

Abstract
Human tumors are composed of diverse malignant and non-malignant cells, generating a complex ecosystem that governs tumor biology and response to treatments. Recent technological advances have enabled the characterization of tumors at single-cell resolution, providing a compelling strategy to dissect their intricate biology. Here we describe recent developments in single-cell expression profiling and the studies applying them in clinical settings. We highlight some of the powerful insights gleaned from these studies for tumor classification, stem cell programs, tumor micro-environment, metastasis, and response to targeted and immune therapies.
The advantage of diversity
Tumors are characterized by genomic instability, limitless replicative potential, deregulation of cellular energetics, tissue invasion and metastasis, sustained angiogenesis, tumor-promoting inflammation and avoidance of immune attack (1). While these hallmarks are shared by most tumors, there is increased recognition that within a single tumor, subsets of cells may be sufficient to enable such phenotypes. Limitless replication may be driven by a subset of undifferentiated cells known as cancer stem cells; tissue invasion and metastasis may be driven by cells that have activated an epithelial-mesenchymal transition (EMT) or other invasion programs; deregulated energetics, as well as sustained angiogenesis, may be driven by cells in particular areas of the tumor, such as those that experience hypoxia and lack of nutrients; inflammation and avoidance of immune attack may be confined to specific regions (e.g. with increased immune infiltration). Such “division of labor” between tumor subpopulations may help to explain a range of tumor phenotypes, most notably, their tendency to either endure through anti-cancer treatments or to recur months or years after tumor regression. The discrepancy between initial tumor response and regression (indicating that cancer cells are being eliminated by treatment) and later tumor recurrence (indicating that not all cancer cells were eliminated) is a testament to the critical significance of intra-tumoral heterogeneity (ITH). Yet, genomic and transcriptomic approaches have traditionally measured tumors as bulk, thus only revealing aggregate cellular profiles and masking important aspects of ITH. The recent emergence of single-cell “omics” profiling is paving the way for a revolution in our ability to comprehensively characterize tumors. Here we review single-cell omics methods and their application to clinical tumor samples. We focus on single cell expression profiling and the insights gained on tumor lineages, stem cell programs, tumor classification, metastasis, microenvironment composition, and response to therapies. Finally, we highlight ongoing developments in single-cell expression profiling that will help tackle unanswered questions in the field.
Profiling tumors at the single-cell level
Advances in chemistry, microfluidics, sequencing and bioinformatics have enabled the development of a number of techniques to profile multiple facets of tumors at the single-cell level (genome, transcriptome, methylome, chromatin accessibility, intracellular and cell surface proteins) (Figure 1), advancing our understanding of ITH. Initially, ITH was considered primarily at the level of the genome, and even prior to single cell approaches, genetic heterogeneity was inferred from bulk tumor profiles, based on the fraction of sequence reads that harbor each mutation (2–6). This approach was significantly empowered by profiling multiple areas of the same tumor (multi-focal sequencing), demonstrating that distinct regions of the same tumor typically harbor both shared and distinct mutations (7,8). Single-cell DNA sequencing studies have further expanded these observations in order to dissect genetic ITH (9–14). Due to the limited sensitivity in detection of individual mutations, most studies of single cell DNA sequencing are often focused on chromosomal copy number aberrations (CNAs) which are detected at much higher accuracy and are associated with considerable ITH in most tumors (15–18). The aggregation of cells sharing CNA profiles has been utilized to enhance phylogenetic analysis at single-nucleotide levels (19).
Figure 1:
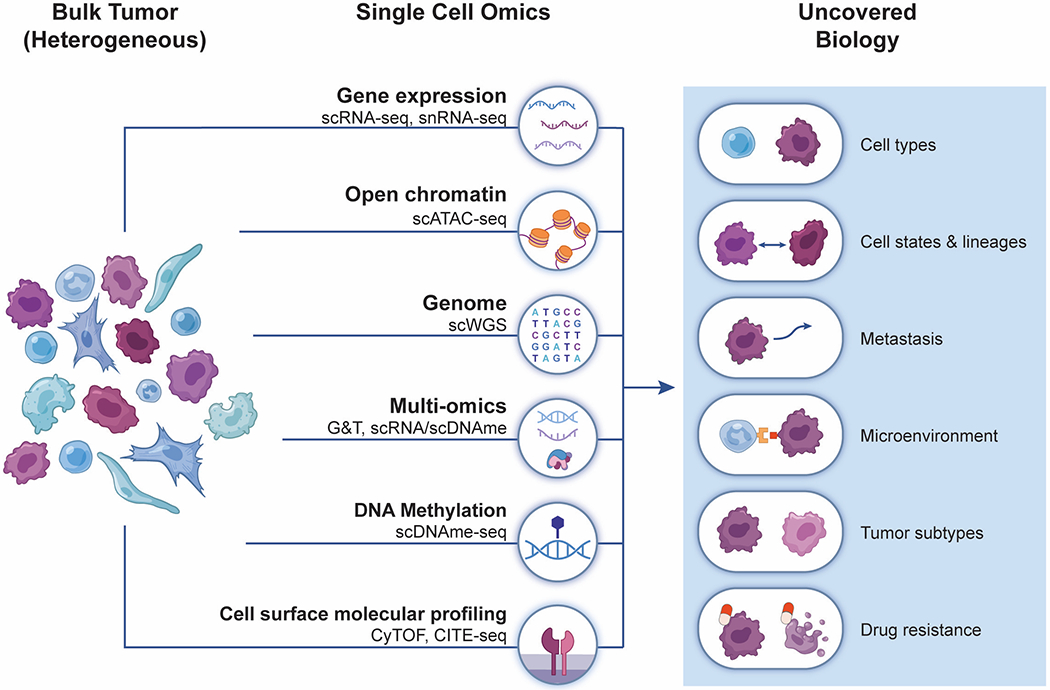
Schematic of the different levels of biological information that can be profiled at single-cell resolution and examples of biology uncovered.
While mapping of genetic heterogeneity is extremely important, it does not reveal the functional state associated with distinct genetic events. Many cancer mutations may reflect passenger events with limited functional implications (20) and even for functional mutations, an exact functional consequence cannot be inferred from sequence information alone. Thus, single-cell RNA-sequencing (scRNA-seq) has recently become a more widely used approach to explore ITH. In principle, scRNA-seq enables analysis of both functional and genetic states, as mutations may be detected within transcripts, especially with protocols that sequence full-length transcripts (as opposed to 3′- or 5′-end sequencing). Yet, the sensitivity of such methods is highly limited (21), primarily due to the low coverage of the transcriptomes in individual cells. New methods aim to improve this coverage by adding locus-specific primers to the scRNA-seq protocol for genes mutated in a given cancer type, as first shown for the BCR-ABL fusion in leukemia (22) and more recently for CALR-mutated myeloproliferative neoplasms (23). However, while these approaches are effective for mutations in highly expressed transcripts, they require prior knowledge of the mutations and are limited to the amplification of few selected loci in each sample. Alternatively, DNA and RNA may both be profiled from the same single cells, and sequenced either for genome and transcriptome (24,25) or alternatively for epigenome DNA methylation and transcriptome, as recently demonstrated for chronic lymphocytic leukemia (26). While these techniques are likely to be applied in larger scale to clinical tumors in the future, integration of multi-omics modalities (27) remains rare, with most studies to date focused on single-cell tumor profiling of either RNA, DNA, or proteins, and accordingly on either the functional or the genetic states of cells.
scRNA-seq has been the most widely adopted approach leveraged for single-cell profiling of clinical tumors. Although scRNA-seq methods have until recently been limited to living cells as the starting material, characterizing individual cell transcriptomes is also possible using frozen tissue via single-nucleus RNA-sequencing (snRNA-seq/sNuc-seq) (28,29). While nuclei have less mRNA content than cells, analyses performed on live cells and nuclei isolated from frozen tissue of the same sample have demonstrated consistent transcriptomic profiles and similar clustering patterns (28). The possibility of profiling a tumor’s transcriptome using frozen samples facilitates the deployment of such approaches to broader clinical contexts and enables the comparison of longitudinal samples, critical to interrogating tumor evolution and response to treatments.
