

Abstract
The correlation between creatine kinase (CK) and blood pressure (BP) was examined prospectively in 120 patients with persistent high CK and 130 individuals with normal CK. Hypertension was defined as systolic BP (SBP) ≥140 mm Hg or diastolic BP (DBP) ≥90 mm Hg or current use of antihypertensive medication. Baseline CK was weakly correlated with SBP (r=0.11, P=.07) and DBP (r=0.16, P=.01) at follow‐up. Persons with persistent high CK had higher SBP (140.8 mm Hg vs 138.2 mm Hg) and DBP (83.2 mm Hg vs 81.0 mm Hg, P=.06) values and were more likely to have hypertension (66.7% vs 55.5%, P=.05) than individuals with normal CK. In age‐ and sex‐adjusted analysis, a 1‐unit change in logCK was associated with a 4.9‐mm Hg higher SBP, a 3.3‐mm Hg higher DBP, and a 2.2‐higher odds for having hypertension at follow‐up (P=.1, .07, and .06, respectively). When including body mass index (BMI) to the model, BMI was a strong and independent predictor for SBP, DBP, and hypertension at follow‐up and the CK effect on blood pressure was substantially attenuated. This study showed that the CK effect on blood pressure is clearly modified by BMI.
Hypertension is a global burden, affecting more than a quarter of the adult population worldwide, which often goes undiagnosed.1 It has been hypothesized, in a biological plausible manner, that high creatine kinase (CK) activity could be a genetic factor responsible for primary hypertension.2 Two large cross‐sectional, population‐based studies have demonstrated an independent dose‐response association between CK and blood pressure (BP).3, 4 High CK has also been associated with failure of antihypertensive therapy.5 In addition, a low CK level was associated with lower BP and an increased prevalence of fainting.6 Cross‐sectional studies, however, make causal inferences impossible because of the lack of temporality between exposure and outcome. Whether the observed relationship between CK and BP is causal or confounded is still unresolved.7, 8 In the present prospective study, we measured CK and BP simultaneously at two points in time and examined whether BP at follow‐up was correlated with CK at baseline. In addition, we examined CK in persons with and without hypertension at follow‐up and whether CK was a long‐term predictor of hypertension.
Methods
Study Population
Figure 1 shows a diagram of the flow of participants. All study participants were recruited from the sixth survey of the Tromsø Study in 2007–2008.9 CK and BP were analyzed cross‐sectionally in 12,828 participants aged 30 to 87 years (mean 58 years), 6834 women and 5994 men, 65% of those eligible. A total of 686 participants had high CK according to references given by the Nordic Reference Interval Project (NORIP)10 and were invited to a second visit for CK control. Of the 686 persons with high CK, 562 attended the second visit (baseline), and CK was measured after 3 days of refraining from alcohol and muscular load, leisure physical activity, muscular training, physiotherapy, acupuncture, or any muscular damage. Of the 562 persons who attended the second visit, 169 had persistent high CK and were invited to the follow‐up study; 120 accepted to participate. Another 130 participants, balanced for sex and 5‐year age groups and CK within the 30% to 50% of CK distribution at screening and after the standardized CK measurement at baseline (persistent normal CK) were included, leaving a total study population of 250 persons (134 women and 116 men). Participants with normal CK were instructed to refrain from use of alcohol and muscular load, leisure physical activity, muscular training, physiotherapy, acupuncture, or any muscular damage 3 days before CK measurement.
Figure 1.
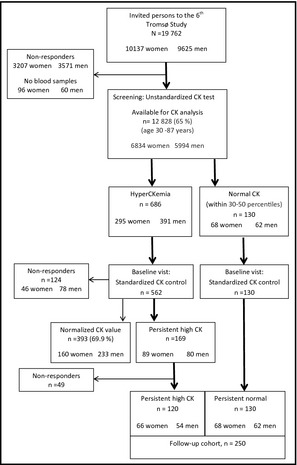
Flow chart of the study population. CK indicates creatine kinase.
Measurement of CK
CK was analyzed by photometry using an enzymatic method (CK‐NAC, Roche Diagnostics, Mannheim, Germany) at the Department of Medical Biochemistry, University Hospital of North Norway, Tromsø. The samples were consecutively analyzed in an automated clinical chemistry analyzer (Modular P, Roche) within 6 hours from withdrawal. The analytical coefficient of variation was ≤1.6%, and the reference interval (2.5%–97.5%) for this method was elaborated by NORIP: women aged 18 years and older, 35 U/L to 210 U/L; men aged 18 to 50 years, 50 U/L to 400 U/L; and men 50 years and older, 40 U/L to 280 U/L.10 When the CK level in a sample was beyond the upper limit, the sample was diluted according to recommendation given by the manufacturer.
