

Hyperaldosteronism (HA) due to nodular adrenal cortical hyperplasia or adrenal adenoma is increasingly recognized as a major cause of resistant or refractory hypertension.1 HA is the most common form of secondary hypertension (HTN), with an estimated prevalence of 4% of hypertensive patients in primary care and around 10% of patients referred to hypertension specialty clinics.1 Patients with HA have higher cardiovascular (CV) disease morbidity and mortality than age‐matched patients with essential HTN and a similar degree of blood pressure (BP) elevation.1 They also have an increased propensity for the development of renal disease characterized early on by tubulointerstitial fibrosis and albuminuria. HA is characterized by an autonomous aldosterone production causing sodium retention, plasma renin activity suppression, CV stiffness and damage, renal fibrosis and albuminuria, and increased potassium excretion, leading to variable degrees of hypokalemia. Aldosterone‐producing adenomas account for around 40% and idiopathic HA for around 60% of HA cases.1
The medical treatment of HA often involves use of at least 3‐4 antihypertensive agents including a mineralocorticoid receptor (MR) antagonist, that generally being spironolactone.1, 2, 3 In this regard, the pathological effects of MR activation cannot be explained alone by its classical renal effects on electrolyte balance and BP.4 Importantly, increased MR activation in vascular tissues also affects vascular inflammation, fibrosis, and stiffness in a BP‐independent manner.3, 4 Vascular stiffness is a consequence of pathophysiological alterations involving endothelial cells (ECs), vascular smooth muscle cells (VSMCs), immune cells, and the extracellular matrix (ECM), and other functional elements of the vessel wall (Figure 1).4, 5, 6, 7, 8 The role of the ECM in modulation of vascular stiffness is well recognized, but recent studies suggest that ECMRs regulate a number of arterial properties such as inflammatory responses and vascular tone, as well as endothelial cortical polymerization/stiffness and ECM stiffness.4, 5, 6, 7, 8
Figure 1.
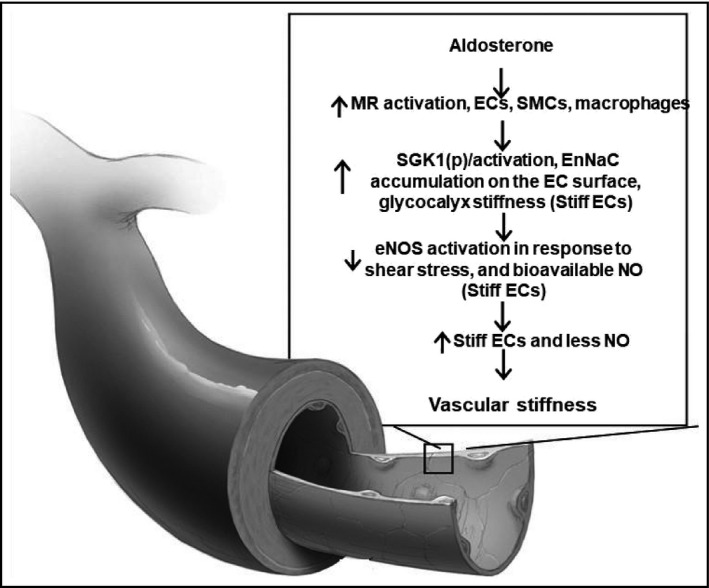
ECMR mediated activation of EnNaC in the pathogenesis of vascular stiffness in hyperaldosteronism. ECMR, endothelial cell; EnNaC, endothelial cell sodium channel; eNOS endothelial cell nitric oxide synthase; NO, nitric oxide; SGK1, serum glucocorticoid kinase; VSMC, vascular smooth muscle cells
Indeed, the use of a MR antagonist such as spironolactone presupposes that, that MR activation is the penultimate pathophysiological process culminating in the resistant hypertension and associated CV stiffness characterizing HA. This antagonism of MRs involves these receptors in CV tissue as well as in the kidney.4, 5, 6, 7, 8, 9 One of the benefits of using a MR antagonist is that this approach reduces CV stiffness.3, 4 Indeed, recent basic research has demonstrated that the deletion of EC MRs prevents the development of vascular and cardiac fibrosis and stiffness.4, 5, 6, 7 This work has also demonstrated that activation of the ECMR increases activity of the endothelial sodium channel (EnNaC), and this is an important mechanism by which CV stiffness is promoted by excess aldosterone (Figure 1).3, 4, 5, 6, 7, 8, 9, 10
In an article in this journal, Izzo et al11 report their long‐term results of treatment with low‐dose amiloride 5‐10 mg daily for 14‐28 years in a small group of patients with HA, including 3 with glucocorticoid‐remediable aldosteronism (GRA), with low‐dose amiloride (a relatively specific epithelial sodium channel [ENaC] blocker). Except for one patient, all had severe resistant hypertension. Substantial BP‐lowering effects of amiloride were seen in 1‐4 weeks after initiation of therapy, and office BPs were well controlled for the next 20 years. Twenty‐four‐hour ambulatory BP monitoring with pulse wave analysis (cardiac output, vascular resistance, augmentation index, reflection magnitude) was assessed after a mean duration of treatment of 18 years, as were regional pulse wave velocities, pulse stiffening ratio, ankle‐brachial index, serum creatinine, estimated glomerular filtration rate, and urinary albumin: creatinine ratio. All indicators were normal in all patients after 18‐years of amiloride, and there were no serious CV events detected during the 20‐year mean follow‐up. The authors concluded that long‐term ENaC blockade normalizes BP and protects macro‐ and micro‐vascular function in HA patients. They then concluded that (a) the adverse vascular effects of aldosterone are mediated via ENaC, not MR activation itself, and are fully preventable or reversible with ENaC blockade and (b) aldosterone may not play a major BP‐independent role in human macro‐ and micro‐ circulatory disease. This is an important observational study that underscores the utility of using low doses of amiloride to treat HA. Perhaps the conclusions about the role of the MR and ENaC are somewhat premature and should be, however, more nuanced. Based on recent basic work iterated below, there appears to be a role for both an MR antagonist such as spironolactone or eplerenone as well as an ENaC inhibitor such as amiloride or a combination of both in the treatment strategy for HA patients.12
