

Abstract
Accumulating evidence indicates that higher levels of salt intake are associated with higher blood pressure levels. The aim of our analysis was to test the hypothesis that the effect of urinary sodium excretion (UNaV) on systolic blood pressure (SBP) is mediated through estimated glomerular filtration rate (eGFR) and arterial stiffness and also to test the direction of the relationship between eGFR and arterial stiffness, in both hypertensive and normotensive patients. We assessed the potential for connection between UNaV and SBP and mediators (eGFR and pulse wave velocity [PWV]) of this relationship using structural equation models of data from 1599 adults ≥18 years of age and without chronic kidney disease who participated in the Third Epidemiologic Study concerning the Prevalence of Arterial Hypertension and Cardiovascular Risk in Romania (SEPHAR III). In hypertensive patients, the indirect effect, mediated through PWV, of UNaV on SBP was 23.9% and 27.7% of the total effect of UNaV on SBP, while in normotensive patients the contribution of PWV to the total effect of UNaV on SBP was slightly lower (15.9% and 18.3% of the total effect of UNaV on SBP). Taken together, our findings support the conclusion that UNaV influences SBP, both directly and indirectly, through the effect on PWV.
Keywords: arterial stiffness, mediation analysis, sodium, systolic blood pressure
1. INTRODUCTION
According to the 2010 Global Burden of Disease Study, high blood pressure is the leading cause, among 67 causes studied, of death and disability‐adjusted life years worldwide.1 Numerous studies have shown a gradual relationship between increased systolic blood pressure (SBP) and a higher cardiovascular risk.2, 3
Accumulating evidence indicates that higher levels of salt intake are associated with higher blood pressure levels.4, 5, 6 The PURE study, which included more than 100 000 adults sampled from the general population from 17 countries, showed that the relation between increased salt intake (assessed by urinary sodium excretion [UNaV]) and increased blood pressure was steeper in patients with hypertension than in those without hypertension.5 Increased salt intake has also been linked to changes in the arterial wall structure and function7 and may determine arterial stiffness.8, 9, 10 Furthermore, arterial stiffness has been shown to cause increased SBP and precede the development of overt hypertension.11, 12, 13, 14, 15 Similarly, the kidneys play an important role in blood pressure regulation, with studies showing that a reduction in the estimated glomerular filtration rate (eGFR) is associated with increased blood pressure levels.16, 17 However, the relationships among salt intake, arterial stiffness, and renal function and their combined impact on blood pressure are still unknown.
The SEPHAR (Epidemiologic Study concerning the Prevalence of Arterial Hypertension and Cardiovascular Risk in Romania) III project was designed to assess the prevalence of hypertension and of other major cardiovascular risk factors, including UNaV, kidney function, and arterial stiffness, in the adult population of Romania.18, 19 Therefore, the aim of our analysis was to test the hypothesis that the effect of UNaV (as a surrogate for salt intake) on SBP is mediated through eGFR and arterial stiffness, as assessed by pulse wave velocity (PWV), and also to test the direction of the relationship between eGFR and arterial stiffness, in both hypertensive and normotensive patients.
2. METHODS
2.1. Study population
Details about the SEPHAR III survey were previously published.18, 19 Briefly, between November 16th 2015 and April 25th 2016, a total number of 2124 Romanian adults, identified through a multistratified sampling procedure from 84 study sites (42 cities and 42 communes), gave written consent to participate in the survey, of which 154 were excluded as they were lost to follow up. Therefore, the SEPHAR III database included only 1970 participants with eligible data for the analysis (complete questionnaires and both study visits).18, 19
From the total 1970 participants of SEPHAR III survey’s database, we further excluded patients who had missing values for the variables of interest (48 and 66 had missing for UNaV and PWV values, respectively).
Because we wanted to assess the relationships in the absence of the interferences of sodium handling due to diuretic use and also in the absence of chronic kidney disease‐induced changes in PWV and sodium handling, we also further excluded these types of patients (159 and 138 patients were on diuretic treatment and were considered to have chronic kidney disease, respectively). Chronic kidney disease was defined as per guidelines.20
As a result, only 1559 patients were considered for further analysis related to this study (Figure 1), number that satisfies the minimum required sample size for the representativity of our adult population (1379 individuals).18
Figure 1.
