

Abstract
Complement factor H (CFH) is the major inhibitor of the alternative pathway of the complement system and is structurally related to beta2-glycoprotein I, which itself is known to bind to ligands including coagulation factor XI (FXI). We observed reduced complement activation when FXI activation was inhibited in a baboon model of lethal systemic inflammation, suggesting crosstalk between FXI and the complement cascade. It is unknown whether FXI or its activated form FXIa directly interacts with the complement system. We explored whether FXI could interact with and inhibit the activity of CFH. We found that FXIa neutralized CFH by cleavage of the Arg341/Arg342 bonds. FXIa reduced the capacity of CFH to enhance the cleavage of C3b by factor I (FI), and the decay of C3bBb. The binding of CFH to human endothelial cells (ECs) was also reduced after incubating CFH with FXIa. The addition of either short- or long-chain polyphosphate enhanced the capacity of FXIa to cleave CFH. FXIa also cleaved CFH that was present on ECs and in the secretome from blood platelets. The generation of FXIa in plasma induced the cleavage of CFH. Moreover, FXIa reduced the cleavage of C3b by FI in serum. Conversely, we observed that CFH inhibited FXI activation by either thrombin or FXIIa. Our study provides a novel molecular link between the contact pathway of coagulation and the complement system. These results suggest that FXIa generation enhances the activity of the complement system, and thus, may potentiate the immune response.
INTRODUCTION
The complement system is the major humoral component of the innate immune system. Activation of complement leads to multiple protective mechanisms that help eliminate intruding bacterial and viral pathogens (1, 2). To protect host cells and tissues from attack by complement, mammals have evolved regulators of the complement cascade including complement factor H (CFH) (3). As the major inhibitor of the alternative pathway of the complement system, CFH acts via two inhibitory mechanisms in solution and on cell surfaces. First, CFH enhances the dissociation of convertase components from C3 convertases (C3bBb) as part of a process termed decay-accelerating activity. Second, CFH provides a cofactor function by binding to C3b, providing a platform for factor I (FI) to bind to the CFH-C3b complex and cleave C3b into fragments that can no longer form the C5 convertase (C3bBbC3b) and the C3 convertase (4).
CFH circulates in plasma at a concentration of 500 μg/ml and has a molecular weight of 150 kDa (5). The liver is the major source of CFH, yet it can be secreted by endothelial cells (6), platelets (7), monocytes (8) and fibroblasts (9). The critical role of CFH in normal physiology is exemplified by the fact that mutations in the CFH gene and autoantibodies against CFH are associated with atypical hemolytic uremic syndrome (10), age-related macular degeneration (11), dense deposit disease and antiphospholipid syndrome (12). CFH is composed of 20 homologous complement control protein (CCP) domains also known as short consensus repeats or sushi domains. The four N-terminal CCP domains bind to C3b and are necessary for cofactor activity (4), whereas the CCP 6-7 domains and the C-terminal CCP 18-20 domains are responsible for binding to glycosaminoglycans/heparan sulfate which facilitates regulation of complement activity on cell surfaces (13).
Interestingly, CFH is structurally related to beta2-glycoprotein I (β2GPI) (14). Although the physiological function of β2GPI in normal individuals remains to be elucidated, β2GPI is the primary target antigen recognized by autoantibodies in patients with the antiphospholipid antibody-syndrome (15). β2GPI binds to several ligands including the coagulation protein factor XI (FXI) and its activated form, FXIa (16). FXI is a member of the plasma contact pathway of coagulation, which is initiated when coagulation factor XII (FXII) is activated on negatively charge molecules (17). Activated FXII (FXIIa) activates the proinflammmatory kallikrein-kinin system and the intrinsic pathway of thrombin and fibrin generation through the activation of FXI (18). It has been suggested that the contact pathway can act as a node connecting coagulation, inflammation and innate immunity, and thus may contribute to the host response to bacterial infections. Our group has shown that inhibition of reciprocal contact activation of FXI and FXII with a monoclonal antibody directed against the A2 domain of FXI, 14E11, decreased inflammatory markers and improved outcomes in murine models of abdominal sepsis and listeriosis (19, 20). Similar results were observed using FXI-deficient mice (21). Moreover, in a lethal nonhuman primate model of systemic inflammation induced by infusion of Staphylococcus (S.) aureus, we observed that pretreatment of animals with a recombinant analog of the anti-FXI antibody, 14E11, improved survival, reducing markers of coagulation, cytokine generation and, complement activation (22). This observation was consistent with early observations that inhibiting FXIIa activity reduced complement activation during systemic inflammation, in vivo (23, 24). Regulation of the complement system by other members of the kallikrein-kinin system (KKS), including FXIIa or kallikrein, has been observed in vitro (25, 26), However, it is not known if FXIa directly interacts with components of the complement system. Based on the fact that CFH is structurally related to β2GPI, we hypothesized that CFH may be a ligand or substrate for FXI(a). Here we demontrate that FXI(a) binds to CFH and that FXIa reduces the inhibitory activity of CFH by cleaving CFH at the Arg341/Arg342 bonds located at the end of the complement control protein (CCP) 6 domain of CFH.
METHODS
Reagents.
Serum-derived CFH, FI, C3b, FB, FD, properdin, polyclonal goat anti-human FB antibody, sheep red blood cells coated with rabbit anti-sheep erythrocyte antiserum, FI-depleted serum, CFH-depleted serum and GVB++ buffer were from Complement Technology (Tyler, TX, USA). CFH-depleted plasma was from assaypro (St Charles, MO, USA). Anti-CFH N-terminal domain antibody was from Santa Cruz Biotechnology (Dallas, TX, USA). Anti-CFH C-terminal domain antibody was from R&D System (Minneapolis, MN, USA). Plasma-derived β2GPI was a kind gift from Rolf Urbanus (Utrecht University, Netherlands). Anti-C3b antibody and normal mouse IgG were from Santa Cruz Biotechnology (Dallas, TX, USA). Soluble short-chain polyP (~70-100 phosphate units in length) and long-chain polyP (>1000 phosphate unit in length) were prepared by size-fractionation of sodium polyphosphate by preparative polyacrylamide gel electrophoresis as previously described (27) and were a kind gift from Jim Morrissey (University of Michigan, USA). PolyP concentrations are reported in terms of phosphate monomer concentration (monomer formula NaPO3). The anti-FXI antibodies, 1A6, 14E11, or 10C9, were generated as previously described (28). rFXIa was generated as previously described (29). Plasma-derived FXI, FXIa, FXa, thrombin and FXI-depleted plasma were from Haematologic Technologies, Inc. (Essex Junction, VT, USA). Plasma-derived FXIIa, prekallikrein (PK), kallikrein, high molecular weight kininogen (HK) and corn trypsin inhibitor (CTI) were from Enzyme Research Laboratories, Inc. (South Bend, IN, USA). Soybean trypsin inhibitor, polybrene, O-phenylenediamine (OPD)substrate, sulfo NHS-LC-biotin, streptavidin and aprotinin were from Sigma-Aldrich (St Louis, MO, USA). Dextran sulfate (average Mr, 500 000) was from Fisher (Pittsburgh, PA, USA). FXIIa/kallikrein chromogenic substrate, Chromogenix S-2302, and FXIa chromogenic substrate, Chromogenix S-2366, were from Diapharma Group, Inc. (West Chester, OH, USA). Activated partial thromboplastin time (aPTT) reagent was from Fisher Diagnostics (Middletown, VA, USA).
