

Abstract
Cell-laden scaffolds of architecture and mechanics that mimic those of the host tissues are important for a wide range of biomedical applications but remain challenging to bioprint. To address these challenges, we report a new method called triggered micropore-forming bioprinting. The approach can yield cell-laden scaffolds of defined architecture and interconnected pores over a range of sizes, encompassing that of many cell types. The viscoelasticity of the bioprinted scaffold can match that of biological tissues and be tuned independently of porosity and stiffness. The bioprinted scaffold also exhibits superior mechanical robustness despite high porosity. The bioprinting method and the resulting scaffolds support cell spreading, migration, and proliferation. The potential of the 3D bioprinting system is demonstrated for vocal fold tissue engineering and as an in vitro cancer model. Other possible applications are foreseen for tissue repair, regenerative medicine, organ-on-chip, drug screening, organ transplantation, and disease modeling.
Graphical Abstract
This work reports a novel approach to 3D-bioprint hierarchical cell-laden scaffolds with an unprecedented combination of porous and viscoelastic properties.
Introduction
Three-dimensional (3D) bioprinting permits cells and biomaterials to be placed in a precise manner within a complex 3D scaffold, and finds broad applications in regenerative medicine, organ-on-chip, drug screening, and organ transplantation.1–3 Recent development has moved toward the goal to recapitulate the architecture and mechanics of biological tissues; in particular, the combination of biomimetic porous structure and viscoelastic response is greatly desired. On the one hand, the porous structure is essential for cell migration, nutrient transport, and waste removal.4 While small pores (<1 µm) constrain cell growth and migration, the motility of cells in a matrix of large pores (>100 µm) is two-dimensional, and thus of less physiological relevance.5 The pores commensurate with cell sizes (~10 µm) facilitate certain cellular activities, as supported by the studies of fibroblast growth and hepatocyte ingrowth.5,6 On the other hand, the viscoelastic response is ubiquitous among soft tissues and tumors. It has been proved to regulate the proliferation, spreading, and differentiation of various cells, including stem cells, cancer cells, fibroblasts, and chondrocytes.7–9 Studies of cellular and tissue engineering have converged upon the idea that a porous viscoelastic scaffold that mimics the host tissue could benefit native and/or transplanted cells for better therapeutic outcomes. However, most of existing bioprinting technologies rely on elastic and nanoporous scaffolds.10 The ways to bioprint cell-laden scaffolds of cell-sized pores and viscoelastic response continue to be explored.
Challenges to fabricate such biomimetic scaffolds are manifold, which have long limited the capacity of manufacturing technologies to tune both the pore size and the viscoelasticity of cell-laden scaffolds (Fig. S1, ESI†).11 First, it is challenging, if not impossible, to extrude cells and biomaterials into cell-sized features, even if small needles and accurate gantries are used. This approach would inevitably suffer from low printing efficiency, high cost, and reduced viability of the cells due to high shear stresses during small needle extrusion. As such, the accessible pore size, defined by the spacing between bioprinted filaments (i.e., feature size P), is typically larger than 100 µm, whereas the intrinsic pore size p of the bioprinted material is much smaller than 1 µm. Second, conventional methods can create 10-µm pores using cryogelation (i.e., gelation occurs below the water freezing temperature) and/or freeze-dry treatment. Those methods have been used in conjunction with 3D printing to fabricate porous scaffolds.12 But such methods are not ideal for bioprinting as cells need to be seeded after scaffold formation to avoid exposure to low temperatures. This often results in a low seeding density and an inhomogeneous cell distribution within the scaffold, especially when the pore size is small.13 Third, the recently reported approaches, based on sacrificial micelle-laden14 or emulsion15 bioinks, are complicated with limited pore interconnectivity and instability of the emulsion of the bioink, limiting the bioprinting time window (Fig. S2, ESI†). In addition, these methods often produce elastic matrices different from the viscoelastic tissues,15 as covalent crosslinks are exploited to stabilize the porous scaffolds. The matrix viscoelasticity depends on crosslink strength, which may be tuned through changes in the type of crosslinks (e.g., physical versus covalent crosslinks). However, for the viscoelastic response, the replacement of the covalent crosslinks with physical crosslinks may further deteriorate the mechanical strength of the porous scaffold.4 These challenges together contribute to a limited bioprinting window, as shown in Fig. S1 and Table S1 (ESI†) that summarize the accessible pore size and viscoelastic response of existing bioprinting technologies.
To address these challenges, here we report a new method designated, “triggered micropore-forming” (TMF) bioprinting, combining the merits of microphase separation, 3D embedding printing, and viscoelastic hydrogels. This method enables the rapid fabrication of porous viscoelastic hydrogel (PVH) scaffolds with hierarchical porous structures and viscoelastic properties, which can be tuned to a great extent to match a wide range of requirements. Different from the previously reported methods, the TMF bioprinting leverages a microphase separation mechanism, triggered by mild stimuli (e.g., body temperature), to create highly interconnected cell-sized pores in a facile and controlled manner (Fig. 1a,b). This mechanism also forms a polymer-concentrated phase, where the polymer content is raised above the initial concentration in the bioink, leading to a mechanically robust scaffold. This effect could compensate for possible reductions in toughness and robustness associated with the creation of porous structure. As proof of principle, we will investigate this idea by using chitosan as a model bioink system. This material, widely used in various biomedical applications, can separate into micro-phases at neutral pH and at body temperature,16 and form hydrogen bonds for gelation. To modulate the viscoelastic response of the bioprinted scaffold, we will exploit the cytocompatible polyethylene glycol (PEG) as crosslink spacers (i.e., competing with chitosan to form hydrogen bonds) to tune the strength of the hydrogen bonds of the chitosan (Fig. 1c). In this work, we will first establish the TMF method and then characterize the mechanics and architecture of the bioprinted PVH scaffold. We will demonstrate the printability of the TMF method for making complex 3D structures and tissue-engineered scaffolds for vocal fold repair and in vitro 3D model for cancer research. We expect that the TMF method would be the first bioprinting method to make cell-laden viscoelastic scaffolds with cell-sized pores and open many possibilities in biomedical applications.
Fig. 1.

