

Abstract
Pentraxin‐3 is a sensitive marker of inflammation that plays dual roles, pathogenic and cardioprotective, in the progression of cardiovascular diseases. Inflammation is intimately involved in salt‐induced hypertension. We investigated the responses of pentraxin‐3 to sodium and potassium supplementation to elucidate the potential role of pentraxin‐3 in salt‐induced hypertension. A total of 48 participants from northwest China were enrolled. All participants were maintained on a 3‐day normal diet, which was sequentially followed by a 7‐day low‐sodium diet, a 7‐day high‐sodium diet, and a 7‐day high‐sodium plus potassium diet. Plasma concentrations of pentraxin‐3 were assessed using ELISA. Plasma pentraxin‐3 decreased significantly during the low‐salt period compared to baseline (0.57 ± 0.19 ng/mL vs 0.72 ± 0.33 ng/mL, P = .012) and increased during the high‐salt period (0.68 ± 0.26 ng/mL vs 0.57 ± 0.19 ng/mL, P = .037). Potassium supplementation inhibited salt‐induced increase in pentraxin‐3 (0.56 ± 0.21 ng/mL vs 0.68 ± 0.26 ng/mL, P = .015). Ln‐transformed pentraxin‐3 at baseline was inversely correlated with BMI (r = −.349, P = .02), DBP (r = −.414, P = .005), MAP (r = −.360, P = .017). We found a positive correlation between the ln‐transformed concentrations of pentraxin‐3 and 24‐hour urinary sodium during low and high Na+ periods (r = .269, P = .012) and a negative relationship with 24 hours urinary potassium excretion during high‐salt and high‐salt plus potassium periods (r = −.246, P = .02). These correlations remained significant after adjusting for confounders. Pentraxin‐3 responses were more prominent in salt‐sensitive individuals than salt‐resistant individuals. Dietary salt and potassium interventions significantly altered circulating pentraxin‐3.
Keywords: inflammation, pentraxin‐3, potassium, salt intake, salt sensitivity
1. INTRODUCTION
Sodium and potassium are 2 important environmental factors in the progression of hypertension. Previous studies by our group and other researchers have revealed that high salt intake contributed to elevated blood pressure (BP) and target organ damage, and high potassium intake protected against salt‐induced injuries.1, 2, 3 However, the response to salt was variable between individuals, which may be attributed to salt sensitivity. Salt‐induced damage was more prominent in salt‐sensitive (SS) individuals than salt‐resistant (SR) individuals.1, 4, 5
Hypertension is a low‐grade inflammatory process. Inflammatory markers, such as C‐reactive protein (CRP), procalcitonin, and interleukin (IL)‐6, are up‐regulated in hypertensive patients.6, 7 Inflammation also plays a pivotal role in salt‐induced hypertension and target organ damage.2, 8 High sodium intake is associated with increased systemic inflammation and boosts T cells and macrophages activation to a proinflammatory phenotype.9 Clinical studies demonstrated that high salt intake was independently and positively associated with serum levels of CRP, tumor necrosis factor‐α, and monocyte chemoattractant protein‐1.10 Potassium supplementation somewhat alleviated the excessive salt‐induced inflammatory process.11
Pentraxin‐3 (PTX‐3), a classic prototype of long pentraxins, is a sensitive inflammatory marker. PTX‐3 is mainly synthesized at the local sites of inflammation by various cell types during exposure to primary inflammatory signals, such as endothelial cells, smooth muscle cells, and monocyte/macrophages.12 Therefore, PTX‐3 effectively reflects localized inflammation and damage. Increased PTX‐3 is associated with cardiovascular diseases (CVD). Elevated PTX‐3 was reported in patients with atherosclerosis, acute myocardial infarction, unstable angina pectoris, and vasculitis.13, 14
Besides, PTX‐3 is higher in newly diagnosed hypertensive and preeclampsia patients and elevated PTX‐3 parallels increase in systolic blood pressure (SBP) and diastolic blood pressure (DBP).6, 7, 15 Carrizzo and colleagues15 demonstrated that PTX‐3 contributed to endothelial dysfunction and impaired endothelium‐dependent vasodilation. Exogenous PTX‐3 administration increased BP in mice in a P‐selectin‐dependent manner. A single nucleotide polymorphism of PTX‐3 was also associated with BP. Chronic kidney disease patients with a PTX‐3 AA genotype of SNP rs3816527 on exon 2 exhibited a higher BP.16 PTX‐3 is involved in inflammation‐associated diseases, but the exact role of PTX‐3 remains controversial. Compelling evidence demonstrated anti‐inflammatory and cardioprotective roles of PTX‐3.17, 18 PTX‐3 deficient mice exhibited exacerbated heart and renal damage after ischemic‐reperfusion injury. High‐density lipoproteins and IL‐10, known as atheroprotective molecules, could enhance the expression of PTX‐3.19 Deban and colleagues20 found that PTX‐3 could regulate inflammatory process through binding with P‐selectin and attenuating neutrophil recruitment at sites of inflammation. Therefore, the role of PTX‐3 in CVD merits further considerations.
