survival of worms exposed to high temperatures for 3 and 4 h (means ± SDs of 3 independent assays of 100 animals).
(F) Fecundity in atf-6 mutants over the first 3 days of adulthood (means ± SDs of n = 5–19 worms per day).
To determine how atf-6 affects the aging process, we asked whether atf-6 loss alters the C. elegans lifespan. Counterintuitively for loss of a homeostatic regulator, we found that the atf-6 deletion mutant was significantly long-lived (57% increase in median, p < 0.0001; Figure 1B), as had been suggested previously (Henis-Korenblit et al., 2010; Wang et al., 2015b). Confirming that this lifespan effect was due to the loss of function of atf-6, we created a second null allele of atf-6 through an early CRISPR-Cas9-generated frameshift, and this also extended lifespan (43%, p < 0.0001; Figure 1B). Furthermore, a subset of healthspan markers were improved in aged atf-6 mutants, including pharyngeal pumping (Figure 1C) but not rhythmicity of the defecation cycle (Figure 1D), suggesting that normal atf-6 function somehow promotes age-onset dysfunction and deterioration. We investigated whether the loss of atf-6 protected animals from heat, a more generalized form of proteotoxic stress that affects all intracellular compartments, but we found no differences from wild-type animals (Figure 1E). Because the ability to withstand extrinsic stress from either tunicamycin (Shen et al., 2005; Wu et al., 2007) or heat treatment (Figures 1A and 1E) was unaffected in atf-6 mutants, we hypothesized that atf-6 may instead play a role in basal ER functions, such as the biosynthetic demands associated with growth and reproduction. When we examined atf-6 mutants for developmental or brood-size effects, however, we observed no developmental delay (data not shown) or defects in reproductive rate relative to wild-type animals (Figure 1F). These findings suggest that in contrast to IRE1 and PERK, ATF-6 is pro-aging under basal conditions in C. elegans, providing an opportunity to discover longevity mechanisms downstream of this conserved transcription factor.
Atf-6 Regulates ER Function and Lifespan through Its Conserved Target, Calreticulin
Given the lack of obvious phenotypes and putative mechanisms for atf-6 longevity, we performed unbiased transcriptomic analyses to understand the physiological roles of ATF-6 in C. elegans. In agreement with previous analysis of atf-6-dependent transcripts in early development (Shen et al., 2005), we found that relatively few genes were differentially expressed (DE) in day 1 atf-6(ok551) adults when compared with wild-type N2 animals: only 26 genes were downregulated (Figure 2A; Table S1), while 101 genes were upregulated (Table S2) using cutoff criterion α < 0.01. Confirming previous studies in C. elegans and our hypothesis that atf-6 may specialize in roles that are separable from canonical proteostasis functions, we found virtually no signature markers of ire-1/xbp-1 activation among these DE genes (Bischof et al., 2008; Shen et al., 2005; Springer et al., 2005) (Tables S1 and S2). This finding argues strongly against one possible model of atf-6 longevity in which compensatory activation of an alternative UPR branch is responsible for the lifespan extension. Because constitutive activation of the ire-1/xbp-1 branch is sufficient to extend lifespan in C. elegans (Taylor and Dillin, 2013), we attempted to further rule out this compensatory model by inhibiting the proximal sensors of ER stress, ire-1 and pek-1, via RNAi and performing lifespan analyses in atf-6 mutants (Figure S1). We found that neither ire-1 nor pek-1 is fully required for atf-6 mutant longevity, although it is technically difficult to completely rule out contributions from the alternative branches considering the synthetic lethality of ire-1/xbp-1 and atf-6 mutants during development (Shen et al., 2005).
Figure 2. Atf-6 Regulates ER Function and Lifespan through Its Conserved Target, Calreticulin.

(A) Volcano plot of differentially expressed transcripts in atf-6(ok551) relative to N2.
(B) Relative transcript abundance of conserved UPR target genes in 1-day old atf-6 mutants (means ± SDs of RNA sequencing [RNA-seq] read counts normalized to wild-type, n = 4).
(C and D) qRT-PCR analysis of changes in UPR transcripts in wild-type worms between days 1 and 7 of adulthood (C), contrasted with changes in UPR transcripts in aging atf-6 mutants (D) (means ± 95% confidence intervals of 3 independent samples of ~100 worms; *p < 0.05, **p < 0.01, and ***p < 0.001).
(E) Lifespan analysis of atf-6(ok551) and crt-1(bz29) mutants (n = 100 animals per curve).
The relatively small pool of DE genes suggests that atf-6-dependent phenotypes arise from its regulation of single or small groups of transcriptional targets, as opposed to a large network effect. We therefore set out to identify the most promising candidate mediators. Among the most significantly affected transcripts, we found two genes previously implicated in ER homeostasis and stress responses: calreticulin/crt-1 (Park et al., 2001), which was downregulated in atf-6 mutants, while choline kinase b-2/ckb-2 (Caruso et al., 2008) was upregulated (Figure 2B). Because atf-6 mutants exhibit both extended lifespan and health-span benefits after 1 week of adulthood, we reasoned that candidate aging factors may show the greatest differences in expression at later time points. We aged worms to 7 days and harvested RNA from wild-type and atf-6(ok551) mutants. While ckb-2 levels appeared more dramatically altered than crt-1 in young mutants, its expression declined similarly during aging in both wild-type and atf-6 backgrounds. However, whereas crt-1 levels are significantly upregulated with age in wild-type animals (Figure 2C), these transcripts are even further reduced with age in atf-6 mutants (Figure 2D), leading us to focus on calreticulin/crt-1 as the most promising candidate for an atf-6-dependent aging factor. If atf-6 deletion promotes lifespan through its effects on crt-1, then, we hypothesized, the loss of crt-1 may be sufficient to reproduce the longevity phenotype. We used a null mutant of calreticulin, crt-1(bz29) (Xu et al., 2001), and, consistent with our hypothesis, crt-1(bz29) mutants are long-lived to a similar extent as atf-6(ok551) (38%, p < 0.001 versus wild-type; Figure 2E). These results highlight calreticulin as a mediator of atf-6 functions in the C. elegans lifespan and are consistent with its evolutionary conservation as a direct target of Atf6 transcription in mammals (Yoshida et al., 1998).