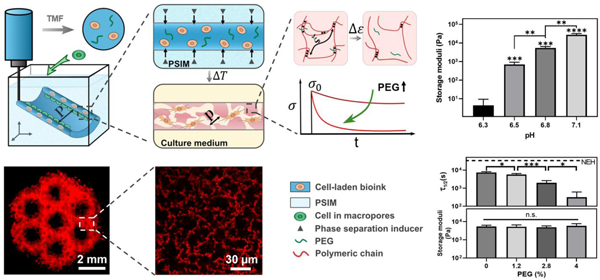
Triggered micropore-forming (TMF) bioprinting. (a) TMF bioprinting of a cell-laden bioink (dark blue) into a scaffold of defined architecture (feature size P). (b) Porous viscoelastic hydrogel (PVH) formed within a phase-separation inducing matrix (PSIM; light blue), which supports the extruded bioink and supplies a phase separation inducer (grey triangles) to form cell-sized pores (pore size p) at elevated temperature. (c) Viscoelastic response of the PVH is modulated with polyethylene glycol (PEG) as a crosslink spacer. The viscoelastic response is manifested with stress relaxation under a constant strain. (d) Fluorescent image of a hierarchical porous scaffold; scale bar 2 mm. (e) Confocal image of the micropores within the bioprinted scaffolds labeled with rhodamine-B (red); scale bar 30 µm.
Results and discussions
Triggered micropore-forming bioprinting
In this study, the base bioink was a mixture of 1.5 wt% chitosan of high deacetylation ratio (95%), 0.04 M phosphate salts, and 0.12 M acetic acid. The mixture with a final pH of 6.2 (below the pKa of chitosan 6.5)17 remained liquid-like and stable at room temperature for at least 24 hours before and during use (Fig. S3a, ESI†). The TMF bioprinting consists of two steps: (i) embedding 3D printing of the cell-laden bioink within a phase-separation inducing matrix (PSIM); (ii) reinforcement of the bioprinted PVH scaffold and removal of PSIM at elevated temperature (37°C). The PSIM was made of sodium bicarbonate-laden gelatin slurries to support and trigger the micropore formation. The extruded bioink reacted with the diffusive sodium bicarbonate from the PSIM (i.e., the amine groups [NH2] of chitosan became deprotonated and neutral) to form bicontinuous micro-phases of water and chitosan due to the change of pH (Fig. S3b, ESI†).18 The reaction resulted in a pores and chitosan-concentrated phases (Fig. 1d,e). After deposition, the scaffold was heated at 37°C to melt and remove the gelatin slurries without dissolving or distorting the bioprinted scaffolds. The rise in temperature also strengthened the hydrogen bonds between the chitosan chains.19 As a result, the scaffold was stiffened (Fig. 2a). To examine the role of hydrogen bonds, we also tested chitosan of lower deacetylation ratio 75–85% (i.e., the lower capacity to form hydrogen bonds), and observed no gelation at 37°C (Fig. S4, ESI†). The importance of embedding printing was confirmed with the attempt to bioprint without the PSIM, which resulted in nozzle clogging and collapse of the printed structures due to the low yielding stress of the gel (Fig. 2b).
Fig. 2.

Mechanical and structural properties of the bioink and the resulting scaffold. (a) Thermal gelation kinetics of the mixture of the bioink and sodium bicarbonate when the temperature is raised from 20 to 37°C. (b) Yield stress measurements for the gelatin-based PSIM (red), the pristine bioink (blue), and the bioink with the gelling agent (sodium bicarbonate, SC; green). (c) Storage moduli of the PVH vary with the pH. (d) Orthogonal control over viscoelasticity and stiffness with the PEG content. The stress relaxation timescale (τ1/2; Top) and the storage modulus (Bottom) as a function of the PEG content (%). NEH: Nanoporous elastic hydrogels made of glyoxal/glycol-chitosan. (e) Average pore sizes of NEH and PVHs with varying PEG content (%). Sample size, N=4. (f) Confocal images of NEH and PVHs containing rhodamine B-labeled chitosan. NEH is used as control and PVHs contain 0 or 2.8% PEG. (g) Frequency distributions of rhodamine B-labeled chitosan intensity. * represents p < 0.05, ** represents p < 0.01, *** represents p < 0.001, **** represents p < 0.0001, n.s. represents p ≥ 0.05.
Tunable stiffness and viscoelasticity
The TMF bioprinting method allows tuning of the matrix microporous hydrogel containing interconnected water-filled stiffness over a wide range through a slight change of the pH. The bioink requires no additional crosslinkers because of the self-crosslinking capacity of chitosan. Raising the pH above the pKa of chitosan, 6.5, causes the amine groups [NH2] of chitosan to deprotonate. This mechanism can elicit microphase separation due to increased hydrophobicity, as well as the formation of hydrogen bonds to stabilize the micropores in PVH.17 Indeed, our results showed that a small change of pH (∆pH<1.0) within a physiological range resulted in variations of the storage modulus encompassing three orders of magnitude (Fig. 2c). The storage moduli (0.5–27 kPa) of the resulting hydrogels spanned the range of most soft tissues.20 Consistently with the proposed pH-mediated crosslinking mechanism, no microphase separation or gelation was observed when the pH was below 6.3.
To further tune the viscoelastic response of the PVH, we exploited the cytocompatible polyethylene glycol (PEG) capable of forming hydrogen bonds with chitosan. We hypothesized the PEG could serve as crosslink spacers to intervene the self-crosslinking of chitosan for tuning the strength of the hydrogen bonds, thereby the matrix viscoelasticity. To verify this mechanism, we varied the amount of PEG in the bioink for a series of PVH scaffolds, denoted as “PEGx” (x referred to the PEG concentration of the final hydrogels). In this series, the default pH was 6.8 for a stiffness level commonly used in many tissue engineering applications unless stated otherwise. Nanoporous elastic hydrogels (NEHs) made of glyoxal crosslinked chitosan or glycol-chitosan were used as the control, which contained covalent crosslinks. We assessed the matrix viscoelasticity with a stress relaxation timescale (τ1/2), over which the stress of a matrix relaxed to one-half of its peak value at a constant shear strain (10%).8 It should be noted that the stress relaxation was measured under shear loading instead of compression to avoid any poroelastic effect. Fig. 2d shows that all the PVHs relaxed faster than the NEH (16500s) and the relaxation time (τ1/2) of PVH decreased substantially as the PEG concentration increased. Interestingly, we found that the elastic modulus of the PVH was independent of the PEG concentration, and thus decoupled from the matrix viscoelasticity. We attributed the orthogonal control of stiffness and viscoelasticity to the fact that the triggered micropore formation and the modulation of bonding strength processes are largely independent. The simple addition of PEG can enable independent tuning of the stiffness and viscoelastic response of the bioprinted scaffolds. The relaxation time did not change for PVHs with different PEG concentrations after 24 hrs of immersion inside complete cell culture medium (Fig. S5, ESI†). To secure the PEG within the network, one can covalently conjugate the PEG with the chitosan by using carbodiimide chemistry and PEG with hydroxysuccinimide ending groups (PEG-NHS).21
Interconnected cell-sized pores
Besides highly tunable mechanical properties, the PVH contains highly interconnected micropores of cell size. To characterize the structure of PVH in details, we synthesized the hydrogels with rhodamine B-conjugated chitosan and imaged them with confocal microscopes. We also used scanning electron microscopy (SEM) to image the hydrogels prepared with a CO2 supercritical dryer to minimize the processing artifact. Both the confocal and SEM images revealed highly interconnected micropores of average pore size 17.8±7.5 µm, comparable to the size of cells such as fibroblasts and stem cells (Fig. 2e,f).22 Higher pore interconnectivity was observed compared to hydrogels fabricated using existing porous bioprinting methods (Fig. S6, ESI†). The porosity of the PVH was greater than 60%. In contrast, no micropores were identified in the covalently crosslinked NEHs as they contain nanoscale pores much smaller than the resolution of the microscopes. The contrast of porosity and pore interconnectivity was manifested under compression tests (Fig. S7, ESI†), where a substantial amount of water was squeezed rapidly out of the PVH, a characteristic phenomenon mimicking that of sponges but unseen in the NEH. By further analyzing the fluorescent signals from the confocal imaging, we confirmed two intensity peaks corresponding to the two phases within the scaffold (Fig. 2g). While microphase separation was observed for all the tested pH conditions, both the porosity and the pore size increased slightly when pH was increased from 6.5 to 7.1. Interestingly, varying the PEG content did not appear to alter the porous structure of the resulting hydrogels. To our knowledge, such combination of structural and viscoelastic properties is unique among the bioprintable scaffolds. The new method significantly expands the bioprinting window of pore size and matrix viscoelasticity, which is difficult to access before.
