

Abstract
Alpha-synuclein (αSyn) preformed fibrils (PFF) induce endogenous αSyn aggregation leading to reduced synaptic transmission. Neuronal activity modulates release of αSyn; however, whether neuronal activity regulates the spreading of αSyn pathology remains elusive. Here, we established a hippocampal slice culture system from wild type (WT) mice and found that both Ca2+ influx and the uptake of αSyn PFF were higher in the CA3 than in the CA1 sub-region. Pharmacologically enhancing neuronal activity substantially increased αSyn pathology in αSyn PFF-treated hippocampal or midbrain slice cultures and accelerated dopaminergic neuron degeneration. Consistently, neuronal hyperactivity promoted PFF trafficking along axons/dendrites within microfluidic chambers. Unexpectedly, enhancing neuronal activity in LRRK2 G2019S mutant slice cultures further increased αSyn pathology, especially with more Lewy body (LB) forming than in WT slice cultures. Finally, following injection of αSyn PFF and chemogenetic modulators into the dorsal striatum of WT mice, both motor behavior and αSyn pathology were exacerbated likely by enhancing neuronal activity since they were ameliorated by reducing neuronal activity. Thus, a greater understanding of the impact of neuronal activity on αSyn aggregation and spreading, as well as dopaminergic neuronal vulnerability, may provide new therapeutic strategies for patients with LB disease (LBD).
Keywords: Organotypic brain slice culture, Neuronal activity, Alpha-synuclein aggregation, Parkinson’s disease
Introduction
Misfolded alpha-synuclein (αSyn) forms neuronal inclusions known as Lewy bodies (LB) and Lewy neurites (LN), which are the pathological hallmarks of Parkinson’s disease (PD) and dementia with Lewy bodies (DLB), while glial cytoplasmic inclusions (GCI) in multiple system atrophy (MSA) also contain fibrillar αSyn but are found in oligodendroglia rather than in neurons [52, 53]. αSyn pathology is also commonly abundant in the brains of Alzheimer’s disease (AD) patients [28], and neurodegenerative diseases with αSyn pathology are commonly referred to as synucleinopathies.
PD is predominantly recognized for its motor symptoms (tremor, rigidity, bradykinesia and postural instability), but patients affected by this disease commonly experience a wide range of non-motor symptoms such as dementia that impact their quality of life. Due to deficiency of dopaminergic (DA) neurons in PD, the subthalamic nucleus (STN) become over activated [43], leading to excessive production of the neurotransmitter, glutamate [45]. Excessive glutamate binds to its ionotropic receptors (NMDA or AMPA) and open the voltage-gated calcium channels, which causes Ca2+ excitotoxicity. Excess Ca2+ load can damage the mitochondria and produce reactive oxygen species (ROS), leading to oxidative stress [13]. Though PD mainly occurs sporadically and is referred to as idiopathic PD, environmental toxins can cause increased production of glutamate, leading to Ca2+ excitotoxicity which makes DA neurons vulnerable to neurodegeneration [4, 13, 42]. And environmental stress is emerging as PD risk factors [8, 30, 40, 41]. However, how and to what extent these environmental exposures modulate the onset and progression of PD remains largely unknown.
Organotypic brain slice cultures (OBSCs) provide a highly suitable preparation of central nervous system (CNS) tissues for exploring the dynamics and physiological features of neuronal and glial cells within their native three-dimensional environments, and OBSCs have been widely applied in neurodegenerative disease research in recent decades [11, 27, 39]. Brain slices can be maintained for many months, offering the possibility for genetic manipulation using viral infection to induce tau and synuclein pathology [10, 15], biochemical analyses of tau and amyloid β (Aβ) aggregation [12, 37], long-term live imaging [21, 49], electrophysiological recordings [22], and therapeutic compound or antibody assessment [36]. In addition, OBSCs can also be used to track the uptake of extracellular toxic amylogenic “seeds” such as those formed by pathological αSyn [31].
Aberrant neuronal hyperactivity has been observed in most neurodegenerative diseases and perturbations of neuronal activity have been implicated as important driving forces of disease progression including the progression of PD [6, 65, 70]. Increasing neuronal activity elevated extracellular tau release and accelerated endocytosis of tau “seeds” in vivo [67], thereby potentiating tau pathology in vivo [65]. In a recent study, chronic sleep deprivation was shown to increase neuronal activity and facilitate the release of Aβ and tau [26]. Moreover, chronic chemogenetic attenuation of the hyperactivity of somatosensory cortex regions reduced Aβ palques [70]. Consistent with this, elevating neuronal activity rapidly increased, while blocking activity decreased, αSyn release in primary mouse hippocampal cultures which was confirmed by in vivo microdialysis [68]. Activity- and Ca2+-dependent αSyn secretion has also previously been reported in mouse striatum [17]. However, whether the cell-to-cell transmission of extracellular αSyn leading to propagation of αSyn pathologies is accelerated by hyperactivity in vivo remains to be determined.
To investigate whether αSyn pathology is influenced by neuronal activity, we applied αSyn PFF prepared from recombinant αSyn monomers that when utilized as seeds can reproducibly induce fibrillar PD-like αSyn pathology in vitro and in vivo [32, 61, 66]. The PFF were either transduced into ex vivo brain slice cultures or stereotaxically injected into WT mice in vivo. We hypothesized that enhancing neuronal activity would significantly potentiate the pathogenesis and spread of αSyn pathology in these PFF seeding systems, and as described below the results of these studies support this hypothesis.
Materials and methods
Mice
The animals were treated in accordance with the NIH Guide for the Care and Use of Experimental Animals approved by the University of Pennsylvania Institutional Animal Care and Use Committee. Pregnant CD1 (embryonic 16-18 days old) mice were used for the isolation of hippocampal neurons. C56BL/6J, LRRK2 G2019S transgenic (Tg) or αSyn knockout (KO) mice have been previously described [1, 35] and were used for preparation of hippocampal or midbrain slices. Where indicated, slices were also prepared from αSyn-GFP knock-in (SGKI) mice in which GFP was inserted between exons-5 and −6 of the Snca locus resulting in the expression of wildtype αSyn fused to a C-terminus fluorescent tag [7]. Heterozygous SGKI animals were used. All mice were housed under a 12 h light-dark cycle with free access to food and water. Both male and female mice were used in this study.
Wire hang behavior test
The wire hang test of motor function was conducted by following a modified protocol previously described [48]. The mice were placed on the top of a standard wire cage lid. The lid was lightly shaken to cause the animals to grip the wires and then turned upside down. The latency of mice to fall off the wire grid was measured, and average values were computed from three trials (15 min apart). Trials were stopped if the mouse remained on the lid after 5 min.
Recombinant αSyn purification and in vitro fibrillization
Full-length human αSyn (1-140) proteins were expressed in BL21 (DE3) RIL cells and purified as previously described [20]. Fibrillization was conducted by diluting recombinant αSyn to 5 mg/mL in sterile Dulbecco’s PBS (Cellgro, Mediatech; pH adjusted to 7.0, without Ca2+ or Mg2+) followed by incubating this recombinant αSyn at 37 °C with constant agitation at 1,000 r.p.m. for 7 days. Successful αSyn fibrillization was verified by electron microscopy, sedimentation studies and a thioflavin T-binding assay as described [60]. Assembled αSyn PFF were aliquoted and stored at −80 °C.
Organotypic hippocampal and midbrain slice cultures for αSyn PFF transduction
Organotypic hippocampal slice cultures were prepared from postnatal (P) 10-12 C56BL/6J, LRRK2 G2019S Tg, αSyn KO mice and littermate control WT mice as previously described [14, 27] with minor modifications. Briefly, pups were sacrificed, and the cerebellum, thalamus and brainstem were discarded to leave the cortex, hippocampus and connecting areas for the studies described below. These fresh samples were kept in ice-cold Krebs buffer (in mM): 1.2 NaH2PO4, 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.5 CaCl2, 25 NaHCO3, 10 D-glucose, 10 HEPES, 1% (v/v) penicillin/streptomycin in ultrapure sterile filtered (0.22 μm) H2O, pH 7.4. Next, 350 μm thick coronal slices were cut using a McIlwain Tissue Chopper (Stoelting, VWR) and 5-6 consecutive slices were positioned on Millicell culture inserts (MilliporeSigma, US) for culture in 6-well plates (ThermoFisher Scientific, US) containing 1 mL of sterile slice culture medium (in 100 mL: 77.5 mL Neurobasal-A, 2 mL B27 supplement, 20 mL heat-inactivated horse serum, 0.5 mL GlutaMAX, 1% (v/v) penicillin/streptomycin). The brain tissues rapidly attach to the membranes and receive nutrition from the slice culture medium through the membrane via capillary action.
For midbrain slice culture preparations, prefrontal cortex and cerebellum were carefully removed and tissues containing midbrains were embedded in 1% low melting agar (Difco, Fisher). Then, midbrain slices (350 μm thickness) were coronally sectioned with Compresstome VF-310 (Precisionary, US) and collected into sterile ice-cold Krebs buffer in a sterile Petri dish on ice. After collecting all of the brain sections, regions rich in dopaminergic neurons (ventral tegmental area/ substantia nigra, or VTA/SN) were selected with forceps, extra cortex/thalamus regions were removed and transferred onto the membrane inserts placed in serum–supplemented medium in culture plates. The dissecting Krebs buffer and culture medium were the same as those used for hippocampal slice cultures.
One day after plating the slices, the culture medium was removed by aspiration and replaced with 1 mL of pre-warmed fresh sterile culture medium. Brain slices were incubated at 37 °C with 5% CO2 and the culture medium was changed by removing it from the bottom of each well twice a week. Contamination and evaporation of medium was monitored each day. Slices are maintained for a minimum of 7 days in vitro (DIV) prior to treatment. On DIV 7, sonicated recombinant αSyn PFF were diluted to the desired concentration in sterile Dulbecco’s PBS (dPBS) and mixed thoroughly in fresh medium by vortexing, then added back to the bottom of each well. In some experiments, either KCl (6 mM), bicuculline (Bic, 10 μM) or picrotoxin (PTX, 25 μM) were added together with PFF to increase neuronal activity. Two days later, the medium was changed completely with pre-warmed fresh culture medium. Thereafter, slices were continuously cultured for different purposes as indicated.
Immunocytochemistry of brain slices
Slices were fixed for 20 mins in 4% paraformaldehyde (PFA) in dPBS (applied both above and below the membrane insert). To reduce the volumes required for subsequent steps, the membranes were cut free of the plastic inserts and the sections of membrane containing the slices were transferred with forceps to individual wells. Slices were washed twice in dPBS, blocked for 1 h in blocking solution (Ca2+/Mg2+-free dPBS with 0.5% Triton X-100 and 3% fetal bovine serum) then incubated in 200 μL primary antibody diluted in blocking solution overnight at 4 °C with shaking. Slices were washed 3 times in dPBS before being incubated for 2 h at room temperature (RT) in the dark with Alexa 488, 568 or 647-conjugated secondary antibodies (Life Technologies-diluted 1:1000 in blocking solution). After the final three dPBS wash, slices were counterstained with DAPI. Images were captured on an LSM 710 inverted confocal microscope (Carl Zeiss). Background subtraction and fluorescence intensity analysis were acquired with ImageJ software (National Institutes of Health, USA).