ER Calcium Flux Functions Downstream of atf-6 in Regulating Lifespan
Calreticulin functions with calnexin as a quality control checkpoint during the glycosylation of ER proteins, but it plays a more outsized role as a calcium-buffering protein, capable of binding and sequestering up to half of the total ER Ca2+ pool (Michalak et al., 2009). Mirroring the lack of a proteostasis defect in atf-6 mutants here, calnexin can effectively compensate for the loss of calreticulin to maintain protein quality control in mammalian cells (Molinari et al., 2004). However, manipulating calreticulin levels causes overt alterations in calcium handling that ultimately drive changes in cell physiology (Michalak et al., 2009). These prior studies and our data suggest a model whereby atf-6 maintains a primary role in the regulation of changes in ER calcium handling in parallel to ire-1/xbp-1 and pek-1 control of ER proteostasis. To test this concept, we placed synchronized L1 larval nematodes on bacterial lawns in the presence of thapsigargin, an inhibitor of ER calcium uptake, and measured their ability to grow and develop during chronic ER calcium stress. In contrast to what we observed with the protein-folding inhibitor tunicamycin (Bischof et al., 2008; Shen et al., 2005; Springer et al., 2005), growth of atf-6 mutants is highly sensitive to thapsigargin and is reduced to the lowest levels among proximal UPR sensors (Figure 3A).
Figure 3. ER Calcium Flux Functions Downstream of atf-6 in Regulating Lifespan.

(A) Relative growth of L1 larval worms exposed to 5 mg/mL of the SERCA inhibitor thapsigargin for 48 h (means ± SDs of n = 88–164 worms combined over 2 independent trials).
(B and C) Representative traces of GCaMP3 imaging in the spermatheca of worms beginning with oocyte entry and ending with spermathecal contraction.
(D and E) Area under the curve (AUC) (D) and time-to-maximum (E) summary measurements of the GCaMP3 calcium traces recorded in the spermatheca of wild-type and atf-6 mutant worms (means ± SDs of 11–16 animals combined over 2 repeats).
(F) Lifespan analysis of atf-6 mutants in the temperature-sensitive IP3R/itr-1(sa73) background at the semi-permissive temperature of 20°C (n = 100 per condition).
(G) Lifespan analysis of the atf-6 interaction with itr-1(sy290) gain-of-function mutation (n = 100 per condition). NS = p > 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 by t test.
These data suggest that atf-6 may indeed play a role in maintaining ER calcium homeostasis, prompting us to determine whether there is evidence that in vivo calcium signaling is perturbed in these animals. One of the best-characterized calcium signaling paradigms in C. elegans is ovulation, in which oocytes entering the smooth muscle-like spermatheca trigger oscillatory ER calcium release via the IP3R, ultimately driving myosin-dependent contraction (Kovacevic et al., 2013). By expressing the genetically encoded calcium indicator GCaMP3 in the spermatheca, we observed the cytosolic calcium oscillations and contraction events in vivo in atf-6 mutants (Figures 3B and 3C). These experiments revealed a number of aberrant phenotypes in atf-6 mutants, including altered calcium oscillatory patterns, delayed calcium release and contraction, and greater total calcium release per contraction (Figures 3B–3E). Thus, atf-6 appears to be a regulator of calcium homeostasis in C. elegans.
Due to the differences observed in the spermathecal calcium signaling of atf-6 mutants and the central role for the IP3R in that process, we hypothesized that ER calcium release via the IP3R may also play a role in the atf-6-dependent lifespan effects. We crossed atf-6 deletion mutants with both gain-of-function (sy290) and temperature-sensitive reduction-of-function (sa73) alleles of the sole C. elegans IP3R ortholog, itr-1. At the sa73 semi-permissive temperature of 20°C, atf-6 longevity is fully suppressed in the itr-1(sa73) reduction-of-function mutants (p = 0.108; Figure 3F). Consistent with a model in which enhanced IP3R-dependent calcium release promotes longevity (Iwasa et al., 2010), the gain-of-function allele, itr-1(sy290), is sufficient to extend lifespan (p = 0.0005), but its effects are not additive when crossed to the atf-6 mutant (p = 0.292; Figure 3G). These results suggest a model in which reduced ER calcium retention in atf-6 mutants functions to extend lifespan.
ER-Mitochondrial Communication via Calcium Regulates Lifespan
We aimed next to better understand the mechanisms by which ER calcium release may be linked to longevity. Because excess cytosolic Ca2+ is more closely linked to pathology than to longevity (Arruda and Hotamisligil, 2015; Mattson and Arumugam, 2018), we hypothesized that another intracellular compartment may compensate for enhanced ER calcium release. Studies have shown that calreticulin not only regulates ER calcium storage and release through the IP3R (Michalak et al., 2009) but it also affects mitochondrial calcium levels (Arnaudeau et al., 2002). Specifically, enhanced ER Ca2+ storage promoted by calreticulin overexpression is counterbalanced by a loss of Ca2+ from mitochondrial pools (Arnaudeau et al., 2002), suggesting an inverse relationship between calcium storage in these two compartments. Furthermore, mild ER stress can result in organelle reorganization to increase communication between ER and mitochondria (Arruda et al., 2014; Bravo et al., 2011). To determine how altered ER calcium handling via atf-6 and itr-1 can affect mitochondrial behavior, we performed in vivo imaging of mitochondrial networks in the nematode intestine. While young adult atf-6 mutants exhibited mild if any alterations in gross mitochondrial morphology, itr-1(sa73) mutants promoted dramatic reorganization of the networks, which is consistent with enhanced mitochondrial fusion (Figures 4A and 4B). Because itr-1(sa73) both suppresses atf-6-mediated longevity and causes mitochondrial networks to become hyperfused, we next asked whether this change in mitochondrial morphology was sufficient to block lifespan extension in this context. We fed atf-6(ok551) mutants Escherichia coli-generating double-stranded RNA (dsRNA) to inhibit the conserved fission factor, dynamin-related protein 1 (drp-1), and this genetically imposed hyperfusion was sufficient to block the effects of atf-6 on the lifespan (p = 0.1212; Figure 4C). Conversely, enhancing mitochondrial fragmentation by feeding nematodes dsRNA to inhibit the ortholog of mammalian mitofusins, fzo-1, has no inhibitory effect on the longevity of atf-6 mutants (p < 0.0001 versus fzo-1(RNAi) alone; Figure 4D). Thus, reducing IP3R function causes mitochondrial hyperfusion, which is sufficient to suppress lifespan extension in atf-6 mutants.