Mechanical robustness
As high porosity concerns the mechanical strength, the mechanical behavior of the PVH under large compression was next investigated. Compression tests showed that PVHs sustained large compressive strains (85%) without rupture, whereas NEH ruptured at 61% strains (Fig. S7c, ESI†). As indicated by the large hysteresis loop, the polymer network of PVH was capable of dissipating energy effectively via the breakup of physical crosslinks. Remarkably, the resulting hydrogel’s structural integrity was not impeded by the highly porous network, even for a polymer concentration of only 1.5 wt%. This property was attributed to the microphase separation, which substantially concentrated the chitosan in the solid phase for a strong polymer network.23 The swelling properties of the PVH were quantified by measuring the weight change of the hydrogels. The swelling ratio was found to be independent of the pH and of the PEG content (Fig. S8, ESI†). Additional observations confirmed the physical stability of the PVHs and small volume change over 1 week. These results conclude that the PVH is mechanically robust and physically stable despite the high porosity and low polymer content.
Biodegradability
The PVH exhibited a slow degradation profile over an extended time period, which was consistent with high physical stability found in swelling tests. Biodegradation assays showed that the PVH degraded slowly over 7 weeks when exposed to lysozyme at a physiological concentration (Fig. S9, ESI†).24 The initial weight loss might be due to the leach out of incomplete crosslinked components in the gels. The negligible dependence of the degradation rate on the pH and the PEG content implied that the degradation may mainly occur upon the chitosan backbone. Such degradation profile is useful for tissue repair and regeneration, as a period of 2–8 weeks is needed for the embedded cells to deposit sufficient matrix to form new tissue.25 If needed, the degradation rate can be accelerated by using oxidized chitosan that undergoes hydrolysis.26
Cell compatibility and motility
Given the biocompatibility of the polymers and the interconnections of the porous structure, it is reasonable to assume that the PVH could support cell viability, migration, and growth. Cell compatibility was evaluated using immortalized human vocal fold fibroblasts (hVFFs), frequently used in previous studies of vocal folds wound healing and repair.27,28 Live/Dead assays were performed to assess the viability during a 7-day culture. The results showed that the PEG concentrations below 4% did not introduce cytotoxicity. Cell viability for all the tested conditions was greater than 90% throughout the culture period (Fig. 3a,b and Fig. S10, ESI†). Cell compatibility of the PVHs was evidenced by substantial hVFFs proliferation in all the PVHs of varying PEG contents (Fig. 3c). In contrast, the cell density decreased in the NEH control group over time.
Fig. 3.

Cytocompatibility of PVHs. (a) Fluorescent images of live/dead cells cultured within hydrogels on Day 7. Live cells are in green and dead cells in red. (b) Cell viability over time. (c) Normalized cell density over time. (d) Fluorescent images of live/dead cells cultured within the cubes (6*6*6 mm3) of NEH and PVH hydrogels on Day 5. Fluorescence-distance profiles show the live/dead signals across the hydrogel cubes. Live cell signals are in green and dead cell signals in red. (e) Live imaging of hVFFs migrating inside PVH. Two mobile cells are marked with blue and green arrows, respectively, while a stationary cell is marked with a yellow arrow for reference. (f) Cell motility within PVHs and NEH. NEH: Glyoxal/glycol-chitosan. * represents p < 0.05, ** represents p < 0.01. Sample size N=5.
To test whether the PVH supports mass transportation over distance for cell culture, we further scaled up the bioprinted scaffold to be 6-mm-width cubes and cultured hVFFs inside. We also included cell-laden NEH cubes for comparison. Thanks to the interconnected micropores, the PVH cubes contained open channels to enable rapid convection of culture media, whereas the NEH cubes were essentially nonperfusable (i.e., no open channel for convection). Accordingly, it was found that the thick PVH cube maintained much higher cell viability than the NEH control. Fluorescent intensity distributions of live (green) and dead (red) signals along the center of cubic hydrogels indicated that the cells stayed alive across the 6-mm width of the PVH cube, while the NEH cube developed a necrotic core inside (Fig. 3d). The results demonstrated that the highly porous structure of PVH enabled efficient mass exchange for cell culture with no need for additional embedded channels.
Besides the mass transport, the porous scaffolds can impose minimal steric constraints over cells, and thus promote cellular activities. Due to the low polymer content, the cells were found to mainly reside in the micro-pores. Cell motility was quantified by tracking the motion of cell nuclei within a horizontal plane. The highly porous and viscoelastic PVHs allowed cells to remodel and migrate through the matrix at a speed of 27.6±8.4 µm/hour (Fig 3e,f). Mitosis was also observed during a 48-hour live-cell imaging (Movie S1, ESI†). In contrast, hVFFs were barely moving in the NEHs, due to the lack of pores and the inability of cells to remodel the elastic matrix. The results suggested that cells can thrive in the porous viscoelastic matrix of PVHs.