While amiloride is an ENaC inhibitor, it should be appreciated that ENaC can be activated via a receptor‐mediated process. Aldosterone acting via MR engagement activates ENaC and thus promotes inward sodium currents through a MR‐dependent process in both renal epithelial cells and in vascular ECs.5, 6, 7 This is apparent as investigators have demonstrated that use of a MR antagonist or use of MR KOs (renal or vascular) prevents aldosterone‐mediated ENaC activation.5, 6, 7, 8 Classically, the ENaC, located in the apical membrane of renal aldosterone‐responsive epithelial cells, plays an essential role in controlling Na+ balance and BP.8 ENaC consists of three subunits, α, β, and γ. Although the α‐subunit is essential for proper channel function, the β/γ subunits act as amplifiers. Recent data have also shown that ENaC exists in the vascular endothelium (EnNaC) and that activation of this sodium channel promotes CV stiffness and dysfunction.5, 6, 7, 8, 9 In particular, our recent research indicates that aldosterone enhances ECMR signaling‐mediated increases in EnNaC activation, in part, by inducing translocation of EnNaC to the plasma membrane of the vascular ECs (Figure 1).5, 6, 9 Further, serum glucocorticoid kinase (SGK1) activation via ECMR signaling is involved in the aldosterone‐induced ENaC expression and activation. Activated SGK1 increases EnNaC production and inhibits its internalization and9 destruction, thereby leading to increased membrane localization of this sodium channel and associated increases in inward sodium currents which, in turn, leads to EC actin polymerization and associated endothelial and vascular stiffness.7, 8, 9 Collectively, recent work from our laboratory has thus demonstrated that activation of the ECMR promotes endothelial stiffness through increased EnNaC synthesis/activation and enhanced EC membrane localization. Endothelial stiffness, in turn, promotes remodeling of the vasculature and a subsequent increase in vascular stiffness. Stiffness of the coronary arteries, in turn, leads to cardiac fibrosis, stiffness, and impaired diastolic relaxation5 leading to heart failure with preserved systolic function. Based on this accumulating body of evidence, one can prevent the CV stiffening effects of excess aldosterone either by blocking vascular MRs with a drug like spironolactone or inhibiting EnNaC activation with amiloride.10, 11 The renal effects of excess aldosterone, such as tubulointerstitial fibrosis and albuminuria, would likewise be attenuated by either an MR antagonist or by the ENaC/EnNaC inhibitor amiloride.7, 8, 9
The article by Izzo et al11 raises a number of additional questions. For example, the authors also point out that despite inhibiting ENaC with amiloride, plasma aldosterone levels would have remained chronically high.7 Further, as none of the subjects received an MR antagonist after starting amiloride, aldosterone was presumably able to chronically activate MR receptors in other cells associated with the vascular wall including VSMCs13 and immune cells, such as monocytes and macrophages.8, 14 As the VSMC MR has been implicated in vascular aging, fibrosis, and stiffness,12, 14 it is interesting that the subjects treated with amiloride did not appear to show changes in CV stiffness nor did they have any detectable CV events during the period for which they were studied.10 Interestingly, VSMCs also express ENaCs which has been implicated as a mechano‐transducer,9, 15 but it is not known whether the VSMC sodium ion channel is directly linked to MR‐mediated vascular dysfunction and CV fibrosis and stiffness. Similarly, ENaC is expressed in dendritic cells and has been suggested to contribute to links between immune cell activation, inflammation and development of hypertension, and CV fibrosis and stiffness.10, 14
In their manuscript, Izzo et al11 also discuss their observation of rapid changes in arterial stiffness perhaps not being consistent with the reversal of slowly occurring remodeling processes (eg, involving collagen turnover). Rather, they suggest that a rapid decline in stiffness more likely reflects the lowering of BP by amiloride. While this may to some extent be the case, it will also be of interest to understand whether more acute/active components of stiffness (eg, vascular cell adhesion, cytoskeletal dynamics, and contractile activity12, 14, 16) are affected by amiloride treatment as seen in animal models.7 In this regard, amiloride may ultimately exert multiple actions through the vascular ENaCs and the renal ENaC that differ in their temporal relationships.7, 9, 11, 17
Overall, the study described by Izzo et al11 provides novel and important observations in the utility of amiloride treatment in HA patients, and their results suggest a considerable degree of concordance between recent experimental animal studies and HA‐induced hypertension and CV stiffness in man. It will be of interest to follow the clinical use of amiloride treatment, perhaps in combination with an MR antagonist11, 17 in larger and more diverse populations of HA. Similarly, the role of such treatment protocols in other disorders that are associated with elevated aldosterone levels will also be of considerable interest. Clearly, more mechanistic studies are also required in animals and cellular models to better understand the exact roles played by vascular and immune cell MRs and associated ENaC activation in hypertension and CV remodeling leading to fibrosis and stiffness.