Follow‐Up Visit and Measurement of BP
The follow‐up examination took place 69 (mean) weeks later. Information about statin use, diabetes mellitus, and leisure physical activity was obtained through personal interview and medical records. The alcohol use disorders identification test (AUDIT) was used to evaluate alcohol consumption.11 Strenuous leisure physical activity was defined as activity with presence of sweating or breathlessness ≥3 hours per week in the past month. Height was measured and weight was registered by a validated digital scale (Weighingblock VB3‐200‐EC, Class III; Vetek, Väddö, Sweden) to calculate body mass index (BMI). BMI was defined as weight (kg) divided by the square of height (m2).
BP was measured with the A&D Model UA‐779 device (A&D Instruments Ltd, Abingdon, Oxon, UK).12 BP was taken while the participants were sitting in a resting position. The cuffs were chosen after measurement of the circumference of the upper arm. After the participants rested for 2 minutes, three readings on the upper right arm were taken, separated with a 1‐minute interval. The average of the last two measurements was used in the analyses.13 Hypertension was defined as systolic BP (SBP) ≥140 mm Hg or diastolic BP (DBP) ≥90 mm Hg or current use of antihypertensive medication. Uncontrolled hypertension was defined as SBP ≥140 mm Hg or DBP ≥90 mm Hg in spite of antihypertensive treatment.
The study was approved by the Regional Ethical Committee for Research and the Norwegian Data Inspectorate (approval number: REK NORD 11/2008). Written consent was obtained from all participants.
Statistical Analyses
As a result of the skewed distribution, CK was log transformed in the analyses. First SBP and DBP at follow‐up were plotted against baseline logCK values (Figure 1) and Pearson correlation coefficients were calculated. Mean values of SBP, DBP, proportions of hypertension, and possible confounders in persistent high CK and persistent normal CK were calculated and the differences were tested by analysis of variance (Table 1). Similarly, age‐ and sex‐adjusted mean values of CK levels and proportions of persistent high CK were estimated in persons with and without hypertension at follow‐up (Table 2). Age‐ and sex‐adjusted means were then calculated in quartiles of SBP and DBP at follow‐up (Table 3). Linear trends across quartiles were tested by linear and logistic regression. Physical activity and BMI were included in the regression analyses for adjustment (Table 4). The SAS System version 9.2 (SAS Institute, Inc, Cary, NC) was used for all statistical analyses. A two‐sided P value of at least .05 was considered statistically significant.
Table 1.
Clinical Characteristics in Persistent High CK and Persistent Normal CK (N=250)
| Persistent High CK (n=120) | Persistent Normal CK (n=130) | P Value | |
|---|---|---|---|
| Baseline | |||
| logCK, U/L | 2.55 | 1.94 | |
| Age, y | 61.0 | 61.3 | .8 |
| Men, % | 45.0 | 47.7 | .8 |
| Weight, kg | 81.8 | 77.7 | .009 |
| Height, cm | 170.2 | 169.5 | .4 |
| Body mass index, kg/m2 | 28.1 | 27.0 | .03 |
| Waist‐hip ratio | 0.92 | 0.92 | .3 |
| Systolic blood pressure, mm Hg | 139.8 | 140.0 | .9 |
| Diastolic blood pressure, mm Hg | 80.2 | 78.4 | .1 |
| Hypertension, %a | 68.5 | 60.1 | .1 |
| Use of antihypertensive drugs, % | 40.1 | 30.0 | .08 |
| Follow‐up | |||
| Systolic blood pressure, mm Hg | 140.8 | 138.2 | .2 |
| Diastolic blood pressure, mm Hg | 83.2 | 81.0 | .06 |
| Hypertension, %a | 66.7 | 55.5 | .05 |
| Use of antihypertensive drugs, % | 44.0 | 45.0 | .7 |
| Uncontrolled hypertension, %b | 66.7 | 66.0 | .9 |
| Body mass index, kg/m2 | 28.0 | 27.1 | .06 |
| AUDIT score | 2.8 | 2.7 | .8 |
| Use of statins, % | 23.2 | 23.8 | .9 |
| Diabetes mellitus, % | 1.7 | 3.1 | .5 |
| Strenuous leisure physical activity, %c | 38.6 | 30.9 | .2 |
Abbreviations: AUDIT, alcohol use disorder identification test; CK, creatine kinase. Values are age‐ and sex‐adjusted. Age was adjusted for sex and sex was adjusted for age. aHypertension was defined as a systolic blood pressure value ≥140 mm Hg or diastolic blood pressure value ≥90 mm Hg or use of antihypertensive medication. bOf patients taking antihypertensive drugs. cStrenuous leisure physical activity was defined as activity with the presence of sweating or breathlessness ≥3 hours per week in the past month.