Figure 4. ER-Mitochondrial Communication via Calcium Regulates Lifespan.

(A) Representative fluorescence z stack projections of intestinal mitochondrial networks via TOMM-20(1–49)::EGFP in day 1 adult animals. Scale bars: 10 μm.
(B) Quantification of mitochondrial morphology (n = 14–27 worms combined over 2 trials; p < 0.0001 comparing control versus itr-1(sa73) and p = 0.001 comparing control versus itr-1(sy290)).
(C and D) Lifespan analysis of worms fed dsRNA for the mitochondrial fission (drp-1, C) and fusion (fzo-1, D) machineries in control and atf-6 mutant animals (n = 100 per condition).
(E) Quantification of western blots for P-AMPK normalized to wild-type animals (means ± SDs of n = 3 independent experiments using lysate from ~600 young adult worms; *p = 0.024 by ANOVA).
(F) Quantification of the ratio of GCaMP7:mKate2 fluorescence as an indicator of mitochondrial calcium content (means ± SDs of n = 96 and 45 combined from 3 independent repeats; p < 0.0001 by t test).
(G) Lifespan analysis of atf-6 mutants harboring a deletion in mcu-1 to ablate acute mitochondrial calcium uptake (n = 100 per condition).
To coordinate functions and respond to stress, the ER can transfer calcium to the mitochondrial matrix via a series of transporters, including the IP3R/itr-1 on the ER surface, and voltage-gated anion channels (VDAC) and mitochondrial calcium uniporters (MCU)-1 in the mitochondrial outer and inner membranes, respectively (Bravo et al., 2011). While this communication between organelles can occur at a distance, ER and mitochondrial membranes also form relatively stable contact sites to mediate more direct and efficient transfer of calcium, lipids, and other metabolites (Csordás et al., 1999; Szabadkai et al., 2006). Once in the matrix, Ca2+ stimulates metabolic and bioenergetic function through the activation of one of several dehydrogenases (Denton, 2009; Griffiths and Rutter, 2009). Given that atf-6 mutants show signs of a mild ER stress and that ER calcium efflux via itr-1 is required for atf-6 effects on the lifespan, we hypothesized that ER-mitochondrial coordination of calcium homeostasis may be an important factor in this longevity mechanism.
While ER-mitochondrial Ca2+ flux is an established mechanism for bioenergetic control in mammalian cells, it is unclear whether the conserved mediators perform similar roles in C. elegans. To determine whether the IP3R regulates bioenergetic functions in C. elegans, we performed western blots to measure the phosphorylation of AMP-activated protein kinase (P-AMPK), an AMP/ATP sensor and surrogate readout for mitochondrial ATP production (Burkewitz et al., 2016; Cárdenas et al., 2010). IP3R/itr-1(sa73) reduction-of-function mutants exhibit elevated P-AMPK, indicating an increase in AMPK activation through enhanced signaling and/or upregulation of expression. This compensatory AMPK activation in itr-1(sa73) mutants is consistent with impaired ATP production and bioenergetic competence downstream of IP3R dysfunction, as previously reported in mammalian models (Cárdenas et al., 2010). Conversely, IP3R/itr-1(sy290) gain-of-function mutants exhibit no change (Figure 4E). While we expected to observe a decline in P-AMPK in the gain-of-function mutants representative of enhanced mitochondrial function, we hypothesize that the lack of effect is due to the already robust mitochondrial metabolic functions in young adults. These findings support conservation of the link between ER Ca2+ release and energetic status, so we set out to determine how organelle Ca2+ dynamics may be perturbed in aging animals. To visualize mitochondrial Ca2+ levels and begin testing the hypothesis that hallmark age-dependent mitochondrial declines are driven by altered Ca2+ homeostasis, we co-expressed mitochondrial matrix-targeted and Ca2+-responsive GCaMP7 along with matrix-targeted and Ca2+-insensitive mKate2 as a normalization factor. Imaging of these reporters in intestinal cells revealed a ~33% decline (p < 0.0001; Figure 4F) in the ratio of mito-GCaMP7 to mito-mKate2 between days 1 and 7 of adulthood, supporting our hypothesis by indicating that reductions in mitochondrial [Ca2+] occur during normal aging that may represent a mechanism of mitochondrial decline. Finally, to test whether the final component of an ER-to-mitochondrial Ca2+ signaling pathway, mitochondrial import, is important in extending lifespan in this context, we crossed animals with mcu-1 deletion into the long-lived atf-6(ok551) background. Mcu-1 deletion alone did not affect lifespan, but supporting a role for interorganellar calcium flux in atf-6-dependent longevity, loss of the mitochondrial calcium importer, mcu-1, reduces atf-6-mediated lifespan extension by 67% (p < 0.0001 compared to atf-6(ok551); Figure 4G).