Lifestyle interventions, such as energy restriction or physical activity, could exert influence on the levels of PTX‐3,18, 21 but the impact of dietary sodium and potassium interventions on PTX‐3 levels is not known. The present intervention study performed a comprehensive assessment of the effects of Na+ and K+ supplementation on plasma levels of PTX‐3 and examined the potentiate correlation between PTX‐3 and BP.
2. METHODS
2.1. Participants
In Liquan, XianYang of ShaanXi province, which is a rural county of northern China, 48 participants were enrolled following completion of a detailed questionnaire and examination. Participants with a history of severe cardiovascular diseases (BP ≥ 160/100), diabetes mellitus, abnormal liver or renal function, malignant diseases, secondary hypertension, or alcohol abuse were all excluded. This research adhered to the guidelines laid down in the Declaration of Helsinki. The institutional ethics committee of Xi'an Jiaotong University Medical School approved the study protocol (code: 2015‐047). All patients were fully informed of the study procedures and provided their consent.
2.2. Dietary intervention
The protocol for intervention was performed as described previously.22 As shown in Figure 1, all participants underwent a 3‐day normal sodium diet, during which demographic information and anthropometric data (height, weight, waist circumference) were collected by trained staff. After that, participants sequentially received a 7‐day low‐salt diet, a 7‐day high‐salt diet, and a 7‐day high‐salt plus potassium supplement diet. All participants were given detailed dietary instructions to avoid additional salt consumption during the baseline investigation. All foods were prepared without salt in a research kitchen. Participants were required to gather together and eat under the supervision of the investigators with prepackaged salt and potassium tablets.
Figure 1.
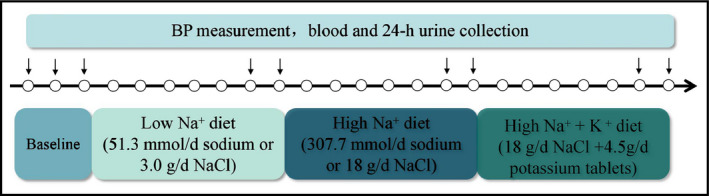
The protocol of dietary intervention
2.3. BP measurements
Office BP was measured by certified physicians using a mercury sphygmomanometer following the protocol recommended by the American Heart Association. BP was measured 3 times with 1‐minute intervals each day of baseline and on the 6th and 7th day of each intervention period. Participants were asked to remove all clothing covering the cuff location and were instructed to sit in a resting position for more than 10 minutes with their legs uncrossed and back and arm supported. The means of 9 measurements at baseline and 6 measurements at each intervention period were recorded as SBP and DBP. Mean arterial pressure (MAP) was calculated using the formula: (SBP − DBP)/3 + DBP. Participants with SBP ≥ 140 mm Hg, DBP ≥ 90 mm Hg, and/or a history of hypertension with current use of antihypertensive medications were defined as hypertensive patients. Participants whose MAP increased > 10 mm Hg while changing from a low Na+ to a high Na+ diet were defined as SS individuals, and the other participants were defined as SR individuals.
2.4. Laboratory analyses
Venous blood samples were obtained after an overnight fasting on the last day of each period. Blood samples were centrifuged at 2500 g and 4°C for 15 minutes within 2 hours of collection, packaged in aliquots, and stored at −80°C until the end of the study for analysis. Basic biochemistry parameters, including lipid profiles, fasting serum glucose, serum creatinine, were analyzed using an automatic biochemical analyzer (Hitachi, Ltd., Tokyo, Japan). Plasma concentrations of PTX‐3 were determined using commercial enzyme‐linked immunosorbent assay kits (ELISA) (R&D Systems Inc., Minneapolis, MN, USA).The sensitivity for PTX‐3 was 0.116 ng/mL.
2.5. Analysis of 24‐hour urine
Under staff guidance 24‐hour urine was collected at 8:00 am on the 2nd day of baseline and the 6th day of each intervention period. Participants were asked to discard the first morning urine and collect all urine secretions for the next 24 hours. Urine sodium and potassium concentrations were analyzed using ion‐selective electrodes (Hitachi, Ltd., Tokyo, Japan). The sodium and potassium excretions in 24‐hour urine were calculated as the 24‐hour total urine volume multiplied by sodium or potassium concentrations.