Quantification of αSyn PFF uptake and degradation in brain slices
Hippocampal slices were cultured for 7 days and equal amounts of human αSyn PFF (5 μg) were added into each slice culture medium followed 24 h later with a complete medium change. Slices were collected at 1-, 3- and 5-day post transduction and homogenized in lysis buffer containing protease and phosphatase inhibitor cocktails. Protein concentrations for all treatments were determined using the BCA assay (Fisher Cat#23223 and 23224) with bovine serum albumin as a standard (Thermo Fisher Cat#23210). Samples were normalized for total protein content and prepared for western blot analysis. Samples (20 μg total protein) were separated on SDS-polyacrylamide gels (4%-20% gradient) and transferred onto 0.22 μm nitrocellulose membranes. Blots were blocked in 5% non-fat milk in TBS, probed with Syn211 (CNDR, human αSyn specific, 1:1,000) and loading control GAPDH (Cell signaling, 1:2,000), and incubated overnight at 4 °C. The blots were further incubated with IRDye-labeled secondary antibodies (IRDye 800 (Li-Cor 925-32210) or IRDye 680 (Li-Cor 925-68071)) for 1 h and scanned using Li-Cor Odyssey Imaging System. After target antigens were detected, the optical densities (ODs) were measured with Image Studio software (Li-Cor Biosciences). Proteins were normalized to GAPDH.
Ca2+ imaging in brain slices
After preparation, hippocampal slices were cultured for 3 days in vitro for recovery. Then, 1.5 μL of neurotropic adeno-associated virus (AAV) particles (pAAV.Syn.NES.jRCaMP1a.WPRE.SV40, Addgene, Cat#100848-AAV1, titer>1×1013 vg/mL) were added into culture medium as a Ca2+ indicator. Three days later, the medium was completely changed to fresh culture medium. Slices were cut out of membrane inserts 7 days later, flipped over and placed in an imaging chamber. The extracellular imaging buffer in mM contained: 119.0 NaCl, 2.5 KCl, 2.0 CaCl2, 2.0 MgCl2, 25.0 HEPES, and 30.0 D-glucose. Laser scanning confocal microscopy (Olympus, FV-1000) equipped with Ar-ion laser (488 nm) and HeNe laser (532 nm) was used to capture Ca2+ dynamics. During live imaging, slices were continuously perfused with O2. Confocal time-lapse images were taken every 5 s during recording. Images were analyzed with ImageJ software (National Institutes of Health).
Microfluidic chamber culture of primary neurons during αSyn PFF transduction
Microfluidic neuronal culture devices with 2 somal compartments connected by a series of microgrooves were obtained from Xona Microfluidics (Temecula, CA). Glass coverslips (Corning Inc.) were coated with poly-d-lysine and affixed to neuronal chambers as per the manufacturer’s instructions. A total of 100,000 dissociated hippocampal mouse neurons were plated onto one chamber of the device. On DIV 7-9, either sonicated human WT αSyn PFF (0.5 μg) were added to the somal compartment and fixed 5 days later for immunocytochemistry (ICC), or sonicated pHrodo red-labeled αSyn PFF (0.5 μg) were added into the somal chamber and uptake was captured 2 days later.
Stereotaxic injections of αSyn PFF and AAV virus
αSyn PFF, which were generated at a concentration of 5 mg/mL, were vortexed and diluted with Dulbecco’s PBS to 2 mg/mL. They were then sonicated at 10 cycles of 30 s on, 30 s off (Diagenode Biorupter UCD-300 bath sonicator). Then, 3-4 months old WT B6C3F1/J mice were deeply anesthetized with ketamine-xylazine-acepromazine and immobilized in a stereotaxic frame (David Kopf Instruments, USA) as described [23, 33]. Mice were injected unilaterally with αSyn PFF by insertion of a single needle into the right forebrain (coordinates: +0.2 mm relative to bregma, +2.0 mm from midline) targeting the dorsal striatum (2.6 mm beneath the dura) with PBS or 2.5 μg of αSyn PFF (2.5 μl) together with or without 2x109 GC AAV virus particles that were used to modulate neuronal activity or control, i.e. pAAV-hSyn-hM3D(Gq)-mCherry (Addgene, Plasmid #50474), pAAV-hSyn-hM4D(Gi)-mCherry (Addgene, Plasmid #50475) and pAAV-hSyn-mCherry (Addgene, Plasmid #114472). Injections were performed using a 10-μL syringe (Hamilton) at a rate of 0.4 μL/min. During the incubation, clozapine-N-oxide (CNO, 0.5 mg/kg; catalog #4936; Tocris Bioscience) was injected at 7 days post virus injection via an intraperitoneal route (i.p.) to activate those chemogenetic receptors. The wire hang motor behavior test was performed at 1 month and 3 months. After the final behavior test at 3 months, mice were euthanized and perfused transcardially with PBS, brains were removed and underwent overnight immersion fixation in 70% ethanol in 150 mM NaCl (pH 7.4) for histopathology.
Immunohistochemistry
After perfusion and fixation, brains were embedded in paraffin blocks, cut into 6-μm sections and mounted on glass slides. The tissue sections were then stained on the slides using standard immunohistochemistry (IHC) as described elsewhere [58]. For IHC staining, phosphorylated αSyn antibody (81a, 1:8,000, CNDR; or EP1536Y, 1:20,000, Abcam, ab51253), or c-fos antibody (1:2,000, Abcam, ab208942) was diluted in blocking buffer. 81a antibody was used to generate typical images, while EP1536Y antibody was used to map p-αSyn pathology throughout whole brain sections. After IHC, sections were scanned with a 3DHISTECH Laminar Scanner (Perkin Elmer) at 20x magnification and quantification of the percentage area occupied by IHC labeled pathologies were performed with HALO software (Indica Labs). Section selection, annotation and quantification were all done blinded to the nature of the injected materials. For quantification of αSyn pathology, coronal sections were selected to closely match the following coordinates, relative to bregma: 2.10 mm, 0.98 mm, −1.22 mm, −2.92 mm and −4.48 mm. The digitized images were imported into the software HALO to allow annotation and quantification of the percent area occupied by phosphorylated αSyn pathology. Standardized annotations were drawn to allow independent quantification of 172 different regions throughout the brain. Each set of annotations was imported onto the desired section and modified by hand to match the designated brain regions.
Quantification and statistical analysis
All experiments were performed with control and experimental treatments side-by-side and in random order with replication of these studies at least three times. The number of samples or animals analyzed in each experiment, the statistical analysis performed, as well as the p values for all results are reported in the figure legends. For all in vivo experiments, the reported “n” represents the number of animals. The data was first tested for normality using D’agostino-pearson test. For comparison between two groups, a t test was applied. A one-way ANOVA was applied when multiple groups were compared with one variable followed by Tukey’s multiple comparison test. If the data did not pass normality, a Kruskal-Wallis test was used followed by Dunn’s multiple comparison test. A two-way ANOVA was applied to compare values between the PFF and PFF plus virus-injected groups of mice followed by Tukey’s multiple comparison test. Statistical analysis was performed with Prism V6.0 software (GraphPad Software, Inc., USA). Data are presented as mean ± SEM. Statistical tests were two-tailed and the level of significance was set at p < 0.05.
Results
αSyn PFF induced phosphorylated αSyn pathology in organotypic hippocampal slice cultures
Previously, we have shown that αSyn PFF compromised synaptic activity in primary cultured hippocampal neurons [66]. To study the initiation and spread of αSyn pathology in a more synaptically-interconnected system, we established an organotypic hippocampal slice culture system (Fig. 1a, b), which has been previously used to study neurodegenerative diseases [11, 27]. After slices were maintained for 7 days in vitro (DIV 7), sonicated αSyn PFF were added into culture medium in a chamber below the insert and can be homogeneously diffused via capillary action from medium onto brain slices on the top at dosages from 1 to 10 μg (Fig. 1c). From measurement of phosphorylated αSyn (p-αSyn) staining with 81a, which labels pathological αSyn [19, 64], we observed a PFF dose-dependent increase in αSyn pathology. Indeed, the highest quantities of αSyn PFF (10 μg) resulted in slice-wide αSyn pathology (Fig. 1c and supplementary Fig. 1a, online resource). However, the number of neurons identified by NeuN-positive nuclei was significantly decreased at 10 μg of PFF, presumably due to neurotoxicity (Supplementary Fig. 1b, online resource). Accordingly, we used 5 μg of αSyn PFF in this study unless otherwise stated.
Fig. 1.