In summary, we set out to identify the specialized roles that atf-6 plays in parallel to the PERK and Ire1/XBP-1 UPR pathways. We found in our characterization of the mutants that atf-6 is a pro-aging factor and that deletion of this gene confers longevity in nematodes. Exploring the mechanism of atf-6 longevity revealed ER calcium homeostasis as a critical downstream function, and highlighted calreticulin/crt-1 as the proximal mediator of altered ER calcium storage and signaling. Calreticulin remains one of the only common and most potently activated targets of ATF-6 between C. elegans and mammals (Shoulders et al., 2013), suggesting conservation of this pathway. However, mammalian Atf6α plays a more pronounced role in promoting the transcription of some canonical protein chaperones (e.g., glucose-regulated protein-75 [GRP75/BiP]) than we observe here in C. elegans (Figure 2B; Shoulders et al., 2013). In fact, because defective proteostasis could mask the effects of any lifespan-extending intervention, we surmise that the diminished role of atf-6 in proteostasis enabled us to identify this role for altered ER calcium signaling in promoting longevity downstream of the UPR. Notably, the respective duties of Atf6 and Xbp1, the former a regulator of a small subset of targets and the latter a more global and dominant regulator of ER function and proteostasis, remain consistent between nematodes and mammals (Shen et al., 2005; Shoulders et al., 2013). This more focused role for atf-6 relative to PERK and Ire1/Xbp1 branches may be useful in differentiating atf-6 or its effectors as a more amenable therapeutic target for age-onset diseases.
Our finding that atf-6 ablation causes few overt phenotypes in young animals yet extends lifespan is both exciting and surprising, but perhaps prompts questions as to why atf-6 would be maintained through evolution if its loss is generally beneficial. atf-6 mutants are clearly sensitive to thapsigargin, a potent sarcoplasmic/endoplasmic reticulum calcium-ATPase (SERCA) inhibitor produced by plants (Figure 3A), indicating its function is important during contexts in which ER calcium homeostasis is threatened. We also observe calcium signaling defects specifically related to reproduction in young animals (Figures 3B–3E); although relatively mild, these changes could rapidly result in competitive disadvantages if cultured among wild-type animals. We also found unexpectedly that calreticulin expression increased dramatically during normal aging in a manner completely dependent on atf-6 (Figures 2B–2D). This finding suggests the existence of an atf-6-mediated chronic ER stress response in aging C. elegans and warrants further investigation in mammals, given that unmitigated UPR signaling occurs in several chronic disease contexts and instigates “inflammaging” (Franceschi et al., 2018; Hotamisligil and Erbay, 2008). It remains unclear why or how ATF-6 would be increasingly activated in older animals, but based on these data, we hypothesize that its effects on aging may occur later in life. It will be important to determine in future studies whether enhanced ATF-6 activity is sufficient to drive mitochondrial calcium levels downward. Our findings suggest that atf-6 represents an example of antagonistic pleiotropy in aging.
Pursuing additional insights into how ER calcium homeostasis may link to lifespan regulation, we demonstrated requirement and sufficiency for the highly conserved ER calcium release channel, the IP3R/itr-1 in the atf-6 pathway. The IP3R is an outlet for both passive leak and active release of Ca2+, and understanding which of these routes of efflux are affected by crt-1 and atf-6 will be an important future goal. Both stress-induced Ca2+ leak and constitutive, low-level activation of IP3R release are linked to enhanced mitochondrial calcium uptake and bioenergetic activation (Bravo et al., 2011; Cárdenas et al., 2010). We have shown here that the C. elegans IP3R also appears to regulate the cell energy state (Figure 4E), and this turns out to be critical in resolving an apparent paradox—that enhancing ER calcium release extends lifespan, yet impaired retention of ER Ca2+ is associated with metabolic pathology (Arruda and Hotamisligil, 2015). Our data suggest that spatiotemporal context is necessary to determine whether modulation of Ca2+ flux proves to be beneficial or detrimental, highlighting especially that the destination compartment of the ion is critical (Figure 4G). These findings support a model in which Ca2+ storage and flux between compartments must be maintained within an optimal window for signaling and bioenergetic tone. This model is apparent in cancer biology, in which too much or too little transport of the ion leads to toxicity or functional deficits (Lovy et al., 2016). We now extend this to the aging field by showing that targeting the age-onset dysregulation of Ca2+ transport is sufficient to promote longevity and health span.
Overall, these findings reveal the importance of understudied, non-canonical UPR outputs, namely ER calcium handling, in the aging process. Furthermore, we identified ER-mitochondrial coordination of calcium as a mechanism in lifespan regulation, raising new questions about the roles of interorganelle signaling in age-related mitochondrial declines. These data open exciting avenues for research into understanding how calcium dyshomeostasis is not simply a marker for age-related disease, but a hallmark and driver of the aging process itself.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, William Mair (wmair@hsph.harvard.edu).