Coomassie blue staining, Western blot and N-terminal sequencing.
Complement factor H (400 nM) was incubated with FXIa (10 nM) at 37°C over a time interval of 0-2 hrs in HBS. Samples were separated by SDS-PAGE under reducing conditions and analyzed by Coomassie blue staining, or they were transferred to PVDF membrane and immunoblotted with an anti-CFH C-terminal domain antibody or an anti-CFH N-terminal domain antibody followed by an HRP-conjugated secondary antibody. Proteins were detected using ECL (GE Healthcare, Piscataway, NJ, USA). For aminoterminal sequencing, bands were excised from the PVDF membrane and analyzed by automated Edman degradation using a Procise 494HT amino acid sequencer (Applied Biosystems, Carlsbad, CA, USA). In selected experiments CFH was incubated with α-thrombin, FXa, FXIIa, or kallikrein for indicated times prior to analysis.
Cell surface Western blotting.
Human umbilical vein endothelial cells (HUVECs, ATCC, Manassas, VA, USA) were grown to confluence in a 24-well plate using an endothelial cell basal medium-2 enriched with supplements (Lonza, Walkersville, MD, USA). HUVECs were incubated with CFH or vehicle for 1 hr in serum-free medium (SFM, Thermo Fisher Scientific) with 0.3% BSA. HUVECs were then washed with serum-free medium and incubated with FXIa (0-30 nM) or vehicle for 30 min. After washing, HUVECs were lysed and subjected to SDS-PAGE in a reducing sample buffer, transferred to PVDF membranes and immunoblotted with an anti-CFH C-terminal domain antibody and an HRP-conjugated secondary antibody. Proteins were detected using ECL (GE Healthcare, Piscataway, NJ, USA).
Blood collection and preparation of plasma.
Human venous blood was drawn by venipuncture from healthy male and female adult volunteers into sodium citrate (0.32% w/v sodium citrate unless otherwise noted) in accordance with the OHSU Institutional Review Board (IRB #1673). Informed consent was received from all human blood donors. Platelet-poor plasma (PPP) was prepared by centrifugation of citrated whole blood from three separate donors at 2150×g for 10 min. Further centrifugation of the plasma fractions at 2150×g for 10 min yielded PPP, which was pooled and stored at −80°C until use. In selected experiments plasma (50 ul) was incubated with an aPTT reagent (50 ul) for 1 hour at 37°C. Plasma CFH was analyzed by Western blot.
Preparation of supernatant from activated platelets.
Human venous blood was drawn in accordance with an IRB-approved protocol from healthy donors and platelets purified as previously described (30). 2×108 platelets/ml were stimulated with 1nM thrombin for 15 min at 37°C. Subsequently, 10 μM hirudin was added to neutralize thrombin. Platelets were then removed by centrifugation and the supernatant was used to measure CFH by Western blot.
CFH cofactor activity.
C3b (300 nM), FI (20 nM) and CFH (50 nM) were incubated in solution in HBS buffer at 37°C for selected times (10, 20, 40 and 80 min). Samples were separated by SDS-PAGE under reducing conditions and the generation of iC3b was analyzed by Coomassie blue staining. In selected experiments CFH was incubated with FXIa for 2 hrs prior to aprotinin being added to stop the reaction. Aprotinin alone was added as a negative control.
To measure CFH cofactor activity in serum, C3b (300 nM) and FI (5 nM) were incubated in FI- depleted serum (1/5 dilution) at 37°C for selected times (10, 20, 40 and 80 min). Samples were separated by SDS-PAGE under reducing conditions and the generation of iC3b was analyzed by Western blotting using an anti-human C3b antibody and an HRP-conjugated secondary antibody. Proteins were detected using ECL. In selected experiments FI-depleted serum was incubated with FXIa 30 nM for 3 hrs prior to aprotinin being added to stop the reaction. Aprotinin alone was added as a negative control.
Decay acceleration assay.
100μl of C3b (5μg/ml) was added to 96-well plates and incubated overnight at 4°C, followed by blocking with 1% BSA, 0.1% Tween 20 in PBS for 1 h at 37°C, and washed with wash buffer (PBS, 0.1% Tween 20, and 25 mM NaCl) supplemented with 10 mM MgCl2. Samples were then incubated at 37°C for 2 h with 50 ng/ml FB, 25 ng/ml FD, and 1000 ng/ml properdin diluted in assay buffer (PBS, 4% BSA, 0.1% Tween 20, and 75 mM NaCl) containing 10 mM MgCl2. The wells were then rinsed and incubated with either buffer or increasing concentrations of CFH (0.25-2 ug/ml) for 30 min in dissociation buffer (PBS, 4% BSA, 0.1% Tween 20, and 25 mM NaCl). After washing, the C3bBbP complexes were detected by ELISA using a polyclonal goat anti-human FB antibody followed by HRP-conjugated anti-goat antibody, both diluted in antibody buffer (PBS, 4% BSA, 0.1% Tween 20, and 25 mM NaCl) supplemented with 10 mM MgCl2. Color was developed using an OPD substrate and absorbance was measured at 450 nm. In selected experiments CFH was incubated with FXIa for 2 hrs prior to aprotinin being added to stop the reaction. Aprotinin alone was added as a negative control.
Erythrocyte lysis assay.
CFH was incubated with FXIa for 2 hrs in GVB++ buffer at 37°C. Aprotinin (50 μM) was added to all samples to stop the reaction. Aprotinin alone was added as a negative control. CFH-depleted human serum (1/100 dilution) was added at increasing concentrations of CFH (100 μl) following by the addition of 5×108 sheep red blood cells coated with rabbit anti-sheep erythrocyte antiserum (100 μl). After 1 hr of incubation at 37°C, samples were centrifuged at 500×g for 5 minutes, supernatants were collected and the erythrocyte lysis was measured at 541 nm.
Measurement of CFH binding to cell surface.
CFH (200 nM) was incubated with FXIa for 2 hrs in serum-free medium with BSA (0.3%) followed by the addition of aprotinin (50μM) to stop the reaction. Aprotinin alone was added as a negative control. Samples were then incubated with confluent HUVECs in a 96-well for 1 hr at 37°C. HUVECs were then washed with HBS, fixed with paraformaldehyde (2%), blocked in PBS with Tween (0.05%) and BSA (3%), and probed with an anti-CFH C-terminal domain antibody for 1 hr. This was followed by incubation with an HRP-coupled secondary anti-rabbit antibody (1:2000) or anti-goat antibody (1:2000), and then measured with an OPD substrate at an absorbance of 450 nm.
Measurement of FXI activation.