Table 2.
CK Levels and Persistent High CK in Persons With and Without Hypertension at Follow‐Up (N=250)
| Hypertension at Follow‐Upa | P Value | ||
|---|---|---|---|
| Yes (n=153) | No (n=97) | ||
| Systolic blood pressure, mm Hg | 147.7 | 126.6 | |
| Diastolic blood pressure, mm Hg | 85.8 | 76.3 | |
| Baseline logCK, U/L | 2.27 | 2.19 | .06 |
| Follow‐up logCK, U/L | 2.23 | 2.13 | .009 |
| Persistent high CK, % | 54.0 | 40.3 | .05 |
| Age, y | 63.8 | 57.1 | <.0001 |
| Male sex, % | 53.6 | 35.7 | .003 |
| Body mass index, kg/m2 | 28.4 | 26.2 | <.0001 |
| Statin use, % | 24.6 | 21.7 | .6 |
| Diabetes, % | 3.8 | 0.0 | .9 |
| AUDIT score | 2.8 | 2.5 | .4 |
| Strenuous leisure physical activity, %b | 34.0 | 35.7 | .8 |
Abbreviations: AUDIT, alcohol use disorder identification test; CK, creatine kinase. All values were age‐ and sex‐adjusted. Age was adjusted for sex and sex was adjusted for age. aHypertension was defined as a systolic blood pressure value ≥140 mm Hg or diastolic blood pressure value ≥90 mm Hg or current use of antihypertensive medication. bStrenuous leisure physical activity was defined as activity with the presence of sweating or breathless ≥3 hours per week in the past month.
Table 3.
CK and Possible Confounders in Quartiles of Blood Pressure (N=250)
| Quartile 1 | Quartile 2 | Quartile 3 | Quartile 4 | P Trend | |
|---|---|---|---|---|---|
| SBP range, mm Hg | 90.5–127.5 | 128.5–137.5 | 138.5–149.5 | 150.0–209.0 | |
| Baseline logCK, U/L | 2.14 | 2.28 | 2.25 | 2.29 | .07 |
| Persistent high CK, % | 33.1 | 56.1 | 49.3 | 55.5 | .08 |
| Body mass index, kg/m2 | 25.6 | 27.6 | 27.9 | 29.0 | <.0001 |
| Statin use, % | 17.3 | 32.3 | 21.3 | 23.0 | .9 |
| Diabetes, % | 0.4 | 1.5 | 4.7 | 2.9 | .3 |
| AUDIT score | 2.5 | 2.5 | 2.9 | 3.0 | .2 |
| Strenuous leisure physical activity, % | 34.0 | 42.3 | 34.1 | 28.4 | .3 |
| DBP range, mm Hg | 59.0–76.0 | 76.5–81.0 | 81.5–87.5 | 88.0–122.0 | |
| Baseline logCK, U/L | 2.19 | 2.22 | 2.25 | 2.30 | .06 |
| Persistent high CK, % | 39.5 | 47.9 | 47.6 | 59.2 | .04 |
| Body mass index, kg/m2 | 26.7 | 26.7 | 28.4 | 28.3 | .005 |
| Statin use, % | 30.9 | 20.1 | 24.7 | 18.3 | .3 |
| Diabetes, % | 2.0 | 3.0 | 3.4 | 1.2 | .8 |
| AUDIT score | 2.6 | 2.4 | 3.2 | 2.7 | .4 |
| Strenuous leisure phys. activity, % | 39.4 | 31.6 | 36.1 | 31.7 | .5 |
Abbreviations: AUDIT, alcohol use disorder identification test; CK, creatine kinase; DBP, diastolic blood pressure; SBP, systolic blood pressure. Values are age‐ and sex‐adjusted.
Table 4.