Whether applied to single cells or nuclei, clinical tumor profiling by RNA-seq requires four main steps. (I) fresh or frozen samples are obtained and disaggregated into a single-cell or single-nucleus suspensions using combinations of mechanical and enzymatic digestion protocols; (II) individual cells or nuclei are separated either by flow cytometry into 96- or 384-well plates or by microfluidic devices into distinct droplets and then lysed; (III) a reverse transcription and RNA-seq protocol (either full length or the 3′ or 5’ends) is applied to individual cells or nuclei, which are barcoded and pooled; (IV) the pooled sample is sequenced and the reads mapped to the transcriptome to assess gene expression and perform additional analyses (see below). Many of these steps need to be optimized for each application, and the methods available offer a trade-off between coverage per cell and transcript (optimized by plate-based methods such as Smart-seq (30,31)) versus number of profiled cells, which is significantly higher with droplet-based platforms (32–34). Alternative methods to label individual cells by combinatorial barcoding, such as SPLiT-seq (split-pool ligation-based transcriptome sequencing), do not require the separation of individual cells into compartments but rather leverage the cells or nuclei themselves as compartments (35–38). Depending on the desired application and biological question, specific single-cell methods may thus be leveraged.
Of Cell Types and Cell States
When performing unbiased expression profiling of cells in a tumor (i.e., without pre-selecting for any particular cell type), the first step in the analysis (after quality controls and filtering of low-quality cells) is to assign the remaining cells into distinct cell types. This is typically performed by clustering the cells and annotating clusters by the identity of upregulated genes. This approach typically works well, yet the tumor introduces an additional task - the distinction between malignant (i.e. with genetic alterations) versus non-malignant cells (such as stromal and immune cells). Combining gene expression clustering with genetic information helps address this challenge. However, as discussed above, the ability of scRNA-seq to detect single point mutations suffers from technical limitations that recent developments are attempting to address (23–25). Importantly, malignant cells may also be identified by CNAs that can be inferred from the average expression of large sets of genes in each chromosomal region. This approach has been used by most recent studies to resolve malignant from non-malignant cells, and in some cases also to identify distinct genetic subclones among the malignant cells (21,39–41).
Relying on the identification of CNAs rather than point mutations highlights a key feature of scRNA-seq datasets that influences all computational analyses: the sensitivity of detection and the accuracy of estimating exact levels for individual genes or mutations is hindered by “dropouts” and the limited efficiency of single-cell profiling; thus, one should not rely on any individual mutation or gene to infer cell identities. Rather, the combined analysis of many related genes (i.e., adjacent genes in the case of CNAs) or a larger gene-set known to reflect a certain biological function or process, enables cells to be annotated with increased accuracy. The choice of gene-sets for such analyses is critically important, and caution should be taken when gene-sets are derived from systems distinct from the one analyzed. Ideally, a gene-set should be derived de novo from the data-driven analysis of a scRNA-seq dataset and then applied to the same or to an extended/related data set to derive cell scores for the corresponding biological process (17,18,21,39–42).
Analysis of the heterogeneity within tumors typically enables two layers of distinction: first, cells are grouped into cell types (such as malignant cells, immune cells, etc.) that typically present as very distinct clusters; each cell type is then associated with additional diversity reflective of distinct cell states, such as context-specific expression programs. Cell cycle phases represent the most common example of distinct cell states, with cells that are not actively cycling, and cycling cells that may be assigned to distinct phases based on relative expression of cell cycle gene-sets. “Cell states” refers here to distinct expression programs with presumed functional implications that are typically continuous and potentially dynamic (e.g., progression along the cell cycle), while cell types are more stable and unique. While these definitions are admittedly imprecise, they reflect important distinctions between two common layers of diversity. In the following section, we primarily focus on cell states that have been identified in scRNA/snRNAseq profiling studies (Table 1) and that exemplify the power of single-cell analysis in decoding tumor biology.
Table 1.
Select studies using single-cell methods to study human intra-tumoral heterogeneity.
| Reference | Sample / tumor type | Number of cells / nuclei (number of samples) |
|---|---|---|
| Miyamoto et al., 2015 (102) | Prostate CTCs | 77 (13) |
| Jordan et al., 2016 (101) | Breast adenocarcinoma CTCs | 74 (16) |
| Patel et al., 2014 (17) | Glioblastoma | 431 (5) |
| Tirosh et al., 2016a (18) | Metastatic melanoma | 4645 (19) |
| Tirosh et al., 2016b (21) | IDH-mutant oligodendroglioma | 4347 (6) |
| Venteicher et al., 2017 (48) | IDH-mutant astrocytoma | 9879 (10) |
| Li et al., 2017 (70) | Colorectal cancer | 590 (11) |
| Chung et al., 2017 (115) | Breast cancer | 515 (11) |
| Puram et al., 2017 (42) | Head and neck cancer | 6000 (18) |
| Savage et al., 2017 (116) | Breast cancer | 3483 |
| Giustacchini et al., 2017 (22) | Chronic myeloid leukemia | 2070 (20) |
| Brady et al., 2017 (117) | Breast cancer ascites | 428 (8) |
| Filbin et al., 2018 (39) | H3 K27M-mutant glioma | 3,321 (6) |
| Kim et al., 2018 (118) | Triple-negative breast cancer | 6,862 (8) |
| Young et al., 2018 (119) | Wilms tumor, Clear cell renal carcinoma, Papillary renal cell carcinoma | 72,501 (23) |
| Livnat-Jerby et al., 2018 (80) | Melanoma | 7,186 (33) |
| Karaayvaz et al., 2018 (120) | Triple-negative breast cancer | 1,189 (6) |
| Gaiti et al., 2019 (26) | Chronic lymphocytic leukemia | 2,652 (18) |
| Neftel et al., 2019 (40) | Glioblastoma, IDH-wildtype | 24,192 (28) |
| Peng et al., 2019 (121) | Pancreatic ductal adenocarcinoma | 57,530 (35) |
| Nam et al., 2019 (23) | CALR-mutated myeloproliferative neoplasms | 38,290 (5) |
| Hovestadt et al., 2019 (41) | Medulloblastoma | 8,734 (25) |
| Laks et al., ,2019 (19) | Breast cancer PDXs, synovial sarcoma (mouse model), follicular lymphoma samples | 51,926 (62) |
| Van Galen et al., 2019 (122) | Acute myeloid leukemia | 38,410 (40) |
| Ji et al., 2020 (123) | Squamous cell carcinoma | 48,164 (22) |
| Maynard et al., 2020 (55) | Non-small cell lung cancer | 23,261 (49) |
| Kim et al., 2020 (62) | Primary and metastatic lung adenocarcinoma, normal lung | 208,506 (44) |
| Lee et al., 2020 (83) | Colorectal cancer | 91,103 (29) |
| Chen et al., 2020 (124) | Nasopharyngeal carcinoma | 47,866 (16) |
| Izar et al., 2020 (71) | High-grade serous ovarian cancer ascites | 10,906 (22) |
| Gojo et al., 2020 (49) | Ependymoma | 74,927 (30) |
| Ireland et al., 2020 (54) | Small cell lung cancer | 15,434 (21) |
CALR: Calreticulin
CTC: Circulating tumor cell
H3 K27M: Histone H3 K27M mutation
IDH: Isocitrate-dehydrogenase
PDX: Patient-derived xenograft
Insights into cancer biology from single-cell genomics studies
Single-cell genomics experiments have generated multiple insights into the biology of different classes of tumors. Below, we highlight different salient findings grouped by biological or clinical questions.