FXI (30 nM) was incubated with 5 nM α-thrombin or 5 nM α-FXIIa in absence or presence of dextran sulfate (2 ug/ml) at 37°C in 25 mM Hepes, pH 7.4, 150 mM NaCl, and 0.1% BSA. Samples were quenched by addition of polybrene (6 μg/ml) to neutralize dextran sulfate and hirudin (10 U/ml) to inactivate thrombin or CTI (50 μg/ml) to inactivate FXIIa, after which the generation of FXIa was quantified by measuring rates of S-2366 hydrolysis at 405nm and converted to FXIa concentrations using a standard curve.
Measurement of FXII activation.
FXII (200nM) was incubated at 37°C with PK (50nM) and HK (50nM) in the presence or absence of long-chain polyP (10 μM), dextran sulfate (2 μg/ml) or aPTT reagent in 25 mM Hepes, pH 7.4, 150 mM NaCl, and 0.1% BSA. After 30 min, samples were quenched by addition of polybrene (6 μg/ml) to neutralize polyP, dextran sulfate and aPTT reagent and by the addition of soybean trypsin inhibitor (50 μg/ml) to inactivate kallikrein, after which the generation of FXIIa was quantified by measuring rates of Chromogenix S-2302 hydrolysis at 405 nm and converted to FXIIa concentrations using a standard curve.
Characterization of FXI binding to CFH.
100μl of streptavidin (5μg/ml) was added to 96-well plates and incubated overnight at 4°C, followed by blocking with 2% BSA for 2 hrs prior to addition of biotinylated CFH or β2GPI (100 nM). The plate was washed and FXI in 100 μl 25mM Hepes, pH 7.4, 150 mM NaCl (HBS) and 0.5% BSA buffer or FH−/− depleted plasma was added. After 1 hr, FXI binding was detected with an anti-FXI antibody (2μg/ml) followed by an HRP-coupled secondary anti-mouse antibody; FXI binding was quantified with an OPD substrate at 450 nm.
RESULTS
CFH is a ligand for FXIa
FXIa is known to bind and cleave β2GPI at the end of the C-terminal domain (Lys317/Thr318), abolishing its binding to anionic phospholipids (31); we tested the hypothesis that FXIa can interact and cleave CFH in a similar manner. Based on SDS-PAGE analysis, we observed that incubation of CFH with plasma-derived FXIa for 2 hrs led to the disappearance of the ~170 kDa CFH band and the appearance of lower molecular weight bands at ~130 kDa and 35~ kDa under reducing conditions (Figure 1A and B). The presence of aprotinin or the anti-FXI antibody 10C9, which binds near the FXIa active site, reduced the ability of FXIa to proteolyze CFH (Figure 1C) as implied by persistence of a single band at ~170 kDa. In contrast, proteolysis of CFH by FXIa was unaffected by the anti-FXI antibody 1A6, which binds the FXI A3 domain and inhibits FXI activation by FXIIa and blocks FIX activation by FXIa, or by the anti-FXI antibody 14E11 which binds the FXI A2 domain and inhibits FXI activation by FXIIa (Figure 1C). Moreover, incubation of CFH with recombinant FXIa (rFXIa) led to the proteolysis of CFH in a similar manner to that of plasma-derived FXIa.
Figure 1:
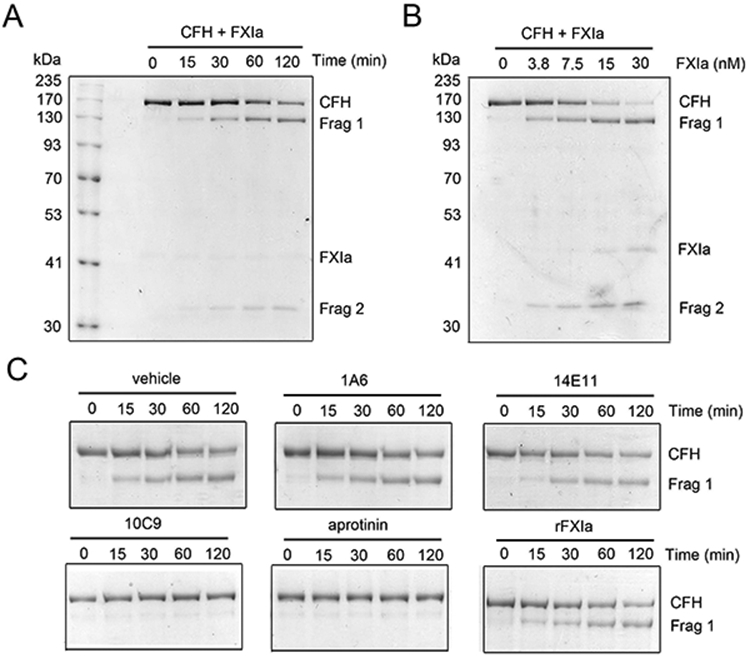
Characterization of the interaction between FXIa and CFH. (A) CFH (400 nM) was incubated with FXIa (10 nM) for selected times (0-2 hrs) at 37°C or (B) with increasing concentrations of FXIa (0-30 nM) for 2 hrs before being separated by SDS-PAGE under reduced conditions and analyzed by Coomassie blue staining. Data are mean ± SD (n = 3). (C) CFH (400 nM) was incubated with FXIa (10 nM) for selected times (0-2 hrs) at 37°C in the presence or absence of 1A6, 14E11, 10C9 (20 μg/ml) or aprotinin (50μM). In selected experiments CFH was incubated with rFXIa
Characterization of the cleavage of CFH by FXIa
Analysis of the CFH fragments generated by FXIa by Western blot using distinct anti-CFH antibodies against the CCP 1-4 domain (anti-N terminal antibody; Figure 2A) or against the CCP 15-20 domain (anti-C terminal antibody; Figure 2B), revealed that the ~35 kDa band contained the CFH N-terminal domain (Figure 2A) while the ~130 kDa band contained the CFH C-terminal domain (Figure 2B). Aminoterminal sequencing of the bands under reducing conditions revealed that the cleavages occurred at the end of the CCP 6 domain (Arg341/Arg342; Figure 2C). Next, we determined if other coagulation proteases also cleave CFH. As shown in Figure 2D, neither FXIIa, FXa or thrombin cleaved CFH. The presence of Zn2+ or Ca2+ did not promote the cleavege of CFH by FXIIa, FXa or thrombin (data not shown). However, kallikrein was able to cleave CFH but at a lower rate than FXIa (Figure 2D).