Baseline LogCK and Persistent High CK as Predictors for Blood Pressure and Hypertension at Follow‐Up (N=250)
| Model I | Model II | Model III | ||||
|---|---|---|---|---|---|---|
| βa (SE) | P Value | βa (SE) | P Value | βa (SE) | P Value | |
| Systolic blood pressure | ||||||
| Baseline logCK | 4.9 (3.2) | .1 | 5.0 (3.2) | .1 | 3.1 (3.2) | .3 |
| Physical activity | – | – | −1.0 (2.3) | .7 | −0.7 (2.2) | .8 |
| Body mass index | – | – | – | – | 1.1 (0.3) | <.0001 |
| Persistent high CK | 2.6 (2.1) | .2 | 2.6 (2.1) | .2 | 1.6 (2.1) | .4 |
| Physical activity | – | – | −0.9 (2.3) | .7 | −0.6 (2.2) | .8 |
| Body mass index | – | – | – | – | 1.2 (0.3) | <.0001 |
| Diastolic blood pressure | ||||||
| Baseline logCK | 3.3 (1.8) | .07 | 3.2 (1.8) | .07 | 2.6 (1.8) | .15 |
| Physical activity | – | – | 0.3 (1.3) | .8 | 0.4 (1.2) | .7 |
| Body mass index | – | – | – | – | 0.4 (0.2) | .01 |
| Persistent high CK | 2.2 (1.2) | .06 | 2.1 (1.2) | .07 | 1.8 (1.2) | .1 |
| Physical activity | – | – | 0.3 (1.3) | .8 | 0.4 (1.2) | .7 |
| Body mass index | – | – | – | – | 0.4 (0.2) | .009 |
| ORb (95% CI) | P Value | ORb (95% CI) | P Value | OR (95% CI) | P Value | |
| Hypertension | ||||||
| Baseline logCK | 2.2 (0.9–5.1) | .06 | 2.3 (1.0–5.3) | .06 | 1.9 (0.8–4.5) | .2 |
| Physical activity | – | – | 0.9 (0.5–1.6) | .7 | 0.9 (0.5–1.6) | .7 |
| Body mass index | – | – | – | – | 1.2 (1.1–1.3) | .0002 |
| Persistent high CK | 1.7 (1.0–3.1) | .05 | 1.8 (1.0–3.1) | .048 | 1.6 (0.9–2.8) | .1 |
| Physical activity | – | – | 0.9 (0.5–1.6) | .7 | 0.9 (0.5–1.6) | .7 |
| Body mass index | – | – | – | – | 1.2 (1.1–1.3) | .0002 |
Abbreviations: CI, confidence interval; OR, odds ratio. Model I was adjusted for age and sex. Model II was adjusted for age, sex, and physical activity. Model III was additionally adjusted for BMI. aValues are regression coefficients (SE) expressed in mm Hg for a 1‐unit change in logCK, physical activity or body mass index (BMI), or for having persistent high creatine kinase (CK). bOdds for hypertension for a 1‐unit change in logCK, physical activity or BMI, or for having persistent high CK.
Results
In Figure 2, the correlations between baseline logCK and SBP and DBP at follow‐up are shown in scatter plots. Baseline CK was significantly correlated with DBP at follow‐up (r=0.16, P=.01). For SBP, the correlation was borderline significant.
Figure 2.
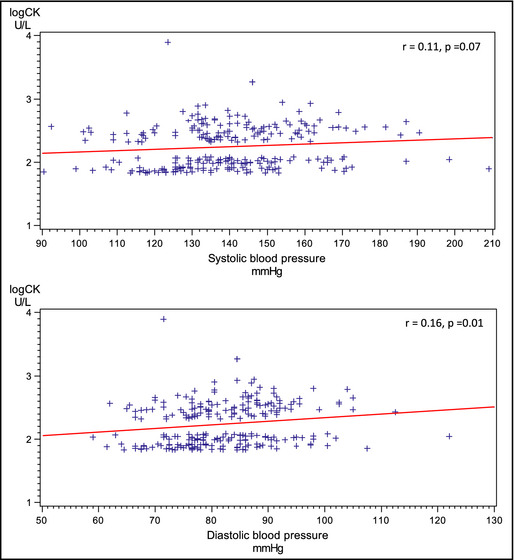
Correlation between creatine kinase (CK) at baseline and blood pressure at follow‐up.
Table 1 shows clinical characteristics of the study participants stratified by CK level. All participants reported to be of Caucasian ethnicity. Persons with persistent high CK used more antihypertensive drugs at baseline and had higher BMI levels, and had higher systolic and diastolic BP values at follow‐up than patients with normal CK. Among patients with persistently high CK, 66.7% were hypertensive at follow‐up, compared with 55.5% among patients with normal CK (P=.05).