2.6. Statistical analysis
Statistical analyses were performed using IBM SPSS Statistics 22.0 (IBM Corp., Chicago, IL, USA). Variable distribution was examined using the Kolmogorov‐Smirnov test. Continuous variables are presented as the means ± standard deviation (SD) for normal distributions and as medians (interquartile range) for skewed distribution. Categorical variables are expressed as frequencies with percentages (n [%]). PTX‐3 concentrations were right skewed, and natural log (ln) transformation was performed during correlation analyses. Differences in biochemical parameters between different intervention periods were assessed using 1‐way ANOVA with repeated measures. Differences between SS and SR participants were assessed using unpaired Student's t test or chi‐square test as appropriate. Pearson's and Spearman's correlation coefficients were calculated to evaluate the relationships between PTX‐3 and related variables. Integrated trapezoidal area under curve with respect to ground (AUCg) and area under curve with respect to increase (AUCi) were calculated as previously reported to compare the differences of the responses of parameters to interventions between SS and SR participants.18, 23 A 2‐sided P value of .05 was used to evaluate statistical significance.
3. RESULTS
3.1. Clinical characteristics of participants
A total of 48 participants completed all the interventions. Table 1 summarizes the general characteristics and biochemical parameters of all participants divided by salt sensitivity. Four hypertensive patients were found, and none of these patients was receiving medication. There were 13 SS participants and 35 SR participants. SS participants were older than SR participants (P = .008). The gender distribution, SBP, DBP, lipid profiles, fast blood glucose, and creatinine showed no obvious difference between SS and SR groups.
Table 1.
Baseline characteristics of participants by salt sensitivity
| Parameters | Total (n = 48) | SS (n = 13) | SR (n = 35) |
|---|---|---|---|
| Age (y) | 55 (48‐62) | 62 (55‐65) | 54 (47‐59) |
| Sex (male, %) | 47.9 | 46.2 | 48.6 |
| BMI (kg/m2) | 23.58 ± 8.62 | 23.76 ± 2.79 | 23.51 ± 3.02 |
| SBP (mm Hg) | 110 ± 15 | 113 ± 18 | 109 ± 13 |
| DBP (mm Hg) | 73 ± 9 | 72 ± 7 | 73 ± 9 |
| TC (mmol/L) | 3.99 (3.38, 4.78) | 4.42 (3.43, 4.42) | 3.79 (3.37, 4.87) |
| TG (mmol/L) | 1.31 ± 0.44 | 1.48 ± 1.00 | 1.25 ± 0.48 |
| HDL‐C (mmol/L) | 1.19 ± 0.23 | 1.17 ± 0.27 | 1.20 ± 0.22 |
| LDL‐C (mmol/L) | 2.20 (1.73, 2.80) | 2.42 (1.47, 2.78) | 2.06 (1.74, 2.81) |
| FBG (mmol/L) | 3.87 ± 0.68 | 4.01 ± 1.03 | 3.82 ± 0.51 |
| CRE (μmol/L) | 56.33 ± 8.65 | 56.58 ± 6.64 | 56.23 ± 9.37 |
BMI, body mass index; CRE, creatinine; DBP, diastolic blood pressure; FBG, fasting blood glucose; HDL‐C, high‐density lipoprotein cholesterol; LDL‐C, low density lipoprotein cholesterol; SBP, systolic blood pressure; TC, total cholesterol; TG, triglyceride.
3.2. BP, 24‐hour urinary sodium and potassium variations during interventions
Systolic blood pressure and MAP in the high‐salt period increased significantly compared to the low‐salt diet (P = .013 and .023, respectively; Table 2). DBP was slightly but not significantly elevated while changing from the low Na+ to the high Na+ diet (P = .056). Potassium supplementation greatly inhibited the salt‐induced elevation in SBP, DBP, MAP (P = .004, .005 and .003, respectively). At the end of each intervention period, 24‐hour sodium and potassium excretions were measured, and the results well reflected the dietary changes. Results showed that 24‐hour urinary sodium excretion decreased significantly during the low‐salt diet (P < .001), increased during high salt intake (P < .001) and further increased after potassium supplementation compared to high‐salt diet (P < .001). Results indicated that 24‐hour urinary potassium decreased from baseline to the low‐salt diet (P = .002) and significantly increased after potassium supplement (P < .001; Table 2). These results strongly indicated that participants complied with the intervention protocol.
Table 2.