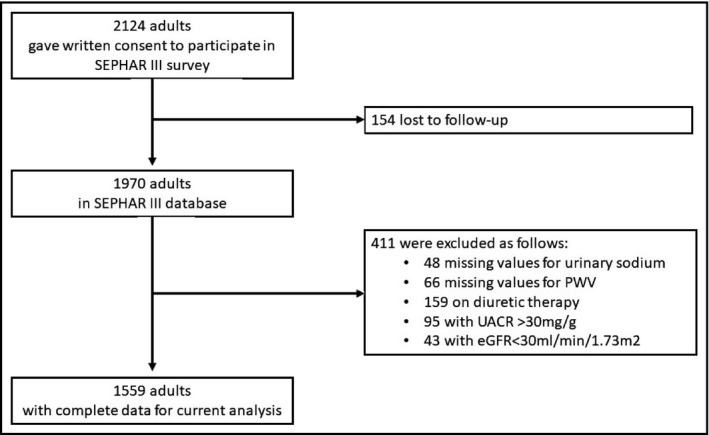
Flow‐chart of the study population. eGFR, estimated glomerular filtration rate; PWV, pulse wave velocity; UACR, urinary albumin/creatinine ratio
2.2. Blood pressure measurement and definition of hypertension
At the study visit, three blood pressure (BP) measurements were performed at 1‐minute interval using an automatic BP measuring device certified by Association for the Advancement of Medical Instrumentation, European Society of Hypertension, and British Society of Hypertension—model OMRON M6 with an adjustable cuff for arm circumferences from 24 to 42 cm, respecting the current guideline recommendations.21 Before performing BP measurements, arm circumference was measured (using a tailor’s tape measure with a maximum deviation of 0.5 cm, at the widest level of the arm) to check if the cuff size is adequate (if arm circumference was more than 42 cm, a cuff for obese study participants was used). BP was measured in both arms and, after that, two additional measurements were performed in the arm with the highest BP value on the first measurement.
As previously described,19 hypertension was defined as a SBP of at least 140 mmHg and/or diastolic BP of at least 90 mmHg at both study visits, using the arithmetic mean of the second and third BP measurement of the study visit (without taking into consideration the first BP measurement), or previously diagnosed hypertension under treatment during the previous 2 weeks, regardless of BP values.
2.3. Urinary sodium excretion, arterial stiffness, and renal function assessment
Urinary sodium excretion was estimated by Kawasaki formula,22 using sodium excretion values determined from the morning spot urine. PWV was determined using an oscillometric device (model Arteriograph TensioMed, TensioMed Kft., Budapest, Hungary) with the patients in supine position, on the dominant arm and leg, after at least 10 minutes rest.18 eGFR was determined using the Chronic Kidney Disease Epidemiology Collaboration (CKD‐EPI) equation.23
2.4. Statistical analysis
Non‐normally distributed variables were expressed as median (with interquartile range [IQR]) and normally distributed variables were as mean ± SD, as appropriate. Categorical variables were presented as percent frequency.
Logarithmic conversion was performed for non‐normally distributed variables, including urinary albumin/creatinine ratio (log urinary albumin/creatinine ratio), serum glucose (log serum glucose), HbA1c (log HbA1c), and PWV (log PWV). Pearson product moment coefficient was used to determine the correlation between paired variables.
Mediation is the process through which an exposure causes disease. We hypothesized that some of the total effect of urinary sodium on systolic blood pressure is mediated by an increase in PWV and/or a decrease in eGFR (the mediators). When a mediator is hypothesized, the total effect can be divided into two parts: the direct and indirect effect. The direct effect is the effect of exposure (urinary sodium) on the outcome (blood pressure) in the absence of the mediators (PWV or eGFR). The indirect pathway is the effect of exposure on the outcome that works through the mediator(s). We performed this analysis using structural equation modeling, in both hypertensive and normotensive patients. We tested two different models to address the putative bidirectional relationship between PWV and eGFR, as mediators for the effect of UNaV on SBP (Figure 2). We used as confounders all the variables that were available in our database (see Table 1). As eGFR estimation algorithm includes age and sex, we did not include these variables as a confounder in the structural equation models. We initially tested all the pathways showed in Figure 2; if one of these pathways was non‐significant, it was removed and the model was reanalyzed.
Figure 2.
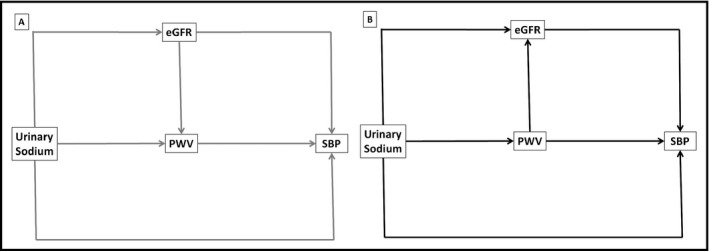
Hypothetical structural equations models outlining two possible pathways between urinary sodium and systolic blood pressure. A, Model 1 (in grey): eGFR→PWV; B, Model 2 (in black): PWV→eGFR. All models will be adjusted for body mass index, serum uric acid, LDL cholesterol (or HDL cholesterol), urinary albumin/creatinine ratio, serum glucose (or HbA1c), and antihypertensive treatment (only in hypertensive patients). eGFR, estimated glomerular filtration rate; PWV, pulse wave velocity; SBP, systolic blood pressure
Table 1.