Figure 2:
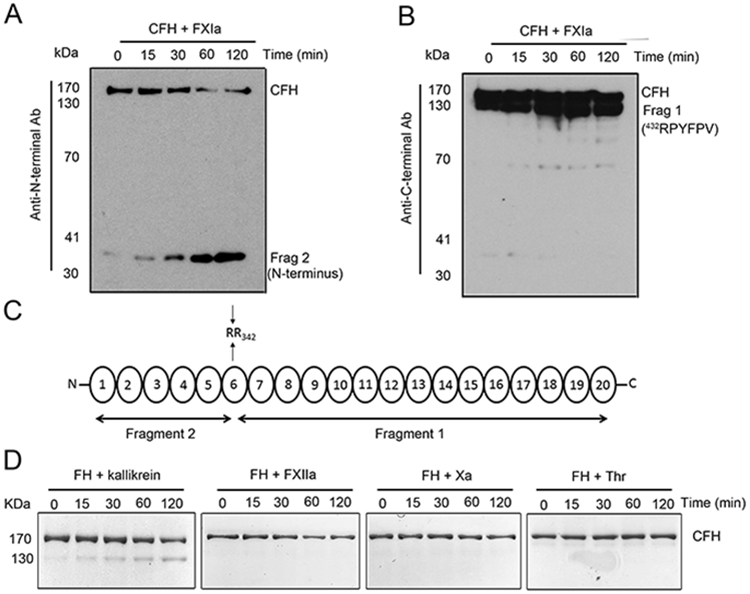
Characterization of complement factor H proteolysis by FXIa. (A) Complement factor H (200 nM) was incubated with FXIa (10 nM) for selected times (0-2 hrs) at 37°C before being analyzed with two different antibodies: anti-CFH N-terminal domain antibody or (B) anti-CFH C-terminal domain antibody. Protein bands were subjected to aminoterminal sequencing, using an automated Edman sequencer. Numbers refer to position of amino acid in full-length CFH protein (C). The identity of the fragments formed upon cleavage of CFH by FXIa are shown in a schematic overview. (D) Complement factor H (400 nM) was incubated with kallikrein, FXa, XIIa or thrombin (30 nM) for selected times (0-2 hrs) at 37°C before being separated by SDS-PAGE under reducing conditions and analyzed by Coomassie blue staining.
Long- and short-chain polyphosphates accelerate the cleavege of CFH by FXIa
Both CFH and FXIa have anion binding sites that are able to interact with polyanions such as heperan sulfate or polyphosphate (32, 33). Moreover, polyphosphate (polyP) enhances the proteolysis of TFPI by FXIa and the activation of FV by FXIa (29, 34). We designed experiments to study whether polyP enhanced the rate of CFH proteolysis by FXIa. SDS-PAGE analysis of CFH incubated with FXIa and short-chain polyP as found in platelets or long-chain polyP as found in bacteria, demonstrated that the rate of CFH cleavage by FXIa was accelerated in the presence of either short-chain or long-chain polyP (10 or 50 μM polyP; Figure 3A-B).
Figure 3:
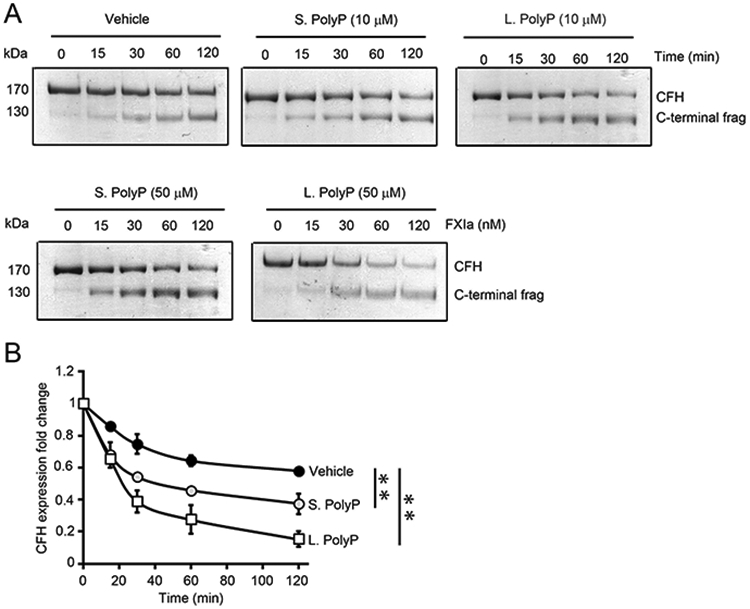
Effect of polyphosphates on factor H proteolysis by FXIa. (A) Complement factor H (400 nM) was incubated with FXIa (10 nM) for selected times (0-2 hours) at 37°C in the presence or absence of 10 μM or 50 μM short or long polyP. (B) CFH cleavage in the absence or presence of short or long polyP (50 μM) was quantified by densitometry.
* indicates between-groups differences with P < 0.01. Two-way ANOVA was used for statistical comparisons. Data are mean ± SD (n = 3).
FXIa decreases the cofactor activity and the decay acceleration activity of CFH
We tested whether the complement cofactor activity of CFH was affected by cleavage of CFH by FXIa. First, we quantified CFH cofactor activity by analyzing the cleavage of C3b by FI in the presence of CFH. The addition of CFH induced the cleavage of C3b by FI (Figure 4A-B). Under these conditions, in the absence of CFH, FI alone was unable to cleave C3b (data not shown). We observed that the incubation of CFH with FXIa decreased the capacity of CFH to promote the cleavage of the alpha chain of C3b (Figure 4A-B). FXIa alone in the absence of CFH or aprotinin alone did not modify the rate of cleavage of C3b by FI (data not shown).
Figure 4:

Evaluation of CFH cofactor activity assay, decay acceleration assay and binding to endothelial cells. (A) C3b (300 nM), FI (20 nM) and CFH (50 nM) or (B) CFH (25 nM) were incubated in solution and the generation of iC3b at specific time points (10, 20, 40 and 80 min) was monitored by SDS-PAGE analysis. In selected experiments CFH was preincubated with FXIa for 2 hrs. Aprotinin was added to all samples to stop the reaction. The disappearance of the αC3b-chain (114 kDa) and the appearance of three new bands (iC3bα) indicate proteolytic cleavage of C3b. (C) 96 well plates were coated with 5 μg/ml C3b and then incubated at 37°C for 2 h with 50 ng/ml factor B, 25 ng/ml factor D, and 1000 ng/ml properdin. The wells were then rinsed and incubated with increasing concentrations of CFH (0.25-2 ug/ml) for 30 min. In selected experiments CFH was incubated with FXIa for 2 hrs. Aprotinin was added to all samples to stop the reaction. The C3bB and C3bBb complexes were detected by ELISA using polyclonal goat anti-human FB antibody. (D) CFH was incubated with FXIa for 2 hrs. Aprotinin was added to all samples to stop the reaction and the decay acceleration assay was measure using sheep red blood cells coated with rabbit anti-sheep erythrocyte antiserum. CFH-depleted human serum in the presence of increasing concentrations of CFH were added and the erythrocyte lysis was measure at 541 nm. (E) CFH (200 nM) was incubated with FXIa for 2 hrs, aprotinin was added to all samples to stop the reaction, and the binding of CFH to HUVECs was measured by cell surface detection of CFH by using an anti-CFH C-terminal domain antibody. * indicates between-groups differences with P < 0.01. Two-way ANOVA was used for statistical comparisons. Data are mean ± SD (n = 3).
We next quantified the decay acceleration activity of CFH by measuring C3bBb complexes using an ELISA-based system. The decay of C3bBb was enhanced by the presence of an increasing concentration of CFH. The incubation of CFH with FXIa reduced the capacity of CFH to enhanced the decay of C3bBb (Figure 4C). Moreover, we observed that CFH prevented the lysis of sheep red blood cells coated with rabbit anti-sheep erythrocyte antiserum in a concentration-dependent manner (Figure 4D). The incubation of CFH with FXIa reduced the capacity of CFH to prevent the lysis of erythrocytes. The addition of FXIa and aprotinin alone did not affect the lysis of sheep erythrocytes (data not shown).