Materials Availability
All unique and stable reagents generated in this study are available either from public repositories (e.g., Caenorhabditis Genetics Center for C. elegans strains) or from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
Source transcriptomic data are available through GEO: GSE156037, and other original data are available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
C. elegans strains and husbandry
N2 wild-type, RB772 [atf-6(ok551)], SJ30 [ire-1(zc14); zcIs4], ZB1028 [crt-1(bz29)], CZ19982 [mcu-1(ju1154)], JT73 [itr-1(sa73)], RB545 [pek-1(ok275)], PS1631 [itr-1(sy290); dpy-20(e1282)] C. elegans strains were obtained from the Caenorhabditis Genetic Center, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). High-throughput deletion strains (e.g., RB772 and RB545) were outcrossed 4–8 times with N2 before use, and PS1631 was backcrossed to N2 to isolate itr-1(sy290) from the dpy-20 co-marker. Additional strains include WBM1093; UN1108; WBM926; WBM947; WBM973; WBM974; WBM1046; WBM1103; WBM1104; WBM1134; WBM1158; BUZ26 (see Key Resources Table for details). Worms were grown and maintained on standard nematode growth media (NGM) seeded with E. coli (OP50–1). E. coli bacteria was cultured overnight in LB at 37°C, after which 100 μL of liquid culture was seeded on plates to grow for 2 days at room temperature. RNAi experiments alternatively employed E. coli (HT115) from the Ahringer library (Source Bioscience) expressing dsRNA against the gene noted or an empty vector control. Experiments with HT115 were performed identically except LB and NGM contained 100 μg ml−1 Carbenicillin, and dsRNA expression was induced by addition of 100 μL IPTG (100 mM) at least 2 hours before worms were introduced to the plates.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| phospho-AMPK (Thr172) | Cell Signaling | Cat# 2535; RRID:AB_331250 |
| Anti rabbit IgG, HRP linked Antibody | Cell Signaling | Cat# 7074S; RRID:AB_2099233 |
| Bacterial and Virus Strains | ||
| E. coli OP50-1 | CGC | WBStrain00041971 |
| E. coli HT115(DE3) | Source Bioscience | WBStrain00041079 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Thapsigargin | Sigma Aldrich | Cat# T9033 |
| Tunicamycin | EMD Millipore | Cat# 5.04570.0001 |
| Critical Commercial Assays | ||
| Taqman Universal Master Mix | Thermo Fisher | Cat# 4440040 |
| RNeasy Mini Kit | QIAGEN | Cat# 74104 |
| TruSeq Stranded mRNA kit | Illumina | Cat# 20020594 |
| Pierce ECL Western Blot substrate | Thermo Fisher | Cat# 32209 |
| Taqman crt-1 assay | Thermo Fisher | Ce02475941_g1 |
| Taqman ckb-2 assay | Thermo Fisher | Ce02445484_m1 |
| Taqman hsp-4 assay | Thermo Fisher | Ce02434878_g1 |
| Taqman xbp-1 s assay | Thermo Fisher | Ce04931896_g1 |
| Taqman xbp-1 assay | Thermo Fisher | Ce02421281_g1 |
| Taqman hsp-3 assay | Thermo Fisher | Ce02495956_g1 |
| Taqman pdi-6 assay | Thermo Fisher | Ce02496946_g1 |
| Deposited Data | ||
| Raw and analyzed RNA-Seq data | This study | GEO: GSE156037 |
| Experimental Models: Organisms/strains | ||
| C. elegans strain: N2 Bristol | CGC | WB: N2 |
| C. elegans strain: RB772: atf-6(ok551) | CGC | WB: RB772 |
| C. elegans strain: SJ30: ire-1(zc14); zcIs4 | CGC | WB: SJ30 |
| C. elegans strain: ZB1028: crt-1(bz29) | CGC | WB: ZB1028 |
| C. elegans strain:CZ19982: mcu-1(ju1154) | CGC | WB: CZ19982 |
| C. elegans strain: JT73: itr-1(sa73) | CGC | WB: JT73 |
| C. elegans strain: RB545: pek-1(ok275) | CGC | WB: RB545 |
| C. elegans strain: PS1631: itr-1(sy290); dpy-20(e1282) | CGC | WB: PS1631 |
| C. elegans strain: WBM926: wbmEx367[ges-1p::tomm20 (aa1-49)::GFP::unc54 3′UTR] | Weir et al., 2017. | N/A |
| C. elegans strain: WBM947 atf-6(ok551); mcu-1(ju1154) | This study | N/A |
| C. elegans strain: WBM1093: atf-6(ok551); xbIs1101[fln-1p::GCaMP3 + pRF4(rol-6)] II | This study | N/A |
| C. elegans strain: WBM973: atf-6(ok551); itr-1(sa73) | This study | N/A |
| C. elegans strain: WBM974: atf-6(ok551); itr-1(sy290) | This study | N/A |
| C. elegans strain: WBM1046: atf-6(wbm27) | This study | N/A |
| C. elegans strain: WBM1103: atf-6(ok551); wbmEx367 [ges-1p::tomm20 aa1-49::GFP::unc54 3′UTR)] | This study | N/A |
| C. elegans strain: WBM1104: itr-1(sy290); wbmEx367 [ges-1p::tomm20 aa1-49::GFP::unc54 3′UTR] | This study | N/A |
| C. elegans strain: WBM1134: itr-1(sa73); wbmEx367 [ges-1p::tomm20 aa1-49::GFP::unc54 3′UTR] | This study | N/A |
| C. elegans strain: WBM1158: crt-1(bz29) V; atf-6(ok551) X | This study | N/A |
| C. elegans strain: UN1108: xbIs1101[fln-1p::GCaMP3 + pRF4(rol-6)] | Kovacevic et al., 2013 | N/A |
| C. elegans strain: WBM933: itr-1(sy290) | This study | N/A |
| C. elegans strain: BUZ26: bugEx2[vha-6p::MLS::GCaMP7b::unc-54 UTR + vha-6p::MLS::mKate2::unc-54 UTR] | This study | N/A |
| Oligonucleotides | ||
| Primer: GCaMP7b F: CATCATGGTATGGCTAGCGTCGACTCATCACGTC | This study | N/A |
| Primer: GCaMP7b R: CGACCGGCGCTCAGTTGGAATTCTCACTTCGCTGTCATC | This study | N/A |
| Primer: mKate2 F CTAGC GTCTCCGAGCTCATTAAAG | This study | N/A |
| Primer: mKate2 R GAATTCTTAACGGTGTCCGAGCTTGG | This study | N/A |
| Primer: MLS F: TCGACTCTAGAGGATCCCCGGGATGTCCGTCCTGAC | This study | N/A |
| Primer: MLS R: GACGTGATGAGTCGACGCTAGCCATACCATGATG | This study | N/A |
| Recombinant DNA | ||
| vha-6p::MLS::GCaMP7b::unc-54 3′UTR | This study | N/A |
| vha-6p::MLS::mKate2::unc-54 3′UTR | This study | N/A |
| pGP-CMV-jGCaMP7b | Dana et al., 2019 | Addgene #104484 |
| Software and Algorithms | ||
| bcbioRNASeq | Steinbaugh et al., 2018 | http://bioinformatics.sph.harvard.edu/bcbioRNASeq |
METHOD DETAILS
Transgene Construction
In order to generate mitochondrial targeted GCaMP7b and mKate2, GCaMP7b was first amplified from pGP-CMV-jGCaMP7b (gift from Douglas Kim & GENIE Project). GCaMP7b was fused to COX8 mitochondrial localization sequence (MLS) and vector backbone with unc-54 3′ untranslated region (pSD2) via Gibson Assembly (NEB). Subsequently, vha-6 intestinal promoter was cloned and inserted upstream of MLS::GCaMP7b via HindIII and BamHI restriction digest and ligation. To generate the complementary vha-6p::MLS::mKate2::unc-54 UTR construct, GCaMP7b was removed and mKate2 sequence inserted after NheI and EcoRI digest and subsequent ligation.