Cellular lineages, their differentiation and plasticity
Developmental programs play a critical role in many malignancies (43). This observation underlies the cancer stem cell model, which suggests that tumors contain subsets of self-renewing stem-like cells underlying tumor-propagation (and in some instances resistance to therapies), along with a majority of more differentiated cells (44,45). This model has traditionally been supported by functional approaches in animal models. Single-cell expression profiling offers an orthogonal approach to revisit the role of stemness in cancer biology and to provide a more granular definition of cellular states that encompasses the continuum of cellular programs found in tumors and their resemblance to normal development (Table 1).
Initial pioneering studies used single-cell quantitative PCR (qPCR) to target selected genes (~50-100), and identified subpopulations that recapitulate normal lineages of the gut epithelium (including LGR5+ stem-like cells) in colon adenocarcinoma (46), and differentiation of stem-like basal cells into luminal cells in triple-negative breast cancer metastases, recapitulating mammary gland architecture (47). Subsequent scRNA-seq studies in isocitrate dehydrogenase (IDH) mutant gliomas uncovered a cellular architecture reminiscent of neural development, with a neural-progenitor-like (NPC-like) compartment enriched for actively cycling cells, as well as non-proliferative compartments differentiated along astrocyte-like (AC-like) or oligodendrocyte-like (OC-like) lineages (21,48). The distinct compartments did not correlate with genetic heterogeneity of those tumors, as evaluated by CNAs, consistent with a cellular hierarchy dictated by differentiation rather than genetic changes, thereby mimicking developmental processes.
Similarly, cellular hierarchies and differentiation patterns were subsequently described in a large number of other cancers, including additional classes of glioma (39,49), melanoma (50), neuroblastoma (51), medulloblastoma (41,52,53), and multiple types of lung cancer (54,55). Importantly, although these studies demonstrate an overall similarity between normal development and cancer hierarchies, they also begin to illustrate the deviations between cancer-related hierarchies and normal differentiation.
In histone H3 lysine27-to-methionine (H3K27M)-mutant diffuse midline glioma, a cellular hierarchy similar to that of IDH-mutant was identified (39). However, in H37K27M tumors the proliferating stem-like compartment comprised the majority rather than the minority of malignant cells, suggesting a differentiation block and correlating with the aggressive clinical behavior observed in this type of glioma (39). Impaired differentiation trajectories may be a common feature of pediatric cancers, as they were also revealed by scRNA-seq in neuroblastoma (51), ependymoma (49) and medulloblastoma (41,52,53), where they are prognostically important and appear to underlie tumor subgroups (see section below on tumor subtypes). In IDH-wildtype glioblastoma, where numerous scRNA-seq studies demonstrated diverse cell states with similarities to neuro-development (17,40,56–60), lineage-tracing experiments demonstrate plasticity with malignant cells that can frequently transition between states (40). Plasticity was also highlighted in small cell lung cancer (SCLC), in a recent scRNA-seq study demonstrating that tumor cells differentiated towards a neuroendocrine lineage can dedifferentiate to a non-neuroendocrine fate via the action of Notch signaling (54). This plasticity explains transitions between previously described subtypes of SCLC (61), and may account for the ITH (54).
Cellular plasticity with transitions to earlier states in developmental lineage may also occur under therapeutic pressure. Indeed, single-cell profiling of patient-derived BRAF V600E-mutant melanoma cell lines after treatment with the BRAF inhibitor vemurafenib reveals a transition toward a primitive neural crest-like phenotype, with high expression of neural growth factor receptor (NGFR) and loss of and reduced expression of the melanocyte transcription factor MITF, after only three days of treatment (50). This transition towards an early developmental phenotype is thought to represent a drug-resistance adaptation (50).
Finally, even in the absence of therapeutic pressure, the cellular architecture of a given entity may drift during tumor progression: in untreated NSCLC samples, tumor cells have been noted to progressively deviate from normal differentiation towards a cancer cell state that becomes dominant as disease progresses and metastasizes (62); in IDH-mutant gliomas, higher grade lesions have decreased fractions of differentiated cells and increased pools of progenitors (48); and in IDH-wildtype glioblastoma, subsets of malignant cells adopt mesenchymal-like programs that are not anchored in neurodevelopment (40,63).
While the determinants of such deviations from normal differentiation in cancer remain difficult to address, these might be related to the frequent cancer-related alterations in chromatin regulation. Pioneering single-cell multi-omics studies that combine RNA and DNA methylation profiling (or chromatin accessibility assays) are beginning to unravel potential mechanisms. In hematological malignancies, mutations in genes involved in DNA methylation were recently shown to disrupt normal hematopoietic differentiation causing shifts in the frequencies of erythroid or myelomonocytic progenitors, due to disruption of methylation-sensitive lineage-specific transcription factors binding sites (64). Altered DNA methylation profiles were also associated with aberrant programs in chronic lymphocytic leukemia (26) and colorectal cancer (65), as suggested by concurrent single-cell DNAme and transcriptomic profiling.
Tumor Subtypes
One of the main goals of cancer genomics in the last two decades has been to identify tumor subtypes that potentially represent distinct disease subsets and may justify further patient stratification. Expression profiling of bulk tumor samples has played a major role in defining tumor classifications in many cancers, including in breast cancer, melanoma, brain tumors and others (3–6,66). Single-cell analyses are providing at least two critical insights into such tumor classification: (I) in some instances, they reveal heterogeneity of these bulk inter-tumoral classifications even among individual cells from a single sample, indicating that bulk classification reflects the relative abundance of co-existing states, rather than unique states of subsets of tumors; (II) they provide a renewed understanding of the biological underpinnings of bulk classifications, by delineating the contributions of specific cell types and their interactions.
Analysis of glioblastoma showed that distinct cells from a single tumor resemble different bulk subtypes (so-called proneural, classical, and mesenchymal glioblastoma subtypes) (17,67). More recently, scRNA-seq analysis of a much larger number of cells and samples related the subtypes to the abundance of distinct malignant cellular states and of immune cells in the micro-environment (40). In medulloblastoma and ependymoma, single-cell analyses provided key insights on the distinction between tumor subgroups by showing that they differ in the differentiation state of the malignant cells they are composed of (41,49). In small cell lung cancer, recently subclassified into four distinct subtypes based on expression of lineage-defining transcription factors – ASCL1 (SCLC-A), NEUROD1 (SCLC-N), POU2F3 (SCLC-P) and YAP1 (SCLC-Y) (61) – scRNA-seq identified the co-existence of several of these subtypes in distinct cells within individual tumors (54). Expression of these transcription factors might actually account for cells in different stages of tumor evolution (54). In melanoma, bulk studies classified tumors as MITF-high or AXL-high, but single-cell analysis showed that both cellular states are identified in individual tumors (18). Interestingly, AXL-high tumors are additionally confounded by the abundance of cancer-associated fibroblasts (CAFs) that highly express AXL and the other genes associated with the AXL-high signature. Similarly, bulk and single-cell studies of colorectal cancer, head-and-neck squamous cell carcinoma (HSNCC), and ovarian cancer have all suggested that mesenchymal subtypes, thought to reflect epithelial-to-mesenchymal transition of malignant cells, primarily reflect the abundance of CAFs (42,68–71).
Overall, these studies highlight the profound impact of single cell analysis on the definition and understanding of tumor classification. Existing classifications by bulk expression profiling can be interpreted as a combination of three effects: the relative frequencies of malignant cell states that co-exist within individual tumors (e.g., glioblastoma subtypes and medulloblastoma subgroups); the relative frequencies of non-malignant cell types (e.g., CAFs in head-and neck and colorectal cancer); and finally, expression programs that reflect unique genetic and epigenetic states of the malignant cells in subsets of tumors. Single cell studies are now providing the ability to distinguish between these cases and will likely serve as the basis for renewed tumor classifications.