In Table 2, the study participants are stratified according to hypertension at follow‐up. Hypertensive persons were older and more likely to be male and had higher BMI levels. CK levels at both baseline (P=.06) and follow‐up (P=.009), as well as the proportion of persistent high CK (P=.05), were higher compared with those in normotensive persons. In Table 3, baseline CK, proportions of persistent high CK, and possible confounders are displayed in quartiles of SBP and DBP. Age‐ and sex‐adjusted estimates of baseline CK increased linearly across strata of both SBP and DBP and the P values for linear trends were borderline significant. The proportion of patients with persistent high CK increased significantly across quartiles of DBP (P=.04). BMI was the only additional risk factor associated with BP.
In age‐ and sex‐adjusted analysis (Table 4, model I), a 1‐unit change in logCK was associated with a 4.9 mm Hg higher SBP, a 3.3 mm Hg higher DBP, and a 2.2 higher odds for having hypertension at follow‐up (P=.1, .07, and .06, respectively). Having persistent high CK was associated with a 2.6 mm Hg higher SBP, a 2.2 mm Hg higher DBP, and a 1.7 higher odds for having hypertension at follow‐up (P=.2, .06, and .05, respectively). When including physical activity (model II) and BMI (model III), BMI was a strong and independent predictor for SBP, DBP, and hypertension. Whereas introduction of physical activity had no impact on CK estimate, adding BMI to the model substantially weakened the CK effect on BP.
A total of 86 persons (35%) were treated with at least one antihypertensive drug at follow‐up. The baseline logCK level among these was 2.29 U/L, compared with 2.21 U/L among the untreated (P=.08). When restricting the analyses to the untreated subgroup only, there were significant attenuations of all CK estimates.
Among the 153 persons with hypertension at follow‐up, baseline logCK was 2.27 U/L compared with 2.19 U/L in patients without hypertension (age‐and sex‐adjusted P=.06). There were 89 patients who currently used antihypertensive medication. Among these, 59 (66%) were still hypertensive in spite of treatment. Their logCK was 2.32 U/L, similar to those who were normotensive.
Discussion
In the present study, baseline CK was weakly correlated with SBP and DBP at follow‐up. In multivariable analyses, the CK effect on BP was about the same as demonstrated in the cross‐sectional study.4 We also found a higher proportion of persistent high CK in hypertensive vs normotensive persons (P=.05). However, after adjustment for BMI, an independent long‐term effect of CK on BP or hypertension was no longer demonstrated. This conflicts with findings from previous cross‐sectional reports.3, 4
Serum CK activity is thought to reflect CK activity from striated skeletal muscle, in particular type II fibers (fast‐twitch), which are less densely vascularized and have a higher vascular resistance than muscles rich in slow‐twitch fibers (type I), postulating that fiber type in skeletal muscles might be of importance in hypertension.14, 15 Patients with high CK activity have been reported to display increased vascular contractility.16 Most recent, the first and most direct evidence suggesting that resistance artery CK mRNA levels relate to BP has been published.17 High CK activity is proposed to enhance pressor responses via increased adenosine triphosphate (ATP) availability for cardiovascular contractility.2 In the arteries, CK is tightly bound near vascular smooth muscle contractile proteins, including myosin adenosine triphosphatase (ATPase) and myosin light chain kinase, where the enzyme provides ATP for smooth muscle contraction.18 The resultant vasoconstriction of small arteries and arterioles increases the total peripheral resistance of blood vessels. In the kidney, CK is functionally coupled with renal Na+/K+‐ATPase and the ATP produced by co‐localized CK is preferentially used for the high and fluctuating ATP demand of sodium transport across the tubular epithelial cells.19 Thus, high CK activity in the kidney tubule cells may lead to increased availability of ATP and greater sodium retention. In addition, increased sodium retention caused by increased renal vascular resistance will raise BP. The heart comprises 20% to 40% of skeletal muscle CK activity,20 and the CK system is of particular importance to maintain local ATP levels constant and contribute to myocardial contractile capacity. Myofibrillar CK, functionally coupled with myosin ATPase, maintains high ATP/ADP ratios and limits the rate of ADP release, which prevents a decline in maximum shortening velocity of the myofibrils. Increased CK activity has been demonstrated in the left ventricle and aorta of spontaneous hypertensive rats.21, 22
In the present study, when BMI was adjusted for, the association between CK and BP was substantially weakened. It has been argued that the observed association between CK and BP is not a causal one, but caused by different physiological and metabolic effects between the different types of muscle fibers.7 Skeletal muscle accounts for about 75% of insulin‐stimulated glucose uptake and is the major site of insulin resistance.23 High CK activity is typically found in type II fibers with cytosolic CK tightly coupled with glycolysis, whereas mitochondrial fatty acid oxidation and glucose uptake are limited. Type II fibers tend to be less insulin‐sensitive than type I fibers.24 This is thought to lead to storage of fatty acids and glucose as lipids in adipose tissue instead of utilization in skeletal muscle, making individuals with type II fiber dominance more prone to gain weight, to be obese, and to develop insulin resistance and type 2 diabetes.25, 26, 27 It is well known that the prevalence of high BP and mean levels of SBP and DBP increases as BMI increases.28 On the other hand, BMI and obesity are also strongly and independently associated with CK, and it has been proposed that a high CK phenotype (muscle fiber type II) is the biological factor that predisposes to both hypertension and obesity.29 In animal studies it has been demonstrated that inhibition of the CK system leads to a shift from type II to type I fiber predominance, with mitochondrial proliferation and increased oxidative phosphorylation, weight loss, and increased insulin sensitivity.30, 31, 32 However, a recent review evaluating existing therapeutic strategies that target the CK system in hypertension and cardiovascular diseases was inconclusive.33 Given the heterogeneity and small sample size of the trials, the authors stated that larger clinical studies are needed to confirm these observations.