Versatile printability
The printability of the TMF method to make constructs with hierarchical structures and viscoelastic gradients was next demonstrated with an extrusion bioprinter (BioAssemblyBot, Advanced Solutions, KY). The bioink system exhibits shear-thinning behavior desirable for the printing process (Fig. S11, ESI†) and can print features of various sizes using a single fixed-size nozzle. Due to the negligible yield stress of the bioinks, a low pneumatic pressure suffices bioprinting. Rheological measurements show that the bioinks have a low viscosity at low shear rate (<1 Pa·s at 1 s−1), which can substantially reduce shear stress and the associated damage to cells. The filament size was tunable through the adjustment of the pneumatic dispenser pressure P and the writing speed v (Fig. 4a); a good agreement was obtained between the experimental data and theoretical estimation (Fig. 4b and Fig. S12, ESI†). Filaments of size 120–1500 µm were successfully printed using one single nozzle (Fig. S13, ESI†); the obtained printing resolution was comparable with other embedding printing methods.29,30 3D lattice structures were also successfully fabricated, and imaged with SEM to show the hierarchical porous structure (Fig. 4c). The TMF method allows bioink composition variations and the fabrication of structures with viscoelastic gradients. A rectangular piece was printed containing three regions of different viscoelastic properties that were seamlessly bonded together. A transition zone of 82±14 µm was clearly identified from the overlapping of fluorescent signals at the boundary (Fig. 4d,e). Micropores were observed in all the gradient regions (Fig. 4g). The printed constructs can also be customized to have certain structural and viscoelastic gradients for specific applications such as wound healing and vascularization. Idealized miniature renditions of complex porous human tissues, such as vocal folds, kidneys, and intervertebral discs with viscoelastic gradient were fabricated to demonstrate the potential of the proposed bioprinting method (Fig. 4f). The results show the versatile printability of PVHs.
Fig. 4.

Printability of PVH for complex constructs of varying compositions. (a) Schematics of embedded bioprinting inside a gelatin supportive matrix. (b) Filament diameters as a function of writing speed and pneumatic pressure. Sample size, N=3. (c) Optical and SEM images of a lattice construct with hierarchical structures. (d) Printed construct of PVHs with varying PEG contents for viscoelastic gradients. White: PEG0; Greenish yellow: FITC-PEG1.2; Red: Rhodamine-PEG2.8. (e) Confocal image and fluorescence-distance profiles at the interface of PVH with viscoelastic gradients. (f) Printed scaffolds of vessel structure, vocal fold, intervertebral disc (IVD), and kidney. The IVD construct contains PVHs of two PEG contents (PEG0 and PEG2.8). (g) Fluorescent images showing printed viscoelastic gradient construct, with confocal images showing the micropores in PEG1.2 and PEG2.8.
Vocal fold engineering
To illustrate the possible application of our approach for tissue engineering, we utilized the TMF bioprinting to deposit multi-layered cell-laden scaffolds for vocal fold tissue engineering. Tissue engineered vocal folds featured a complex layered structure, with fine features (i.e., as small as 1 mm in thickness) and various cell types.31 The layer covering the laryngeal muscle, i.e. lamina propria, contains mainly vocal fold fibroblasts and itself is covered by a dense layer of epithelial cells. To recapitulate this structure, we mixed hVFFs and 0.02% collagen monomers within the bioink and fabricated a bilayer vocal fold construct, shown in Fig. 4f. According to previous studies on hVFFs, a very low amount of collagen was added to provide cell binding ligands,32 which was found not to change the overall mechanical properties of PVHs (Fig. S14, ESI†). We then seeded human bronchial epithelial cells (hBEpCs) on top of the basement membrane, as illustrated in Fig. 5a. As there is yet no readily available vocal fold epithelial cell line,31 hBEpCs were used in lieu of vocal fold epithelial cells. Fig. 5b shows that the epithelial cells adhered and formed a dense and connected epithelium layer on the surface of the construct. The porous construct was able to substantially improve the spreading of the fibroblasts inside the scaffold, relative to the nonporous NEH. The structural integrity of the hydrogel constructs was maintained after the co-culture of cells. The result proves the feasibility of the TMF bioprinting approach for making cell-laden scaffolds of predefined shapes for the co-culture of cells. It illustrates possible applicability to other systems, although further work would be required to demonstrate a bioprinted functional vocal fold.
Fig. 5.

Bioprinting for soft tissue engineering and in vitro cancer modeling. (a) Bioprinting of tissue-engineered vocal fold. (b) Human vocal fold fibroblasts (hVFFs) are bioprinted inside the scaffold (II) with human Bronchial Epithelial Cells (hBEpCs) seeded on top (I). Cyan: F-actin; Blue: DAPI. The circularity of hVFFs within the porous viscoelastic hydrogel (PVH) is compared that in nonporous elastic hydrogels (NEH). (c) Bioprinted in vitro cancer model. MDA-MB-231 cancer cells labeled with green fluorescent protein are bioprinted inside NEH and PVH of varying PEG content for different viscoelasticity. The cell density (d) and the volume of cell aggregates (e) are monitored over time. P values were determined by ANOVA test; *P ≤ 0.05; ***P ≤ 0.001; ****P ≤ 0.0001. Sample size, N=3.
In vitro cancer model
To further demonstrate the versatility of the TMF method, we also bioprinted a breast cancer cell (MDA-MB-231) within PVHs for the use of in vitro cancer models. We demonstrated the bioprinting of cell-laden PVH constructs while varying viscoelastic properties by tuning the PEG content as described above. Consistent with the aforementioned study, the TMF bioprinting was compatible with the cancer cells and the resulting PVH construct promoted the proliferation of cancer cells compared to the nonporous NEH construct (Fig. 5c–e). Interestingly, we found that the more elastic construct (i.e., PEG0) upregulated the proliferation and aggregation of cancer cells, indicative of invasiveness. This finding is consistent with those in recent reports showing increased cancer cell migration and proliferation in more elastic matrices.33,34 This study implies that mechanotransduction is mediated jointly by the structural and viscoelastic properties of the bioprinted scaffolds.