Blood pressure levels and 24‐h urinary sodium and potassium excretions at baseline and during dietary interventions
| Parameters | Baseline | LS | HS | HS+K | P value |
|---|---|---|---|---|---|
| SBP (mm Hg) | 110 ± 15 | 109 ± 12 | 116 ± 17 | 108 ± 13 | .020 |
| DBP (mm Hg) | 73 ± 9 | 74 ± 8 | 77 ± 9 | 72 ± 9 | .018 |
| MAP (mm Hg) | 85 ± 10 | 86 ± 9 | 90 ± 11 | 84 ± 10 | .014 |
| UNa (mmol/24 h) | 153.5 ± 48.7 | 81.2 ± 34.3 | 167.6 ± 60.5 | 246.9 ± 76.6 | <.001 |
| UK (mmol/24 h) | 43.1 ± 21.4 | 29.3 ± 12.4 | 31.7 ± 18.1 | 70.5 ± 26.3 | <.001 |
DBP, diastolic blood pressure; HS + K, high‐salt plus potassium diet; HS, high‐salt diet; LS, low‐salt diet; MAP, mean arterial pressure; SBP, systolic blood pressure; UK, 24 h urinary potassium excretion; UNa, 24 h urinary sodium excretion.
3.3. PTX‐3 responses to interventions
Plasma PTX‐3 significantly decreased during the low‐salt period compared to baseline (0.57 ± 0.19 ng/mL vs 0.72 ± 0.33 ng/mL, P = .012). While changing to high Na+ period, circulating PTX‐3 elevated (0.68 ± 0.26 ng/mL vs 0.57 ± 0.19 ng/mL, P = .037). Potassium supplementation inhibited the high‐salt‐induced increase of PTX‐3 (0.56 ± 0.21 ng/mL vs 0.68 ± 0.26 ng/mL, P = .015) (Figure 2A).
Figure 2.
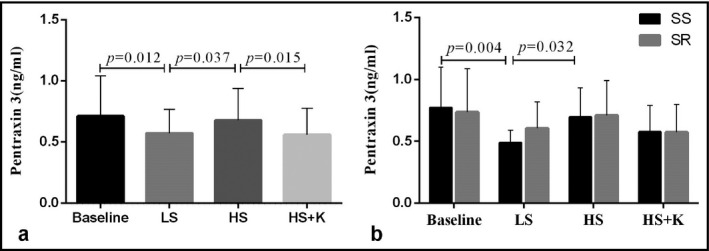
The plasma levels of pentraxin‐3 of all participants (A) and by salt sensitivity (B) in each period. Data were analyzed by using 1‐way ANOVA with repeated measures. HS, high‐salt period; HS + K, high‐salt plus potassium period; LS, low‐salt period; SS, salt‐sensitive participants; SR, salt‐resistant participants
3.4. Correlations of PTX‐3 with metabolic parameters and 24‐hour urinary sodium and potassium
Pentraxin‐3 is intimately correlated with metabolic diseases. Therefore, we examined the correlations between ln‐transformed PTX‐3 and various metabolic parameters at baseline in this group. Correlation analyses revealed that ln‐transformed PTX‐3 inversely correlated with BMI (r = −.349, P = .02), DBP (r = −.414, P = .005), and MAP (r = −.360, P = .017; Table 3). The correlations with SBP, total cholesterol (TC), triglycerides (TG), high‐density lipoprotein cholesterol (HDL‐C), low‐density lipoprotein cholesterol (LDL‐C), and fasting blood glucose (FBG) were not statistically significant.
Table 3.
The correlations between pentraxin 3 and metabolic parameters
| Parameters | r | P value |
|---|---|---|
| BMI | −.349 | .020* |
| SBP | −.297 | .500 |
| DBP | −.414 | .005* |
| MAP | −.360 | .017* |
| TC | −.226 | .141 |
| TG | −.159 | .303 |
| HDL‐C | −.007 | .965 |
| LDL‐C | −.238 | .121 |
| FBG | −.257 | .093 |
BMI, body mass index; CRE, creatinine; DBP, diastolic blood pressure; FBG, fasting blood glucose; HDL‐C, high‐density lipoprotein cholesterol; LDL‐C, low‐density lipoprotein cholesterol; MAP, mean arterial pressure; SBP, systolic blood pressure; TC, total cholesterol; TG, triglyceride.
*P < .05.
We found a positive correlation between the ln‐transformed concentrations of PTX‐3 and 24‐hour urinary sodium during low and high Na+ periods (r = .269, P = .012; Figure 3A). This correlation remained significant after adjusting for age, gender, BMI, and DBP (adjusted r = .225, P = .041). The ln‐transformed concentrations of PTX‐3 was negatively correlated with 24‐hour urinary potassium excretion during the high Na+ and high Na+ plus K+ periods (r = −.246, P = .02; Figure 3B). It was still significant after confounders were adjusted (adjusted r = −.263, P = .015).
Figure 3.