Tumor microenvironment and response to immunotherapy
Tumor microenvironment (TME) composition impacts tumor biology and therapeutic response through various mechanisms. The most common TME cell types include T cells, macrophages, endothelial cells and fibroblasts. Each of these components, as well as less common components, contribute to tumor phenotypes through their direct functions as well as by other mechanisms such as ligand-receptor interactions influencing other cell types. Single cell profiling has been used to characterize these components, including unbiased studies that characterize the entire tumor ecosystem as well as many focused studies that zoom in on particular components such as fibroblasts, macrophages and T cells. Not surprisingly, T cells have attracted the most attention due to their critical role in the efficacy of immunotherapies (72).
Effector CD8+ T cells can lyse malignant cells, but at least two mechanisms may result in less effective responses: first, chronic antigen stimulation may result in T cell exhaustion and loss of effector programs; second, tumor-infiltrating lymphocytes (TILs) may be excluded from the tumor core through a complex set of mechanisms, including interactions with additional TME components such as CAFs, endothelial cells, and macrophages. scRNA-seq offers a compelling approach to dissect the TME components, interrogate dysfunction-specific expression signatures and identify potential mechanisms of immune exclusion. Additionally, scRNA-seq reads that cover the T cell receptor (TCR; either through full-length or V(D)J tailored protocols) enable the reconstruction of TCR sequences and the identification of clonal T cell subsets, and enable linking the expression state of cells to their clonal and functional properties (18,73–77). The power of such approaches to dissect immune responses is illustrated by several single-cell studies of melanoma that identified dysfunction signatures confounded by T cell activation (18,78), distinct states of CD8+ T cells associated with patient tumor regression or progression (79) and a T cell exclusion program expressed by malignant cells and that can predict response to treatment with immune checkpoint inhibitors (ICIs) (80).
Other examples include NSCLC, in which scRNA-seq studies have identified populations of pre-exhausted T cells associated with better prognosis (81), and liver cancer, where increased LAYN expression is associated with cytotoxic T cell exhaustion and worse overall prognosis (75). In colorectal cancer, scRNA-seq highlighted subsets of tumor-associated macrophages (TAMs) with distinct inflammatory (inhibited by anti-CSF1R antibodies) and pro-angiogenic transcriptional profiles (82). Interestingly, differentiation of colorectal cancer cells along post-absorptive (characterized by TP53 and APC mutations) versus a non-absorptive lineage (characterized by KRAS and BRAF mutations) influenced the composition of the tumor microenvironment, with the post-absorptive lineage cells promoting proliferation of cytotoxic T cells immunity and the non-absorptive lineage promoting myofibroblasts and proinflammatory macrophages (83). In gliomas and brain metastases, scRNA-seq and mass cytometry efforts have refined our understanding of the immune composition of these tumors and suggested mechanisms promoting immunosuppression (84–86). These studies have shown that the TME of gliomas differs according to tumor type, with IDH-mutant tumors’ TME composed primarily of activated microglia while IDH-wildtype tumors are enriched for monocyte-derived macrophages (84,85). In brain metastasis, downregulation of Cx3cr1 and upregulation of Cxcl10 in myeloid cells has been suggested as mechanism promoting immunosuppression (86).
Another interesting application of scRNA-seq in glioblastoma has been to leverage the approach to assess the technical success of a neoantigen vaccine trial: by coordinately interrogating the TCR sequence of vaccine-reactive T cells in blood and in the tumor, a study was able to demonstrate the presence of functionally active, neoantigen-specific T cells in a glioblastoma sample (87). Other studies have leveraged mass cytometry to describe the immune infiltration of high-grade ER+ and ER− breast cancer(88), hepatocellular carcinoma (89), renal (90) and lung carcinomas (91). Finally, in basal cell carcinoma, scATAC-seq has helped highlight key regulatory programs underlying T cell exhaustion (92). Going forward, complementing the expression state of TILs and other TME components with their spatial localization and direct physical interactions will yield additional insights into the ecosystem of tumors.
Metastasis
Metastatic progression remains a formidable challenge in the management of systemic cancers, with metastases often being treatment-refractory and accounting for most cancer-related deaths. As further described below, at least three distinct approaches are utilizing single cell profiling to improve our understanding of metastasis: (I) the characterization of cell states that facilitate metastasis, (II) the comparison of matched primary and metastatic samples, and (III) the analysis of circulating tumor cells.
Single cell profiling of primary tumors may uncover cellular states that facilitate metastasis and that are enriched in patients with, versus those without, metastases. In particular, epithelial-to-mesenchymal transition (EMT) is the most cited mechanism for facilitating metastasis, yet it remains controversial and extensively debated, primarily due to the focus of EMT research on model systems and the difficulty in characterizing EMT in patient samples. ScRNA-seq studies now provide an improved capacity to evaluate the exact patterns of EMT directly in clinical specimen. In HNSCC, scRNA-seq identified an EMT-like program at the invasive edge of the tumor, which was modulated by CAFs and enriched in patients with metastases (42). Notably, this program lacked certain features of traditional EMT programs, such as expression of core EMT transcription factors and loss of epithelial markers, and it was found only in two of the four HNSCC subtypes. Such context-specificity of EMT is supported by other recent studies and may help to resolve conflicting reports, thereby underscoring the significance of directly defining cellular states in diverse human tumors.
Single-cell profiling also enables the characterization of circulating tumor cells (CTCs), providing a window into early stages of the metastatic process. CTCs often express EMT programs, consistent with the role of EMT in promoting metastasis (93,94). In addition, analysis of breast cancer CTCs demonstrated an abundance of CTC clusters, with higher metastatic potential than individual CTCs (95). Profiling of the CTC clusters revealed increased expression of the adherens junction protein, plakoglobin, which maintained cell clusters and thereby increased the rate of metastases.
Finally, by comparing matched primary and metastatic samples at the single-cell level, it is possible to identify differences in genetics, cell type composition and cellular states. Genetic differences can help to trace the evolutionary process that underlie metastasis, for example by revealing which genetic clone has seeded the metastasis. Interestingly, one study demonstrated that multiple clones co-migrated and established the invasive lesions in breast cancer (9). In addition to genetics, the differences in cell type composition and cellular states between primary and metastatic samples provide insight into the adaptations of metastatic cells as they colonize different organs or anatomical compartments (42,95–99). For example, a study of leptomeningeal metastasis profiled malignant cells from cerebrospinal fluid (CSF) , and identified upregulation of LCN2 and other genes that enable iron scavenging from inflammatory cells, which promotes survival of the metastatic cells in the iron-poor CSF environment (97).
Response to targeted therapy
Response to therapy may be expected to reflect a combination of clonal selection and cellular adaptation that influence both the malignant cells and non-malignant cells in the TME. When probed in bulk post-treatment tissues, these effects are difficult to tease apart, but scRNA-seq may offer the appropriate resolution to dissect these effects. In BRAF-mutant melanoma, scRNA-seq identified cells in a resistance-associated state already in pre-treated samples (18). Further in vitro single cell studies showed that this program is increased upon treatment, not only due to an expected clonal selection, but also due to a drug-induced epigenetic reprogramming, which might help to explain the widespread resistance to BRAF inhibition in melanoma (100). In NSCLC, tyrosine kinase inhibitors (TKIs) were shown to induce the expression of an alveolar-regenerative cell signature in residual tumor cells, consistent with a therapy-induced cell state transition (55). In ER+/HER2− breast cancer, scRNA-seq demonstrated increased expression of HER2 after multiple courses of therapy with a phenotype that is less sensitive to targeted therapy than in primary HER2+ breast cancer (101). In prostate cancer CTCs, scRNA-seq implicated the activation of noncanonical Wnt signaling as a mechanism of resistance to antiandrogen therapy (102).