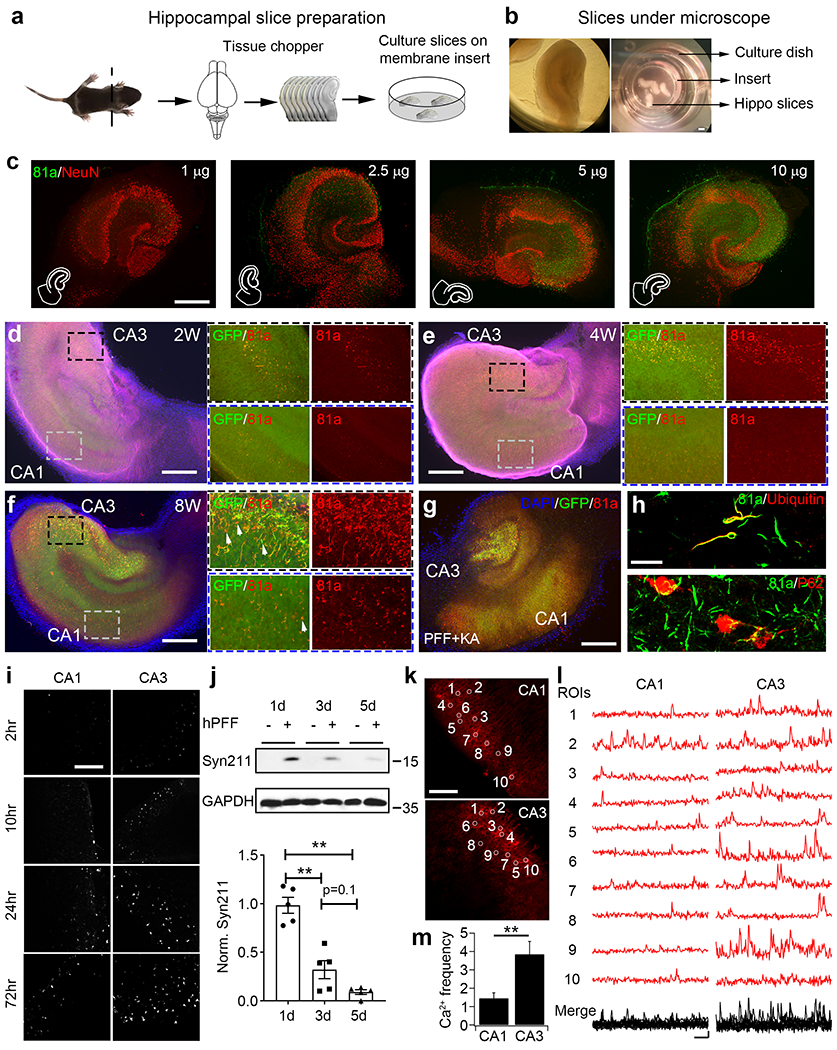
αSyn PFF induced phosphorylated αSyn pathology in organotypic hippocampal slice cultures. a These cartoons illustrate the preparation and culture of hippocampal slices on membrane inserts. b Images of hippocampal slices under the microscope and in culture dish. Please note that slices are cultured on the top of membrane inserts, culture medium and sonicated αSyn PFF were added on the bottom of the membrane. c Dose dependence of αSyn PFF induced p-αSyn pathology in hippocampal slices. αSyn PFF were diluted in Ca2+/Mg2+-free dPBS, sonicated, and then added into culture medium of hippocampal slices (1-10 μg per 6-well dish as indicated in the panels). Two weeks post-PFF addition, slices were fixed with PFA and followed by ICC as indicated in Methods. Green color: αSyn Serine-129 phosphorylation antibody, 81a; red color: neuronal marker, NeuN. Experiments were independently repeated for 3 times. Scale bar: 250 μm. d-f Time dependence of αSyn PFF induced p-αSyn pathology in hippocampal slices. αSyn PFF were pre-sonicated and added into culture medium at 5 μg per 6-well plate, then slices were cultured for 2 (2W, panel d), 4 (4W, panel e) and 8 weeks (8W, panel f) as indicated. Black dashed rectangles represent the zoom in of the CA3 regions, where blue dashed rectangles represent the zoom in of the CA1 regions. Please note that magenta color was removed in panel (f) to show the details of 81a and green colors. Arrows in panel (f) indicate the GFP signals that do not colocalize with 81a. Blue color: DAPI; green color: endogenous αSyn GFP knock in (SGKI); red color: 81a; magenta color: dendritic marker, MAP2. Experiments were independently repeated for 4 times. Scale bar: 250 μm. g 81a staining of αSyn PFF-treated SGKI slices. In this experiment, slices were pre-treated with kainic acid (KA, 5μM) to induce neuronal degeneration in CA3 pyramidal cells as indicated by the loss of GFP and 81a signals in CA3 region. Experiments were independently repeated for 2 times. Scale bar: 250 μm. h Co-localization of 81a with ubiquitin (upper panel) or p62 (bottom panel). Experiments were independently repeated for 3 times. Scale bar: 20 μm. i Time-dependent uptake of pHrodo-red labeled αSyn PFF in CA1 and CA3. pHrodo-red signal was saturated at around 24 h post-PFF addition. Experiments were independently repeated for 3 times. Scale bar: 100 μm. j Upper panels: Endocytosis and degradation of human αSyn PFF as measured by western blot. Bottom panel: quantification of Syn211 signals from above panels. Syn211: human-specific αSyn antibody; GAPDH: loading controls for different groups. Experiments were independently repeated for 5 times. k and l Typical graphs and spontaneous Ca2+ imaging traces from cultured hippocampal slices. Ca2+ sensor RCaMP1 was overexpressed with AAV virus, and Ca2+ signals were taken 10 days later after AAV addition. White circles in images of CA1 and CA3 in panel (k) indicate the cells randomly selected to generate the typical Ca2+ signal traces in panel (l). Scale bar in (k): 100 μm; Scale bar in (l): 10 s and 0.05 ΔF/F0. For Ca2+ signal analysis in panel (l), intensity of 10 s baseline before peak signal was averaged and used as F0 [63]. m Quantification of Ca2+ frequency from panel (l). Experiments were independently repeated for 4 times. **: p<0.01. Data are presented as mean ± SEM. See also Supplementary Fig. 1–9 and movie 1, online resource
Next, the time-dependence of αSyn PFF-induced pathology in cultured slices was examined by adding PFF at DIV 7 and fixing the slices 2-, 4- and 8-weeks post-treatment (Fig. 1d–f and supplementary Fig. 2, online resource). In this experiment, we used heterozygous αSyn-GFP transgenic knock in (SGKI) mice [7] to directly monitor the morphological changes of cultured slices as well as the aggregation of αSyn over time. At 2 weeks post-PFF addition, only a small number of 81a-positive (p-αSyn) puncta were observed and they colocalized with endogenous GFP-labeled αSyn aggregates (Fig. 1d). At 4 and 8 weeks post-PFF treatment, more p-αSyn pathology was found and the neuritic p-αSyn staining was more extensive at 8 weeks (Fig. 1e, f). However, some of the p-αSyn pathology was not fully co-localized with the αSyn-GFP aggregates (Fig. 1f, zoom in panels of CA1 and CA3) and some αSyn PFF-treated slices were not able to survive for 8 weeks (data not shown). Thus, in most of the PFF studies reported hereafter, we cultured hippocampal slices up to 4 weeks before fixation. More p-αSyn pathology was always found in CA3 than CA1 within the same slices at all the time points we investigated (Fig. 1d–f, right zoom in panels). Note that, some PFF seeds were taken up intracellularly early during treatment, but for unknown reasons it takes longer to develop aggregates and spreading. In our recent paper on neuronal activity and αSyn spread [66], we used patch clamp pipette to infuse sonicated αSyn PFF into the whole cell patched primary cultured hippocampal neurons and found that, the direct infusion of αSyn PFF induced 81a-positive αSyn pathology as soon as 20 hours after treatment. This is quite different from introducing αSyn PFF into the culture media which takes 3-4 days to develop detectable pathologies [66]. Thus, unlike PFF transduction in the medium, the infused αSyn PFF do not need to undergo uptake through the plasma membrane, trafficking along the endo-lysosomal pathway, processing and release from the lysosome into the cytosol before recruiting endogenous αSyn monomer into insoluble aggregates [29]. During the long-term culturing of slices, complex processes such as synthesis, degradation and turn over are all occurring simultaneously and we cannot preclude the possibility that some PFF seeds could linger longer inside the lysosomes and were released from ruptured lysosomes later, resulting in a delay in the induction of aggregates.
To investigate the potential role of neuronal activity on αSyn pathology, we pretreated slices with kainic acid (KA) to reversibly depolarize CA3 pyramidal neurons and analyzed αSyn pathology in the context of αSyn PFF treatment of the cultures (KA plus PFF, Fig. 1g and supplementary Fig. 3, online resource). Two weeks post-KA plus αSyn PFF treatment, both GFP and 81a signals were diminished in the CA3 region, but the CA1 and other regions remained unchanged (Fig. 1g), which confirmed our observations that more p-αSyn pathology was indeed present in CA3 pyramidal neurons. In addition, we co-stained p-αSyn pathology with two other commonly used disease markers, ubiquitin and p62 (Fig. 1h), which are present in LB and LN of PD and DLB brains [47, 51]. Moreover, some of the p-αSyn staining partially co-localized in neurons with the CA pyramidal neuron marker, Math2, but not with the DG neuron marker, Prox1 (Supplementary Fig. 4, online resource), which is consistent with previous in vivo data where less endogenous αSyn was expressed in DG sub-regions [34]. To exclude the possibility of off-target effect of phosphorylation dependent Ser129 (81a) antibody in slice cultures, αSyn knock out (KO) hippocampal slices were cultured and treated with PFF for 1-month period. No obvious 81a signals were detected in αSyn KO slices (Supplementary Fig. 5, online resource). Additionally, the 81a signals colocalized quite well with the amyloidogenic β-sheet structure binding dye, pFTAA and ThioS, in slices (Supplementary Fig. 6, online resource). What’s more, no obvious cross reaction of 81a with other activity-activated phosphorylated proteins, like c-fos, MAPK, pCREB or DARP32, can be detected (Supplementary Fig. 7, online resource).
Since more αSyn pathology was found in the CA3 than the CA1 in this slice culture system, we further hypothesized that neurons in the CA3 region are more active and have higher capacity to endocytose extracellular αSyn PFF and propagate αSyn pathology in hippocampal slices. To test this hypothesis, we first performed uptake studies of pHrodo red-labeled αSyn PFF (pHRed-PFF) in cultured slices (Fig. 1i). pHrodo red is a fluorogenic dye that dramatically increases in fluorescence in acidic surroundings and can be used as endocytic indicator [29]. As in the above experiments, sonicated pHRed-PFF were added into the slice culture medium. pHrodo red signal became visible at around 2 h post-transduction and saturated at around 24 h (Fig. 1i and supplementary Fig. 8, online resource). Consistent with the localization of αSyn pathology (Fig. 1d–f), more pHrodo red signal was observed in the CA3 than CA1 regions, which confirmed that more PFF were taken up by CA3 neurons (Fig. 1i). This was also confirmed in αSyn knock out (KO) hippocampal slices, where more pHRed-PFF were taken up in the CA3 than CA1, indicating that the uptake of PFF is independent of αSyn expression in these regions (Supplementary Fig. 9, online resource). Next, we investigated the degradation of αSyn PFF after uptake in slices. Human αSyn PFF were added into slice culture medium for 1, 3 and 5 days and slices were collected for western blot experiments. The amount of human αSyn as detected with Syn211 antibody peaked at 1-day post transduction and decreased at 3 days and 5 days, indicating degradation of the internalized PFF (Fig. 1j).
Finally, to further investigate the neuronal activity of the CA1 and CA3 regions in cultured hippocampal slices, human synapsin-promoter driven Ca2+ reporter RCaMP1 was overexpressed after AAV infection of hippocampal slices (Fig. 1k). Spontaneous Ca2+ signals were captured from both the CA1 and CA3 within the same slice 10 days after AAV addition (Fig. 1l). The Ca2+ spike frequency was 2.5 times higher in the CA3 than CA1 (Fig. 1m and supplementary movie 1, online resource), which further supported the hypothesis that the greater neuronal activity in the CA3 regions promoted αSyn PFF uptake and resulted in more αSyn pathology in cultured hippocampal slices.
Enhancing neuronal activity facilitates the phosphorylation of αSyn in hippocampal slices
To further investigate the effect of neuronal activity on the initiation of αSyn pathology and subsequent propagation in hippocampal slices, we co-treated slices with PFF and compounds that are known to depolarize or hyperactivate neurons (Fig. 2a, b). Compared with PFF treatment alone, KCl depolarization, or bicuculline (Bic) or picrotoxin (PTX) activation, of hippocampal slices induced more αSyn pathology in CA3, CA2 and CA1 regions, but not in DG neurons (Fig. 2c), which do not express abundant αSyn for recruitment of pathology to occur under this condition [34]. To exclude the possibility of compound toxicities affecting neuronal activity, we performed western blots and found no significant changes of αSyn or GAPDH expression with any of the treatment modalities here (Supplementary Fig. 10a, online resource). Similarly, no significant cell death was identified with propidium iodide (PI) staining (Supplementary Fig. 10b, online resource).
Fig. 2.