CONFLICT OF INTEREST
None.
Funding information
Drs. Hill and Sowers' research has been funded by the NIH.
ACKNOWLEDGMENTS
The authors wish to thank Meg Duffy for her excellent assistance in the preparation and submission of this commentary.
REFERENCES
- 1. Monticone S, D'Ascenzo F, Moretti C, et al. Cardiovascular events and target organ damage in primary aldosteronism compared with essential hypertension: a systematic review and meta‐analysis. Lancet Diabetes Endocrinol. 2018;6:41‐50. [DOI] [PubMed] [Google Scholar]
- 2. Bauersachs J, Jaisser F, Toto R. Mineralocorticoid receptor activation and mineralocorticoid receptor antagonist treatment in cardiac and renal diseases. Hypertension. 2015;65:257‐263. [DOI] [PubMed] [Google Scholar]
- 3. Tarjus A, Amador C, Michea L, Jaisser F. Vascular mineralocorticoid receptor and blood pressure regulation. Curr Opin Pharmacol. 2015;21:138‐144. [DOI] [PubMed] [Google Scholar]
- 4. DeMarco VG, Habibi J, Jia G, et al. Low‐dose mineralocorticoid receptor blockade prevents western diet‐induced arterial stiffening in female mice. Hypertension. 2015;66:99‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jia G, Habibi J, DeMarco VG, et al. Endothelial mineralocorticoid receptor deletion prevents diet‐induced cardiac diastolic dysfunction in females. Hypertension. 2015;66:1159‐1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jia G, Habibi J, Aroor AR, et al. Endothelial mineralocorticoid receptor mediates diet induced aortic stiffness in females. Circ Res. 2016;118(6):935‐943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martinez‐Lemus LA, Aroor AR, Ramirez‐Perez FI, et al. Amiloride improves endothelial function and reduces vascular stiffness in female mice fed a western diet. Front Physiol. 2017;30(8):456‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jia G, Habibi J, Aroor AR, et al. Epithelial sodium channel in aldosterone‐induced endothelial stiffness and aortic dysfunction. Hypertension. 2018;72(3):731‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kleyman TR, Kashlan OB, Hughey RP. Epithelial Na channel regulation by extracellular and intracellular factors. Annu Rev Physiol. 2018;80:263‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li Q, Fung E. Multifaceted functions of epithelial Na+ channel in modulating blood pressure. Hypertension. 2019;73:273‐281. [DOI] [PubMed] [Google Scholar]
- 11. Izzo JL Jr., Hong M, Hussain T, Osmond PJ. Long‐term BP control and vascular health in patients with hyperaldosteronism treated with low‐dose, amiloride‐based therapy. J Clin Hypertens. 2019;21:922‐928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jia G, Aroor A, Martinez‐Lemus L, Sowers JR. Potential role of antihypertensive medications in preventing excessive arterial stiffening. Curr Hypertens Rep. 2018;20(9):76‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McCurley A, Pires PW, Bender SB, et al. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med. 2012;18(9):1429‐1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barbaro NR, Foss JD, Kryshtal DO, et al. Dendritic cell amiloride‐sensitive channels mediate sodium‐induced inflammation and hypertension. Cell Rep. 2017;21(4):1009‐1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Drummond HA, Jernigan NL, Grifoni SC. Sensing tension: epithelial sodium channel/acid‐sensing ion channel proteins in cardiovascular homeostasis. Hypertension. 2008;51:1265‐1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hill MA, Davis MJ, Meininger GA, Potocnik SJ, Murphy TV. Arteriolar myogenic signaling mechanisms: implications for local vascular function. Clin Hemorheol Microcirc. 2006;34:67‐79. [PubMed] [Google Scholar]
- 17. Jia G, Aroor AR, Hill MA, Sowers JR. Role of renin‐angiotensin‐aldosterone system activation in promoting cardiovascular fibrosis and stiffness. Hypertension. 2018;72(3):537‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]