Demographic, clinical and biological characteristics of the study population
| All (N = 1559) | Hypertensive (N = 600) | Normotensive (N = 959) | P | |
|---|---|---|---|---|
| Demographic characteristics | ||||
| Age, y | 47.3 ± 17.1 | 55.8 ± 15.0 | 42.0 ± 16.2 | <0.001 |
| Male, N (%) | 729 (46.8) | 304 (50.7) | 425 (44.3) | 0.01 |
| BMI, kg/m2 | 28.2 ± 5.7 | 30.3 ± 5.5 | 26.6 ± 5.1 | <0.001 |
| Anti‐hypertensive treatment, N (%) | – | 412 (68.7) | – | NA |
| Systolic blood pressure, mmHg | 133.9 ± 21.4 | 147.9 ± 22.2 | 125.0 ± 15.2 | <0.001 |
| Diastolic blood pressure, mmHg | 84.7 ± 11.9 | 90.9 ± 12.2 | 80.9 ± 10.0 | <0.001 |
| Heart rate, beats/min | 76.7 ± 11.4 | 76.9 ± 11.9 | 76.6 ± 11.0 | 0.59 |
| Laboratory values | ||||
| Serum uric acid, mg/dL | 4.9 ± 1.3 | 5.2 ± 1.4 | 4.7 ± 1.2 | <0.001 |
| eGFR, mL/min per 1.73 m2 | 100.6 ± 16.8 | 94.1 ± 15.2 | 104.6 ± 16.6 | <0.001 |
| HDL‐Cholesterol, mg/dL | 55.4 ± 16.5 | 54.1 ± 16.9 | 56.1 ± 16.3 | 0.02 |
| LDL‐Cholesterol, mg/dL | 128.8 ± 40.2 | 134.4 ± 40.9 | 125.3 ± 39.4 | <0.001 |
| Urinary albumin/creatinine ratio, mg/g | 3.9 (2.5‐6.9) | 4.5 (2.7‐7.7) | 3.6 (2.4‐6.4) | <0.001 |
| Serum glucose, mg/dL | 97.0 (90.0‐106.0) | 101.0 (94.0‐114.5) | 94.0 (88.0‐102.0) | <0.001 |
| HbA1c | 5.3 (5.1‐5.7) | 5.5 (5.2‐5.9) | 5.2 (5.0‐5.5) | <0.001 |
| Urinary sodium, mmol/d | 209.4 ± 67.4 | 227.3 ± 69.7 | 198.2 ± 63.4 | <0.001 |
| Pulse wave velocity, m/s | 8.2 (7.1‐9.9) | 9.4 (8.0‐11.0) | 7.6 (6.6‐8.9) | <0.001 |
Data are expressed as mean ± SD, median with IR, or percent frequency, as appropriate.
The goodness‐of‐fit test was performed using absolute fit indices and incremental fit measures. The absolute fit indices included the root mean square residual and the goodness of fit index. Incremental fit measures included the non‐normed fit index and the comparative fit index.
We considered a two‐tailed P value of <0.05 to be significant. All statistical analyses were performed using Stata SE software, version 12 (StataCorp. Stata Statistical Software: Release 12. College Station, TX: StataCorp LP).
3. RESULTS
3.1. Characteristics of the study population
Our study included 1559 patients (mean age 47.3 years, 46.8% males). The mean SBP and DBP values for the entire population were 133.9 ± 21.4 and 84.7 ± 11.9 mmHg, respectively; the median PWV value was 8.2 (IQR 7.1‐9.9) m/s. There were 691 (40.7%) patients diagnosed with hypertension, of which 467 (67.6%) were on antihypertensive treatment (Table 1).