Overall, these and other studies are highlighting the formidable adaptive potential of malignant cells under therapeutic pressure. We expect that single cell profiling of post-treatment samples will dramatically expand in the next few years and may even be integrated with clinical trials in an attempt to identify the cellular responses to treatments, and the identity and mechanisms of residual malignant cells. However, in many clinical contexts, the residual cells immediately following partially successful treatments are undetected. These cells may remain dormant for some time until they resume growth and give rise to tumor relapse. Yet, by that time, the resulting tumor have undergone further genetic and epigenetic evolution, which masks the mechanisms of initial drug resistance. Hence, it is unclear to what extent resistance mechanisms can be gleaned from analysis of recurring tumors, which is motivating attempts to directly isolate and profile single cells from minimal residual disease (MRD). This can be achieved in hematological malignancies (103) or through sequential isolation of CTCs following treatments. Alternatively, this is possible with animal models in which MRD can be detected and analyzed (47,104). This approach was used initially to assess MRD in a melanoma model after response to BRAF inhibition (104), and future profiling of MRD in a range of animal models, after response to clinically-relevant treatments, may advance our understanding of resistance mechanisms.
Emerging technologies and future developments
Single-cell genomics methods are ushering a new area in the study of cancer biology, improving our understanding of tumor heterogeneity, evolution, interactions with the tumor microenvironment and response to therapy. Technological developments are enabling multiple new avenues to tackle questions of biological and clinical relevance (Figure 2). Increased throughput of scRNA-seq platforms, where thousands of cells are routinely profiled (32–34), is enabling the discovery of small but clinically significant tumor subpopulations, including those with adaptations underlying drug resistance or metastatic progression. The possibility of profiling frozen samples by sNuc-seq (28) enables the characterization of samples stored at biobanks, facilitating cross-center collaborations and the profiling at scale of rare tumor entities or matched primary and recurrent samples for the study of tumor evolution. Experiments that concurrently evaluate transcriptional and epigenomic states (DNA methylation or chromatin accessibility) in individual cells (27,105) are providing deeper insights into mechanisms governing state diversity and transitions in tumors. Spatially-resolved single-cell profiling methods, such as spatial transcriptomics of tissue sections (106,107), enable better characterization of interactions within the tumor microenvironment (108–110). Advances in mass spectrometry methods are also beginning to allow the characterization of thousands of proteins at single-cell resolution without the need of using tagging antibodies (111,112), as well as to quantify increasing numbers of metabolites in cancer cells (113,114). The studies we have reviewed mark the beginning of the era of single-cell profiling of human tumors, which is rapidly broadening in resolution, scale and scope. While the complexity uncovered may appear daunting, the deeper understanding gained from these approaches offers unprecedented opportunities for renewed attempts at improving the management of cancer patients.
Figure 2:
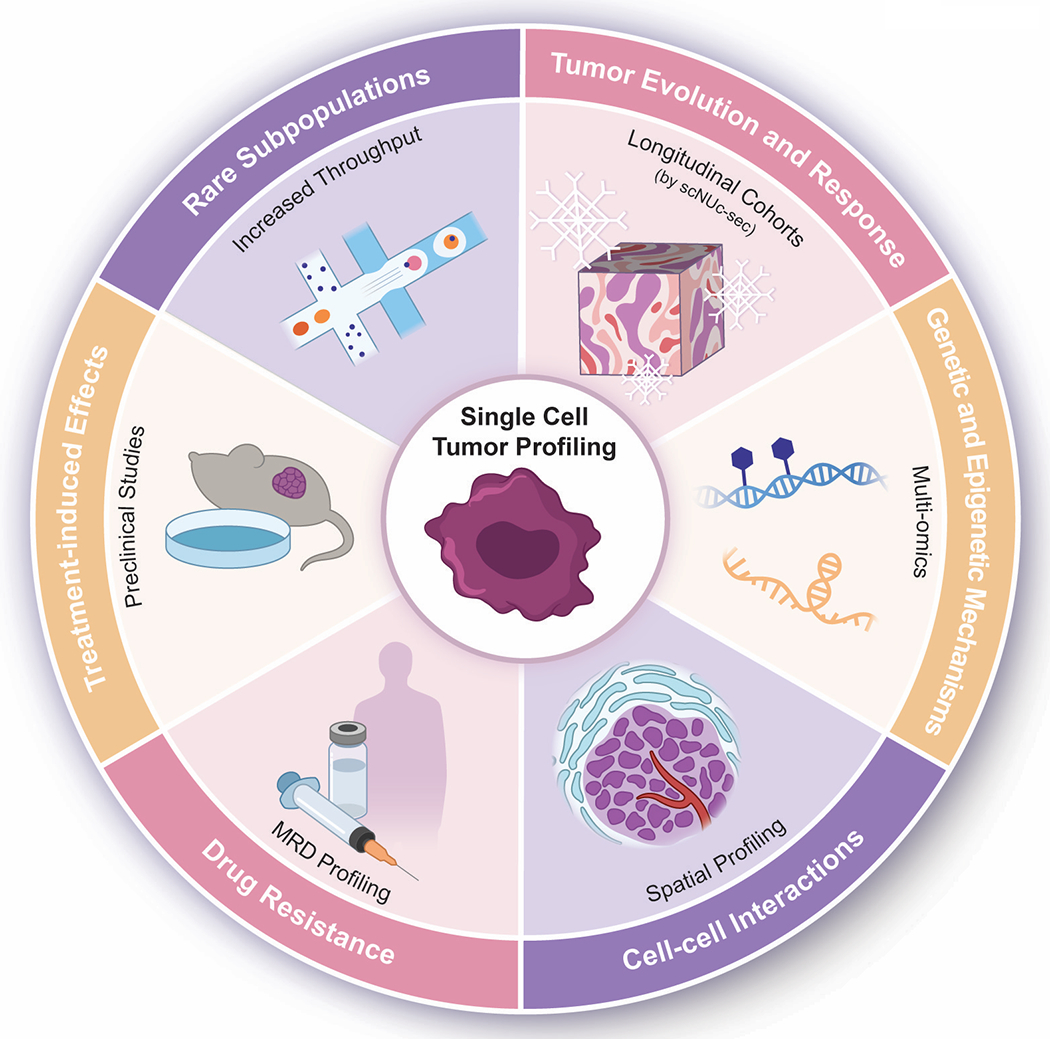
Recent developments in single-cell genomics approaches and the different biological and clinical questions in oncology they will inform.
Significance.
Intra-tumor heterogeneity (ITH) has been a major barrier to our understanding of cancer. Single-cell genomics is leading a revolution in our ability to systematically dissect ITH. In this review we focus on single cell expression profiling and insights gleaned on stem cell programs, tumor classification, metastasis, microenvironment composition, and tumor response to therapies.
Acknowledgements
We thank SciStories LLC for figure schematics. This work was supported by a Broad Institute–Israel Science Foundation Collaborative Project Award (I.T. and M.L.S.), grants from the Mark Foundation Emerging Leader Award (M.L.S.), the Sontag Foundation Distinguished Scientist Award (M.L.S.) and N.I.H. R37CA245523 (M.L.S). I.T. is the incumbent of the Dr. Celia Zwillenberg-Fridman and Dr. Lutz Zwillenberg Career Development Chair, and is supported by the Zuckerman STEM Leadership Program, the Mexican Friends New Generation, the Benoziyo Endowment Fund, and grants from the Human Frontiers Science Program. L.N.G.C. was supported by NIH K12CA090354.
Footnotes
Conflict of Interest
M.L.S. is equity holder, scientific co-founder and advisory board member of Immunitas Therapeutics. I.T. is an advisory board member of Immunitas Therapeutics.