Extrachromosomal transgenic arrays were created by microinjection of the two plasmids into N2 animals.
Survival Analyses
Lifespan experiments were performed on standard nematode growth media plates at 20°C. Animals were fed with OP50–1 E. coli except in the case of RNAi experiments, which employed dsRNA-expressing HT115 bacteria. Worms were synchronized by picking 1 day-old adult worms onto plates for short, timed egg lays, after which the adult worms were removed. When these progeny reached late L4/young adulthood, 100 worms were transferred to 5–10 fresh plates at 10–20 worms per plate and this was considered time = 0. Worms were transferred to fresh bacterial lawns every other day to separate from progeny until the first deaths (10–14 d). Survival was scored every 1–2 days and a worm was deemed dead when unresponsive to 3 taps on the head and tail. Worms were censored due to contamination on the plate, worms leaving the agar media, progeny hatching inside the adult, or loss of vulval integrity during reproduction. To test survival on tunicamycin, toxin in DMSO (EMD Millipore) or DMSO control was added to NGM to a final concentration of 30 mg/mL before pouring 6-well plates. 10 mL of OP50–1 culture was spotted in each well 1 day before worms. Twelve day-1 adult worms were picked into each well for a total of 72 worms per sample. Worms were scored as above. Animals on tunicamycin plates were not transferred to separate progeny, as this concentration of tunicamycin effectively sterilizes the adults. Statistical significance for survival assays was determined through Mantel-Cox Log-rank (GraphPad Prism). Survivorship figures depict independent trials (combined n = 72 or 100 animals from 5–10 plates run in parallel), and each of these experiments were repeated again independently 1–11 times. Summary results and statistics for all survival experiments are presented in Table S3.
Healthspan Assays
Worms were synchronized by egg lay and maintained as normal on OP50–1 lawns. After day-1 measurements, worms were transferred to fresh lawns at least every other day to separate progeny. To measure the oscillatory period of the defecation motor program, the timings of 8 consecutive pBocs were scored for 4–5 undisturbed worms of each genotype; after day 6 pBocs become severely irregular and each worm was measured for 20 minutes. Pharyngeal pumping was counted manually over 20 s intervals in undisturbed animals within the OP50–1 lawn. Worms which left the lawn during either assay were not recorded. Data were analyzed with t tests (GraphPad Prism).
Heat Stress
Worms synchronized by egg-lay were allowed to develop at 20°C for ~72 hours to adulthood. Once adults, 25 worms from each genotype were transferred to each of 4 fresh NGM plates with small OP50–1 lawns, for a total of 100 animals. Alternating by genotype, plates were arranged in a single layer in a 37°C incubator until the respective time point, at which point plates were removed and returned to 20°C to recover overnight. Survival was scored 24 h after stress by responsiveness to touch.
Thapsigargin Stress
To test for growth during ER calcium stress, OP50–1 lawns were grown for 1 day before spotting thapsigargin in DMSO (Sigma) or DMSO control directly on to lawns to reach a final concentration of 5 mg/mL in the plate. Thapsigargin was given 24 hours to diffuse into the NGM before ~30–60 synchronized L1s were introduced. After 48 hours, worms were washed off plates in M9 buffer, anesthetized in 2 mM sodium azide (Sigma), returned to unseeded NGM plates, and imaged at 4x magnification with a Zeiss Imager.M2 microscope. A binary mask was applied to images and the area of each worm was recorded (ImageJ). Data were analyzed with t tests (GraphPad Prism).
Transcriptomic Analysis
In each of 4 independent biological replicates, roughly 1000 hypochlorite-synchronized L1s were washed onto OP50–1 lawns and allowed to develop for 72 h to adulthood, before washing in M9 to remove bacteria, resuspending in Qiazol (QIAGEN) and snap-freezing in liquid nitrogen. Total RNA was isolated after disrupting worms with 5 freeze-thaws with Qiazol extraction and RNeasy mini kit (QIAGEN). cDNA libraries were prepared through TruSeq Stranded mRNA kit (Illumina) and sequenced with an Illumina NextSeq500 over 75 cycles by the Dana Farber Molecular Biology Core. Reads were analyzed through the bcbio pipeline and bcbioRNASeq R package (http://bioinformatics.sph.harvard.edu/bcbioRNASeq). Quality control with MultiQC was performed in parallel to differential expression with counts aligned to WBcel235 using STAR/featureCounts, revealing mapping rates > 95% for ~14–25 million total reads per replicate. Differential expression analysis was performed with DESeq2 via the bcbioRNASeq R package with counts generated by salmon (https://combine-lab.github.io/salmon). Significance was defined with adjusted P value (alpha; 1% false discovery rate) cutoff of 0.01.
Gene Expression Assays
Total RNA was isolated from ~100 L4 stage animals. RNA was extracted using Qiazol reagent (QIAGEN) then column purified by RNeasy micro kit (QIAGEN). cDNA was generated using SuperScript VILO master mix (Invitrogen). Taqman real-time qPCR experiments were performed on a StepOne Plus instrument (Applied Biosystems) following the manufacturer’s instructions. Data were analyzed with the comparative ΔΔCt method using Y45F10D.4 as endogenous control. ExpressionSuite software (Applied Biosystems) was used to generate average fold-change and 95% confidence intervals relative to day 1 adults and t test for statistical significance.