As CFH recognizes host surfaces through its interactions with select polyanions, such as glycosaminoglycan or heparan sulfate, through its CCP 6-7 and 19-20 domains (13), we investigated whether the cleavage of CFH by FXIa at the CCP 6 domain affects the binding of CFH to endothelial cells. We observed that CFH was able to bind to the surface of endothelial cells in a concentration-dependent manner (Figure 4E). After incubation of CFH with FXIa, the binding of CFH to endothelial cells was significantly decreased (Figure 4E). The addition of aprotinin alone did not affect the binding of CFH to endothelial cells (data not shown)
FXIa cleaves CFH present on the surface of endothelial cells, in platelet releasate, in plasma and in serum
To determine whether FXIa is also able to cleave CFH in a more complex system, we first studied whether FXIa can cleave CFH that is bound to the surface of endothelial cells (HUVECs). We observed that after incubating HUVECs with FXIa for 30 min, the levels of CFH that was bound to the surface of ECs diminished in a FXIa concentration-dependent manner (Figure 5A). Moreover, we observed that FXIa is also able to cleave CFH present in the releasate of activated platelets (figure 5B).
Figure 5:

Characterization of CFH proteolysis by FXIa in serum. (A) HUVECs incubated with CFH (200 nM) for 1 hr at 37°C, washed and treated with FXIa (0-30 nM) for 30 min at 37°C. Aprotinin was added to all samples to stop the reaction. Cells lysates were analyzed by Western blotting with an anti-CFH C-terminal domain antibody. (B) Supernatant from activated platelets (2.5 x 108) was incubated with FXIa (30 nM) for selected times (0-180 min). CFH was analyzed by Western blotting with an anti-CFH C-terminal domain antibody. (C) Human plasma or FXI-depleted plasma (FXI−/− plasma) was incubated with aPTT reagent or FXIa (30 nM) for 1 hr at 3 °C. CFH was analyzed by Western blotting with an anti-CFH C-terminal domain antibody. (D) FI-depleted serum was incubated with FXIa (30 nM) for selected times (0-180 min). CFH was analyzed by Western blotting with an anti-CFH C-terminal domain antibody. (E) C3b (300 nM) and FI (5 nM) were incubated in FI-depleted serum (1/5) at 37°C for selected times (10, 20, 40 and 80 min). Samples were separated by SDS-PAGE under reducing conditions and the generation of iC3b was analyzed by Western blotting using an anti-human C3b antibody. In selected experiments FI-depleted serum was incubated with FXIa (30 nM) for 3 hrs. Aprotinin was added to all samples to stop the reaction.
We next determined whether CFH can be cleaved in a plasma system by FXIa generated by the activation of the contact pathway of coagulation. Human plasma was incubated with aPTT reagent (ellagic acid) for 1 hour to induce the generation of FXIa by FXIIa in plasma. As shown in Figure 5C, the addition of aPTT reagent to human plasma induced the generation of the 130 kDa CFH fragment. In the sample where the aPTT reagent was incubated with FXI-depleted plasma, the appearance of the 130 kDa fragment was dramatically reduced. The reconstitution of FXI-depleted plasma with recombinant FXIa once again induced the appearance of the CFH fragment.
Lastly, experiments were designed to characterize the ability of FXIa to decrease the cofactor activity of CFH in serum using FI-depleted serum. As shown in Figure 5D, FXIa was able to cleave CFH present in FI-depleted serum sample in a time-dependent manner. The addition of C3b and FI to FI-depleted serum induced the cleavage of the alpha chain of C3b. Incubation of FI-depleted serum with FXIa for 3 hrs decreased the cleavage of C3b by FI (Figure 5E). The addition of aprotinin alone did not affect the cleavage of C3b by FI in FI-depleted serum.
Inhibition of FXI activation by CFH
It has been shown that the binding of β2GPI to FXI inhibits the activation of FXI by either FXIIa or thrombin (16). Moreover, as CFH has been implicated to play a role in impinging upon the activation of coagulation factors (12), we examined whether CFH is able to inhibit the activation of FXI. We first measured the activation of FXI by FXIIa or thrombin in the presence of dextran sulfate, which is known to act as a cofactor for this reaction. We observed that the generation of FXIa by FXIIa or thrombin in the presence of dextran sulfate (DXS) was decreased by the addition of β2GPI or CFH in a concentration-dependent manner (Figure 6A-B). In contrast, the activation of FXI by FXIIa or thrombin was not affected by the addition of β2GPI or CFH in the absence of DXS (Figure 6C). It was shown that β2GPI does not bind to DS (16), suggesting that β2GPI does not inhibit FXI activation by competing for binding to DS. We also tested whether CFH is able to inhibit the activation of FXII. As figure 5D shows, β2GPI or CFH did not inhibit the generation of FXIIa by negatively charge molecules such as DS, long-chain polyP or aPTT reagent (ellagic acid; Figure 6D). FXI is known to bind to β2GPI with an estimated binding constant of 11.7±4.9×10−9 M (16). Binding studies were performed to characterize the interaction between FXI and biotinylated CFH or β2GPI. FXI bound to immobilized CFH or β2GPI in a concentration-dependent manner (Figure 6E). FXI binding to CFH was unaffected by the presence of either Ca2+ or Zn2+ (data not shown). Moreover, to determine whether FXI present in plasma also can bind to CFH, we designed experiment using FH-depleted plasma. As shown in Figure 6F, FXI present in plasma was able to interact with biotinylated CFH (Figure 6F).