Bibliography
- 1.Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 2.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akbani R, Akdemir KC, Aksoy BA, Albert M, Ally A, Amin SB, et al. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abou-El-Ardat K, Seifert M, Becker K, Eisenreich S, Lehmann M, Hackmann K, et al. Comprehensive molecular characterization of multifocal glioblastoma proves its monoclonal origin and reveals novel insights into clonal evolution and heterogeneity of glioblastomas. Neuro Oncol. 2017;19:546–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leong TL, Gayevskiy V, Steinfort DP, De Massy MR, Gonzalez-Rajal A, Marini KD, et al. Deep multi-region whole-genome sequencing reveals heterogeneity and gene-by-environment interactions in treatment-naive, metastatic lung cancer. Oncogene [Internet]. Springer US; 2019;38:1661–75. Available from: 10.1038/s41388-018-0536-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casasent AK, Schalck A, Gao R, Sei E, Long A, Pangburn W, et al. Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing. Cell [Internet]. Elsevier Inc.; 2018;172:205–217. e12. Available from: 10.1016/j.cell.2017.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Francis JM, Zhang CZ, Maire CL, Jung J, Manzo VE, Adalsteinsson VA, et al. EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov. 2014;4:956–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao R, Davis A, McDonald TO, Sei E, Shi X, Wang Y, et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat Genet. 2016;48:1119–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leung ML, Davis A, Gao R, Casasent A, Wang Y, Sei E, et al. Single-cell DNA sequencing reveals a late-dissemination model in metastatic colorectal cancer. Genome Res. 2017;27:1287–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Navin NE, Chen K. Genotyping tumor clones from single-cell data. Nat Methods. Nature Publishing Group; 2016;13:555–6. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. Nature Publishing Group; 2014;512:155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell. 2018;33:676–689. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan J, Lee HO, Lee S, Ryu DE, Lee S, Xue C, et al. Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell RNA-seq data. Genome Res. 2018;28:1217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science [Internet]. 2014. [cited 2015 Sep 9];344:1396–401. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4123637&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science [Internet]. 2016. [cited 2016 Oct 7];352:189–96. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27124452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laks E, McPherson A, Zahn H, Lai D, Steif A, Brimhall J, et al. Clonal Decomposition and DNA Replication States Defined by Scaled Single-Cell Genome Sequencing. Cell. 2019;179:1207–1221. e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams MJ, Werner B, Barnes CP, Graham TA, Sottoriva A. Identification of neutral tumor evolution across cancer types. Nat Genet [Internet]. Nature Publishing Group; 2016;48:238–44. Available from: 10.1038/ng.3489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. Nature Publishing Group; 2016;539:309–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giustacchini A, Thongjuea S, Barkas N, Woll PS, Povinelli BJ, Booth CAG, et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat Med [Internet]. Nature Publishing Group; 2017;23:692–702. Available from: 10.1038/nm.4336 [DOI] [PubMed] [Google Scholar]
- 23.Nam AS, Kim KT, Chaligne R, Izzo F, Ang C, Taylor J, et al. Somatic mutations and cell identity linked by Genotyping of Transcriptomes. Nature [Internet]. Springer US; 2019;571:355–60. Available from: 10.1038/s41586-019-1367-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Macaulay IC, Haerty W, Kumar P, Li YI, Hu TX, Teng MJ, et al. G&T-seq: Parallel sequencing of single-cell genomes and transcriptomes. Nat Methods. 2015;12:519–22. [DOI] [PubMed] [Google Scholar]
- 25.Macaulay IC, Teng MJ, Haerty W, Kumar P, Ponting CP, Voet T. Separation and parallel sequencing of the genomes and transcriptomes of single cells using G&T-seq. Nat Protoc. 2016;11:2081–103. [DOI] [PubMed] [Google Scholar]
- 26.Gaiti F, Chaligne R, Gu H, Brand RM, Kothen-Hill S, Schulman RC, et al. Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature [Internet]. Springer US; 2019;569:576–80. Available from: 10.1038/s41586-019-1198-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nam AS, Chaligne R, Landau DA. Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat Rev Genet [Internet]. Springer US; 2020;1–16. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32807900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slyper M, Porter CBM, Ashenberg O, Waldman J, Drokhlyansky E, Wakiro I, et al. A single-cell and single-nucleus RNA-seq toolbox for fresh and frozen human tumors. Nat Med. 2020;26:792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding J, Adiconis X, Simmons SK, Kowalczyk MS, Hession CC, Marjanovic ND, et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat Biotechnol. 2020;1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc [Internet]. 2014. [cited 2016 Sep 26];9:171–81. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24385147 [DOI] [PubMed] [Google Scholar]
- 31.Hagemann-Jensen M, Ziegenhain C, Chen P, Ramsköld D, Hendriks GJ, Larsson AJM, et al. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat Biotechnol [Internet]. Springer US; 2020;38:708–14. Available from: 10.1038/s41587-020-0497-0 [DOI] [PubMed] [Google Scholar]
- 32.Picelli S Single-cell RNA-sequencing: The future of genome biology is now. RNA Biol [Internet]. Taylor & Francis; 2017;14:637–50. Available from: 10.1080/15476286.2016.1201618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baran-Gale J, Chandra T, Kirschner K. Experimental design for single-cell RNA sequencing. Brief Funct Genomics. 2018;17:233–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.See P, Lum J, Chen J, Ginhoux F. A single-cell sequencing guide for immunologists. Front Immunol. 2018;9:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science (80- ). 2018;360:176–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin Y, Jiang Y, Lam KWG, Berletch JB, Disteche CM, Noble WS, et al. High-Throughput Single-Cell Sequencing with Linear Amplification. Mol Cell [Internet]. Elsevier Inc.; 2019;76:676–690. e10. Available from: 10.1016/j.molcel.2019.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao J, Zhou W, Steemers F, Trapnell C, Shendure J. Sci-fate characterizes the dynamics of gene expression in single cells. Nat Biotechnol. 2020;1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Srivatsan SR, McFaline-Figueroa JL, Ramani V, Saunders L, Cao J, Packer J, et al. Massively multiplex chemical transcriptomics at single-cell resolution. Science (80- ). 2020;367:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Filbin MG, Tirosh I, Hovestadt V, Shaw ML, Escalante LE, Mathewson ND, et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science (80- ). 2018;360:331–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell. 2019;178:835–849. e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hovestadt V, Smith KS, Bihannic L, Filbin MG, Shaw MKL, Baumgartner A, et al. Resolving medulloblastoma cellular architecture by single-cell genomics. Nature [Internet]. Springer US; 2019;572:74–9. Available from: 10.1038/s41586-019-1434-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell [Internet]. Elsevier Inc.; 2017;171:1611–1624. e24. Available from: 10.1016/j.cell.2017.10.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat Rev Clin Oncol. Nature Publishing Group; 2017;14:611–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275–91. [DOI] [PubMed] [Google Scholar]
- 45.Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentin CLL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dalerba P, Kalisky T, Sahoo D, Rajendran PS, Rothenberg ME, Leyrat AA, et al. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat Biotechnol. Nature Publishing Group; 2011;29:1120–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. 2015;526:131–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science (80- ). 2017;355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gojo J, Englinger B, Jiang L, Hübner JM, Shaw ML, Hack OA, et al. Single-Cell RNA-Seq Reveals Cellular Hierarchies and Impaired Developmental Trajectories in Pediatric Ependymoma. Cancer Cell. 2020;38:44–59. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Su Y, Wei W, Robert L, Xue M, Tsoi J, Garcia-Diaz A, et al. Single-cell analysis resolves the cell state transition and signaling dynamics associated with melanoma drug-induced resistance. Proc Natl Acad Sci U S A. 2017;114:13679–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong R, Yang R, Zhan Y, Zheng S, Li K, Wang J, et al. Single-Cell Characterization of Malignant Phenotypes and Developmental Trajectories of Adrenal Neuroblastoma Article Single-Cell Characterization of Malignant Phenotypes and Developmental Trajectories of Adrenal Neuroblastoma. Cancer Cell. 2020;38:1–18. [DOI] [PubMed] [Google Scholar]