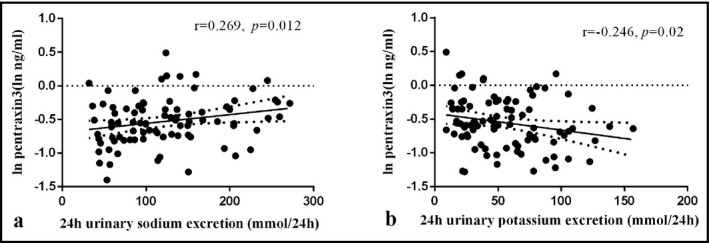
The correlations between 24 h urinary sodium excretions during low‐ and high‐salt periods (A), 24 h urinary potassium excretions during high‐salt and high‐salt plus potassium periods (B) and ln‐transformed PTX‐3. Pearson correlation tests were done while correlation analyses
3.5. The influence of salt sensitivity on the response of PTX‐3
Finally, we examined the influence of salt sensitivity on the responses of plasma PTX‐3. At baseline, PTX‐3 was nonsignificantly higher in SS than in SR participants (0.75 ± 0.30 ng/mL vs 0.70 ± 0.34 ng/mL, P > .05). Plasma PTX‐3 was lower in SS participants than the SR individuals during the low‐salt period (0.48 ± 0.12 ng/mL vs 0.60 ± 0.21 ng/mL, P = .025), but no significant differences were observed during the high Na+ and high Na+ plus K+ period. Furthermore, the responses of PTX‐3 to interventions were more prominent in SS than in SR individuals. Plasma PTX‐3 of SS individuals declined from 0.75 ± 0.30 ng/mL to 0.48 ± 0.12 ng/mL (P = .004) after changing to low salt intake and increased to 0.68 ± 0.22 ng/mL during the high‐salt period (compared with low Na+ period, P = .032). The concentrations of PTX‐3 did not significantly decrease (0.58 ± 0.18 ng/mL) after potassium supplementation. However, the serum PTX‐3 of SR participants exhibited no obvious differences during interventions (Figure 2B).
To further assess the influence of salt sensitivity on the interventions, we calculated the AUCg and AUCi of SBP, DBP, 24‐hour urinary Na+, 24‐hour urinary K+, and plasma PTX‐3 in SS and SR participants. However, the AUCg and AUCi of indexes exhibited no obvious differences between the 2 groups, except for SBP (Figure 4A‐E). The separate effects of sodium and potassium on PTX‐3, assessed by AUCg from baseline to high‐salt period and AUCg from the high‐salt diet to high‐salt plus potassium period, were not significantly different between SS and SR participants (Figure 4F).
Figure 4.
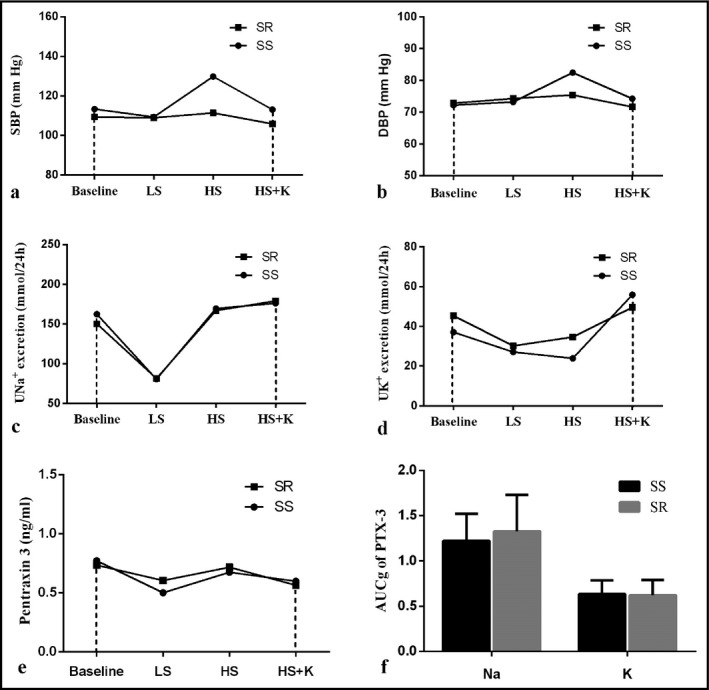
Delineate the AUCg of SBP (A), DBP (B), 24 h urinary sodium (C) and potassium (D) excretion, plasma PTX‐3 (E) during interventions by salt sensitivity. F, Showed the separate effects of sodium and potassium on the plasma level of PTX‐3 divided by salt sensitivity. Differences between SS and SR group were compared via unpaired Student's t test. AUCg, area under curve with respect to ground; DBP, diastolic blood pressure; LS, low‐salt period; HS, high‐salt period; HS + K, high‐salt plus potassium period; PTX‐3, pentraxin 3; SBP, systolic blood pressure; SR, salt‐resistant participants; SS, salt‐sensitive participants
4. DISCUSSION
To the best of our knowledge, this report is the first study to specifically demonstrate the influence of salt and potassium interventions on plasma levels of PTX‐3. Circulating PTX‐3 declined during a low‐salt diet and increased after high salt intake. Dietary potassium supplementation blunted the high‐salt‐induced increase in PTX‐3. 24‐hour urinary sodium excretion positively correlated with PTX‐3 but 24‐hour urinary potassium excretion was negatively associated with PTX‐3. The responses of PTX‐3 to dietary interventions were more pronounced in SS participants. Results from this study further strengthen the need for salt restriction and potassium supplementation, especially in SS participants.