Discussion
The extracellular matrix (ECM) of biological tissues is often highly porous and viscoelastic. To repair or model the ECM, an ideal bioprinted scaffold should provide a familiar and supportive microenvironment for the native and/or transplanted cells.4,10 Bioprinting technologies are still needed to recapitulate the architecture and mechanics of many soft biological tissues. In this work, the TMF bioprinting method is developed to combine the merits of embedding bioprinting, microphase separation, and viscoelastic hydrogels. Different from the previously reported methods based on cryogelation or emulsion bioinks, the TMF method exploits a new mechanism based on stimuli-triggered microphase separation to form interconnected cell-sized pores. The PSIM not only enables embedding printing, but also triggers microphase separation and partial crosslinking of the bioink after extrusion. The post-extrusion thermal treatment (to 37°C) is to bond each filament in the sequential printing process and to stiffen the whole structure, as evidenced by the physical integrity of the bioprinted scaffolds. The TMF method can successfully mitigate the complication of nozzle clogging or structure collapse, and enable the use of low-viscosity bioinks to minimize the shear-induced damage on laden cells during extrusion.35
One key advantage of the TMF method is to allow the rapid fabrication of homogeneous hierarchical hydrogels laden with cells. The resulting scaffolds encompass pores at the macro-(>100 µm), micro- (~20 µm) and nano-scale (<100 nm) within a single structure. The macroscopic pores are created through precise position control of a robotic extrusion dispenser. The micropores comparable to the size of cells are formed through the triggered micropore formation, while the nanopores are the intrinsic meshes of the polymer network. While the micropores are largely determined by the phase separation behavior of the bioink, the macropores are intrinsically tunable in size as they are defined by the motion of the bioprinter nozzle during the printing process. Our results demonstrate that the microporous structure can promote cellular activities and facilitate mass exchange. The controllable macropores or channels can help mimic macroscopic features of certain tissues, promote vascularization in tissue scaffolds, and enhance perfusion in applications within bioreactors and microfluidic devices. When compared to the emulsion15 or sacrificial micelle-based 14 bioprinting reported recently, the bioink used in our method is thermodynamically stable and can extend the operation window and shelf life (more than 24 hours), by excluding the use of unstable liquid emulsions or potential nozzle-clogging particles. In addition, the scaffolds fabricated with our method exhibit remarkable mechanical toughness in spite of the interconnected porous structure. It is attributed to the concentration effect of microphase separation, which is unseen in other bioprinting systems.
Another advantage of the method is its unique ability to decouple viscoelasticity, stiffness, and porosity by design. It is realized through tuning of the pH and the addition of PEG. To better understand the viscoelastic response, we further analyzed the stress relaxation curves with the Maxwell-Wiechert model and found two distinct viscoelastic time constants on the orders of 10 and 104 seconds across the tested conditions (Fig. S15 and Table S2, ESI†). The small time constant, τ1, can be related to breaking of physical cross-links, while the large time constant, τ2, is linked to entanglement disassociation and viscous flows of the chitosan chains. Both time constants decreased with increasing PEG inputs, indicative of a reduction in the bonding strength of physical crosslinks. Notably, the stress-relaxation moduli of the PVHs decayed linearly as a function of the Logarithm of time (Fig. S13a, ESI†). Such responses better mimic the viscoelastic behavior of human tissues undergoing stress relaxation than those obtained with ionically-crosslinked alginate hydrogels reported previously (Fig. S13b, ESI†).8 The high-level control over the viscoelastic properties allows tailoring their mechanics to mimic biological tissues.
Compared to planar two-dimensional (2D) culture and 3D cultures using nanoporous hydrogels, the bioprinted scaffolds made with the TMF method can fully support cell growth and migration without imposing many physical constraints. The pore size allows cells laded within the PVH scaffolds to sense and respond to the 3D matrix, in contrast with scaffolds with very large pores (>100 µm) where cells are essentially cultured in 2D. Our result shows that the cell-sized porous structure of PVH is particularly beneficial to tissue engineering applications with a medium-range thickness as it eliminates the need and the complexity of implanting vascularization during scaffold fabrication. The concept was illustrated with bioprinted scaffolds laden with two types of cells for vocal fold tissue engineering. Moreover, the use of PVH allows one to vary the viscoelasticity of the matrix exposed to cells, a feature potentially useful for the mechanotransduction study. As an exemplar, we cultured breast cancer cells in PVH and NEH and found that both the porous structure and the matrix viscoelasticity mediated their activities. Recent mechanotransduction studies of the effects of viscoelasticity on cellular activity have used nanoporous hydrogels and conventional hydrogel synthesis.8 With the TMF bioprinting, one can deposit more than one cell types and spatially control the microenvironment they are exposed to, as both the structural and viscoelastic properties are tunable. The PVH could be further leveraged to mimic the pore gradients found in cartilage or bone, or to manufacture thicker functional tissue constructs that maintain viability over time. This work could open many opportunities in regenerative medicine, tissue engineering, disease modelling and drug screening.
Conclusions
To sum up, we developed a new bioprinting strategy to make tissue-mimicking scaffolds with a unique combination of structural and mechanical properties. We demonstrated its versatile printability for the fabrication of hierarchical porous scaffolds, and the ability to decouple viscoelasticity, stiffness, and porosity by design. A wide range of elastic modulus and viscoelastic responses were demonstrated. The micropores were formed by stimuli-triggered microphase separation in a biocompatible manner and stabilized by physical crosslinks of chitosan without additional cross-linkers. By combining more than one microphase separation systems, one could produce bimodal micro-pores or other pore size distributions, in addition to macroscopic pores controlled by the deposition of the printhead. The method could print scaffold materials that are auspicious in promoting cell spreading and proliferation. We thus anticipate that the same or similar ideas can be applied to other tissue engineering and biomedical applications. This work has the potential to find new applications in tissue engineering, regenerative medicine, organ transplantation, and disease modeling.
Materials and methods
Triggered micropore-forming bioprinting
Unless otherwise specified, the chemicals used in the current work were purchased from Sigma-Aldrich and used without further purification. Chitosan (DDA: 95%, medium and high molecular weight) was purchased from Xi’an Lyphar Biotech (Shanxi, China). 2.5 wt% chitosan powder was dissolved in 0.2 M acetic acid to form a chitosan solution. A phosphate solution was prepared by mixing 0.1 M sodium phosphate dibasic (Na2HPO4, Sigma S7907) and 0.1 M sodium phosphate monobasic (NaH2PO4, Sigma S8282) with a volume ratio of 50:3. Bioink PEG0 was obtained by adding 2 units of phosphate solution into 3 units of chitosan solution on a vortex mixer drop by drop. As a result, the final concentration for chitosan was 1.5% and 0.12 M for acetic acid. To make PEG1.2, PEG2.8, and PEG4 bioinks, 3, 7, and 10 wt% of polyethylene glycol (PEG, Mn=4,000, Sigma 81240) was added to the phosphate solution before mixing with chitosan.
The phase-separation inducing matrix (PSIM) was formed with gelatin slurries following a modified protocol.30 Briefly, 4.5 wt% type A gelatin from porcine skin (G2500) was dissolved in an washed with cold SC solution at least 3 times. The supportive sodium bicarbonate (SC) solution (Fisher, S233–500) containing 0.04M sodium phosphate monobasic and 0.094 M SC. The pH was adjusted with 1 M HCl to desirable values. The mixture was heated at 60°C under vigorous magnetic stirring until gelatin was fully dissolved. 250 ml of SC-laden gelatin was transferred to a 500 ml mason jar and kept at 4°C overnight. Cold SC solution was used to fill up the mason jar and capped with a blender blade (Oster, 4127–33A). The mixture was blended for 120 seconds to produce gelatin particles. The particles were matrix was obtained by centrifuging the gelatin slurry at 225g. Kimwipes was used to remove the excessive SC solution in the matrix before bioprinting.