As compared with normotensive patients, hypertensive patients were older (55.8 ± 15.0 vs 42.0 ± 16.2 years, P < 0.001), had a higher BMI (30.3 ± 5.5 vs 26.6 ± 5.1 kg/m2, P < 0.001) and were more frequently male (50.7% vs 44.3%, P < 0.001). These patients showed also increased serum uric acid (5.2 ± 1.4 vs 4.7 ± 1.2 mg/dL, P < 0.001), LDL cholesterol (134.4 ± 40.9 vs 125.3 ± 39.4 mg/dL, P < 0.001), UACR (median 4.5 [IQR 2.7‐7.7] vs median 3.6 [IQR 2.4‐6.4] mg/g, P < 0.001), serum glucose (median 101.0 [IQR 94.0‐114.5] mg/dL vs median 94.0 [IQR 88.0‐102.0] mg/dL, P < 0.001), HbA1c (median 5.5 [IQR 5.2‐5.9]% vs median 5.2 [IQR 5.0‐5.5] mg/dL, P < 0.001), urinary sodium (227.3 ± 69.7 vs 198.2 ± 63.4 mmol/d, P < 0.001), and PWV values (median 9.4 [IQR 8.0‐11.0] vs 7.6 [IQR 6.6‐8.9] m/s, P < 0.001), but lower eGFR (94.1 ± 15.2 vs 104.6 ± 16.6 mL/min per 1.73 m2, P < 0.001) and HDL cholesterol (54.1 ± 16.9 vs 56.1 ± 16.3 mg/dL, P = 0.02) than normotensive patients (Table 1).
3.2. Correlations among urinary sodium, pulse wave velocity, eGFR, and systolic blood pressure
Significant associations were found among UNaV, PWV, eGFR, and SBP in both hypertensive and normotensive patients. Specifically, UnaV was positively associated with PWV and SBP, and negatively with eGFR (Table 2).
Table 2.
Univariable correlation analysis of urinary sodium with PWV, eGFR and systolic blood pressure, in hypertensive and normotensive patients
| Urinary sodium, mmol/d | ||||
|---|---|---|---|---|
| Hipertensive | Normotensive | |||
| Coeff | P | Coeff | P | |
| PWVa, m/s | 0.21 | <0.001 | 0.22 | <0.001 |
| eGFR, mL/min per 1.73 m2 | −0.13 | 0.002 | −0.14 | <0.001 |
| Systolic blood pressure, mmHg | 0.21 | <0.001 | 0.26 | <0.001 |
eGFR, estimated glomerular filtration rate; PWV, pulse wave velocity.
Using logarithmic transformation.
3.3. Structural equation models
The use of this statistical approach adds important information to the relationship between UNaV (as a marker of sodium intake) and SBP because it permits us to assess more complex pathways, with direct and indirect effects of UNaV on blood pressure. We designed two structural equation models to test this relationship, taking into consideration also PWV and eGFR as possible mediators and the possible bidirectional relationship between these two variables (Figure 2A,B).
3.4. Hypertensive patients
Figure 3 depicts the structural equation models with the calculated magnitude of the direct and indirect effects of UNaV on SBP in hypertensive patients. In Model 1, where the putative effect of eGFR on PWV is taken into account, UNaV has significant direct and indirect effects on SBP. Specifically, UNaV has a direct effect on PWV which in turn has a direct effect on SBP (the largest of all the effects tested in this model). Therefore, the indirect effect mediated by PWV accounts for a significant proportion of 23.9% of the total effect of UNaV on SBP (Table 3). In addition, while UNaV does not directly impact eGFR, the model indicates that eGFR has a significant inverse effect on SBP, which is mainly mediated via PWV.
Figure 3.
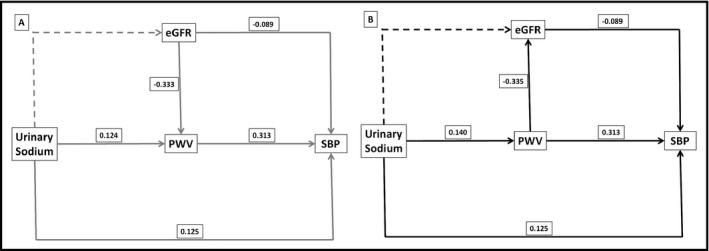
Path analysis illustrated by the structural equation model with standardized coefficients of direct effects for hypertensive patients. A, Model 1 (eGFR→PWV) is represented in grey lines; B, Model 2 (PWV→eGFR) in black lines. Dashed lines represent non‐significant pathways (tested, but not included in the model) and solid lines represent significant pathways (included in the model). All models are adjusted for body mass index, serum uric acid, LDL cholesterol, urinary albumin/creatinine ratio, serum glucose, and antihypertensive treatment. Using HDL Cholesterol and HbA1c instead of LDL cholesterol did not change the size or the direction of the effects. eGFR, estimated glomerular filtration rate; PWV, pulse wave velocity; SBP, systolic blood pressure
Table 3.