Inflammation is an important mediator of salt‐induced hypertension. High‐salt conditions promote the secretion of several inflammatory cytokines and prompt cells to differentiate to a proinflammatory rather than anti‐inflammatory phenotype.2, 9, 10 Clinical data demonstrated that high salt intake was associated with higher levels of monocytes, IL‐6, and IL‐23 and lower IL‐10 level.24 Experimental studies demonstrated that high salt boosted the activation of proinflammatory Th17 cells and M1 macrophages and blunted the activation of M2 macrophages.9 As speculated, we found PTX‐3 was positively associated with dietary salt intake. High salt intake contributed to increased plasma PTX‐3. High potassium intake alleviated the detrimental effects of high salt via enhanced sodium excretion.4 An animal study also demonstrated that potassium played a prominent role in suppressing renal inflammation and relieving renal injuries independent of salt intake.25 Potassium supplementation exhibited a protective role against salt‐induced increase in BP and PTX‐3 in the present study. A further increase in 24‐hour sodium excretion after potassium supplementation may explain the effects of potassium.
However, Gijsbers and colleagues11 observed no significant effect of sodium and potassium interventions on other inflammatory markers, including IL‐1β, IL‐6, IL‐8, and CRP during sensitivity analysis. The following reasons may explain the divergent results. First, participants in the Gijsbers study were all hypertensives with a fasting supine SBP between 130 and 159 mm Hg, and the response to dietary intervention may be aberrant in hypertensive patients. We excluded stages 2 and 3 hypertensive patients, and only 4 hypertensive patients were included in our study. Besides, the Gijsbers study used 2.4 g/d and 5.4 g/d sodium as the low‐ and high‐salt diet. We followed the protocol of the GenSalt study and used 1.2 g/d and 7.1 g/d sodium during the low‐ and high‐salt periods. The bigger difference in salt intake from the low‐ to high‐salt diets may have contributed to a more obvious result.
Salt sensitivity is an intermediate phenotype of hypertension that greatly correlates with severe target organ damage and poor clinical outcomes. In accordance, we found the responses of PTX‐3 were pronounced only in SS participants, which suggested more severe salt‐induced inflammation. Our group found that high salt failed to elevate the plasma level of adiponectin that could inhibit the expression of PTX‐3 in SS participants, which was concordant with present results. However, there were no obvious differences in the AUCg and AUCi of various parameters between SS and SR participants, except SBP.
Pentraxin‐3 increases after high‐salt intervention, but whether this increase is simply a result of inflammation or involved in the alleviation of inflammatory processes is still controversial. PTX‐3 is actively synthesized during inflammation and it is a cardiovascular risk factor. However, accumulating studies have revealed a cardioprotective role of PTX‐3, especially in acute ischemia‐reperfusion injury of heart and kidneys.18, 26 PTX‐3 may also regulate the inflammatory process via binding with P‐selectin and attenuating neutrophil recruitment at sites of inflammation.20 PTX‐3 deficiency in ApoE−/− mice contributed to larger atherosclerotic lesions, which was accompanied with increased proinflammatory genes expression and macrophage accumulation within the plaque.27 HDL and IL‐10 could enhance PTX‐3 expression but interferon‐γ is an inhibitor of PTX‐3.19 One clinical study reported that PTX‐3 was inversely associated with fat mass and CRP and positively associated with IL‐10.21 This evidence strengthened the cardioprotective role of PTX‐3. The potential effects of apoptotic cells clearance and modulation of innate immunity may also contribute to the cardioprotective role of PTX‐3.17
We found that PTX‐3 was negatively correlated with DBP and MAP at baseline in the present study, which was in contrast to previous studies that reported a positive correlation between PTX‐3 and BP.6, 7 There was also a negative correlation between PTX‐3 and BMI, which was supported by previous studies.21 The correlation coefficients of PTX‐3 with other metabolic parameters were negative but did not reach statistical significance. BP and PTX‐3 were synergistically altered during the intervention periods, but a weak but significant correlation between DBP and PTX‐3 remained (r = −.219, P = .011). These results indicated a possible protective role of PTX‐3 in response to salt intervention rather than a simple marker of inflammation. The increase in PTX‐3 may be a feedback mechanism involved in the dampening of excessive inflammation.