Study Strengths and Limitations
To our knowledge, this is the first prospective study to describe the relationship between CK and BP in humans. Baseline CK was measured standardized after 3 days to avoid bias caused by muscular damage of other causes. In the absence of overt tissue damage, serum CK activity after 3 days of rest is thought to mainly reflect skeletal muscle cytosolic CK activity.34 Three days may still be too short an interval since the half‐life of CK is considered to be more than 7 days.35, 36 For SBP, DBP, and hypertension, the associations with CK were all high but did not reach significance. However, the study number was somewhat limited and this may be one explanation for the negative results. Another weakness of the study is that BP was measured after only 2 minutes of resting, and the average BP was obtained from only two recordings. Especially for SBP, which has a wider range of variability than DBP,37 random error in BP monitoring may have attenuated the associations. We used only BMI for assessment of body composition. However, BMI does not distinguish lean mass from fat mass and it may not accurately reflect total body fat in all individuals, particularly in those who are very muscular, have high bone density, or are elderly. Even highly correlated with BMI,38 inclusion of skinfold thickness would improve the relative contribution of lean mass and fat mass to BP level.
Although we lack a distinct clinical measure of muscle mass, waist‐hip ratio might help to differentiate abdominal (central) adiposity from muscle mass. There was no difference in waist‐hip ratio between patients with persistent high CK and normal CK. In a previous study in the same population, we could neither find any difference in waist‐hip ratio or difference in muscle strength for dominant handgrip or knee extension in men,39 which may indicate that muscle mass was relatively equal in the groups.
In line with previous reports,3, 4 we found that hypertensive individuals had significantly higher CK levels than normotensive individuals both at baseline and at follow‐up. However, the CK level in uncontrolled hypertensive persons was equal to treatment‐responsive patients. This contrasts with previous findings.5 An explanation for this could be a drug effect on CK. It is known that β‐blockers, angiotensin‐converting enzyme inhibitors, and angiotensin II antagonists can increase CK level. If high CK activity is a genetic factor for primary hypertension, it would be of interest to know whether CK falls or remains high when BP is lowered. This should be addressed in future studies.
Conclusions
Our study provides some new information on the temporal correlation between CK and BP. However, the CK effect is clearly modified by BMI. The proposed biological link between muscle fiber phenotype and CK activity with hypertension and obesity are intriguing but need further confirmation. Based on these findings and previous observational studies, future research should focus on causal modeling in animal studies to assess whether vascular CK overexpression leads to hypertension and whether CK inhibition lowers BP independently of BMI. Another interesting hypothesis is whether CK may be a prognostic marker in antihypertensive treatment.
Acknowledgments and disclosure
This study was supported by grants from the Norwegian Neuromuscular Disorders Foundation. There are no conflicts of interest concerning this paper.
J Clin Hypertens (Greenwich). 2014;16:820–826. © 2014 Wiley Periodicals, Inc.