Two pneumatic-controlled bioprinters were used to fabricate 3D scaffolds. For printing tasks with BioScaffolder 3.1 (GeSiM, Germany), G-code was generated by scripting in Matlab and Slic3r (https://slic3r.org/). For printing tasks with BioAssemblyBot (Advanced Solutions, KY), TSIM software was used for the geometry modeling and fabrication. 30-gauge cylindrical printing nozzles (150 µm inner diameter, 12.7 mm nozzle length, Nordson EFD, RI) were used for all the printing tasks. Bioprinting was conducted inside 6- and 12-well culture plates (Eppendorf, Germany) filled with a supportive SC-laden gelatin matrix.
To prepare glycol-chitosan NEHs, glycol-chitosan (G7753) was first dissolved in PBS 1X to form a 5% solution. The crosslinker solution was prepared by diluting 40% glyoxal solution (128465) in PBS 1X to form a 0.01% glyoxal solution.27,28 Covalently crosslinked NEHs was obtained by mixing a glycol-chitosan solution, a diluted glyoxal solution, and a cell suspension in culture medium with a volume ratio of 4:5:1. The final polymer concentration was 2%. To prepare chitosan NEHs, a 2.5% chitosan solution was mixed with a 0.02% glyoxal solution with a 3:2 volume ratio. The mixture was incubated at 37°C for over 40 mins to form covalently crosslinked hydrogels.
Mechanical characterization
All mechanical characterizations were conducted using a torsional rheometer (HDR-2, TA Instruments, DE) with parallel plates (upper plate diameter: 20 mm). The yield stress of the materials was determined by applying an amplitude sweep from 0.001% to 1000% of shear strain at 1 Hz. The storage and loss moduli vs. shear stress were plotted to determine the yield point. The shear moduli of PVHs were obtained from isothermal time sweeps at a frequency of 1 Hz and 0.1% strain at 37°C for 1 hour. Relaxation moduli were obtained by holding a step shear strain of 10% and measuring the shear stress-time history; the mechanical response reflected only the viscoelastic property of the tested scaffolds, while the poroelastic response (i.e., water migration) was avoided. Compression tests were conducted by applying an 85% compressive strain with a rate of 10 µm/s on cylindrical hydrogels prepared using a silicone mold. Loading and unloading axial forces were measured as a function of displacement.
Rhodamine B/FITC-labeled chitosan
Rhodamine-B isothiocyanate (Cayman Chemical, 20653) or Fluorescein-5 isothiocyanate (Thermo Fisher, F1907) was conjugated to chitosan polymeric chains following established protocols.36,37 Briefly, 1 wt% chitosan was first dissolved in 80 mM acetic acid and sterilized through 0.22 µm PES filters (Thermo Scientific, 13100106). Anhydrous methanol (Fisher Scientific, A412–1) was added to the filtered chitosan solution with a volume ratio of 1:1. The mixtures were stirred for 3 hours at room temperature and degassed before use. Rhodamine B and FITC were dissolved in methanol at 2 mg/ml and 1 mg/ml, respectively. The staining solution was added to the chitosan/methanol mixture drop-by-drop under stirring. The final concentration of fluorescent dyes in the reaction medium was controlled to give the label to D-glucosamine residue at a ratio of 1:50. The reaction lasted for 18 hours for rhodamine B-labeled chitosan and 1 hour for FITC-labeled chitosan under dark at room temperature. Then, 1 N NaOH solution (S2770) was used to precipitate chitosan from the solution. The precipitates were collected and dialyzed with DI water until no fluorescent signal was detectable in the water.
Cell culture in 2D
Immortalized hVFFs were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Corning, NY) containing sodium pyruvate and supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 1% MEM non-essential amino acids. hBEpC were cultured in Eagle’s Minimum Essential Medium (EMEM) (Corning, NY) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, 1% MEM non-essential amino acid, and 1% L-glutamine. Cells were incubated at 37°C, in 5% CO2 humidified atmosphere. The media were changed every three days for 2D cultures. Cells were disassociated using 0.25% trypsin-EDTA when the cell confluency reached 70%.
Vocal fold bioprinting and co-culture
0.1 ml collagen monomer (RatCol 4 mg/mL, Advanced BioMatrix) were first added to 1 mL 2.5% chitosan in acetic acid. Then 2 units of buffer solution containing 0.1 M PB and 0.114 M SC were added into 3 units of chitosan solution on a vortex mixer drop by drop to raise the pH of Bioink0 to 6.2. Next, hVFFs were homogeneously suspended in Bioink0 at a final concentration of 0.5 million/mL and homogenized with a gentle vortex. The hVFF-laden bioink was printing inside PSIM (pH=6.8) to form scaffolds with predefined shapes (vocal fold M5 model, size: ~15x10x2 mm). A mixture of 50% complete DMEM and 50% EMEM (both with 20% FBS) was used as the co-culture medium. On the next day, a drop of collagen monomer was dropped on top of the printed scaffolds to give a thin layer of collagen coating. 0.75 million/cm2 hBEpCs were seeded on top of each scaffold one day after the coating. The co-culture medium was changed every other day. Samples were co-cultured for 9 days before immunostaining.
MDA-MB-231 in vitro cancer model
GFP-labeled MDA-MB-231 cancer cells were suspended inside bioinks containing different concentrations of PEG with the same method described previously. The cell density of the bioinks was 1.75 million/mL. All the bioinks were printed inside PSIM (pH=6.8) to form hydrogel discs (10 mm in diameter and 1 mm in thickness). Completed DMEM with 10% FBS was used for 3D culture. The medium was changed every 3 days. GFP signals showing the cell nuclei were used to visualize cancer aggregates at 1, 11, 21, and 31 days.
Staining and imaging
To evaluate the cytocompatibility of PVHs, hVFFs were suspended in chitosan bioinks with different PEG contents at a cellular concentration of 4 million/mL. Disk-shaped PVH scaffolds with 10 mm in diameter and 1 mm in thickness were fabricated. Complete DMEM with 10% FBS was used as cell culture medium and changed every day. HVFFs were stained by a Live/Dead viability kit (Invitrogen, L3224) inside 3D chitosan matrices on Day 0, 3, 7. Imaging of fixed hVFFs was conducted using a confocal laser scanning microscope (LSM710, Zeiss, Germany). Live cells were shown in green fluorescence and dead cells were shown in red. For assessing viability in thick hydrogel scaffolds, hVFFs were suspended in bioinks with a concentration of 5 million/mL and printed into 6×6×6 mm3 hydrogel cubes. NEH hydrogel cubes were fabricated with mold casting. The hydrogel cubes were cultured for 5 days and sectioned using a micro-dissection blade before being transferred into a 35-mm petri dish with glass bottom (Matsunami Glass, D35–14-1.5-U) for viability staining and imaging (Axiovert A1, Zeiss).