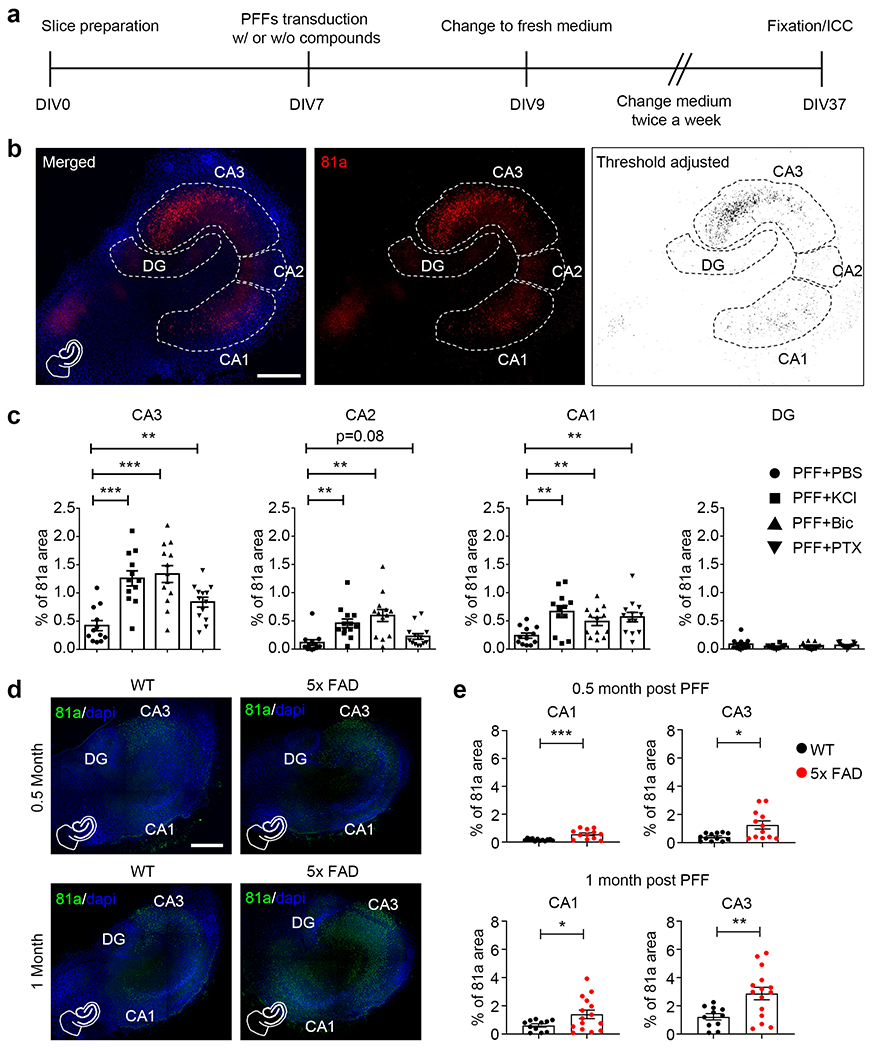
Enhancing neuronal activity facilitates the phosphorylation of αSyn in hippocampal slices. a Experimental protocol indicates the preparation and treatment of hippocampal slices. b Typical 81a staining with segmentation and threshold adjustment of hippocampal slices. On DIV 7, slices were treated with αSyn PFF at 5 μg per well. At the same time, different compounds were added together with PFF into the culture media to modulate neuronal activity. Left panel: merge of 81a and DAPI. Middle panel: 81a staining with white dashed lines to indicate the borders of DG, CA3, CA2 and CA1 for further quantification. Right panel: ImageJ software was used to adjust threshold to reduce background from middle panel. Scale bar: 250 μm. c Quantification of percentage of 81a area from CA3, CA2, CA1 and DG treated with αSyn PFF together with PBS, potassium chloride (KCl), bicuculline (Bic) or picrotoxin (PTX) to enhance neuronal activity. Experiments were independently repeated for 3 times. d Same as panel B, but slices were prepared from WT and 5xFAD littermate pups, αSyn PFF were added at DIV 7 for 0.5 month and 1 month, respectively. Green color: 81a; blue color: DAPI. Scale bar: 250 μm. e Quantification of percentage of 81a area in WT and 5xFAD slices. Experiments were independently repeated for 4 times. *, p<0.05; **, p<0.01; ***, p<0.001. Data are presented as mean ± SEM. See also Supplementary Fig. 10–12 and movie 2, online resource
Since higher neuronal activity also correlated with increased uptake of PFF (Fig. 1i), we wanted to investigate whether neuronal activity might also enhance αSyn pathology through increased recruitment of endogenous αSyn or via enhancement of cell-to-cell spread of αSyn pathology. From previous studies, we know that αSyn PFF will be degraded by about 90% 5 days after addition to slices (Fig. 1j). We thus added αSyn PFF to slices at DIV 7, and 7 days later (DIV 14), PBS or PTX were added into the culture medium. After 2 days (DIV 16), culture medium was changed back to normal fresh medium and slices were cultured for another 21 days prior to immunostaining for αSyn pathology (DIV 37) (Supplementary Fig. 11a, online resource). Using this modified protocol, more αSyn pathology was found in PTX-treated slices (Supplementary Fig. 11b, c, online resource), indicating that neuronal activity alone promotes the formation and/or propagation of αSyn pathology in slice cultures after the initial uptake of PFF seeds. However, it remains possible that the thickness of the brain slices decreased overtime which could possibly confound the interpretation of the increase in immunofluorescence signals. Although slices were treated with either PFF alone or PFF plus KCl or PTX, it is hard to estimate whether KC or PTX has any effect on the reduction in thickness of the slices during culture. While we did not observe obvious defects in the morphology of slices or loss of NeuN-positive neuronal markers with the dose of PFF we used (Fig. 1d, e and supplementary Fig. 1, online resource), we believe that the αSyn aggregates induced by PFF alone or PFF plus KCl should have similar effects on the thickness of slices. However, factors such as KCl or PTX, could have other unknown effects on the thickness of the slices that could eventually affects the intensity of the immunofluorescence signals.
It has recently been reported that neuronal hyperactivity as measured by two-photon Ca2+ imaging precedes Aβ plaque formation in the hippocampus of an AD mouse model [6]. To investigate whether Aβ-mediated effects on neuronal activity may alter the formation or spread of αSyn pathology, we utilized the commonly used 5xFAD mouse model of Aβ plaque formation [38]. These mice express human APP and PSEN1 transgenes with five AD-linked mutations and develop an abundance of Aβ plaques within their brains. Hippocampal slices were prepared from 5xFAD heterozygous and non-transgenic littermate 10-day old pups, an age when no amyloid plaques are observed [38], and the Ca2+ indicator RCaMP1 was expressed via AAV virus infection to monitor neuronal activity. Notably, increased neuronal activity was observed throughout the slices of heterozygous 5xFAD mice compared to slices from the WT mice (Supplementary movie 2, online resource). As previously observed, more αSyn pathology was found in the CA3 than CA1 at 2 and 4 weeks after PFF addition (Fig. 2d, e). As a control experiment, no obvious αSyn pathology was developed in 5xFAD slices without PFF transduction with or without hyperactivity (Supplementary Fig. 12, online resource).
Since young 5xFAD pups were used for slice preparation, no Aβ plaques were developed in these slices, and the young 5xFAD transgenic slices displayed hyperactivity ex vivo in culture; while in Bassil et al. 2020, they used adult mice, where abundant plaques were present everywhere in the brain [3]. Thus, consistent with pharmacological enhancement in neuronal activity, the spontaneous hyperactivity of neurons in the young 5xFAD slices promoted αSyn PFF endocytosis and then potentiated pathology in brain slice cultures, while in Bassil et al. 2020, the presence of amyloid plaques facilitated the formation and spreading of αSyn aggregates [3].
Enhancing neuronal activity facilitates αSyn PFF “seeds” trafficking along axons
Microfluidic chambers provide a platform for studying cell-to-cell transmission and transport of fluorescent labeled PFF via live imaging at the soma and axon/dendrite level, thus offering certain technical advantages over in vivo and hippocampal slice culture systems [61]. Accordingly, dissociated hippocampal neurons were plated into one chamber of a microfluidic plate and live imaging was performed to analyze the internalization and traffic of pHRed-PFF along distal axons growing in grooves of the chambers (Fig. 3a). The imaging revealed more pHRed-PFF puncta in grooves with neurons that were treated with compounds to increase activity (Fig. 3b). Based on the quantification, both the puncta quantities and the distance travelled were higher in hyperactive chambers (Fig. 3d, left two panels). Additionally, some chambers were treated with αSyn PFF plus bicuculline or picrotoxin for longer times and fixed with PFA to analyze the induced αSyn pathology. Greater 81a staining was found in somatic chambers, grooves and axonal chambers after induction of hyperactivity (Fig. 3c, d, right two panels), indicating that enhanced neuronal activity facilitates αSyn PFF uptake and trafficking along axons and potentiates αSyn pathology both in primary dissociated neuron cultures and hippocampal slice cultures.
Fig. 3.
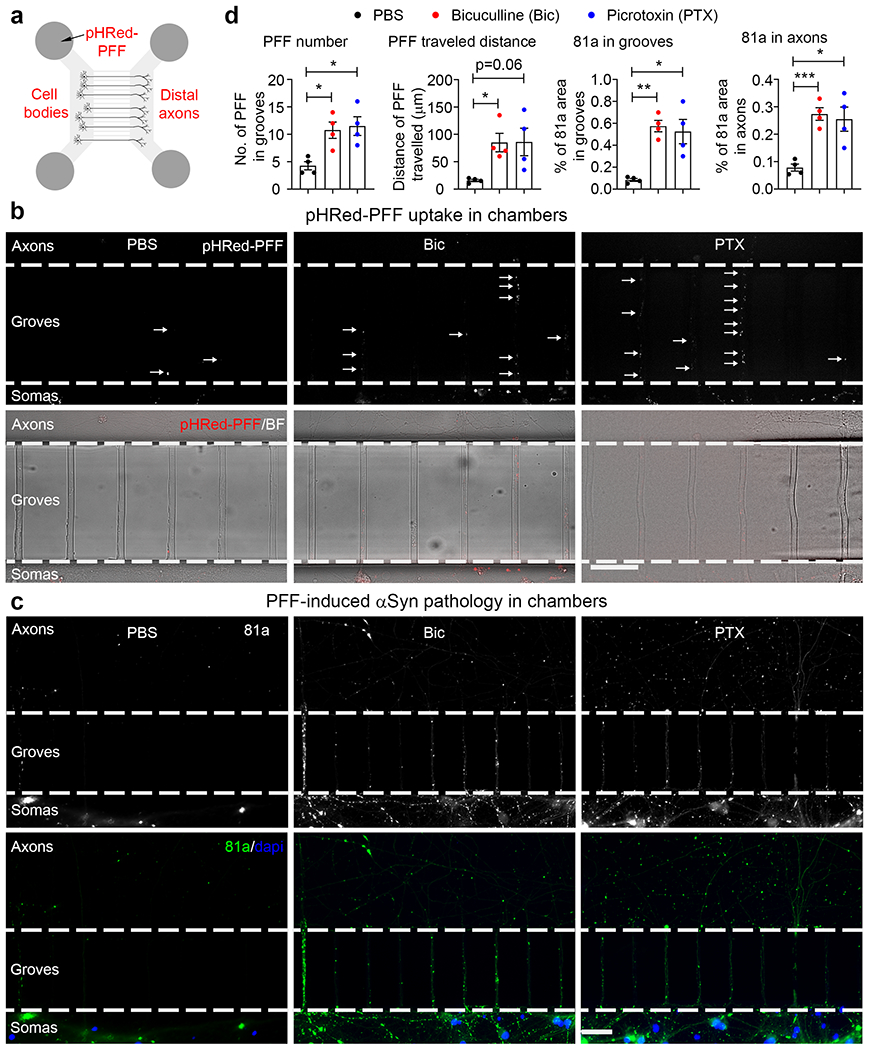
Enhancing neuronal activity facilitates αSyn PFF trafficking along axons in microfluidic chambers. a Schematic cartoon illustrates primary culture of hippocampal neurons and addition of pHRed-PFF in a microfluidic chamber. 0.5 μg PFF were added in the somatic chamber at DIV 10–12 and images were taken 2 days later. pHRed-PFF: pHrodo-red labeled αSyn PFF. b Uptake and trafficking of pHRed-PFF in microfluidic chamber. Upper panels: individual arrows indicate the pHRed-PFF “seeds” located in grooves of microfluidic chambers; bottom panels: merge of pHRed-PFF and bright field (BF) channels. Experiments were independently repeated for 3 times. Scale bar: 50 μm. c ICC staining of 81a signals in microfluidic chambers treated with αSyn PFF together with PBS, Bic or PTX as indicated. After enhancing neuronal activity, more 81a signal was found in both axons and grooves. Upper panels: 81a staining of αSyn PFF with or without compound treatment; bottom panels: merge of 81a (green color) and DAPI (blue color). Experiments were independently repeated for 3 times. d Quantification of pHRed-PFF puncta and 81a pathology from panels (b) and (c). *, p<0.05; **, p<0.01; ***, p<0.001. Data are presented as mean ± SEM.