Figure 6:
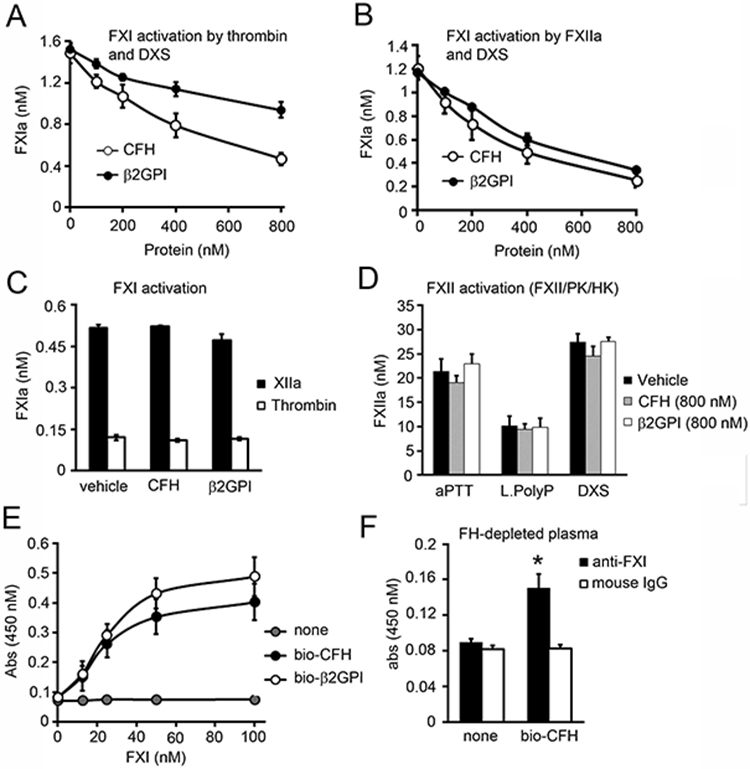
Inhibition of FXI activation by CFH. (A) FXI (30 nM) was incubated with either thrombin (5 nM) or (B) FXIIa (5 nM) in the presence of dextran sulfate (DXS, 2 ug/ml) for 15 min at 37°C in the presence or absence of increasing concentrations of factor H (0-800 nM) or β2GPI. FXIa generation was measured with a chromogenic substrate. (C) FXI (100 nM) was incubated with either thrombin (10 nM) or FXIIa (10 nM) in the absence of DXS for 30 min at 37°C in the presence or absence of factor H (800 nM) or β2GPI (800 nM). FXIa generation was measured with a chromogenic substrate. (D) FXII activation was measured following the addition of 200 nM FXII, 50 nM PK and 50 nM HK in the presence of aPTT reagent, 10 μM long polyP or DXS (2 μg/ml) in the presence or absence of factor H or β2GPI (800 nM). FXIIa generation was measured with a chromogenic substrate. (E) 96-well plates were coated with 5 μg/ml streptavidin prior to addition of vehicle, biotinylated CFH (bio-CFH) or β2GPI (bio- β2GPI) (100 nM). Increasing concentrations of FXI was added and binding was detected with a specific antibody against FXI. (F) 96 well plates were coated with 5 μg/ml streptavidin prior to addition of vehicle or biotinylated CFH (bio-CFH). FH-depleted plasma was added and binding was detected with a specific antibody against FXI or a mouse IgG control. * P < 0.05 with respect to vehicle. Mann-Whitney U test was used for statistical comparisons. Data are mean ± SD (n = 3).
DISCUSSION
FXI plays a significant role in the pathologic host response to some infections in mouse models. In addition to FXI’s requisite role in promoting thrombin generation through activation of FIX and activity towards other coagulation factors (28, 35), FXI has been implicated in regulating several host defense pathways (36). While it is unclear whether FXI activation is a driver, bystander, or victim of inflammation, FXI has been shown to contribute to the activation of the fibrinolytic system, activation of complement system, and potentiation of inflammatory cytokines and bradykinin generation. Here we show for the first time that FXIa can neutralize the inhibitory role of CFH on complement activation.
CFH is a major inhibitor of the alternative pathway of complement activation. The N-terminal CCP1-4 domains of CFH have C3bBb decay accelerating and FI cofactor regulatory functions. The CCP6-7 and CCP19-20 C-terminal domains bind to negatively charged extracellular glycosaminoglycans and anionic heparan sulphate, protecting host tissues from attack by complement (3). Moreover, CCP6-7 seems to bind to C3b and CCP5-6 may stabilize the optimal conformation of CCP 1-4 for interaction with C3b (37). Mutations in, or autoantibodies against, these regions have been associated with defects in the function of CFH, resulting in dysregulation of the complement cascade. Interestingly, a mutation at CFH Arg341 (R341A) at the end of CCP6 domain, which is the FXIa cleavage site identified in this study, has been associated with age-related macular degeneration perhaps in part due to a reduction in the capacity of the mutant form of CFH to bind heparin (11).
Herein we show for the first time that FXIa can enhance complement activation by binding and cleaving CFH. Trypsinization of purified CFH has been investigated by several groups to study the structure and function of CFH (38-40), but trypsin cleaved CFH in multiple sites. FXIa specifically cleaves CFH at the Arg341/Arg342 bonds at the end of CCP6 domain, resulting in a reduction in the cofactor and decay acceleration activity of CFH, while reducing its capacity to bind to the surface of cells. Whether the mutation of CFH Arg341 in patients with macular degeneration alters the potentially physiological or possibly pathological function of FXI in regulating the complement cascade remains to be explored.
In a recent study by our group, we found that antibody inhibition of the interaction between FXI and FXII prevented tissue damage and complement activation following the injection of a lethal dose of heat-inactivated Staphylococcus aureus into baboons (22). Specifically, targeting reciprocal FXI/ FXII activation by this intervention significantly decreased the levels of circulating C3b and C5b-9 as well as the level of C5b-9 deposited in the kidneys. The contact system proteases FXII and PK are known to activate the complement system in vitro. FXIIa can activate the complement cascade via activation of the C1qrs complex (25), while kallikrein has been shown to activate factor B, C3 and C5 (26, 41). Based on our finding that FXIa can also feed back to activate FXII, the effect of inhibiting FXI activation in vivo on complement activation may be explained, at least partly, by a reduction in the generation of FXIIa by FXIa. Complement activation in sepsis is mainly driven by the alternative pathway. Our observation that the interaction of FXIa with CFH directly effects complement activation suggest that direct effect of FXIa on CFH during sepsis may enhance propagation of the alternative pathway and formation of C3b and the C5b-9 terminal complex. Moreover, our findings support the notion that FXIa plays a pathological role in the host defense response to certain bacterial or viral infections. Activation of the contact system in pathologic situations is expected to activate the complement system. This work would posit that the interplay between complement activation and activation or activity of the members of the contact activation system should be considered when evaulating therapeutic agents designed to inhibit complement pathways.
The blood coagulation and complement systems are functionally and evolutionarily related and exhibit crossover activities. For example, complement C1 inhibitor is the main inhibitor of the contact system proteases FXIIa and kallikrein (42). Deficiency or altered function in complement proteins, including CFH, have been associated with inflammatory diseases with thrombotic components, such as atypical haemolytic uremic syndrome, systemic lupus erythematosus, rheumatoid arthritis, anti-phospholipid antibody syndrome and age-related macular degeneration (43). Moreover, CFH may contribute to the presentation of clinical thrombotic microangiopathies through a complement-independent mechanism. It has been reported that CFH enhances cleavage of von Willebrand factor by ADAMTS-13 (44), which incidentally is also a substrate for FXIa (35). CFH is also a ligand for the platelet αIIbβ3 integrin (45), potentially reducing the avidity of platelets for adhesive ligands during thrombus formation. Autoantibodies against β2GPI, which is structurally similar to CFH has been associated with anti-phospholipid antibody syndrome (12). β2GPI binds to negatively charged procoagulant phospholipids expressed on cell surfaces such as phosphatidylserine present on activated platelets, thus inhibiting the assembly of the platelet prothrombinase complex (46). Similarly, CFH has been shown to bind to procoagulant phospholipids (12). Furthermore, β2GPI can neutralize the contact pathway of coagulation thought the inhibition of the activation of FXI by FXIIa and thrombin (16). Again mirroring these effects, CFH has been shown to exhibit anticoagulant properties by prolonging coagulation times in a contact activation system-dependent manner. While CFH-FXIIa complexes have been observed in human plasma (47), it is not clear at this point if CHF is an important regulator of contact activation. Herein we demonstrate that CFH directly inhibits the generation of FXIa by thrombin and FXIIa, suggesting a further connection between the complement pathway and the contact activation system. Whether the activity of CFH plays a physiological role in hemostasis in select tissues or a pathological role in thrombosis in certain diseases remains to be seen.