Direct, indirect and total effects of urinary sodium on endogeneous variables in hypertensive and normotensive patients
| Hypertensive patients (600) | Normotensive patients (959) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Direct effect | Indirect effect | Total effect | Direct effect | Indirect effect | Total effect | |||||||
| Endogenous variables | Coeff | P | Coeff | P | Coeff | P | Coeff | P | Coeff | P | Coeff | P |
| Model 1 | ||||||||||||
| eGFR | – | – | – | – | – | – | – | – | – | – | – | – |
| PWV | 0.124 | <0.001 | – | – | 0.124 | <0.001 | 0.099 | 0.001 | – | – | 0.099 | 0.001 |
| SBP | 0.125 | <0.001 | 0.039 | <0.001 | 0.163 | <0.001 | 0.116 | <0.001 | 0.022 | 0.001 | 0.138 | <0.001 |
| Model 2 | ||||||||||||
| eGFR | – | – | −0.047 | <0.001 | −0.047 | <0.001 | – | – | −0.037 | 0.002 | −0.037 | 0.002 |
| PWV | 0.140 | <0.001 | – | – | 0.140 | <0.001 | 0.105 | 0.001 | – | – | 0.105 | 0.001 |
| SBP | 0.125 | <0.001 | 0.048 | <0.001 | 0.173 | <0.001 | 0.116 | <0.001 | 0.026 | 0.003 | 0.142 | <0.001 |
Adjusted for uric acid, urinary albumin/creatinine ratio, LDL‐Cholesterol and serum glucose. In the hypertensive patients we also adjusted for the use of antihypertensive treatment.
In Model 2, where, in contrast to Model 1, PWV can influence eGFR, both the direct and indirect effects of UNaV on SBP remain highly significant. Furthermore, this model reveals that while the indirect effect of UNaV on SBP is primarily mediated by PWV (as in Model 1), PWV has also a direct effect on eGFR which impacts SBP. Similar to Model 1, the indirect effect mediated by PWV and PWV‐induced changes in eGFR accounted for a significant 27.7% of the total effect of urinary sodium on SBP (Table 3).
Both models showed a close fit, which indicates that the effect of UNaV on SBP is mediated by PWV and that the reciprocal potentiation between eGFR and PWV contributes to this mechanism.
3.5. Normotensive patients
Figure 4 shows the structural equation models for the SBP outcome in the normotensive population. The qualitative relationships between the variables tested were similar to those found in hypertensive patients, for both of the models tested. While still significant, the contribution of PWV to the total effect of UNaV on SBP was slightly lower (15.9% for Model 1% and 18.3% for Model 2). The fit for both models was excellent, indicating that even in normotensive conditions, there is a plausible bi‐directional relationship between eGFR and PWV, which contributes significantly to the overall effect of UNaV on SBP.
Figure 4.

Path analysis illustrated by the structural equation model with standardized coefficients of direct effects for normotensive patients. A, Model 1 (eGFR→PWV) is represented in grey lines; B, Model 2 (PWV→eGFR) in black lines. Dashed lines represent non‐significant pathways (tested, but not included in the model) and solid lines represent significant pathways (included in the model). All models are adjusted for body mass index, serum uric acid, LDL cholesterol, urinary albumin/creatinine ratio, and serum glucose. Using HDL cholesterol and HbA1c instead of LDL cholesterol did not change the size or the direction of the effects. eGFR, estimated glomerular filtration rate; PWV, pulse wave velocity; SBP, systolic blood pressure
4. DISCUSSION
The major findings of this study are as follows: (a) Arterial stiffness partly mediates the effect of salt intake on SBP in both hypertensive and normotensive conditions; (b) There is a reciprocal relationship between renal function and arterial stiffness which ultimately impacts SBP, even in the absence of chronic kidney disease. While the deleterious effects of high salt intake, arterial stiffness, and renal dysfunction on blood pressure have long been recognized, this study provides evidence for a relationship of significant magnitude linking high salt intake with increased arterial stiffness, which in turn leads to elevations in SBP. Moreover, by using a cohort from a national survey, we found that the impact of salt intake on arterial stiffness may still contribute to elevations in SBP, even in normotensive conditions.
Increased salt intake is well recognized as one of the most important and modifiable risk factors for hypertension.24, 25 Beyond the positive relationship observed between salt intake and blood pressure in cross‐sectional trials, there are also numerous interventional studies that reported a decrease in blood pressure levels following reduced dietary sodium intake.26 A meta‐analysis, which included 36 randomized controlled trials indicated that reduced sodium intake was associated with a mean decrease in SBP levels of 3.39 mmHg (95%CI 2.46‐3.39 mmHg). This effect was of greater magnitude in hypertensive as compared to normotensive patients. Consistent with these data, the results of the mediation analysis in our study indicate that both the direct and indirect effects of UNaV on SBP are more pronounced in hypertensive than in normotensive patients.