Enhancing neuronal activity exacerbates dopaminergic neuron vulnerability in the context of elevated αSyn pathology
Since neuronal activity per se implies high-energy dependence of Ca2+ influx, midbrain dopaminergic neurons of the VTA are particularly vulnerable to harmful Ca2+-induced stress [56]. For these reasons, we investigated the effects of neuronal activity on αSyn pathology in midbrain dopaminergic neurons and its degeneration.
To this end, we developed an organotypic midbrain slice culture system (Fig. 4a). Briefly, after αSyn PFF addition to midbrain slice culture medium, the slices were maintained for 1 month and then immunostained with 81a and TH antibodies (Fig. 4b, c). Some TH neurons were 81a-positive (Fig. 4d), recapitulating the pathological features of PD and DLB brains [28]. Moreover, we also identified some 81a-positive nigral neurons that had little or no TH signals (Fig. 4d, right panel), suggestive of dopaminergic neuronal degeneration. Consistent with hippocampal slice culture, more αSyn pathology was found in PFF-treated midbrain slices when hyperactivity was induced by either KCl or PTX (Fig. 4e). In addition, when compared with PBS-treated slices, detectable TH neuron loss (~10%) was identified in PFF-treated slices. Further, there was about a 25% greater TH neuron loss in midbrain slices treated with PFF plus KCl or PTX to depolarize or hyperactivate slices, respectively (Fig. 4f). In contrast, no significant TH neuron loss was seen in slices treated with KCl or PTX in the absence of PFF (Fig. 4f). Thus, enhancing neuronal activity, especially in TH neurons, potentiates not only αSyn pathology but also accelerates neurodegeneration.
Fig. 4.
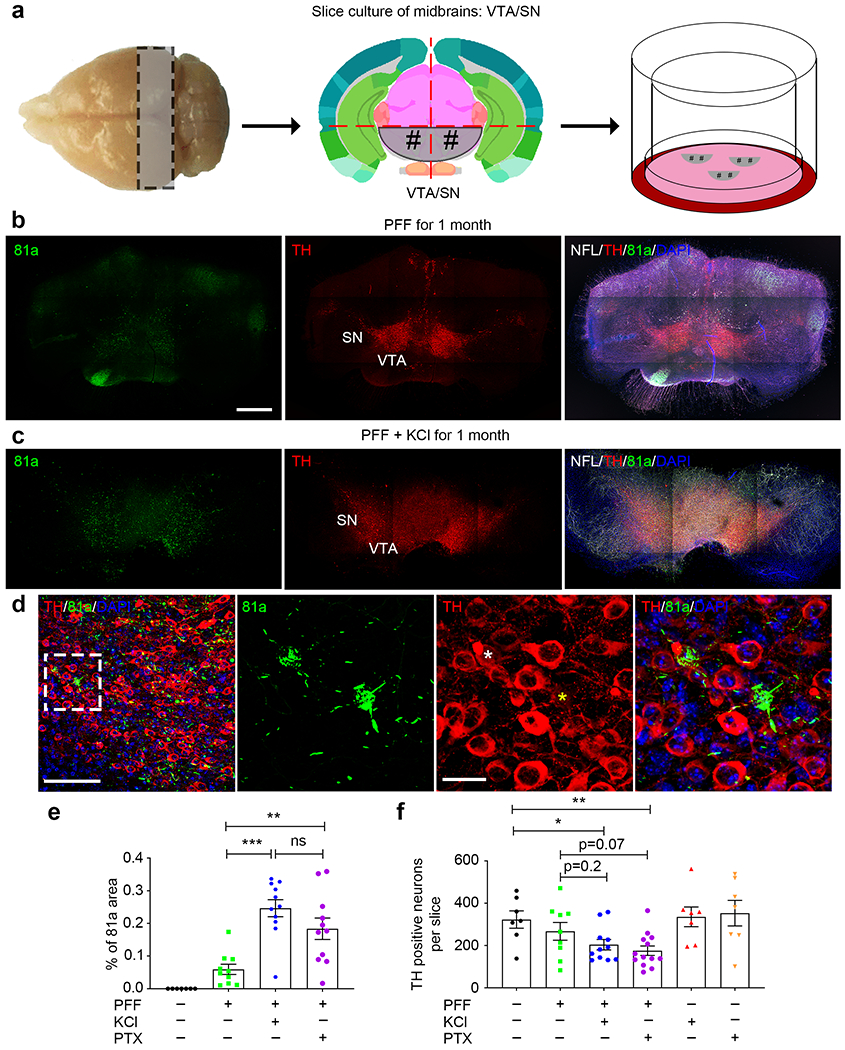
Enhancing neuronal activity exacerbates dopaminergic neuron vulnerability in the context of elevated p-αSyn level. a Schematic cartoon illustrates the preparation and culture of VTA/SN from midbrains. As with hippocampal slice cultures, mouse midbrains were coronally sectioned at a thickness of 350 μm, then VTA/SN were dissected and placed onto membrane inserts for subsequent culture. At DIV 7, αSyn PFF were added into the culture medium and cultured for another 1 month. b and c Typical graphs of 81a and TH positive dopaminergic neurons in midbrain slice cultures with or without KCl to enhance neuronal activity. NFL: neurofilament light chain, TH: dopaminergic neuronal marker. Scale bar: 200 μm. d Zoom in of PFF plus KCl-treated 81a and TH staining, where LB and TH neuron degeneration were observed. White asterisk indicates a TH-positive neuron bearing LB pathology, where yellow asterisk indicates a degenerating TH neuron bearing LB pathology. e and f Quantification of percentage of 81a area and TH positive neurons in midbrain slices with distinct treatments. Experiments were independently repeated for 3 times. *, p<0.05; **, p<0.01; ***, p<0.001; ns, not significant. Data are presented as mean ± SEM.
However, increased neuronal activity results in more dopamine release in midbrains [46], and more dopamine facilitates αSyn aggregation in the context of extracellular αSyn PFF seeds [57]. On the other hand, enhanced neuronal activity opened the Ca2+ channels and raised Ca2+ concentration inside DA neurons. Excess Ca2+ load can damage the mitochondria and produce reactive oxygen species (ROS), leading to oxidative stress and Ca2+ excitotoxicity which makes DA neurons vulnerable to neurodegeneration [4, 13, 42].
Enhancing neuronal activity results in more LB pathology in slices from LRRK2 with gain-of-function mutation
Although most patients have idiopathic PD, 5-15% of patients have PD due to genetic mutations or other risk factors. In the past decades, several PD candidate genes have been identified, including LRRK2 as a PD risk factor [41, 62]. We have recently shown that LRRK2 GTPase gain-of-function mutant G2019S mice exhibit little change in the amount of αSyn pathology after αSyn PFF injection into dorsal striatum when compared with WT littermate mice. However, the spreading pattern of αSyn pathology from the injection site to synaptically-interconnected regions is highly accelerated in the G2019S mice [25]. This led us to ask whether the LRRK2 G2019S mutation has any effect on αSyn pathology under conditions of neuronal hyperactivity.
To answer this question, hippocampal slices from LRRK2 G2019S (LRRK2 GS) mutant littermate mice with distinct genotypes; i.e., WT, heterozygous (HZ) and homozygous knock in (KI) mice, were prepared and treated with αSyn PFF for 1 month with or without KCl to depolarize neurons (Fig. 5a). Again, αSyn pathology was increased in WT slices treated with PFF plus KCl, and interestingly, the αSyn pathology was further increased ~2-fold in LRRK2 GS KI hippocampal slices (Fig. 5b, c). When LRRK2 GS KI slices were pre-treated with the LRRK2 GTPase inhibitor, MLi-2 (either 100 nM or 250 nM), the αSyn pathology was greatly reduced to a level that was comparable to WT slices co-treated with αSyn PFF plus KCl (Fig. 5d). Additionally, when further analyzing the details of αSyn pathology, we noted more LB in LRRK2 GS KI slices co-treated with αSyn PFF plus KCl (Fig. 5b, e). Taken together, we conclude that enhancing neuronal activity had a potentiating effect on LB formation in LRRK2 GS KI slices, which broadens our understanding of the consequences of the LRRK2 GS mutation in conditions of neuronal hyperactivity or environmental stress.
Fig. 5.