There were some limitations in the present study. Participants reported similar dietary habits and exhibited good compliance to the study protocol, but the limited number and restricted region of these participants weakens the reliability of the results. Therefore, large and multiethnic clinical trials are required to determine whether our results may be generalized to populations with diverse backgrounds. Besides, we paid little attention to the underlying mechanisms of alterations in plasma PTX‐3. Therefore, further studies are warranted to elucidate the exact mechanisms.
In summary, this study revealed that a low‐salt diet decreased PTX‐3 concentrations and high sodium intake contributed to increased plasma PTX‐3 from the level observed during a low‐salt period in Chinese adults. Potassium supplementation blunted the salt‐induced increase in PTX‐3. Alterations of PTX‐3 were more pronounced in SS than in SR participants. These findings strengthened the detrimental effects of high salt intake and support the use of salt restriction and potassium supplementation in diets. However, the exact mechanism and significance of the salt‐ and potassium‐induced alterations of PTX‐3 merit further consideration.
DISCLOSURE
The authors declare that there is no conflict of interest.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China No. 81370357, No. 81570381 (J.‐J.M.), No. 81600327 (Y.W.) and No. 81700368 (C.C.); the Clinical Research Award of the First Affiliated Hospital of Xi'an Jiaotong University of China No. XJTU1AF‐CRF‐2015‐006; Grant 2017YFC1307604 from the Major Chronic Non‐communicable Disease Prevention and Control Research Key Project of the Ministry of Science and Technology of the People's Republic of China; and Grant 2017ZDXM‐SF‐107 from the Key Research Project of Shaanxi Province.
Hu J‐W, Wang Y, Chu C, et al. The responses of the inflammatory marker, pentraxin 3, to dietary sodium and potassium interventions. J Clin Hypertens. 2018;20:925–931. 10.1111/jch.13273
Jia‐Wen Hu and Yang Wang equally contributed to this work.
REFERENCES
- 1. Fang Y, Mu JJ, He LC, Wang SC, Liu ZQ. Salt loading on plasma asymmetrical dimethylarginine and the protective role of potassium supplement in normotensive salt‐sensitive Asians. Hypertension. 2006;48:724‐729. [DOI] [PubMed] [Google Scholar]
- 2. Zhou MS, Schulman IH, Raij L. Vascular inflammation, insulin resistance, and endothelial dysfunction in salt‐sensitive hypertension: role of nuclear factor kappa B activation. J Hypertens. 2010;28:527‐535. [DOI] [PubMed] [Google Scholar]
- 3. Zhou MS, Kosaka H, Yoneyama H. Potassium augments vascular relaxation mediated by nitric oxide in the carotid arteries of hypertensive Dahl rats. Am J Hypertens. 2000;13(6 Pt 1):666‐672. [DOI] [PubMed] [Google Scholar]
- 4. Liu Z, Peng J, Lu F, et al. Salt loading and potassium supplementation: effects on ambulatory arterial stiffness index and endothelin‐1 levels in normotensive and mild hypertensive patients. J Clin Hypertens (Greenwich). 2013;15:485‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Franco V, Oparil S. Salt sensitivity, a determinant of blood pressure, cardiovascular disease and survival. J Am Coll Nutr. 2006;25(3 Suppl):247S‐255S. [DOI] [PubMed] [Google Scholar]
- 6. Parlak A, Aydoğan U, Iyisoy A, et al. Elevated pentraxin‐3 levels are related to blood pressure levels in hypertensive patients: an observational study. Anadolu Kardiyol Derg. 2012;12:298‐304. [DOI] [PubMed] [Google Scholar]
- 7. Yavuzer H, Cengiz M, Yavuzer S, et al. Procalcitonin and pentraxin‐3: current biomarkers in inflammation in white coat hypertension. J Hum Hypertens. 2016;30:424‐429. [DOI] [PubMed] [Google Scholar]
- 8. Yilmaz R, Akoglu H, Altun B, Yildirim T, Arici M, Erdem Y. Dietary salt intake is related to inflammation and albuminuria in primary hypertensive patients. Eur J Clin Nutr. 2012;66:1214‐1218. [DOI] [PubMed] [Google Scholar]