- 52.Jessa S, Blanchet-Cohen A, Krug B, Vladoiu M, Coutelier M, Faury D, et al. Stalled developmental programs at the root of pediatric brain tumors. Nat Genet. 2019;51:1702–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vladoiu MC, El-Hamamy I, Donovan LK, Farooq H, Holgado BL, Sundaravadanam Y, et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature [Internet]. Springer US; 2019;572:67–73. Available from: 10.1038/s41586-019-1158-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ireland AS, Micinski AM, Kastner DW, Guo B, Wait SJ, Spainhower KB, et al. MYC Drives Temporal Evolution of Small Cell Lung Cancer Subtypes by Reprogramming Neuroendocrine Fate. Cancer Cell. 2020;38:60–78. e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maynard A, McCoach CE, Rotow JK, Harris L, Haderk F, Kerr DL, et al. Therapy-Induced Evolution of Human Lung Cancer Revealed by Single-Cell RNA Sequencing. Cell [Internet]. Elsevier Inc.; 2020;182:1–20. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0092867420308825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Müller S, Liu SJ, Di Lullo E, Malatesta M, Pollen AA, Nowakowski TJ, et al. Single-cell sequencing maps gene expression to mutational phylogenies in PDGF and EGF driven gliomas. Mol Syst Biol. 2016;12:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bhaduri A, Di Lullo E, Jung D, Müller S, Crouch EE, Espinosa CS, et al. Outer Radial Glia-like Cancer Stem Cells Contribute to Heterogeneity of Glioblastoma. Cell Stem Cell. 2020;26:48–63. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang L, Babikir H, Muller S, Yagnik G, Shamardani K, Catalan F, et al. The phenotypes of proliferating glioblastoma cells reside on a single axis of variation. Cancer Discov. American Association for Cancer Research (AACR); 2019;CD-19-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weng Q, Wang J, Wang J, He D, Cheng Z, Zhang F, et al. Single-Cell Transcriptomics Uncovers Glial Progenitor Diversity and Cell Fate Determinants during Development and Gliomagenesis. Cell Stem Cell. 2019;24:707–723. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Couturier CP, Ayyadhury S, Le PU, Monlong J, Riva G, Allache R, et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal brain development hierarchy. Nat Commun [Internet]. Springer US; 2020;11:1–19. Available from: https://www.biorxiv.org/content/10.1101/449439v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rudin CM, Poirier JT, Byers LA, Dive C, Dowlati A, George J, et al. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer [Internet]. Springer US; 2019;19:289–97. Available from: 10.1038/s41568-019-0133-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim N, Kim HK, Lee K, Hong Y, Cho JH, Choi JW, et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun. 2020;11:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pine AR, Cirigliano SM, Nicholson JG, Hu Y, Linkous A, Miyaguchi K, et al. Tumor Microenvironment Is Critical for the Maintenance of Cellular States Found in Primary Glioblastomas. Cancer Discov. 2020;10:964–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Izzo F, Lee SC, Poran A, Chaligne R, Gaiti F, Gross B, et al. DNA methylation disruption reshapes the hematopoietic differentiation landscape. Nat Genet [Internet]. Springer US; 2020;52:378–87. Available from: 10.1038/s41588-020-0595-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bian S, Hou Y, Zhou X, Li X, Yong J, Wang Y, et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science (80- ). 2018;362:1060–3. [DOI] [PubMed] [Google Scholar]
- 66.Muzny DM, Bainbridge MN, Chang K, Dinh HH, Drummond JA, Fowler G, et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature [Internet]. Nature Publishing Group; 2012;487:330–7. Available from: 10.1038/nature11252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gill BJ, Pisapia DJ, Malone HR, Goldstein H, Lei L, Sonabend A, et al. MRI-localized biopsies reveal subtype-specific differences in molecular and cellular composition at the margins of glioblastoma. Proc Natl Acad Sci U S A. 2014;111:12550–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Calon A, Lonardo E, Berenguer-Llergo A, Espinet E, Hernando-Momblona X, Iglesias M, et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet [Internet]. Nature Publishing Group; 2015;47:320–9. Available from: 10.1038/ng.3225 [DOI] [PubMed] [Google Scholar]
- 69.Isella C, Terrasi A, Bellomo SE, Petti C, Galatola G, Muratore A, et al. Stromal contribution to the colorectal cancer transcriptome. Nat Genet. 2015;47:312–9. [DOI] [PubMed] [Google Scholar]
- 70.Li H, Courtois ET, Sengupta D, Tan Y, Chen KH, Goh JJL, et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat Genet. Nature Publishing Group; 2017;49:708–18. [DOI] [PubMed] [Google Scholar]
- 71.Izar B, Tirosh I, Stover EH, Wakiro I, Cuoco MS, Alter I, et al. A single-cell landscape of high-grade serous ovarian cancer. Nat Med [Internet]. 2020;1–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32572264 [DOI] [PMC free article] [PubMed]
- 72.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–30. [DOI] [PubMed] [Google Scholar]
- 73.Eltahla AA, Rizzetto S, Pirozyan MR, Betz-Stablein BD, Venturi V, Kedzierska K, et al. Linking the T cell receptor to the single cell transcriptome in antigen-specific human T cells. Immunol Cell Biol. 2016;94:604–11. [DOI] [PubMed] [Google Scholar]
- 74.Stubbington MJT, Lönnberg T, Proserpio V, Clare S, Speak AO, Dougan G, et al. T cell fate and clonality inference from single-cell transcriptomes. Nat Methods. 2016;13:329–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell [Internet]. Elsevier; 2017;169:1342–1356. e16. Available from: 10.1016/j.cell.2017.05.035 [DOI] [PubMed] [Google Scholar]
- 76.Zhang SQ, Ma KY, Schonnesen AA, Zhang M, He C, Sun E, et al. High-throughput determination of the antigen specificities of T cell receptors in single cells. Nat Biotechnol. 2018;36:1156–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kula T, Dezfulian MH, Wang CI, Abdelfattah NS, Hartman ZC, Wucherpfennig KW, et al. T-Scan: A Genome-wide Method for the Systematic Discovery of T Cell Epitopes. Cell [Internet]. Elsevier Inc.; 2019;178:1016–1028. e13. Available from: 10.1016/j.cell.2019.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H, et al. A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell [Internet]. Elsevier; 2016;166:1500–1511. e9. Available from: 10.1016/j.cell.2016.08.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell. 2018;175:998–1013. e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jerby-arnon L, Shah P, Cuoco MS, Rodman C, Su M, Melms JC, et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell. 2018;175:984–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med [Internet]. Springer US; 2018;24:978–85. Available from: 10.1038/s41591-018-0045-3 [DOI] [PubMed] [Google Scholar]
- 82.Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell [Internet]. Elsevier; 2020;181:442–459. e29. Available from: 10.1016/j.cell.2020.03.048 [DOI] [PubMed] [Google Scholar]
- 83.Lee HO, Hong Y, Etlioglu HE, Cho YB, Pomella V, Van den Bosch B, et al. Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat Genet [Internet]. Springer US; 2020;52:594–603. Available from: 10.1038/s41588-020-0636-z [DOI] [PubMed] [Google Scholar]
- 84.Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, Nassiri S, et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell [Internet]. Elsevier; 2020;181:1643–1660. e17. Available from: 10.1016/j.cell.2020.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Friebel E, Kapolou K, Unger S, Nunez NG, Utz S, Rushing EJ, et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell. 2020;181:1–17. [DOI] [PubMed] [Google Scholar]