Western Blots
Synchronized populations of 600 young adult worms were collected from bacterial lawns and washed three times in M9. Supernatant was removed and worm pellets were snap-frozen in liquid nitrogen. Worms were lysed by sonication in RIPA buffer with protease inhibitors (Roche Applied Science) and phosphatase inhibitors (Roche). Lysate was centrifuged at 14,000 g for 15 min at 4°C. Protein concentration was quantified by Pierce BCA protein assay kit (Thermo Fisher Scientific). Samples were heated in 95°C for 15 min with 5X Reducing Sample Buffer added. 20 μg protein was loaded for SDS-PAGE and transferred to PVDF membrane. The PVDF membrane was blocked with 5% milk before incubation with phospho-AMPK Thr172 primary antibody (Cell signaling, MA, USA #2535, 1:1000). Antibody signals were developed using ECL Western Blotting Detection Reagent (Thermo Fisher Scientific) and bands were quantified with ImageJ (Figure S2).
Mitochondrial morphology
Synchronized populations were grown to day-1 adults at 20°C, and picked unanesthetized onto 10% agarose pads with 0.05 μm Polybead microspheres (Polysciences) for immobilization. Imaging was performed on a Ti2 CSU-W1 confocal microscope (Nikon) with 488-nm illumination of eGFP through a Plan-Apochromat 100x/1.45 objective. Qualitative assessment of mitochondrial morphology was made by blinded analysis, scoring worms based on three categories: tubular (interconnected mitochondrial network), intermediate (combination of interconnected network and isolated smaller mitochondria) or fragmented (mostly fragmented mitochondria). Differences between strains were tested for significance with Chi-square analysis (GraphPad Prism).
Calcium Imaging
For spermathecal imaging, partially synchronized populations were obtained by egg prep and animals were grown at 23°C for ~54 h, around the time of the first ovulation. Live animals were immobilized with 0.01% tetramisole and 0.1% tricaine in M9 buffer before mounting on 2% agarose pads or with 0.05 μm Polybead microspheres (Polysciences) diluted 1:2 in water and mounted on 5% agarose pads. Confocal microscopy was performed on an LSM 710 confocal microscope (Zeiss) equipped with Zen software (Zeiss) using a Plan-Apochromat 63 × /1.40 oil DIC M27 objective. A 488-nm laser was used for GCaMP3. Live animals were imaged for ~30 min total. For mitochondrial imaging, synchronized populations were grown to day-1 adults at 20°C, and picked unanesthetized onto 10% agarose pads with 1–2 μL of 0.05 μm Polybead microsphere suspension (Polysciences) for immobilization. Images of mitochondrial targeted GCaMP7b and mKate2 were taken in the anterior-most intestinal cells using a Nikon Ti2 with CSU-W1 spinning disk, 488 and 561 nm laser excitation, and Plan-Apochromat 100x/1.49 objective. Using Nikon Elements, mitochondrial networks were identified and masked via mKate2 signal. Subsequently, the ratio of GCaMP7f:mKate2 signal was calculated from the entire mitochondrial network of single intestinal cells. In aged samples, bright spot detection was used to exclude punctate protein aggregates from the calculations.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantitative PCR data were analyzed with Expression Suite software (Applied Biosystems) as described in Methods. All other statistical analyses were performed with Graphpad Prism. Test, p value, and sample sizes are indicated in figure legends. Error bars represent standard deviations unless otherwise noted. See also Table S3 for summary of lifespan assays.
Supplementary Material
Highlights.
Loss of the UPRER mediator ATF-6 in C. elegans extends lifespan
ATF-6 loss reverses age-associated increases in calreticulin, an ER calcium sink
ER calciumefflux via the InsP3R is required to extend lifespan
ER calcium release enhances mitochondrial dynamics and bioenergetics
ACKNOWLEDGMENTS
We thank the Mair and Burkewitz labs for their constructive feedback. We also acknowledge the Hotamisligil lab for their scientific discussion and input. K.B. is supported by NIH/National Institute on Aging (NIA) grant R00AG052666; E.J.C. is supported by NIH/National Institute of General Medical Sciences (NIGMS) grant GM110268. W.B.M. is supported by NIH/NIA grants R01AG044346, R01AG051954, R21AG056930, and R01AG059595.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108125.
REFERENCES
- Arnaudeau S, Frieden M, Nakamura K, Castelbou C, Michalak M, and Demaurex N (2002). Calreticulin differentially modulates calcium uptake and release in the endoplasmic reticulum and mitochondria. J. Biol. Chem. 277, 46696–46705. [DOI] [PubMed] [Google Scholar]
- Arruda AP, and Hotamisligil GS (2015). Calcium Homeostasis and Organelle Function in the Pathogenesis of Obesity and Diabetes. Cell Metab 22, 381–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arruda AP, Pers BM, Parlakgül G, Güney E, Inouye K, and Hotamisligil GS (2014). Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med 20, 1427–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof LJ, Kao C-Y, Los FCO, Gonzalez MR, Shen Z, Briggs SP, van der Goot FG, and Aroian RV (2008). Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo. PLOS Pathog 4, e1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M, Quiroga C, Rodriguez AE, Verdejo HE, Ferreira J, et al. (2011). Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci 124, 2143–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo R, Parra V, Gatica D, Rodriguez AE, Torrealba N, Paredes F, Wang ZV, Zorzano A, Hill JA, Jaimovich E, et al. (2013). Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int. Rev. Cell Mol. Biol 301, 215–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkewitz K, Weir HJM, and Mair WB (2016). AMPK as a Pro-longevity Target. Exp Suppl 107, 227–256. [DOI] [PubMed] [Google Scholar]
- Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, Vais H, Cheung K-H, Yang J, Parker I, et al. (2010). Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso M-E, Jenna S, Bouchecareilh M, Baillie DL, Boismenu D, Halawani D, Latterich M, and Chevet E (2008). GTPase-mediated regulation of the unfolded protein response in Caenorhabditis elegans is dependent on the AAA+ ATPase CDC-48. Mol. Cell. Biol 28, 4261–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordás G, Thomas AP, and Hajnóczky G (1999). Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J 18, 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana H, Sun Y, Mohar B, Hulse BK, Hasseman JP, Tsegaye G, Tsang A, Wong A, Patel R, Macklin JJ, et al. (2019). High-performance calcium indicators for imaging activity in neuronal populations and microcompartments. Nat. Methods 16, 649–657. [DOI] [PubMed] [Google Scholar]