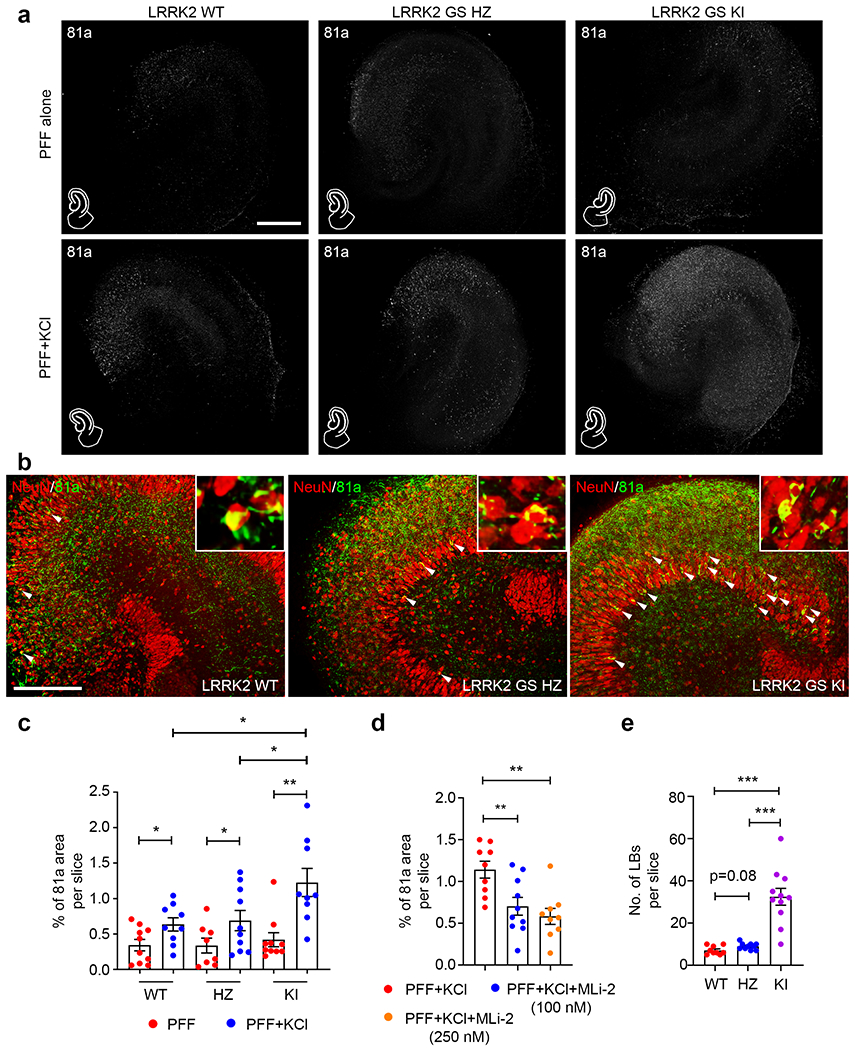
More LB pathologies were found in slices from LRRK2 kinase gain-of-function mutant mice after enhancing neuronal activity. a Typical images of the 81a signal in different genotypes of LRRK2 G2019S mutant hippocampal slice treated with αSyn PFF alone (upper panels) or αSyn PFF plus KCl (bottom panels) for 1 month. Abbreviations: WT, wildtype; HZ, LRRK2 G2019S heterozygous; KI, LRRK2 G2019S homozygous knock in. Scale bar: 250 μm. b Double staining of NeuN and 81a in hippocampal slices with different LRRK2 G2019S genotypes under hyperactivity. Zoom in images are representatives of LB pathology. Whites arrows indicate LB pathology in the whole hippocampal slices. Scale bar: 100 μm. c Quantification of percentage of 81a pathology in hippocampal slices from distinct LRRK2 G2019S genotypes treated with PFF or PFF plus KCl. d Quantification of 81a pathology in slices that were pre-treated with LRRK2 kinase inhibitor, MLi-2. e Quantification of LB pathology in LRRK2 G2019S slices under hyperactivity. *, p<0.05; **, p<0.01; ***, p<0.001. Data are presented as mean ± SEM.
Enhancing neuronal activity facilitates αSyn pathology spread in vivo and exacerbates impairments in motor behavior
Since αSyn PFF efficiently seed the aggregation of endogenous soluble αSyn in primary cultures of WT neurons [61, 66] as well as hippocampal slices (Fig. 1), we asked whether transmission of αSyn pathology might be accelerated in WT mice in vivo under conditions of neuronal hyperactivity. To address this issue, pathogenic αSyn PFF were stereotaxically injected into the dorsal striatum of WT (B6C3F1/J) mice. At the same time, we also injected AAV particles harboring designer receptors that can be exclusively activated by designer drugs (DREADDs). Yuan and Grutzendler demonstrated that chronic attenuation of neuronal activity in vivo with inhibitory (hM4D) DREADDs reduces amyloid deposition, where increase neuronal activity with excitatory (hM3D) DREADDs leads to a significant increase in amyloid plaque numbers [70]. AAV-hSyn-hM3D(Gq)-mCherry, AAV-hSyn-hM4D(Gi)-mCherry or AAV-hSyn-mCherry were used to hyperactivate, hypoactivate or serve as control reagents. Clozapine-N-oxide (CNO) was administrated twice a week via intraperitoneal (i.p.) injections 7 days after virus injection to activate DREADDs in the AAV-infected mice (Fig. 6a).
Fig. 6.
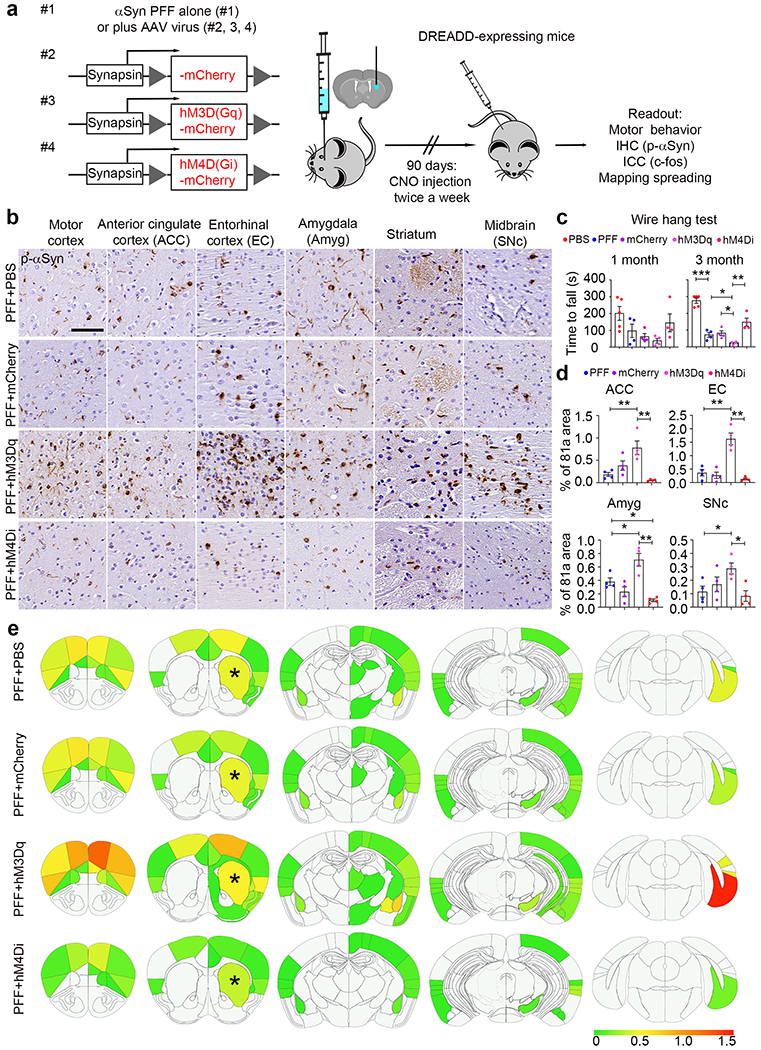
Enhancing neuronal activity facilitates p-αSyn pathology spreading in vivo and exacerbates motor impairments. a Schematic model of unilateral stereotaxic injection of chemogenetic neuronal activity modulators and αSyn PFF into the dorsal striatum. Four groups were investigated: #1, αSyn PFF alone; #2, αSyn PFF plus AAV virus overexpressing human synapsin promotor driven mCherry (control virus); #3, αSyn PFF plus AAV virus overexpressing human synapsin promotor driven hM3D(Gq)-mCherry (hyperactivity); #4, αSyn PFF plus AAV virus overexpressing human synapsin promotor driven hM4D(Gi)-mCherry (hypoactivity). 2×109 GC amounts of AAV virus particles were injected into each mouse. One-week post-surgery, clozapine-N-oxide (CNO) was infused via intraperitoneal injection twice a week to activate or inhibit neuronal activity. During the CNO administration period, motor behavior defects were measured at 1- and 3-month post-surgery. After the final behavior test, mice were euthanized followed by IHC staining to map p-αSyn pathology and spreading throughout the whole brains. 4 mice were used for each treatment group. b Typical IHC graphs showing the p-αSyn pathology (81a) within distinct brain regions under four different treatments. Scale bar: 100 μm. c Quantification of wire hang test. d Quantification of p-αSyn pathology from panel (b). e Heatmap distributions of p-αSyn pathology throughout distinct sections. For quantification of αSyn pathology, coronal sections were selected to closely match the following coordinates, relative to bregma: 2.10 mm, 0.98 mm, −1.22 mm, −2.92 mm and −4.48 mm (from left to right panels). Asterisks indicate the dorsal striatal injections of αSyn PFF with or without virus. Scale bar indicates the percentage of p-αSyn occupied per sub-regions. *, p<0.05; **, p<0.01; ***, p<0.001. Data are presented as mean ± SEM. See also Supplementary Fig. 13, online resource
At 1-month post CNO administration, motor defects were detected by the wire hang test in αSyn PFF-injected mice, and these deficits were exacerbated in the mice with increased neuronal activity, while, the PFF-dependent motor deficits were partially rescued in mice with decreased neuronal activity (Fig. 6c). However, due to the variability of the results, no statistically significant changes were observed. In contrast, more severe motor defects were observed at 3-month post CNO administration and the changes in the mice with altered neuronal activity were statistically significant (Fig. 6c). Additionally, activation of DREADDs was confirmed by c-fos staining in striatum and amygdala (Supplementary Fig. 13, online resource).
Deposits of p-αSyn detected by 81a staining were visible at the injection site (striatum) 3 months after a single unilateral αSyn PFF inoculation (Fig. 6b). Intraneuronal αSyn inclusions were also present in several areas interconnected to the striatum, most prominently in motor cortex, anterior cingulate cortex, entorhinal cortex, amygdala and midbrain SNc (Fig. 6b), recapitulating human LB/LN [19, 28]. There was a marked increase of αSyn pathology in DREADD-expressing mice with increased neuronal activity, and αSyn pathology was dramatically decreased in mice with reduced neuronal activity (Fig. 6b, d). Additionally, when analyzing the p-αSyn throughout the whole brain sections to map pathology spreading in vivo, in mice injected with αSyn PFF and hyperactivation DREADDs, the p-αSyn pathology was not only highly abundant in those “hot spots” listed above (Fig. 6b), but also observed in ipsilateral thalamus, subthalamus, corpus callosum and hippocampus probably via internal capsule of the striatal injection site, and this pattern was abolished in mice injected with αSyn PFF and hypoactivation DREADDs (Fig. 6e), suggesting that cell-to-cell transmission of αSyn pathology not only followed interneural connectivity but also was highly modulated by neuronal activity in vivo.
Discussion
The progressive accumulation of αSyn inclusions in PD patients is correlated with the decline in motor and/or cognitive functions [5, 28], although the events triggering αSyn pathology and how it leads to neurodegeneration in the CNS is poorly understood. Importantly, the progression of αSyn pathology in PD appears to follow a stereotypical pattern that arises first in the brainstem and then spreads rostrally to neocortical regions [5, 18]. This hierarchical pattern of disease progression suggests that cell-to-cell transmission of αSyn pathology is the basis for the pathological spread, and modulating this cell-to-cell transmission could very likely affect the αSyn pathology burden in the brain.