Arterial stiffness represents the distinctive feature of vascular ageing. In their landmark paper, Zheng and colleagues showed that in patients with no medical history of arterial hypertension, ankle‐brachial PWV may predict the onset of arterial hypertension.27 Importantly, this relationship was mostly related to the effect on SBP. However, evidence for the role of high salt intake in promoting arterial stiffness is scarce. Experimental studies suggest that increased salt intake elevates arterial stiffness.28, 29 While the mechanisms responsible for this effect have not been unequivocally identified, chronic alterations in the secretory properties of the vascular smooth muscle and impaired endothelial function appear to underlie arterial stiffening in the presence of high salt intake.30 In a recent meta‐analysis that included 11 randomized controlled trials, lower sodium intake was associated with a significantly lower mean PWV (−2.84%, 95% CI −5.08 to −0.51%) as compared with the higher sodium regimen.31 Although the response of PWV to lowering of salt intake has not been consistently documented in normotensive individuals,32 in the aforementioned meta‐analysis the effect of salt reduction on PWV was not influenced by normo‐/hypertensive status. Our results further reinforce these findings. The direct effect of UNaV on PWV, as a mediator in the relationship between sodium and SBP, was significant in both hypertensive and normotensive patients; however, this effect was larger in the hypertensive patients. Furthermore, the indirect effect of salt intake on SBP (the effect which is mediated mostly through PWV) is also larger in hypertensive patients than in normotensive patients, irrespective of the chosen model.
The mediation analysis in this study was unable to identify any direct relationship between the levels of salt intake, as assessed by UNaV and the prevailing renal function indicated by eGFR. As only patients without chronic kidney disease were included in this study, this finding is not unexpected. Specifically, if the kidneys are capable of excreting the increased sodium load at the same perfusion pressure, volume expansion and attendant increases in blood pressure would not occur.33 This is likely the case in this patient population, where renal functional reserve is still sufficient to preserve sodium balance. Nevertheless, this compensatory mechanism may mask the increased filtration in individual nephrons, resulting in unchanged eGFR in those patients where subtle nephron loss has already occurred. Notwithstanding, when significant reductions in eGFR are present, even below the clinical threshold for inclusion in a CKD category, an increase in blood pressure in response to elevated salt intake is to be expected. Indeed, as discussed below, mediation analysis indicates that eGFR per se does have an impact (albeit quantitatively minor) on SBP.
Another important finding of our analysis is that eGFR and PWV are inversely and reciprocally interconnected. More to the point, arterial stiffness causes a decrease in renal function and conversely, low eGFR may cause increased arterial stiffness. However, it remains unknown whether renal dysfunction, interpreted as a decrease in eGFR values, precedes the increase in arterial stiffness or vice versa. There are numerous studies that showed that a decreased eGFR is associated with an increased PWV.34, 35, 36, 37 There are several pathological mechanisms that could explain how a decline in renal function would determine an increase in arterial stiffness, including the accumulation of advanced glycation end products, elastin fragmentation and replacement with collagen, increased lipid peroxidation, deposition of materials in the arterial wall, hyperplasia of smooth muscle cell, enhanced renin‐angiotensin system activity, increased inflammatory markers, and excess fluid volume.37 On the other hand, excessive aortic stiffness increases transmission of pulsatile power into the peripheral vasculature. In line with this assumption, Woodard et al have shown that higher PWV is negatively related to measured GFR and that this relation is substantially mediated by higher‐flow pulsatility through microvascular constriction or rarefaction and higher vascular resistance in functional kidney tissues.38 Nevertheless, the effect of eGFR in this mediation analysis is rather low, when we consider both the lack of a direct effect of UNaV on eGFR values and also the overall effect in the UNaV→PWV→eGFR→SBP pathway as compared to the direct effect of the PWV in the UNaV→PWV→SBP pathway. Thus, the prominent pathophysiological mechanism explaining the impact of dietary sodium on SBP appears to be related to the UNaV→PWV→SBP pathway.
There are several limitations to our study. First, our study was cross‐sectional and, therefore, cannot show causality. By using a mediation analysis we can support potential causative pathways, but these pathways must be later confirmed in adequately performed studies. Secondly, we used the Kawasaki formula for estimating the 24‐hour urinary sodium excretion and not the actual 24‐hour measurement of urinary sodium. However, this method was already validated and showed a good intraclass coefficient with the actual 24‐hour measurement of urinary sodium.5 Thirdly, our cohort comes from a single country and, as such, our results may not be inferred to other populations. However, our population was large and homogenous, excluding patients with presumed chronic kidney disease (reduced eGFR and/or proteinuria).
5. PERSPECTIVES
This study may represent an important step in understanding the relationship between UNaV (as a surrogate of salt intake) and SBP. Taken together, these findings support the conclusion that UNaV influences SBP, both directly, through the effect on PWV, and indirectly, as well as the bidirectional relationship between PWV and eGFR in this pathway. Additional prospective as well as interventional trials are, however, needed to test the causal inferences presented here.