KEY POINTS.
FXIa cleaves CFH at the Arg341/Arg342 bonds at the end of the CCP6 domain of CFH.
FXIa reduces CFH cofactor and decay-accelerating activity and binding to ECs.
CFH binds FXI and inhibits FXI activation by either thrombin or FXIIa.
Acknowledgements
We would like to thank Dr. Rolf Urbanus for providing β2GPI and Drs. James H. Morrissey and Stephanie Smith for providing long- and short-chain polyP.
Sources of funding
This work was supported by grants from the National Institutes of Health (R01HL101972 and R01AI157037). O.J.T. McCarty is an American Heart Association Established Investigator.
Abbreviations
- CFH
complement factor H
- FXI
coagulation factor XI
- FXII
coagulation factor XII
- FXIa
activated FXI
- rFXIa
recombinant FXIa
- FXIIa
activated FXIIa
- FI
factor I
- FB
factor B
- FD
factor D
- CCP
complement control protein
- DXS
Dextran sulfate
- β2GPI
beta2-glycoprotein I
- HK
high molecular weight kininogen
- PK
prekallikrein
- CTI
corn trypsin inhibitor
- OPD
O-phenylenediamine
- aPTT
activated partial thromboplastin time
- BSA
bovine serum albumin
- HBS
25mM Hepes, pH 7.4, 150 mM NaCl buffer
- HUVECs
Human umbilical vein endothelial cells
Footnotes
Disclosers
A. Gruber, C.U. Lorentz, E.I. Tucker, and Oregon Health & Science University have a significant financial interest in Aronora, Inc., a company that may have a commercial interest in the results of this research. This potential conflict of interest has been reviewed and managed by the Oregon Health & Science University Conflict of Interest in Research Committee. The remaining authors declare no competing financial interests.
REFERENCES
- 1.Bernet J, Mullick J, Singh AK, and Sahu A. 2003. Viral mimicry of the complement system. J. Biosci 28: 249–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sjöberg AP, Trouw LA, and Blom AM. 2009. Complement activation and inhibition: a delicate balance. Trends Immunol. 30: 83–90. [DOI] [PubMed] [Google Scholar]
- 3.Parente R, Clark SJ, Inforzato A, and Day AJ. 2017. Complement factor H in host defense and immune evasion. Cell. Mol. Life Sci 74: 1605–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu J, Wu YQ, Ricklin D, Janssen BJC, Lambris JD, and Gros P. 2009. Structure of complement fragment C3b-factor H and implications for host protection by complement regulators. Nat. Immunol 10: 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perkins SJ, Nan R, Li K, Khan S, and Miller A. 2012. Complement Factor H-ligand interactions: Self-association, multivalency and dissociation constants. Immunobiology 217: 281–297. [DOI] [PubMed] [Google Scholar]
- 6.Brooimans RA, van der Ark AA, Buurman WA, van Es LA, and Daha MR. 1990. Differential regulation of complement factor H and C3 production in human umbilical vein endothelial cells by IFN-gamma and IL-1. J. Immunol 144: 3835–40. [PubMed] [Google Scholar]
- 7.Devine DV, and Rosse WF. 1987. Regulation of the activity of platelet-bound C3 convertase of the alternative pathway of complement by platelet factor H. Proc. Natl. Acad. Sci. U. S. A 84: 5873–5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whaley K 1980. Biosynthesis of the complement components and the regulatory proteins of the alternative complement pathway by human peripheral blood monocytes. J. Exp. Med 151: 501–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katz Y, and Strunk RC. 1988. Synthesis and regulation of complement protein factor H in human skin fibroblasts. J. Immunol 141: 559–63. [PubMed] [Google Scholar]
- 10.Józsi M, Heinen S, Hartmann A, Ostrowicz CW, Hälbich S, Richter H, Kunert A, Licht C, Saunders RE, Perkins SJ, Zipfel PF, and Skerka C. 2006. Factor H and Atypical Hemolytic Uremic Syndrome: Mutations in the C-Terminus Cause Structural Changes and Defective Recognition Functions. J. Am. Soc. Nephrol 17: 170–177. [DOI] [PubMed] [Google Scholar]
- 11.Prosser BE, Johnson S, Roversi P, Herbert AP, Blaum BS, Tyrrell J, Jowitt TA, Clark SJ, Tarelli E, Uhrín D, Barlow PN, Sim RB, Day AJ, and Lea SM. 2007. Structural basis for complement factor H–linked age-related macular degeneration. J. Exp. Med 204: 2277–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferluga J, Kishore U, and Sim RB. 2014. A potential anti-coagulant role of complement factor H. Mol. Immunol 59: 188–193. [DOI] [PubMed] [Google Scholar]
- 13.Morgan HP, Schmidt CQ, Guariento M, Blaum BS, Gillespie D, Herbert AP, Kavanagh D, Mertens HDT, Svergun DI, Johansson CM, Uhrín D, Barlow PN, and Hannan JP. 2011. Structural basis for engagement by complement factor H of C3b on a self surface. Nat. Struct. Mol. Biol 18: 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.PUURUNEN M, JOKIRANTA S, VAARALA O, and MERI S. 1995. Lack of Functional Similarity Between Complement Factor H and Anticardiolipin Cofactor, β2-Glycoprotein I (Apolipoprotein H). Scand. J. Immunol 42: 547–550. [DOI] [PubMed] [Google Scholar]
- 15.McNeil HP, Simpson RJ, Chesterman CN, and Krilis SA. 1990. Anti-phospholipid antibodies are directed against a complex antigen that includes a lipid-binding inhibitor of coagulation: β2-glycoprotein I (apolipoprotein H). Proc. Natl. Acad. Sci. U. S. A 87: 4120–4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi T, Iverson GM, Qi JC, Cockerill KA, Linnik MD, Konecny P, and Krilis SA. 2004. 2-Glycoprotein I binds factor XI and inhibits its activation by thrombin and factor XIIa: Loss of inhibition by clipped 2-glycoprotein I. Proc. Natl. Acad. Sci 101: 3939–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puy C, Rigg RA, and McCarty OJT. 2016. The hemostatic role of factor XI. Thromb. Res 141: S8–S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colman RW, and Schmaier AH. 1997. Contact system: a vascular biology modulator with anticoagulant, profibrinolytic, antiadhesive, and proinflammatory attributes. Blood 90: 3819–43. [PubMed] [Google Scholar]
- 19.Tucker EI, Verbout NG, Leung PY, Hurst S, McCarty OJT, Gailani D, and Gruber A. 2012. Inhibition of factor XI activation attenuates inflammation and coagulopathy while improving the survival of mouse polymicrobial sepsis. Blood 119: 4762–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo D, Szaba FM, Kummer LW, Johnson LL, Tucker EI, Gruber A, Gailani D, S. ST. 2012. Factor XI-deficient mice display reduced inflammation, coagulopathy, and bacterial growth during listeriosis. Infect Immun 80: 91–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tucker EI, Gailani D, Hurst S, Cheng Q, Hanson SR, and Gruber A. 2008. Survival advantage of coagulation factor XI-deficient mice during peritoneal sepsis. J Infect Dis 198: 271–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silasi R, Keshari RS, Lupu C, Van Rensburg WJ, Chaaban H, Regmi G, Shamanaev A, Shatzel JJ, Puy C, Lorentz CU, Tucker EI, Gailani D, Gruber A, McCarty OJT, and Lupu F. 2019. Inhibition of contact-mediated activation of factor XI protects baboons against S aureus–induced organ damage and death. Blood Adv. 3: 658–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jansen PM, Pixley RA, Brouwer M, De Jong IW, Chang ACK, Hack CE, Taylor FB, and Colman RW. 1996. Inhibition of factor XII in septic baboons attenuates the activation of complement and fibrinolytic systems and reduces the release of interleukin-6 and neutrophil elastase. Blood 87: 2337–2344. [PubMed] [Google Scholar]