References
- 1. Kearney PM, Whelton M, Reynolds K, et al. Global burden of hypertension: analysis of worldwide data. Lancet. 2005;365:217–223. [DOI] [PubMed] [Google Scholar]
- 2. Brewster LM, Clark JF, van Montfrans GA. Is greater tissue activity of creatine kinase the genetic factor increasing hypertension risk in African people of sub‐Saharan black descent? J Hypertens. 2000;18:1537–1544. [DOI] [PubMed] [Google Scholar]
- 3. Brewster LM, Mairuhu G, Bindraban NR, et al. Creatine kinase activity is associated with blood pressure. Circulation. 2006;14:2034–2039. [DOI] [PubMed] [Google Scholar]
- 4. Johnsen SH, Lilleng H, Wilsgaard T, Bekkelund SI. Creatine kinase activity and blood pressure in a normal population: the Tromsø study. J Hypertens. 2011;29:36–42. [DOI] [PubMed] [Google Scholar]
- 5. Oudman I, Kewalbansingh PV, van Valkengoed I, et al. Creatine kinase is associated with failure of hypertension treatment. J Hypertens. 2013;31:1025–1031. [DOI] [PubMed] [Google Scholar]
- 6. Brewster LM, Mairuhu G, Ganzeboom K, et al. Low creatine kinase is associated with a high population incidence of fainting. Clin Auton Res. 2009;19:231–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pickering TG. Muscular hypertension: is creatine kinase responsible for hypertension in blacks? J Clin Hypertens (Greenwich). 2008;10:73–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Johnsen SH, Lilleng H, Bekkelund SI. Is the association between creatine kinase and blood pressure causal? J Hypertens. 2011;29:1019. [DOI] [PubMed] [Google Scholar]
- 9. Jacobsen BK, Eggen AE, Mathiesen EB, et al. Cohort profile: the Tromsø study. Int J Epidemiol. 2012;41:961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rustad P, Felding P, Franzson L, et al. The Nordic Reference Interval Project 2000: recommended reference intervals for 25 common biochemical properties. Scand J Clin Lab Invest. 2004;64:271–284. [DOI] [PubMed] [Google Scholar]
- 11. Saunders JB, Aasland OG, Babor TF, et al. Development of the Alcohol Use Disorders Identification Test (AUDIT): WHO collaborative project on early detection of persons with harmful alcohol consumption–II. Addiction. 1993;88:791–804. [DOI] [PubMed] [Google Scholar]
- 12. Daniele L, Olivio B, Gianluca T, et al. Validation of the A&D UA‐631 (UA‐799 Life Source) device for self‐measurement of blood pressure and relationship between its performance and large artery compliance. Blood Press Monit. 2001;7:1–6. [DOI] [PubMed] [Google Scholar]
- 13. Pickering TG, Hall JE, Appel LJ, et al. Recommendations for blood pressure measurement in humans and experimental animals: part 1: blood pressure measurement in humans: a statement for professionals from the Subcommittee of Professional and Public Education of the American Heart Association Council on High Blood Pressure Research. Hypertension. 2005;45:142–161. [DOI] [PubMed] [Google Scholar]
- 14. Juhlin‐Dannfelt A, Frisk‐Holmberg M, Karlsson J, Tesch P. Central and peripheral circulation in relation to muscle‐fiber composition in normo‐ and hypertensive man. Clin Sci (Lond). 1979;56:335–340. [DOI] [PubMed] [Google Scholar]
- 15. Hernandez N, Torres SH, Vera O, et al. Muscle fiber composition and capillarization in relation to metabolic alterations in hypertensive men. J Med. 2001;32:67–82. [PubMed] [Google Scholar]
- 16. Brewster LM, Taherzadeh Z, Volger S, et al. Ethnic differences in resistance artery contractility of normotensive pregnant women. Am J Physiol Heart Circ Physiol. 2010;299:H431–H436. [DOI] [PubMed] [Google Scholar]
- 17. Karamat FA, Oudman I, Ris‐Stalpers C, et al. Resistance artery creatine kinase mRNA and blood pressure in humans. Hypertension. 2014;63:68–73. [DOI] [PubMed] [Google Scholar]
- 18. Dzeja PP, Terzic A. Phosphotransfer networks and cellular energetics. J Exp Biol. 2003;206:2039–2047. [DOI] [PubMed] [Google Scholar]
- 19. Blum H, Balschi JA, Johnson RG Jr. Coupled in vivo activity of creatine phosphokinase and the membrane‐bound (Na+,K+)‐ATPase in the resting and stimulated electric organ of the electric fish Narcine brasiliensis . J Biol Chem. 1991;266:10254–10259. [PubMed] [Google Scholar]
- 20. Dawson DM, Fine IH. Creatin kinase in human tissues. Arch Neurol. 1967;16:175–180. [DOI] [PubMed] [Google Scholar]