For immunostaining, the hydrogels were first washed with pre-warmed PBS twice and then fixed in 3.7% formaldehyde solution for 15 mins followed. The fixed samples were washed with PBS again twice and permeabilized with 0.1% Triton X-100 in PBS for 5 minutes. The samples were blocked in 1% bovine serum albumin (BSA, A1595) for 1 hour. To prepare the staining solution, 10 µL of Alexa Fluor 633 Phalloidin (Invitrogen, A22284) was diluted into 200 µL PBS containing 1% BSA. Next, the samples were incubated inside the staining solution at room temperature for 30 mins followed by three times PBS wash. The nuclei were counterstained with Hoechst 33342 (Invitrogen, H3570) using a 1:2000 dilution for 10 min, followed by rinsing twice with PBS.
For cell motility study, live cells were dyed with orange DNA-selective stains (Invitrogen, V35005) and embedded inside chitosan hydrogels immediately after staining. The cell-laden hydrogels were transferred into an 8-well culture plate without cell adhesion coating (Ibidi, 80821) and monitored using a fluorescent imaging platform under RFP for 48 hours (JuLi Stage, NanoEnTek Inc., South Korea).
The polymer network was imaged with a confocal microscope (LSM 710, Zeiss). Samples were prepared by mixing fluorescent-labeled bioinks and cross-linkers in a vial and transfer 150 µL into 35 mm Petri dish with coverslip bottom (MatTek, P35G-0–10-C). Hydrogels were immersed under PBS and imaged as prepared. Polymer network was imaged with 10x, 20x, and 63x (oil) objective lenses. For imaging of bioprinted constructs, Axiovert A1 inverted microscope (Zeiss) equipped with a motorized stage was used to obtain fluorescent signals at multiple locations.
Macro- and microscopic pores were also imaged using a field emission scanning electron microscope (F50, FEI) under various magnifications. Before SEM imaging, all the samples were dehydrated using a CO2 supercritical point dryer (CPD030, Leica) to preserve the original pore size. The dehydrated samples were coated 4 nm Pt using a high-resolution sputter coater (ACE600, Leica) to increase surface conductivity.
All photographic images were taken under a regular light source with DSLR cameras (Mark III or D70, Canon) with a variety of lenses. All the printed constructs were taken out of the liquid and photographed in ambient conditions.
Imaging analysis
3D reconstruction of polymeric network was performed using Imaris (Bitplane, CT). Pore size was analyzed by measuring 150 pores for each type of PVHs using the measuring tool in ImageJ. Porosity was calculated by first transforming the confocal images into binary images and dividing the number of white pixels by the number of black pixels. Fluorescent intensity was calculated by first transforming the confocal images into grayscale images and then using Matlab to calculate the grayscale value distribution. Cell number was calculated by using Analyze Particles toolbox in ImageJ. The viability rate was calculated by dividing the number of live cells by the total number of cells. Fluorescent images of bioprinted constructs were created by stitching fluorescent images taken at multiple locations using Imaris Stitcher (Bitplane, CT).
Swelling and biodegradation assays
The swelling ratios were determined by immersing the PVHs in PBS (pH=7.4) at 37°C with gentle mechanical stimulation (75 RPM). The excess PBS on the hydrogel surface was removed at pre-determined time intervals using a pipette. The swelling ratio was calculated by dividing the measured wet weight by the initial wet weight.
For biodegradation assays, all hydrogels were lyophilized and measured the initial polymer dry weight. After that, an enzyme solution consisted of 13 µg/ml lysozyme (MP Biomedicals, 100831) in PBS was added to the lyophilized gels. The samples were incubated at 37°C with gentle mechanical stimulation over a period of 49 days. The enzyme solution was changed every other day. At pre-determined time intervals, the enzyme solution was removed. The samples were then washed three times for 5 minutes with PBS. The samples were then lyophilized and measured the remaining polymer dry weight. The remaining ratio of the polymer was calculated by dividing the dry weight of the remaining polymer by the dry weight of the initial gels.
Statistical analysis
A sample size of N≥3 was used for all experiments. Data are shown as Mean ± SD. Statistical analysis was performed using one-way ANOVA and post hoc Tukey tests for multiple comparisons or Student’s t-tests for comparison between two groups (Prism 8). P values ˂0.05 were considered statistically
Supplementary Material
We report a new method and bioink system to bioprint 3D hierarchical cell-laden scaffolds with an unprecedented combination of structural and mechanical properties, which are shown to be a friendly and supportive environment for various cells. While 3D bioprinting has been under extensive studies for many years, few methods can create direct cell-laden scaffolds mimicking the porous and viscoelastic properties of biological tissues. Different from the previously reported approaches relying on the sacrificial materials embedded within the bionks, we propose a new method designated “triggered micropore-forming” (TMF) bioprinting, exploiting a new mechanism based on stimuli-triggered microphase separation to form interconnected cell-sized pores. The substantially concentrated solid phase during microphase separation empowers the bioprinted hydrogels with superior mechanical stability despite high porosity. The decoupled porous, stiffness, and viscoelastic properties enable the orthogonal control of hydrogels for the first time with a wide tunable range. TMF bioprinting is capable to fabricate complex architectures with defined geometries and viscoelastic gradients. The method has been demonstrated to print scaffold materials that are auspicious in promoting cell spreading and proliferation. We thus anticipate that the same or similar ideas can be applied to other tissue engineering and biomedical applications.
Acknowledgements
The authors thank Dr. Susan Thibeault (University of Wisconsin-Madison) for providing the hVFFs and Dr. John Hanrahan (McGill University) for providing the HBEpCs. The authors also thank Qiman Gao and Ling Chen for their help with photography. G.B. thanks the Advanced BioImaging Facility (ABIF) and the Facility for Electron Microscopy Research (FEMR) for providing access to their imaging facilities. G.B., H.R., A.R., M.S., H.W., and L.M. were supported by the National Institutes for Deafness and other Communication Disorders of the National Institutes of Health under awards number R01-DC005788 and R01-DC014461. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Z.M. and J.L. acknowledge funding from Natural Sciences and Engineering Research Council of Canada (grant RGPIN-2018-04146 and DGECR-2018-00294) and the Canada Foundation for Innovation (grant 37719). T.J. thanks the China Scholarship Council (201403170354) and McGill Engineering Doctoral Award (90025) for their support.