CONFLICT OF INTEREST
None.
Siriopol D, Covic A, Iliescu R, et al. Arterial stiffness mediates the effect of salt intake on systolic blood pressure. J Clin Hypertens. 2018;20:1587–1594. 10.1111/jch.13399
REFERENCES
- 1. Lim SS, Vos T, Flaxman AD, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2224‐2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lewington S, Clarke R, Qizilbash N, Peto R, Collins R. Prospective Studies Collaboration. Age‐specific relevance of usual blood pressure to vascular mortality: a meta‐analysis of individual data for one million adults in 61 prospective studies. Lancet. 2002;360:1903‐1913. [DOI] [PubMed] [Google Scholar]
- 3. Rapsomaniki E, Timmis A, George J, et al. pressure and incidence of twelve cardiovascular diseases: lifetime risks, healthy life‐years lost, and age‐specific associations in 1.25 million people. Lancet. 2014;383:1899‐1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Intersalt Cooperative Research Group . Intersalt: an international study of electrolyte excretion and blood pressure — results for 24 hour urinary sodium and potassium excretion. BMJ. 1988;297:319‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mente A, O’Donnell MJ, Rangarajan S, et al. Association of urinary sodium and potassium excretion with blood pressure. N Engl J Med. 2014;371:601‐611. [DOI] [PubMed] [Google Scholar]
- 6. Graudal NA, Hubeck‐Graudal T, Jurgens G. Effects of low sodium diet versus high sodium diet on blood pressure, renin, aldosterone, catecholamines, cholesterol, and triglyceride. Cochrane Database Syst Rev. 2017;4:CD004022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de Cavanagh E, Ferder LF, Ferder MD, Stella IY, Toblli JE, Inserra F. Vascular structure and oxidative stress in salt‐loaded spontaneously hypertensive rats: effects of losartan and atenolol. Am J Hypertens. 2010;23:1318‐1325. [DOI] [PubMed] [Google Scholar]
- 8. Park S, Park JB, Lakatta EG. Association of central hemodynamics with estimated 24‐h urinary sodium in patients with hypertension. J Hypertens. 2011;29:1502‐1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. He FJ, Marciniak M, Visagie E, et al. Effect of modest salt reduction on blood pressure, urinary albumin, and pulse wave velocity in white, black and Asian mild hypertensives. Hypertension. 2009;54:482‐488. [DOI] [PubMed] [Google Scholar]
- 10. Hummel SL, Seymour EM, Brook RD, et al. Low‐sodium dietary approaches to stop hypertension diet reduces blood pressure, arterial stiffness, and oxidative stress in hypertensive heart failure with preserved ejection fraction. Hypertension. 2012;60:1200‐1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liao D, Arnett DK, Tyroler HA, et al. Arterial stiffness and the development of hypertension. The ARIC study. Hypertension. 1999;34(2):201‐206. [DOI] [PubMed] [Google Scholar]
- 12. Dernellis J, Panaretou M. Aortic stiffness is an independent predictor of progression to hypertension in nonhypertensive subjects. Hypertension. 2005;45:426‐431. [DOI] [PubMed] [Google Scholar]
- 13. Najjar SS, Scuteri A, Shetty V, et al. Pulse wave velocity is an independent predictor of the longitudinal increase in systolic blood pressure and of incident hypertension in the Baltimore Longitudinal Study of Aging. J Am Coll Cardiol. 2008;51:1377‐1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takase H, Dohi Y, Toriyama T, et al. Brachial‐ankle pulse wave velocity predicts increase in blood pressure and onset of hypertension. Am J Hypertens. 2011;24:667‐673. [DOI] [PubMed] [Google Scholar]
- 15. Kaess BM, Rong J, Larson MG, et al. Aortic stiffness, blood pressure progression, and incident hypertension. JAMA. 2012;308:875‐881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Coresh J, Astor BC, Greene T, Eknoyan G, Levey AS. Prevalence of chronic kidney disease and decreased kidney function in the adult US population: Third National Health and Nutrition Examination Survey. Am J Kidney Dis. 2003;41:1‐12. [DOI] [PubMed] [Google Scholar]
- 17. Nilsson C, Christensson A, Nilsson PM, Bennet L. Renal function and its association with blood pressure in Middle Eastern immigrants and native Swedes. J Hypertens. 2017;35(12):2493‐2500. [DOI] [PubMed] [Google Scholar]