- Denton RM (2009). Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 1787, 1309–1316. [DOI] [PubMed] [Google Scholar]
- Frakes AE, and Dillin A (2017). The UPRER: Sensor and Coordinator of Organismal Homeostasis. Mol. Cell 66, 761–771. [DOI] [PubMed] [Google Scholar]
- Franceschi C, Garagnani P, Parini P, Giuliani C, and Santoro A (2018). Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol 14, 576–590. [DOI] [PubMed] [Google Scholar]
- Griffiths EJ, and Rutter GA (2009). Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim. Biophys. Acta 1787, 1324–1333. [DOI] [PubMed] [Google Scholar]
- Harnoss JM, Le Thomas A, Shemorry A, Marsters SA, Lawrence DA, Lu M, Chen YA, Qing J, Totpal K, Kan D, et al. (2019). Disruption of IRE1α through its kinase domain attenuates multiple myeloma. Proc. Natl. Acad. Sci. USA 116, 16420–16429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henis-Korenblit S, Zhang P, Hansen M, McCormick M, Lee S-J, Cary M, and Kenyon C (2010). Insulin/IGF-1 signaling mutants reprogram ER stress response regulators to promote longevity. Proc. Natl. Acad. Sci. USA 107, 9730–9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS (2010). Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, and Erbay E (2008). Nutrient sensing and inflammation in metabolic diseases. Nat. Rev. Immunol 8, 923–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasa H, Yu S, Xue J, and Driscoll M (2010). Novel EGF pathway regulators modulate C. elegans healthspan and lifespan via EGF receptor, PLC-gamma, and IP3R activation. Aging Cell 9, 490–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacevic I, Orozco JM, and Cram EJ (2013). Filamin and phospholipase C-ε are required for calcium signaling in the Caenorhabditis elegans spermatheca. PLOS Genet 9, e1003510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovy A, Foskett JK, and Cárdenas C (2016). InsP3R, the calcium whisperer: maintaining mitochondrial function in cancer. Mol. Cell. Oncol 3, e1185563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, and Arumugam TV (2018). Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metab 27, 1176–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalak M, Groenendyk J, Szabo E, Gold LI, and Opas M (2009). Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J 417, 651–666. [DOI] [PubMed] [Google Scholar]
- Molinari M, Eriksson KK, Calanca V, Galli C, Cresswell P, Michalak M, and Helenius A (2004). Contrasting functions of calreticulin and calnexin in glycoprotein folding and ER quality control. Mol. Cell 13, 125–135. [DOI] [PubMed] [Google Scholar]
- Park BJ, Lee DG, Yu JR, Jung SK, Choi K, Lee J, Lee J, Kim YS, Lee JI, Kwon JY, et al. (2001). Calreticulin, a calcium-binding molecular chaperone, is required for stress response and fertility in Caenorhabditis elegans. Mol. Biol. Cell 12, 2835–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, and Walter P (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol 8, 519–529. [DOI] [PubMed] [Google Scholar]
- Shen X, Ellis RE, Sakaki K, and Kaufman RJ (2005). Genetic interactions due to constitutive and inducible gene regulation mediated by the unfolded protein response in C. elegans. PLOS Genet 1, e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, Yates JR 3rd, Su AI, Kelly JW, and Wiseman RL (2013). Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep 3, 1279–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer W, Hoppe T, Schmidt E, and Baumeister R (2005). A Caenorhabditis elegans Parkin mutant with altered solubility couples alpha-synuclein aggregation to proteotoxic stress. Hum. Mol. Genet 14, 3407–3423. [DOI] [PubMed] [Google Scholar]
- Steinbaugh MJ, Pantano L, Kirchner RD, Barrera V, Chapman BA, Piper ME, Mistry M, et al. (2018). bcbioRNASeq: R Package for Bcbio RNA-Seq Analysis [version 2; peer review: 1 approved, 1 approved with reservations]. F1000Res 6, 1976. [Google Scholar]
- Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, and Rizzuto R (2006). Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol 175, 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RC, and Dillin A (2013). XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell 153, 1435–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufanli O, Telkoparan Akillilar P, Acosta-Alvear D, Kocaturk B, Onat UI, Hamid SM, Çimen I, Walter P, Weber C, and Erbay E (2017). Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl. Acad. Sci. USA 114, E1395–E1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Ryoo HD, Qi Y, and Jasper H (2015a). PERK Limits Drosophila Lifespan by Promoting Intestinal Stem Cell Proliferation in Response to ER Stress. PLOS Genet 11, e1005220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Liu J, Xie F, Gao X, Ye J-H, Sun L-Y, Wei R, and Ai J (2015b). miR-124/ATF-6, a novel lifespan extension pathway of Astragalus polysaccharide in Caenorhabditis elegans. J. Cell. Biochem 116, 242–251. [DOI] [PubMed] [Google Scholar]
- Weir HJ, Yao P, Huynh FK, Escoubas CC, Goncalves RL, Burkewitz K, Laboy R, Hirschey MD, and Mair WB (2017). Dietary Restriction and AMPK Increase Lifespan via Mitochondrial Network and Peroxisome Remodeling. Cell Metab 26, 884–896.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, Song B, Yau GD-Y, and Kaufman RJ (2007). ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 13, 351–364. [DOI] [PubMed] [Google Scholar]
- Xu K, Tavernarakis N, and Driscoll M (2001). Necrotic cell death in C. elegans requires the function of calreticulin and regulators of Ca(2+) release from the endoplasmic reticulum. Neuron 31, 957–971. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, and Mori K (2007). Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 13, 365–376. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Haze K, Yanagi H, Yura T, and Mori K (1998). Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem 273, 33741–33749. [DOI] [PubMed] [Google Scholar]
- Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, and Cavener DR (2002). The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol. Cell. Biol 22, 3864–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Wong HN, Song B, Miller CN, Scheuner D, and Kaufman RJ (2005). The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. J. Clin. Invest 115, 268–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source transcriptomic data are available through GEO: GSE156037, and other original data are available upon request.