- 21. Seccia TM, Atlante A, Vulpis V, et al. Mitochondrial energy metabolism in the left ventricular tissue of spontaneously hypertensive rats: abnormalities in both adeninenucleotide and phosphate translocators and enzyme adenylate‐kinase and creatinephosphokinase activities. Clin Exp Hypertens. 1998;20:345–358. [DOI] [PubMed] [Google Scholar]
- 22. Clark JF, Radda GK, Boehm EA. The effects of antihypertensive therapy on the structural, mechanical and metabolic properties of the rat aorta. J Muscle Res Cell Motil. 2000;21:255–267. [DOI] [PubMed] [Google Scholar]
- 23. Julius S, Gudbrandsson T, Jamerson K, et al. The hemodynamic link between insulin resistance and hypertension. J Hypertens. 1991;9:983–986. [DOI] [PubMed] [Google Scholar]
- 24. Lillioja S, Young AA, Culter CL, et al. Skeletal muscle capillary density and fiber type are possible determinants of in vivo insulin resistance in man. J Clin Invest. 1987;80:415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tanner CJ, Barakat HA, Dohm GL, et al. Muscle fiber type is associated with obesity and weight loss. Am J Physiol Endocrinol Metab. 2002;282:E1191–E1196. [DOI] [PubMed] [Google Scholar]
- 26. Wade AJ, Marbut MM, Round JM. Muscle fibre type and aetiology of obesity. Lancet. 1990;335:805–808. [DOI] [PubMed] [Google Scholar]
- 27. Sun G, Ukkola O, Rankinen T, et al. Skeletal muscle characteristics predict body fat gain in response to overfeeding in never‐obese young men. Metabolism. 2002;51:451–456. [DOI] [PubMed] [Google Scholar]
- 28. Brown CD, Higgins M, Donato KA, et al. Body mass index and the prevalence of hypertension and dyslipidemia. Obes Res. 2000;8:605–619. [DOI] [PubMed] [Google Scholar]
- 29. Oudman I, Jegernath Z, Kewalbansing P, et al. Creatine kinase is associated with obesity in the general population. J Hypertens. 2011;29(suppl A):e220. [Google Scholar]
- 30. Shoubridge EA, Chaliss RA, Hayes DJ, Radda GK. Biochemical adaptation in the skeletal muscle of rats depleted of creatine with the substrate analogue beta‐guanidinpropionic acid. Biochem J. 1985;232:125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Clark JF, Khuchua Z, Kuznetsov AV, et al. Actions of the creatine analogue beta‐guanidinpropionic acid on rat heart mitochondria. Biochem J. 1994;300:211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ren JM, Semenkovich CF, Holloszy JO. Adaptation of muscle to creatine depletion: effect on GLUT‐4 glucose transporter expression. Am J Physiol. 1993;264:C146–C150. [DOI] [PubMed] [Google Scholar]
- 33. Horjus DL, Oudman I, van Montfrans GA, Brewster LM. Creatine and creatine analogues in hypertension and cardiovascular disease. Cochrane Database Syst Rev. 2011;9:CD005184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lindena J, Diederichs F, Wittenberg H, Trautschold I. Kinetic of adjustment of enzyme catalytic concentrations in the extracellular space of the man, the dog and the rat. Approach to a quantitative diagnostic enzymology, V. Communication. J Clin Chem Clin Biochem. 1986;24:61–71. [DOI] [PubMed] [Google Scholar]
- 35. Serrao FV, Foerster B, Spada S, et al. Functional changes of human quadriceps muscle injury by eccentric exercise. Braz J Med Biol Res. 2003;36:781–786. [DOI] [PubMed] [Google Scholar]
- 36. Ehlers GG, Ball TE, Liston L. Creatine kinase levels are elevated during 2‐a‐day practices in collegiate football players. J Athl Train. 2002;37:151–156. [PMC free article] [PubMed] [Google Scholar]
- 37. Hansen TW, Thijs L, Li Y, et al. International Database on Ambulatory Blood Pressure in Relation to Cardiovascular Outcomes Investigators . Prognostic value of reading‐to‐reading blood pressure variability over 24 hours in 8938 subjects from 11 populations. Hypertension. 2010;55:1049–1057. [DOI] [PubMed] [Google Scholar]
- 38. Yarnell JWG, Patterson CC, Thomas HF, Sweetnam PM. Central obesity: predictive value of skinfold measurements for subsequent ischaemic heart disease at 14 years follow‐up in the Caerphilly study. Int J Obes Relat Metab Disord. 2001;25:1546–1549. [DOI] [PubMed] [Google Scholar]
- 39. Lilleng H, Abeler K, Johnsen SH, et al. Clinical impact of persistent hyperCKemia in a Norwegian general population: a case‐control study. Neuromuscul Disord. 2013;23:29–35. [DOI] [PubMed] [Google Scholar]