- 24.Pixley RA, De La Cadena R, Page JD, Kaufman N, Wyshock EG, Chang A, Taylor FB, and Colman RW. 1993. The contact system contributes to hypotension but not disseminated intravascular coagulation in lethal bacteremia: In vivo use of a monoclonal anti-factor XII antibody to block contact activation in baboons. J. Clin. Invest 91: 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghebrehiwet B, Randazzo BP, Dunn JT, Silverberg M, and Kaplan AP. 1983. Mechanisms of activation of the classical pathway of complement by Hageman factor fragment. J. Clin. Invest 71: 1450–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Irmscher S, Döring N, Halder LD, Jo EAH, Kopka I, Dunker C, Jacobsen ID, Luo S, Slevogt H, Lorkowski S, Beyersdorf N, Zipfel PF, and Skerka C. 2018. Kallikrein Cleaves C3 and Activates Complement. J. Innate Immun 10: 94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith SA, Choi SH, Davis-Harrison R, Huyck J, Boettcher J, Rienstra CM, and Morrissey JH. 2010. Polyphosphate exerts differential effects on blood clotting, depending on polymer size. Blood 116: 4353–4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Puy C, Tucker EI, Matafonov A, Cheng Q, Zientek KD, Gailani D, Gruber A, and McCarty OJT. 2015. Activated factor XI increases the procoagulant activity of the extrinsic pathway by inactivating tissue factor pathway inhibitor. Blood 125: 1488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puy C, Tucker EI, Ivanov IS, Gailani D, Smith SA, Morrissey JH, Gruber A, and McCarty OJT. 2016. Platelet-Derived Short-Chain Polyphosphates Enhance the Inactivation of Tissue Factor Pathway Inhibitor by Activated Coagulation Factor XI. PLoS One 11: e0165172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aslan JE, Itakura A, Gertz JM, and McCarty OJT. 2012. Platelet shape change and spreading. Methods Mol Biol 788: 91–100. [DOI] [PubMed] [Google Scholar]
- 31.Shi T, Giannakopoulos B, Iverson GM, Cockerill KA, Linnik MD, and Krilis SA. 2005. Domain V of β2-glycoprotein I binds factor XI/XIa and is cleaved at Lys317-Thr318. J. Biol. Chem 280: 907–912. [DOI] [PubMed] [Google Scholar]
- 32.Geng Y, Verhamme IM, Smith SA, Cheng Q, Sun M, Sheehan JP, Morrissey JH, and Gailani D. 2013. Factor XI anion-binding sites are required for productive interactions with polyphosphate. J. Thromb. Haemost 11: 2020–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wat JM, Foley JH, Krisinger MJ, Ocariza LM, Lei V, Wasney GA, Lameignere E, Strynadka NC, Smith SA, Morrissey JH, and Conway EM. 2014. Polyphosphate suppresses complement via the terminal pathway. Blood 123: 768–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi SH, Smith SA, and Morrissey JH. 2015. Polyphosphate accelerates factor V activation by factor XIa. Thromb. Haemost 113: 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garland KS, Reitsma SE, Shirai T, Zilberman-Rudenko J, Tucker EI, Gailani D, Gruber A, McCarty OJT, and Puy C. 2017. Removal of the C-Terminal Domains of ADAMTS13 by Activated Coagulation Factor XI induces Platelet Adhesion on Endothelial Cells under Flow Conditions. Front. Med 4: 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raghunathan V, Zilberman-Rudenko J, Olson SR, Lupu F, McCarty OJT, and Shatzel JJ. 2019. The contact pathway and sepsis. Res. Pract. Thromb. Haemost 3: 331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haque A, Cortes C, Alam MN, Sreedhar M, Ferreira VP, and Pangburn MK. 2020. Characterization of Binding Properties of Individual Functional Sites of Human Complement Factor H. Front. Immunol 11: 1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alsenz J, Schulz TF, Lambris JD, Sim RB, and Dierich MP. 1985. Structural and functional analysis of the complement component factor H with the use of different enzymes and monoclonal antibodies to factor H. Biochem. J 232: 841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alsenz J, Lambris JD, Schulz TF, and Dierich MP. 1984. Localization of the complement-component-C3b-binding site and the cofactor activity for factor I in the 38 kDa tryptic fragment of factor H. Biochem. J 224: 389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koistinen V 1992. Limited tryptic cleavage of complement factor H abrogates recognition of sialic acid-containing surfaces by the alternative pathway of complement. Biochem. J 283: 317–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hiemstra PS, Daha MR, and Bouma BN. 1985. Activation of factor B of the complement system by kallikrein and its light chain. Thromb. Res 38: 491–503. [DOI] [PubMed] [Google Scholar]
- 42.Schoenfeld AK, Lahrsen E, and Alban S. 2016. Regulation of complement and contact system activation via C1 inhibitor potentiation and factor XIIa activity modulation by sulfated glycans - Structure-activity relationships. PLoS One 11: e0165493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferluga J, Kouser L, Murugaiah V, Sim RB, and Kishore U. 2017. Potential influences of complement factor H in autoimmune inflammatory and thrombotic disorders. Mol. Immunol 84: 84–106. [DOI] [PubMed] [Google Scholar]
- 44.Feng S, Liang X, Cruz MA, Vu H, Zhou Z, Pemmaraju N, Dong J-F, Kroll MH, and Afshar-Kharghan V. 2013. The Interaction between Factor H and Von Willebrand Factor. PLoS One 8: e73715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mnjoyan Z, Li J, and Afshar-Kharghan V. 2008. Factor H binds to platelet integrin α IIb β 3. Platelets 19: 512–519. [DOI] [PubMed] [Google Scholar]
- 46.Nimpf J, Bevers EM, Bomans PHH, Till U, Wurm H, Kostner GM, and Zwaal RFA. 1986. Prothrombinase activity of human platelets is inhibited by β2-glycoprotein-I. BBA - Gen. Subj 884: 142–149. [DOI] [PubMed] [Google Scholar]
- 47.Thangaraj SS, Christiansen SH, Graversen JH, Sidelmann JJ, Hansen SWK, Bygum A, Gram JB, and Palarasah Y. 2020. Contact activation-induced complex formation between complement factor H and coagulation factor XIIa. J. Thromb. Haemost 18. [DOI] [PubMed] [Google Scholar]