Footnotes
Publisher's Disclaimer: This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication.
Publisher's Disclaimer: Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. We will replace this Accepted Manuscript with the edited and formatted Advance Article as soon as it is available.
Electronic Supplementary Information (ESI) available. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
References
- 1.Kang H, Lee SJ, Ko IK, Kengla C, Yoo JJ and Atala A, Nat. Biotechnol, 2016, 34, 312–319. [DOI] [PubMed] [Google Scholar]
- 2.Liu X, Yuk H, Lin S, Parada GA, Tang TC, Tham E, de la Fuente-Nunez C, Lu TK and Zhao X, Adv. Mater, 2018, 30, 1704821. [DOI] [PubMed] [Google Scholar]
- 3.Jiang T, Munguia-lopez JG, Flores-torres S, Kort-mascort J and Kinsella JM, Appl. Phys. Rev, 2019, 6, 011310. [Google Scholar]
- 4.Loh QL and Choong C, Tissue Eng. Part B Rev, 2013, 19, 485–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiu YC, Cheng MH, Engel H, Kao SW, Larson JC, Gupta S and Brey EM, Biomaterials, 2011, 32, 6045–6051. [DOI] [PubMed] [Google Scholar]
- 6.Oh SH, Park IK, Kim JM and Lee JH, Biomaterials, 2007, 28, 1664–1671. [DOI] [PubMed] [Google Scholar]
- 7.Wisdom KM, Adebowale K, Chang J, Lee JY, Nam S, Desai R, Rossen NS, Rafat M, West RB, Hodgson L and Chaudhuri O, Nat. Commun, 2018, 9, 4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaudhuri O, Gu L, Klumpers D, Darnell M, Bencherif SA, Weaver JC, Huebsch N, Lee HP, Lippens E, Duda GN and Mooney DJ, Nat. Mater, 2016, 15, 326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee H, Gu L, Mooney DJ, Levenston ME and Chaudhuri O, Nat. Mater, 2017, 16, 1243–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malda J, Visser J, Melchels FP, Jüngst T, Hennink WE, Dhert WJA, Groll J and Hutmacher DW, Adv. Mater, 2013, 25, 5011–5028. [DOI] [PubMed] [Google Scholar]
- 11.Cui H, Nowicki M, Fisher JP and Zhang LG, Adv. Healthc. Mater, 2017, 6, 1601118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Béduer A, Piacentini N, Aeberli L, Da Silva A, Verheyen CA, Bonini F, Rochat A, Filippova A, Serex L, Renaud P and Braschler T, Acta Biomater, 2018, 76, 71–79. [DOI] [PubMed] [Google Scholar]
- 13.Sobral JM, Caridade SG, Sousa RA, Mano JF and Reis RL, Acta Biomater, 2011, 7, 1009–1018. [DOI] [PubMed] [Google Scholar]
- 14.Armstrong JPK, Burke M, Carter BM, Davis SA and Perriman AW, Adv. Healthc. Mater, 2016, 5, 1724–1730. [DOI] [PubMed] [Google Scholar]
- 15.Ying G, Jiang N, Maharjan S, Yin Y, Chai R, Cao X, Yang J, Miri AK, Hassan S and Zhang YS, Adv. Mater, 2018, 30, 1805460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiu YL, Chen SC, Su CJ, Hsiao CW, Chen YM, Chen HL and Sung HW, Biomaterials, 2009, 30, 4877–4888. [DOI] [PubMed] [Google Scholar]
- 17.Yang J, Bai R and Suo Z, Adv. Mater, 2018, 30, 1800671. [DOI] [PubMed] [Google Scholar]
- 18.Monette A, Ceccaldi C, Assaad E, Lerouge S and Lapointe R, Biomaterials, 2016, 75, 237–249. [DOI] [PubMed] [Google Scholar]
- 19.Chenite A, Chaput C, Wang D, Combes C, Buschmann MD, Hoemann CD, Leroux JC, Atkinson BL, Binette F and Selmani A, Biomaterials, 2000, 21, 2155–2161. [DOI] [PubMed] [Google Scholar]
- 20.Vining KH and Mooney DJ, Nat. Rev. Mol. Cell Biol, 2017, 18, 728–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nam S, Stowers R, Lou J, Xia Y and Chaudhuri O, Biomaterials, 2019, 200, 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ge J, Guo L, Wang S, Zhang Y, Cai T, Zhao RCH and Wu Y, Stem Cell Rev. Reports, 2014, 10, 295–303. [DOI] [PubMed] [Google Scholar]
- 23.Bencherif SA, Sands RW, Bhatta D, Arany P, Verbeke CS, Edwards DA and Mooney DJ, Proc. Natl. Acad. Sci, 2012, 109, 19590–19595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nordtveit RJ, Vårum KM and Smidsrød O, Carbohydr. Polym, 1996, 29, 163–167. [Google Scholar]
- 25.Hubbell JA and West JL, Macromolecules, 1999, 32, 241–244. [Google Scholar]
- 26.Vold IMN and Christensen BE, Carbohydr. Res, 2005, 340, 679–684. [DOI] [PubMed] [Google Scholar]
- 27.Ravanbakhsh H, Bao G and Mongeau L, Sci. Rep, 2020, 10, 2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ravanbakhsh H, Bao G, Latifi N and Mongeau LG, Mater. Sci. Eng. C, 2019, 103, 109861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeon O, Bin Lee Y, Jeong H, Lee SJ, Wells D and Alsberg E, Mater. Horizons, 2019, 6, 1625–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hinton TJ, Jallerat Q, Palchesko RN, Park JH, Grodzicki MS, Shue HJ, Ramadan MH, Hudson AR and Feinberg AW, Sci. Adv, 2015, 1, e1500758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lungova V, Chen X, Wang Z, Kendziorski C and Thibeault SL, Nat. Commun, 2019, 10, 4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Latifi N, Asgari M, Vali H and Mongeau L, Sci. Rep, 2018, 8, 1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lang NR, Skodzek K, Hurst S, Mainka A, Steinwachs J, Schneider J, Aifantis KE and Fabry B, Acta Biomater, 2015, 13, 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pathak A and Kumar S, Proc. Natl. Acad. Sci. U. S. A, 2012, 109, 10334–10339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ouyang L, Yao R, Zhao Y and Sun W, Biofabrication, 2016, 8, 035020. [DOI] [PubMed] [Google Scholar]
- 36.Ma O, Lavertu M, Sun J, Nguyen S, Buschmann MD, Winnik FM and Hoemann CD, Carbohydr. Polym, 2008, 72, 616–624. [Google Scholar]
- 37.Qaqish RB and Amiji MM, Carbohydr. Polym, 1999, 38, 99–107. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.