- 18. Dorobantu M, Darabont R, Dimulescu D, et al. New national epidemiological survey for the assessment of trend in hypertension’s prevalence, treatment and control among the adult population of Romania: SEPHAR III: design and methodology. J Hypertens Res. 2016;2:143‐152. [Google Scholar]
- 19. Dorobantu M, Tautu OF, Dimulescu D, et al. Perspectives on hypertension’s prevalence, treatment and control in a high cardiovascular risk East European country: data from the SEPHAR III survey. J Hypertens. 2018;36(3):690‐700. [DOI] [PubMed] [Google Scholar]
- 20. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int Suppl. 2013;3:1‐150. https://kdigo.org/wp-content/uploads/2017/02/KDIGO_2012_CKD_GL.pdf [Google Scholar]
- 21. Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH‐ESC Guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology. J Hypertens. 2013;31:1281‐1357. [DOI] [PubMed] [Google Scholar]
- 22. Kawasaki T, Itoh K, Uezono K, Sasaki H. A simple method for estimating 24 h urinary sodium and potassium excretion from second morning voiding urine specimen in adults. Clin Exp Pharmacol Physiol. 1993;20:7‐14. [DOI] [PubMed] [Google Scholar]
- 23. Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150(9):604‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weinberger MH. Sodium, potassium, and blood pressure. Am J Hypertens. 1997;10:46S‐48S. [PubMed] [Google Scholar]
- 25. Chien KL, Hsu HC, Chen PC, et al. Urinary sodium and potassium excretion and risk of hypertension in Chinese: report from a community‐based cohort study in Taiwan. J Hypertens. 2008;26:1750‐1756. [DOI] [PubMed] [Google Scholar]
- 26. He FJ, Li J, Macgregor GA. Effect of longer term modest salt reduction on blood pressure: Cochrane systematic review and meta‐analysis of randomised trials. BMJ. 2013;346:f1325. [DOI] [PubMed] [Google Scholar]
- 27. Zheng X, Jin C, Liu Y, et al. Arterial stiffness as a predictor of clinical hypertension. J Clin Hypertens. 2015;17:582‐591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Limas C, Westrum B, Limas CJ, Cohn JN. Effect of salt on the vascular lesions of spontaneously hypertensive rats. Hypertension. 1980;2:477‐489. [DOI] [PubMed] [Google Scholar]
- 29. Partovian C, Benetos A, Pommies JP, Safar ME. Effects of a chronic high‐salt diet on large artery structure: role of endogenous bradykinin. Am J Physiol. 1998;274:H1423‐H1428. [DOI] [PubMed] [Google Scholar]
- 30. Safar ME, Thuilliez C, Richard V, Benetos A. Pressure‐independent contribution of sodium to large artery structure and function in hypertension. Cardiovascular Res. 2000;46(2):269‐276. [DOI] [PubMed] [Google Scholar]
- 31. D’Elia L, Galletti F, La Fata E, Sabino P, Strazzullo P. Effect of dietary sodium restriction on arterial stiffness: systematic review and meta‐analysis of the randomized controlled trials. J Hypertens. 2018;36(4):734‐743. [DOI] [PubMed] [Google Scholar]
- 32. Todd AS, Macginley RJ, Schollum JB, et al. Dietary sodium loading in normotensive healthy volunteers does not increase arterial vascular reactivity or blood pressure. Nephrology. 2012;17:249‐256. [DOI] [PubMed] [Google Scholar]
- 33. Hall JE. Renal dysfunction, rather than nonrenal vascular dysfunction, mediates salt‐Induced hypertension. Circulation. 2016;133:894‐906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Briet M, Bozec E, Laurent S, et al. Arterial stiffness and enlargement in mild to moderate chronic kidney disease. Kidney Int. 2006;69:350‐357. [DOI] [PubMed] [Google Scholar]
- 35. Kawamoto R, Kohara K, Tabara Y, et al. An association between decreased estimated glomerular filtration rate and arterial stiffness. Intern Med. 2008;47:593‐598. [DOI] [PubMed] [Google Scholar]
- 36. Tomiyama H, Tanaka H, Hashimoto H, et al. Arterial stiffness and declines in individualswith normal renal function/early chronic kidney disease. Atherosclerosis. 2010;212(1):345‐350. [DOI] [PubMed] [Google Scholar]
- 37. Sengstock D, Sands RL, Gillespie BW, et al. Dominance of traditional cardiovascular risk factors over renal function in predicting arterial stiffness in subjects with chronic kidney disease. Nephrol Dial Transplant. 2010;25:853‐861. [DOI] [PubMed] [Google Scholar]
- 38. Woodard T, Sigurdsson S, Gotal JD, et al. Mediation analysis of aortic stiffness and renal microvascular function. J Am Soc Nephrol. 2015;26(5):1181‐1187. [DOI] [PMC free article] [PubMed] [Google Scholar]