Thus, one of the goals of our study was to establish a new transmission model of PD-related αSyn pathology to allow the investigation of mechanisms by which risk factors and neuronal physiology might contribute to αSyn pathogenesis. To accomplish this, we established OBSCs which were first reported around 30 years ago [54]. However, only recently has the OBSC technique been applied to studies of neurodegenerative diseases [10–12, 14, 15, 27, 39], although most of these prior studies either lacked vital controls or used slices from Tg mutant mouse models. Moreover, studies that used viral overexpression models to mimic PD pathogenesis [10] are limited by the fact that most of PD patients are idiopathic, thus lacking SNCA mutations or duplications with no evidence of αSyn overexpression [55]. In our OBSC system, we added recombinant αSyn PFF directly into the culture medium of WT brain slices, which induced LB/LN throughout the whole brain slice cultures, recapitulating the phenotypes seen in authentic human synucleinopathies, such as PD, PDD, DLB, and AD with concomitant LB [28].
Technically, both WT and Tg animals can be used for slice preparation. Importantly, multiple brain slices could be prepared from one mouse, which minimizes the number of animals that need to be bred per study and also reduces the experimental variations between groups. Although human induced pluripotent stem cell (iPSC)-derived organoids have emerged as another choice for investigating neurodegenerative proteinopathies like PD and AD, these studies still face many technical and conceptual challenges [2, 9, 59]. The complexity of organoid culture system could also limit its wide application.
Aberrant neuronal hyperactivity has been reported in several studies of neurodegenerative disease. Specifically, chemogenetically-induced decreases in neuron activity in cortex reduced amyloid plaques [70], while enhancing neuron activity potentiated tau release and tau pathology in vitro and in vivo [65, 67]. A recent paper from our laboratory indicates that more αSyn pathology developed in the presence of amyloid plaques in the 5xFAD mice [3], which might be due to a hyperactive neuron profile as suggested by our Ca2+ imaging experiment in slice cultures (see also [6]). Moreover, our results are consistent with a prior report showing that elevating neuronal activity rapidly increased, while blocking activity decreased, αSyn release in vitro and in vivo [68]. The αSyn is detected in exosomes and synaptic vesicles, and the secretion of αSyn from cell bodies and axons is Ca2+-dependent and also depends on the integrity of the in vivo neuronal network [16, 17]. Clinically, about 50% of PD patients experience cerebellar hyperactivity and epileptic seizures [50, 69]. Thus, the novel findings in our study linking neuronal activity to disease pathogenesis provides compelling evidence that neuronal activity can facilitate cell-to-cell transmission and spreading of αSyn pathology, further implying the critical role of the known aberrant neuronal activity in PD to the progression and spread of αSyn pathologies.
Additionally, another unique aspect of our study is that we elucidated the effects of neuronal activity on αSyn pathology in conjunction with PD genetic risk factor, LRRK2. Mutations in LRRK2 gene are the most frequently reported monogenic cause of PD and are common in both early and late-onset PD, occurring in both familial and sporadic PD patients with a wide variety of clinical and pathological features [44]. Among these mutations, the glycine to serine substitution (G2019S), is the most common and was estimated by the international LRRK2 consortium to represent 1% of sporadic and 4% of familial PD patients worldwide [24]. Previously, we demonstrated the deleterious function of LRRK2 G2019S mutation in the αSyn PFF seeding model in vivo, where spreading of p-αSyn pathology in synaptically-connected brain regions are highly accelerated in G2019S mice [25]. Here, we investigated the combined effects of LRRK2 G2019S mutation and neuronal activity on p-αSyn pathology, and showed that not only neuritic p-αSyn but also LB were increased about 2.5-fold under hyperactive conditions, providing potential applications of LRRK2 G2019S mutation in modeling LB formation. However, future mechanistic studies are needed to further elucidate the turnover and recruitment of endogenous αSyn into LB in the context of pathogenic αSyn PFF seeds, neuronal hyperactivity as well as LRRK2 GTPase gain-of-function.
Finally, our ex vivo findings were extended and confirmed in an in vivo study, where motor defects and αSyn pathology were modulated by changes in neuronal activity. Although propagation of LB/LN in human PD is postulated to start in the brainstem and ascend toward neocortical regions with disease progression [5], the data presented here and recent studies [61] indicate that the transmission of pathological αSyn can occur bi-directionally within a network of interconnected populations. Nonetheless, our findings clearly suggest a direct relationship between the accumulation of pathological αSyn and neuron pathophysiology.
Thus, neuronal activity should be considered as a driving force that accelerates or exacerbates the spread of αSyn pathology in human PD patients. Indeed, modulating neuronal activity could emerge as a novel therapeutic for the treatment of synucleinopathies.
Supplementary Material
Acknowledgements
We thank Dr. Kurt R. Brunden for reading and editing of this manuscript. We thank Drs. Douglas A Coulter (Children’s hospital of Philadelphia) and Hajime Takano (Children’s hospital of Philadelphia) for valuable discussion and technical support. We thank S. Leight and T. Schuck for technical assistance. We thank Drs. A. Caputo for providing SGKI mice and M. X. Henderson for providing LRRK2 G2019S KI mice and discussion. We thank D. M. Riddle for primary hippocampal neuron cultures. We thank Dr. S. Xie for help with statistical analysis.
Funding
This work was supported by NIH/NIA U19 Center grant (AG062418), the Jeff and Anne Keefer Fund, and the Neurodegenerative Disease Research Fund.
Footnotes
Conflict of interest The authors declare no competing interests.
References
- 1.Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A et al. (2000) Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25: 239–252 [DOI] [PubMed] [Google Scholar]
- 2.Arber C, Lovejoy C, Wray S (2017) Stem cell models of Alzheimer’s disease: progress and challenges. Alzheimers Res Ther 9: 42 Doi 10.1186/s13195-017-0268-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bassil F, Brown HJ, Pattabhiraman S, Iwasyk JE, Maghames CM, Meymand ES, Cox TO, Riddle DM, Zhang B, Trojanowski JQ et al. (2020) Amyloid-Beta (Abeta) Plaques Promote Seeding and Spreading of Alpha-Synuclein and Tau in a Mouse Model of Lewy Body Disorders with Abeta Pathology. Neuron 105: 260–275 e266 Doi 10.1016/j.neuron.2019.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bove J, Prou D, Perier C, Przedborski S (2005) Toxin-induced models of Parkinson’s disease. NeuroRx 2: 484–494 Doi 10.1602/neurorx.2.3.484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24: 197–211 Doi 10.1016/s0197-4580(02)00065-9 [DOI] [PubMed] [Google Scholar]
- 6.Busche MA, Konnerth A (2016) Impairments of neural circuit function in Alzheimer’s disease. Philosophical transactions of the Royal Society of London Series B, Biological sciences 371: Doi 10.1098/rstb.2015.0429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caputo A, Liang Y, Raabe TD, Lo A, Horvath M, Zhang B, Brown HJ, Stieber A, Luk KC (2020) Snca-GFP knock-in mice reflect patterns of endogenous expression and pathological seeding. eNeuro: Doi 10.1523/ENEURO.0007-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen H, Ritz B (2018) The Search for Environmental Causes of Parkinson’s Disease: Moving Forward. J Parkinsons Dis 8: S9–S17 Doi 10.3233/JPD-181493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D’Avanzo C, Chen H, Hooli B, Asselin C, Muffat J et al. (2014) A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515: 274–278 Doi 10.1038/nature13800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Croft CL, Cruz PE, Ryu DH, Ceballos-Diaz C, Strang KH, Woody BM, Lin WL, Deture M, Rodriguez-Lebron E, Dickson DW et al. (2019) rAAV-based brain slice culture models of Alzheimer’s and Parkinson’s disease inclusion pathologies. The Journal of experimental medicine 216: 539–555 Doi 10.1084/jem.20182184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Croft CL, Futch HS, Moore BD, Golde TE (2019) Organotypic brain slice cultures to model neurodegenerative proteinopathies. Mol Neurodegener 14: 45 Doi 10.1186/s13024-019-0346-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Croft CL, Kurbatskaya K, Hanger DP, Noble W (2017) Inhibition of glycogen synthase kinase-3 by BTA-EG4 reduces tau abnormalities in an organotypic brain slice culture model of Alzheimer’s disease. Sci Rep 7: 7434 Doi 10.1038/s41598-017-07906-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong XX, Wang Y, Qin ZH (2009) Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin 30: 379–387 Doi 10.1038/aps.2009.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duff K, Noble W, Gaynor K, Matsuoka Y (2002) Organotypic slice cultures from transgenic mice as disease model systems. J Mol Neurosci 19: 317–320 Doi 10.1385/JMN:19:3:317 [DOI] [PubMed] [Google Scholar]
- 15.Elfarrash S, Jensen NM, Ferreira N, Betzer C, Thevathasan JV, Diekmann R, Adel M, Omar NM, Boraie MZ, Gad S et al. (2019) Organotypic slice culture model demonstrates inter-neuronal spreading of alpha-synuclein aggregates. Acta Neuropathol Commun 7: 213 Doi 10.1186/s40478-019-0865-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K (2010) Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci 30: 6838–6851 Doi 10.1523/JNEUROSCI.5699-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emmanouilidou E, Minakaki G, Keramioti MV, Xylaki M, Balafas E, Chrysanthou-Piterou M, Kloukina I, Vekrellis K (2016) GABA transmission via ATP-dependent K+ channels regulates alpha-synuclein secretion in mouse striatum. Brain 139: 871–890 Doi 10.1093/brain/awv403 [DOI] [PubMed] [Google Scholar]
- 18.Fahn S (2003) Description of Parkinson’s disease as a clinical syndrome. Ann N Y Acad Sci 991: 1–14 Doi 10.1111/j.1749-6632.2003.tb07458.x [DOI] [PubMed] [Google Scholar]
- 19.Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T (2002) alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 4: 160–164 Doi 10.1038/ncb748 [DOI] [PubMed] [Google Scholar]
- 20.Giasson BI, Murray IV, Trojanowski JQ, Lee VM (2001) A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. The Journal of biological chemistry 276: 2380–2386 Doi 10.1074/jbc.M008919200 [DOI] [PubMed] [Google Scholar]
- 21.Gogolla N, Galimberti I, DePaola V, Caroni P (2006) Preparation of organotypic hippocampal slice cultures for long-term live imaging. Nat Protoc 1: 1165–1171 Doi 10.1038/nprot.2006.168 [DOI] [PubMed] [Google Scholar]