- 86.Guldner IH, Wang Q, Yang L, Golomb SM, Zhao Z, Lopez JA, et al. CNS-Native Myeloid Cells Drive Immune Suppression in the Brain Metastatic Niche through Cxcl10. Cell [Internet]. Elsevier Inc.; 2020;183:1234–1248. e25. Available from: 10.1016/j.cell.2020.09.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature [Internet]. Springer US; 2019;565:234–9. Available from: https://www.nature.com/articles/s41586-018-0792-9?utm_source=researcher_app&utm_medium=referral&utm_campaign=MKEF_USG_Researcher_inbound [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wagner J, Rapsomaniki MA, Chevrier S, Anzeneder T, Langwieder C, Dykgers A, et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell. 2019;177:1330–1345. e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chew V, Lai L, Pan L, Lim CJ, Li J, Ong R, et al. Delineation of an immunosuppressive gradient in hepatocellular carcinoma using high-dimensional proteomic and transcriptomic analyses. Proc Natl Acad Sci U S A. 2017;114:E5900–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chevrier S, Levine JH, Zanotelli VRT, Silina K, Schulz D, Bacac M, et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell. 2017;169:736–749. e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lavin Y, Kobayashi S, Leader A, Amir E ad D, Elefant N, Bigenwald C, et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell [Internet]. Elsevier Inc.; 2017;169:750–765. e17. Available from: 10.1016/j.cell.2017.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat Biotechnol [Internet]. Springer US; 2019;37:925–36. Available from: http://www.nature.com/articles/s41587-019-0206-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cheng YH, Chen YC, Lin E, Brien R, Jung S, Chen YT, et al. Hydro-Seq enables contamination-free high-throughput single-cell RNA-sequencing for circulating tumor cells. Nat Commun [Internet]. Springer US; 2019;10:1–11. Available from: 10.1038/s41467-019-10122-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ting DT, Wittner BS, Ligorio M, Vincent Jordan N, Shah AM, Miyamoto DT, et al. Single-cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep. 2014;8:1905–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell [Internet]. Elsevier Inc.; 2014;158:1110–22. Available from: 10.1016/j.cell.2014.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Su Z, Wang Z, Ni X, Duan J, Gao Y, Zhuo M, et al. Inferring the evolution and progression of small-cell lung cancer by single-cell sequencing of circulating tumor cells. Clin Cancer Res. 2019;25:5049–60. [DOI] [PubMed] [Google Scholar]
- 97.Chi Y, Remsik J, Kiseliovas V, Derderian C, Sener U, Alghader M, et al. Cancer cells deploy lipocalin-2 to collect limiting iron in leptomeningeal metastasis. Science. 2020;369:276–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, et al. Tumour evolution inferred by single-cell sequencing. Nature. Nature Publishing Group; 2011;472:90–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rao M, Oh K, Moffitt R, Thompson P, Li J, Liu J, et al. Comparative single-cell RNA sequencing (scRNA-seq) reveals liver metastasis-specific targets in a patient with small intestinal neuroendocrine cancer. Cold Spring Harb Mol case Stud. 2020;6:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C, et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. Nature Publishing Group; 2017;546:431–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jordan NV, Bardia A, Wittner BS, Benes C, Ligorio M, Zheng Y, et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature. Nature Publishing Group; 2016;537:102–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT, et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science (80- ). 2015;349:1351–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ediriwickrema A, Aleshin A, Reiter JG, Corces MR, Kohnke T, Stafford M, et al. Single-cell mutational profiling enhances the clinical evaluation of AML MRD. Blood Adv. 2020;4:943–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rambow F, Rogiers A, Marin-Bejar O, Aibar S, Femel J, Dewaele M, et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell [Internet]. Elsevier Inc.; 2018;174:843–855. e19. Available from: 10.1016/j.cell.2018.06.025 [DOI] [PubMed] [Google Scholar]
- 105.Xu J, Nuno K, Litzenburger UM, Qi Y, Corces MR, Majeti R, et al. Single-cell lineage tracing by endogenous mutations enriched in transposase accessible mitochondrial DNA. Elife. 2019;8:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stahl PL, Salmen F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science (80- ) [Internet]. 2016;353:78–82. Available from: http://www.sciencemag.org/cgi/doi/10.1126/science.aaf2403 [DOI] [PubMed] [Google Scholar]
- 107.Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, et al. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science (80- ). 2019;363:1463–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Thrane K, Eriksson H, Maaskola J, Hansson J, Lundeberg J. Spatially resolved transcriptomics enables dissection of genetic heterogeneity in stage III cutaneous malignant melanoma. Cancer Res. 2018;78:5970–9. [DOI] [PubMed] [Google Scholar]
- 109.Berglund E, Maaskola J, Schultz N, Friedrich S, Marklund M, Bergenstråhle J, et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat Commun. 2018;9:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Moncada R, Barkley D, Wagner F, Chiodin M, Devlin JC, Baron M, et al. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat Biotechnol [Internet]. Springer US; 2020;38:333–42. Available from: 10.1038/s41587-019-0392-8 [DOI] [PubMed] [Google Scholar]
- 111.Budnik B, Levy E, Harmange G, Slavov N. SCoPE-MS: Mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation 06 Biological Sciences 0601 Biochemistry and Cell Biology 06 Biological Sciences 0604 Genetics. Genome Biol. Genome Biology; 2018;19:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Slavov N Unpicking the proteome in single cells. Science (80- ). 2020;367:512–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Evers TMJ, Hochane M, Tans SJ, Heeren RMA, Semrau S, Nemes P, et al. Deciphering Metabolic Heterogeneity by Single-Cell Analysis. Anal Chem. 2019;91:13314–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gilmore IS, Heiles S, Pieterse CL. Metabolic Imaging at the Single-Cell Scale: Recent Advances in Mass Spectrometry Imaging. Annu Rev Anal Chem. 2019;12:201–24. [DOI] [PubMed] [Google Scholar]
- 115.Chung W, Eum HH, Lee HO, Lee KM, Lee HB, Kim KT, et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun [Internet]. Nature Publishing Group; 2017;8:1–12. Available from: 10.1038/ncomms15081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Savage P, Blanchet-Cohen A, Revil T, Badescu D, Saleh SMI, Wang YC, et al. A Targetable EGFR-Dependent Tumor-Initiating Program in Breast Cancer. Cell Rep. 2017;21:1140–9. [DOI] [PubMed] [Google Scholar]
- 117.Brady SW, McQuerry JA, Qiao Y, Piccolo SR, Shrestha G, Jenkins DF, et al. Combating subclonal evolution of resistant cancer phenotypes. Nat Commun [Internet]. Springer US; 2017;8. Available from: 10.1038/s41467-017-01174-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim C, Gao R, Sei E, Brandt R, Hartman J, Hatschek T, et al. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell [Internet]. Elsevier Inc.; 2018;173:879–893. e13. Available from: 10.1016/j.cell.2018.03.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Young MD, Mitchell TJ, Vieira Braga FA, Tran MGB, Stewart BJ, Ferdinand JR, et al. Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science (80- ). 2018;361:594–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Karaayvaz M, Cristea S, Gillespie SM, Patel AP, Mylvaganam R, Luo CC, et al. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat Commun [Internet]. Springer US; 2018;9:1–10. Available from: 10.1038/s41467-018-06052-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Peng J, Sun BF, Chen CY, Zhou JY, Chen YS, Chen H, et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res [Internet]. Springer US; 2019;29:725–38. Available from: 10.1038/s41422-019-0195-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.van Galen P, Hovestadt V, Wadsworth MH, Hughes TK, Griffin GK, Battaglia S, et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell [Internet]. Elsevier Inc.; 2019;176:1265–1281. e24. Available from: 10.1016/j.cell.2019.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ji AL, Rubin AJ, Thrane K, Jiang S, Reynolds DL, Meyers RM, et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. J Clean Prod. 2020;497–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chen YP, Yin JH, Li WF, Li HJ, Chen DP, Zhang CJ, et al. Single-cell transcriptomics reveals regulators underlying immune cell diversity and immune subtypes associated with prognosis in nasopharyngeal carcinoma. Cell Res [Internet]. Springer US; 2020;0:1–19. Available from: 10.1038/s41422-020-0374-x [DOI] [PMC free article] [PubMed] [Google Scholar]