- 9. Binger KJ, Gebhardt M, Heinig M, et al. High salt reduces the activation of IL‐4‐ and IL‐13‐stimulated macrophages. J Clin Invest. 2015;125:4223‐4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu F, Mu J, Yuan Z, et al. High salt intake fails to enhance plasma adiponectin in normotensive salt‐sensitive subjects. Nutrition. 2012;28:422‐425. [DOI] [PubMed] [Google Scholar]
- 11. Gijsbers L, Dower JI, Schalkwijk CG, et al. Effects of sodium and potassium supplementation on endothelial function: a fully controlled dietary intervention study. Br J Nutr. 2015;114:1419‐1426. [DOI] [PubMed] [Google Scholar]
- 12. Rolph MS, Zimmer S, Bottazzi B, Garlanda C, Mantovani A, Hansson GK. Production of the long pentraxin PTX3 in advanced atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2002;22:e10‐e14. [DOI] [PubMed] [Google Scholar]
- 13. Matsui S, Ishii J, Kitagawa F, et al. Pentraxin 3 in unstable angina and non‐ST‐segment elevation myocardial infarction. Atherosclerosis. 2010;210:220‐225. [DOI] [PubMed] [Google Scholar]
- 14. Peri G, Introna M, Corradi D, et al. PTX3, A prototypical long pentraxin, is an early indicator of acute myocardial infarction in humans. Circulation. 2000;102:636‐641. [DOI] [PubMed] [Google Scholar]
- 15. Carrizzo A, Lenzi P, Procaccini C, et al. Pentraxin 3 induces vascular endothelial dysfunction through a p‐selectin/matrix metalloproteinase‐1 pathway. Circulation. 2015;131:1495‐1505; discussion 1505. [DOI] [PubMed] [Google Scholar]
- 16. Badr EA, Hamoda GE, Tayel SI, Elshayeb EI. Association of genetic variants of pentraxin 3 rs3816527 with hypertension in chronic kidney disease patients. Mol Cell Biochem. 2017;425:203‐212. [DOI] [PubMed] [Google Scholar]
- 17. Kunes P, Holubcova Z, Kolackova M, Krejsek J. Pentraxin 3(PTX 3): an endogenous modulator of the inflammatory response. Mediators Inflamm. 2012;2012:920517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Slusher AL, Mock JT, Whitehurst M, Maharaj A, Huang CJ. The impact of obesity on pentraxin 3 and inflammatory milieu to acute aerobic exercise. Metabolism. 2015;64:323‐329. [DOI] [PubMed] [Google Scholar]
- 19. Norata GD, Marchesi P, Pirillo A, et al. Long pentraxin 3, a key component of innate immunity, is modulated by high‐density lipoproteins in endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:925‐931. [DOI] [PubMed] [Google Scholar]
- 20. Deban L, Russo RC, Sironi M, et al. Regulation of leukocyte recruitment by the long pentraxin PTX3. Nat Immunol. 2010;11:328‐334. [DOI] [PubMed] [Google Scholar]
- 21. Bosutti A, Malaponte G, Zanetti M, et al. Calorie restriction modulates inactivity‐induced changes in the inflammatory markers C‐reactive protein and pentraxin‐3. J Clin Endocrinol Metab. 2008;93:3226‐3229. [DOI] [PubMed] [Google Scholar]
- 22. Wang Y, Liu FQ, Wang D, et al. Effect of salt intake and potassium supplementation on serum renalase levels in Chinese adults: a randomized trial. Medicine (Baltimore). 2014;93:e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pruessner JC, Kirschbaum C, Meinlschmid G, Hellhammer DH. Two formulas for computation of the area under the curve represent measures of total hormone concentration versus time‐dependent change. Psychoneuroendocrinology. 2003;28:916‐931. [DOI] [PubMed] [Google Scholar]
- 24. Yi B, Titze J, Rykova M, et al. Effects of dietary salt levels on monocytic cells and immune responses in healthy human subjects: a longitudinal study. Transl Res. 2015;166:103‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang W, Soltero L, Zhang P, Huang XR, Lan HY, Adrogue HJ. Renal inflammation is modulated by potassium in chronic kidney disease: possible role of SMAD7. Am J Physiol Renal Physiol. 2007;293:F1123‐F1130. [DOI] [PubMed] [Google Scholar]
- 26. Xiao Y, Yang N, Zhang Q, Wang Y, Yang S, Liu Z. Pentraxin 3 inhibits acute renal injury‐induced interstitial fibrosis through suppression of IL‐6/Stat3 pathway. Inflammation. 2014;37:1895‐1901. [DOI] [PubMed] [Google Scholar]
- 27. Norata GD, Marchesi P, Pulakazhi VVK, et al. Deficiency of the long pentraxin PTX3 promotes vascular inflammation and atherosclerosis. Circulation. 2009;120:699‐708. [DOI] [PubMed] [Google Scholar]