- 22.Gong W, Sencar J, Bakkum DJ, Jackel D, Obien ME, Radivojevic M, Hierlemann AR (2016) Multiple Single-Unit Long-Term Tracking on Organotypic Hippocampal Slices Using High-Density Microelectrode Arrays. Front Neurosci 10: 537 Doi 10.3389/fnins.2016.00537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ et al. (2013) Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell 154: 103–117 Doi 10.1016/j.cell.2013.05.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S et al. (2008) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol 7: 583–590 Doi 10.1016/S1474-4422(08)70117-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henderson MX, Cornblath EJ, Darwich A, Zhang B, Brown H, Gathagan RJ, Sandler RM, Bassett DS, Trojanowski JQ, Lee VMY (2019) Spread of alpha-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nat Neurosci 22: 1248–1257 Doi 10.1038/s41593-019-0457-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holth JK, Fritschi SK, Wang C, Pedersen NP, Cirrito JR, Mahan TE, Finn MB, Manis M, Geerling JC, Fuller PM et al. (2019) The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 363: 880–884 Doi 10.1126/science.aav2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Humpel C (2015) Organotypic brain slice cultures: A review. Neuroscience 305: 86–98 Doi 10.1016/j.neuroscience.2015.07.086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Irwin DJ, Lee VM, Trojanowski JQ (2013) Parkinson’s disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci 14: 626–636 Doi 10.1038/nrn3549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karpowicz RJ Jr., Haney CM, Mihaila TS, Sandler RM, Petersson EJ, Lee VM (2017) Selective imaging of internalized proteopathic alpha-synuclein seeds in primary neurons reveals mechanistic insight into transmission of synucleinopathies. The Journal of biological chemistry 292: 13482–13497 Doi 10.1074/jbc.M117.780296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, Cormier-Dequaire F, Hassoun SM, Pujol C, Ciura S et al. (2016) Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am J Hum Genet 98: 500–513 Doi 10.1016/j.ajhg.2016.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loria F, Vargas JY, Bousset L, Syan S, Salles A, Melki R, Zurzolo C (2017) alpha-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol 134: 789–808 Doi 10.1007/s00401-017-1746-2 [DOI] [PubMed] [Google Scholar]
- 32.Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM (2012) Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338: 949–953 Doi 10.1126/science.1227157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM (2012) Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med 209: 975–986 Doi 10.1084/jem.20112457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luna E, Decker SC, Riddle DM, Caputo A, Zhang B, Cole T, Caswell C, Xie SX, Lee VMY, Luk KC (2018) Differential alpha-synuclein expression contributes to selective vulnerability of hippocampal neuron subpopulations to fibril-induced toxicity. Acta Neuropathol 135: 855–875 Doi 10.1007/s00401-018-1829-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matikainen-Ankney BA, Kezunovic N, Mesias RE, Tian Y, Williams FM, Huntley GW, Benson DL (2016) Altered Development of Synapse Structure and Function in Striatum Caused by Parkinson’s Disease-Linked LRRK2-G2019S Mutation. The Journal of neuroscience : the official journal of the Society for Neuroscience 36: 7128–7141 Doi 10.1523/JNEUROSCI.3314-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Messing L, Decker JM, Joseph M, Mandelkow E, Mandelkow EM (2013) Cascade of tau toxicity in inducible hippocampal brain slices and prevention by aggregation inhibitors. Neurobiol Aging 34: 1343–1354 Doi 10.1016/j.neurobiolaging.2012.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Novotny R, Langer F, Mahler J, Skodras A, Vlachos A, Wegenast-Braun BM, Kaeser SA, Neher JJ, Eisele YS, Pietrowski MJ et al. (2016) Conversion of Synthetic Abeta to In Vivo Active Seeds and Amyloid Plaque Formation in a Hippocampal Slice Culture Model. J Neurosci 36: 5084–5093 Doi 10.1523/JNEUROSCI.0258-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L et al. (2006) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. The Journal of neuroscience : the official journal of the Society for Neuroscience 26: 10129–10140 Doi 10.1523/JNEUROSCI.1202-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Opitz-Araya X, Barria A (2011) Organotypic hippocampal slice cultures. J Vis Exp: Doi 10.3791/2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pang SY, Ho PW, Liu HF, Leung CT, Li L, Chang EES, Ramsden DB, Ho SL (2019) The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl Neurodegener 8: 23 Doi 10.1186/s40035-019-0165-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pankratz N, Foroud T (2007) Genetics of Parkinson disease. Genet Med 9: 801–811 Doi 10.1097/gim.0b013e31815bf97c [DOI] [PubMed] [Google Scholar]
- 42.Perier C, Vila M (2012) Mitochondrial biology and Parkinson’s disease. Cold Spring Harb Perspect Med 2: a009332 Doi 10.1101/cshperspect.a009332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Remple MS, Bradenham CH, Kao CC, Charles PD, Neimat JS, Konrad PE (2011) Subthalamic nucleus neuronal firing rate increases with Parkinson’s disease progression. Mov Disord 26: 1657–1662 Doi 10.1002/mds.23708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ren C, Ding Y, Wei S, Guan L, Zhang C, Ji Y, Wang F, Yin S, Yin P (2019) G2019S Variation in LRRK2: An Ideal Model for the Study of Parkinson’s Disease? Front Hum Neurosci 13: 306 Doi 10.3389/fnhum.2019.00306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rodriguez MC, Obeso JA, Olanow CW (1998) Subthalamic nucleus-mediated excitotoxicity in Parkinson’s disease: a target for neuroprotection. Ann Neurol 44: S175–188 Doi 10.1002/ana.410440726 [DOI] [PubMed] [Google Scholar]
- 46.Rodriguez PC, Pereira DB, Borgkvist A, Wong MY, Barnard C, Sonders MS, Zhang H, Sames D, Sulzer D (2013) Fluorescent dopamine tracer resolves individual dopaminergic synapses and their activity in the brain. Proc Natl Acad Sci U S A 110: 870–875 Doi 10.1073/pnas.1213569110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sampathu DM, Giasson BI, Pawlyk AC, Trojanowski JQ, Lee VM (2003) Ubiquitination of alpha-synuclein is not required for formation of pathological inclusions in alpha-synucleinopathies. Am J Pathol 163: 91–100 Doi 10.1016/s0002-9440(10)63633-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Santa-Maria I, Diaz-Ruiz C, Ksiezak-Reding H, Chen A, Ho L, Wang J, Pasinetti GM (2012) GSPE interferes with tau aggregation in vivo: implication for treating tauopathy. Neurobiol Aging 33: 2072–2081 Doi 10.1016/j.neurobiolaging.2011.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schatzle P, Kapitein LC, Hoogenraad CC (2016) Live imaging of microtubule dynamics in organotypic hippocampal slice cultures. Methods Cell Biol 131: 107–126 Doi 10.1016/bs.mcb.2015.06.006 [DOI] [PubMed] [Google Scholar]
- 50.Son AY, Biagioni MC, Kaminski D, Gurevich A, Stone B, Di Rocco A (2016) Parkinson’s Disease and Cryptogenic Epilepsy. Case Rep Neurol Med 2016: 3745631 Doi 10.1155/2016/3745631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M (1998) Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett 251: 205–208 Doi 10.1016/s0304-3940(98)00504-7 [DOI] [PubMed] [Google Scholar]
- 52.Spillantini MG, Goedert M (2016) Synucleinopathies: past, present and future. Neuropathol Appl Neurobiol 42: 3–5 Doi 10.1111/nan.12311 [DOI] [PubMed] [Google Scholar]
- 53.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388: 839–840 Doi 10.1038/42166 [DOI] [PubMed] [Google Scholar]
- 54.Stoppini L, Buchs PA, Muller D (1991) A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37: 173–182 Doi 10.1016/0165-0270(91)90128-m [DOI] [PubMed] [Google Scholar]
- 55.Su X, Fischer DL, Li X, Bankiewicz K, Sortwell CE, Federoff HJ (2017) Alpha-Synuclein mRNA Is Not Increased in Sporadic PD and Alpha-Synuclein Accumulation Does Not Block GDNF Signaling in Parkinson’s Disease and Disease Models. Mol Ther 25: 2231–2235 Doi 10.1016/j.ymthe.2017.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Surmeier DJ, Obeso JA, Halliday GM (2017) Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 18: 101–113 Doi 10.1038/nrn.2016.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thakur P, Breger LS, Lundblad M, Wan OW, Mattsson B, Luk KC, Lee VMY, Trojanowski JQ, Bjorklund A (2017) Modeling Parkinson’s disease pathology by combination of fibril seeds and alpha-synuclein overexpression in the rat brain. Proc Natl Acad Sci U S A 114: E8284–E8293 Doi 10.1073/pnas.1710442114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uemura N, Uemura MT, Lo A, Bassil F, Zhang B, Luk KC, Lee VM, Takahashi R, Trojanowski JQ (2019) Slow Progressive Accumulation of Oligodendroglial Alpha-Synuclein (alpha-Syn) Pathology in Synthetic alpha-Syn Fibril-Induced Mouse Models of Synucleinopathy. Journal of neuropathology and experimental neurology 78: 877–890 Doi 10.1093/jnen/nlz070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Velasco S, Kedaigle AJ, Simmons SK, Nash A, Rocha M, Quadrato G, Paulsen B, Nguyen L, Adiconis X, Regev A et al. (2019) Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 570: 523–527 Doi 10.1038/s41586-019-1289-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Volpicelli-Daley LA, Luk KC, Lee VM (2014) Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat Protoc 9: 2135–2146 Doi 10.1038/nprot.2014.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM (2011) Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72: 57–71 Doi 10.1016/j.neuron.2011.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang C, Kang X, Zhou L, Chai Z, Wu Q, Huang R, Xu H, Hu M, Sun X, Sun S et al. (2018) Synaptotagmin-11 is a critical mediator of parkin-linked neurotoxicity and Parkinson’s disease-like pathology. Nat Commun 9: 81 Doi 10.1038/s41467-017-02593-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang Y, Wu Q, Hu M, Liu B, Chai Z, Huang R, Wang Y, Xu H, Zhou L, Zheng L et al. (2017) Ligand- and voltage-gated Ca(2+) channels differentially regulate the mode of vesicular neuropeptide release in mammalian sensory neurons. Sci Signal 10: 484 Doi 10.1126/scisignal.aal1683 [DOI] [PubMed] [Google Scholar]
- 64.Waxman EA, Giasson BI (2008) Specificity and regulation of casein kinase-mediated phosphorylation of alpha-synuclein. J Neuropathol Exp Neurol 67: 402–416 Doi 10.1097/NEN.0b013e31816fc995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, Sanders DW, Cook C, Fu H, Boonen RA et al. (2016) Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci 19: 1085–1092 Doi 10.1038/nn.4328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu Q, Takano H, Riddle DM, Trojanowski JQ, Coulter DA, Lee VM (2019) alpha-Synuclein (alphaSyn) Preformed Fibrils Induce Endogenous alphaSyn Aggregation, Compromise Synaptic Activity and Enhance Synapse Loss in Cultured Excitatory Hippocampal Neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 39: 5080–5094 Doi 10.1523/JNEUROSCI.0060-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamada K, Holth JK, Liao F, Stewart FR, Mahan TE, Jiang H, Cirrito JR, Patel TK, Hochgrafe K, Mandelkow EM et al. (2014) Neuronal activity regulates extracellular tau in vivo. J Exp Med 211: 387–393 Doi 10.1084/jem.20131685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yamada K, Iwatsubo T (2018) Extracellular alpha-synuclein levels are regulated by neuronal activity. Molecular neurodegeneration 13: 9 Doi 10.1186/s13024-018-0241-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu H, Sternad D, Corcos DM, Vaillancourt DE (2007) Role of hyperactive cerebellum and motor cortex in Parkinson’s disease. Neuroimage 35: 222–233 Doi 10.1016/j.neuroimage.2006.11.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yuan P, Grutzendler J (2016) Attenuation of beta-Amyloid Deposition and Neurotoxicity by Chemogenetic Modulation of Neural Activity. The Journal of neuroscience : the official journal of the Society for Neuroscience 36: 632–641 Doi 10.1523/JNEUROSCI.2531-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.